

Abstract
Polymers functionalized with azlactone (or oxazolone) functionality have become increasingly useful for the rapid and modular design of functional materials. Because azlactones can react via ring-opening reactions with a variety of different nucleophilic species (e.g., primary amines, hydroxyl groups, and thiol functionality), azlactone-functionalized materials can serve as convenient ‘reactive’ platforms for the post-synthesis or post-fabrication introduction of a broad range of chemical functionality to soluble polymers, insoluble supports, and surfaces/interfaces. The last decade has seen an increase in both the number and the variety of reports that exploit the properties and the reactivities of azlactone-functionalized polymers. Here, we highlight recent work from several different laboratories, including our own, toward the design and characterization of azlactone-functionalized polymers, with a particular emphasis on: (i) new synthetic approaches for the preparation of well-defined azlactone-functionalized polymers using living/controlled methods of polymerization, (ii) the design and modular synthesis of side-chain functionalized polymers and block copolymers via post-polymerization modification of azlactone-functionalized polymers, (iii) the development of reactive polymeric supports useful in the contexts of separations and catalysis, and (iv) methods for the fabrication of reactive thin films and other approaches to the immobilization of azlactone functionality on surfaces and interfaces. Examples discussed herein reveal a growing awareness of azlactone functionality as a useful tool for polymer chemists, and highlight several ways that the unique reactivity of these materials can both complement and provide useful alternatives to other reactive polymers currently used to design functional materials.
TOC image
Polymers bearing azlactone groups are useful as ‘reactive’ platforms for the design, fabrication, and functionalization of soluble polymers, insoluble supports, and reactive surfaces/interfaces.
I. Introduction: Importance of Reactive Polymers and the Reactivity of Azlactones
Polymers and other macromolecular materials bearing chemically reactive functional groups provide versatile and powerful tools for polymer chemists and serve as reactive/modular platforms for the subsequent elaboration of a broad range of functional materials. Reactive polymers are often synthesized directly from monomers containing functionality that is inert to the polymerization conditions, but that can be used post-polymerization to introduce additional functionality and/or modify further the physicochemical properties of the resulting material.1–4 This approach to materials synthesis and design brings with it a number of potential practical advantages. First, it can provide straightforward routes to the synthesis of functional polymers that may be difficult to access by direct polymerization of a given functional monomer.1–2 Second, the use of these materials as ‘reactive’ templates can provide rapid access to a diverse range of polymer structures and obviate the need to synthesize and polymerize independently new classes of functional monomers. In this context, synthetic approaches based on the post-polymerization functionalization of a common reactive template can also provide opportunities to design libraries of polymers with similar molecular weights and molecular weight distributions, and can thus facilitate identification of structure-property relationships that govern the behaviors of new materials.5–7 Finally, the incorporation of reactive polymers into other macromolecular architectures (e.g., crosslinked assemblies, thin films, etc.) provides opportunities to design surfaces, interfaces, and other materials that might be difficult (or impossible) to fabricate by direct incorporation of other functional polymers (e.g., by post-fabrication treatment with functionality that might hinder or otherwise not survive conditions used during fabrication).
The continued development of new methods of polymerization, including living/controlled methods, has provided many approaches to the synthesis of functional polymers having a range of different reactivities and a variety of different well-defined architectures. Several helpful reviews discuss the range of different reactive polymers, and the chemical transformations that can be used for post-polymerization modification of these materials, that have been developed and investigated to date.1–4 Here, we focus our attention on one particular class of reactive polymer that has become an increasingly useful tool for the synthesis of side-chain functionalized polymers and the fabrication of reactive surfaces and interfaces – polymers bearing azlactone (or oxazolone) functionality.
Azlactones are lactone-based functional groups that undergo ring-opening reactions in the presence of nucleophiles such as primary amines, alcohols, or thiol functionality (an example showing the reaction of a typical azlactone group with a primary amine is shown in Scheme 1).8 Depending on their structures, ring-opening reactions of azlactones can be efficient and may be regarded, in some respects, as proceeding with several of the practical advantages associated with conventional click-type organic reactions described by Sharpless and coworkers9 (although, as noted recently, these types of efficient reactions are not explicitly ‘click’ reactions10). For example, polymers bearing azlactone functionality can react rapidly, at room temperature, and in the absence of a catalyst (or the generation of byproducts), with primary amine-functionalized molecules, making this approach useful for the rapid synthesis of side-chain functionalized poly(acrylamide)-type polymers.8,11–15 Relative to some other functional groups used for the design of other classes of reactive polymers (e.g., activated esters containing N-hydroxysuccinimide or pentafluorophenyl groups, etc.), azlactone functionality is relatively stable to hydrolysis and functionalization of these materials can thus be performed under aqueous conditions without significant competitive hydrolysis.16 In addition, the solubility of many azlactone-functionalized polymers in many different organic solvents, including DMSO and DMF, makes possible the functionalization of these reactive scaffolds with a diverse range of small and large molecules (e.g., hydrophobic, hydrophilic, or amphiphilic small molecules, as well as peptides, proteins, and other biomacromolecules).8
Scheme 1.
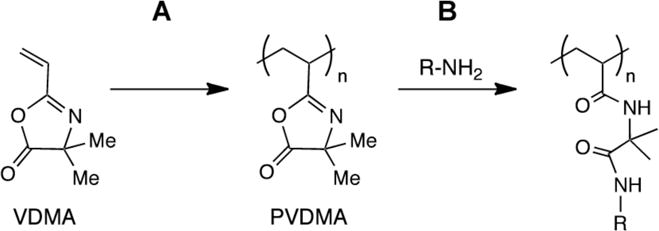
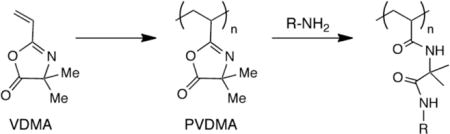
Schematic showing (A) polymerization of 2-vinyl-4,4-dimethylazlactone (VDMA) to yield poly(2-vinyl-4,4-dimethylazlactone) (PVDMA), and (B) the post-polymerization reaction of pendant azlactone functionality with a nucleophilic species (here, a primary amine) to yield a side-chain functionalized poly(acrylamide)-type polymer.
The general reactivity of azlactone functionality is well documented, and azlactone-functionalized polymers have been investigated in numerous past studies, in both fundamental and applied contexts, as a platform for the design of reactive materials.8 This past work has included the investigation of these materials as reactive templates for the synthesis of soluble polymers8,11 and the design of reactive, insoluble polymer supports useful for the immobilization of proteins and other molecules.8,17–21 From a historical perspective, it is important to note that a significant amount of the efforts to understand and exploit the chemistry of azlactones in the context of polymer science has been conducted in industrial settings, most notably by groups at 3M, Polaroid, and Rohm Gmbh. Analysis of literature reports over the last 10-15 years, however, reveals a broader and growing awareness of azlactone functionality as a tool for polymer chemists, and highlights many ways that the unique reactivity of these materials can both complement and provide alternatives to other, more conventionally-used reactive polymers.
Here, we provide an overview and account of recent progress toward the development and application of azlactone-functionalized polymers. In this context, we note that Heilmann and coworkers published a comprehensive and very useful review describing the chemistry and technology of azlactone-based monomers and polymers in 2001.8 Interested readers will find additional useful details on numerous aspects of the reactivity of azlactones as well as historical perspectives on the development of azlactone-functionalized polymers in that review. The sections below are focused primarily on discussions of progress toward the development of azlactone-based materials over the last 10 years.
II. Advances in the Synthesis of Azlactone-Functionalized Polymers
Conventional Free Radical Polymerization
Much of the early work on the design of azlactone-functionalized polymers made use of conventional free radical polymerization (CFRP) methods for the solution, bulk, dispersion, or suspension polymerization of azlactone-functionalized vinyl monomers. These approaches generally lead to reactions that proceed to high conversions under a range of conditions.8,22 Although several different types of vinyl monomers have been investigated (Table 1), polymerization of 2-vinyl-4,4-dimethylazlactone (VDMA) has been particularly well-studied and has become the most common model monomer for the design of azlactone-functionalized materials. Heilmann and coworkers at 3M have investigated the conventional free radical polymerization of VDMA and the copolymerization of VDMA with a range of different vinyl monomers (e.g., styrene, methyl methacrylate, and various acrylates).8,22 The polymerization behaviors and reactivity ratios for VDMA and several of these co-monomers have been determined experimentally.22 In this latter context, 3M has reported extensively on the copolymerization of VDMA with methyl methacrylate (MMA), motivated, at least in part, by the potential utility of these copolymers for the design of reactive biomaterials.23
Table 1.
Examples of polymerization and co-polymerization of azlactone-functionalized monomer(s) using controlled/living polymerization techniques.
| Polymerization Method | Azlactone Monomer(s) | Comonomers | Reference(s) |
| CFRP |
|
styrene/methyl methacrylate methyl acrylate/n-butyl acrylate i-octyl acrylate/vinyl pyrrolidone |
8, 22 |
| NMP |
|
styrene/4-acetoxy styrene methyl acrylate/methyl methacrylat n-butyl acrylate/t-butyl acrylate acrylonitrile/vinyl pyrrolidone N,N-dimethylacrylamide ethylene glycol methyl ether acrylate |
27 |
| ATRP |
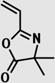
|
styrene/methyl acrylate methyl methacrylate |
28, 30, 31 |
| RAFT |
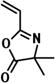
|
N-isopropylacrylamide/styrene methyl methacrylate/methyl acrylate |
13,26,32 |
| ROMP |
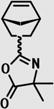
|
– | 25, 29 |
Living/Controlled Methods of Polymerization
Living/controlled methods of polymerization have provided polymer chemists with powerful tools for the synthesis of polymers having well-defined molecular weights and molecular weight distributions.24 Over the last decade, several of these methods have been successfully extended to the polymerization of azlactone-based monomers for the design of well-defined, azlactone-functionalized polymers and copolymers.13,25–32 Azlactone-functionalized monomers (and any relevant co-monomers) used in each of the studies described below are summarized in Table 1.
Nitroxide-mediated polymerization (NMP)
Tully et al. reported one of the first demonstrations of the living/controlled polymerization of VDMA,27 using an α-hydrido alkoxyamine initiator and the corresponding nitroxide to initiate nitroxide-mediated polymerization (NMP) (Scheme 2). These reactions proceed with high conversions, good control over molecular weight, and enable the synthesis of azlactone-functionalized polymers having narrow polydispersities (e.g., PDI <1.10). This early study also characterized the NMP of two additional azlactone-functionalized monomers 2-(4′-vinyl)phenyl-4,4-dimethyl-5-oxazolone (VPDMO) and 2-isopropenyl-4,4-dimethyl-5-oxazolone (IDMO) (Table 1). Bulk homopolymerization of VPDMO was demonstrated to proceed to high conversion and resulted in polymers having low molecular weight distributions (e.g., PDIs ranging from 1.2 to 1.3). In contrast, bulk homopolymerization of IDMO was reported to proceed without appreciable control over molecular weight or polydispersity (e.g., PDIs typically > 1.5). The polymerization behavior of IDMO was similar to that of MMA, likely due to the structural and electronic similarities of these two monomers.27 The addition of styrene as a co-monomer during the polymerization of either IDMO or MMA provided reasonable control over polymerization and afforded statistical copolymers with PDIs of ~1.4.
Scheme 2.
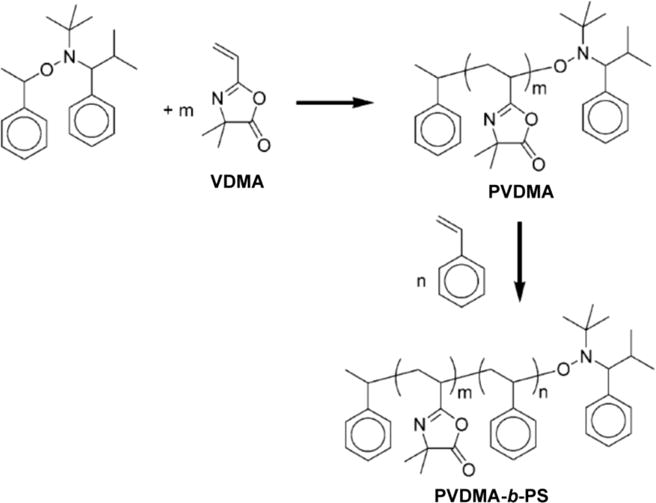
Synthesis of PVDMA and PVDMA-b-PS using NMP. Adapted with permission from reference [27].
Tully et al. also prepared reactive statistical copolymers with controlled molecular weights and low PDIs by NMP of VDMA and a series of acrylate, acrylamide, and N-vinylamide comonomers (Table 1).27 Well-defined azlactone-functionalized block copolymers were also synthesized using NMP, however, the success of these approaches generally depended on the nature of the initiating block used. For example, it was possible to use an alkoxyamine-functionalized poly(n-butyl acrylate) macroinitiator to synthesize poly(butyl acrylate-co-VDMA), but attempts to polymerize other blocks from poly(VDMA) macroinitiators were not successful.27 Finally, these investigators demonstrated the ability to functionalize these reactive polymers and copolymers to produce new copolymer structures by post-polymerization reaction of well-defined PVDMA homopolymers with benzylamine, and the block copolymer PVDMA-b-PS with morpholine.
Atom transfer radical polymerization (ATRP)
Fontaine and coworkers reported the polymerization of VDMA using copper-catalyzed ATRP both in solution and on solid polymer supports (Scheme 3).28,31 In a first demonstration of the ATRP of VDMA, Fournier et al. surveyed a range of amine-based ligands, initiators, copper halides, and initiator/copper/ligand ratios used to promote controlled polymerization.28 Catalyst formed from copper bromide (CuBr) and the ligand tris[2-(dimethylamino)ethyl]amine (Me6TREN) at a ratio of 1:1 was determined to provide the best control over molecular weight while maintaining low polydispersity and good conversion. These investigators subsequently demonstrated that ATRP could be used to synthesize well-defined statistical copolymers and block copolymers of VDMA, styrene, and MA.28
Scheme 3.
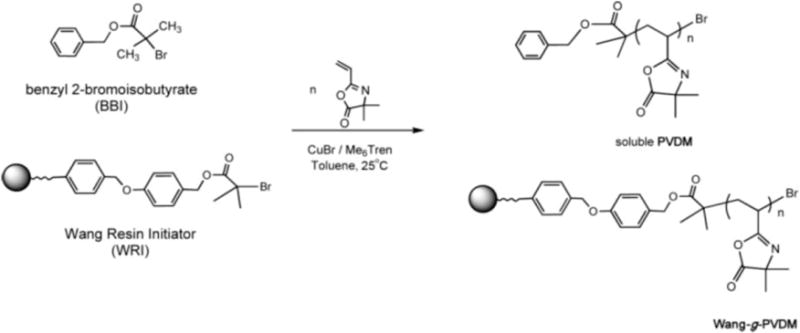
Synthesis of PVDMA using ATRP and a Wang resin functionalized with a sacrificial initiator. Reproduced with permission from reference [30].
Fontaine and coworkers also used ATRP to promote the polymerization of VDMA from the surfaces of insoluble solid supports (Scheme 3).31 As one example of this approach, a Wang resin halide initiator was synthesized and used to initiate copolymerization of VDMA and styrene using CuBr/Me6TREN as the catalytic system. Homopolymers and VDMA-co-S statistical and block copolymers with low PDIs (1.1-1.4) and controlled molecular weights (as determined by characterization of cleaved polymer) were prepared using this approach. These azlactone-functionalized supports were subsequently investigated as insoluble scavengers of small-molecule amines (of potential interest, for example, in the contexts of purification and separations).31 Supports containing high azlactone loadings (e.g., 6 mmol/g) were demonstrated to remove amine-containing compounds such as benzylamine efficiently from solutions mixed with the polymer-decorated resins. Additional work demonstrated that these insoluble azlactone-functionalized resins could be used to immobilize copper catalysts and mediate ATRP.30 For example, reactive immobilization of N,N,N′,N′-tetraethyldiethylenetriamine (TEDETA) on azlactone-functionalized supports created sites for complexation with CuBr; these immobilized complexes then served as supported catalysts for the ATRP of MMA and styrene and facilitated purification of the resulting polymers. Additional discussion of other approaches used to fabricate azlactone-functionalized solid supports is included in the sections below.
Reversible addition-fragmentation chain transfer (RAFT) polymerization
Polymerization of VDMA using RAFT was first demonstrated in a preliminary report by Schilli et al., in which these authors surveyed the synthesis of a series of reactive polymers.26 RAFT polymerization of VDMA using 2-cyanoisopropyl dithiobenzoate (CPDB) as a chain transfer agent yielded polymers with narrow polydispersities (PDIs ≤1.10) and molecular weights that closely matched predicted molecular weights. Thermoresponsive and reactive block copolymers were subsequently synthesized by RAFT copolymerization of VDMA and N-isopropyacrylamide (NIPAAm). Lokitz et al. and Pascual et al. have characterized the synthesis and solution properties of azlactone-functionalized homopolymers and block copolymers synthesized by RAFT polymerization (Scheme 4).13,32 Lokitz and coworkers demonstrated that PVDMA could be synthesized with low PDIs and good control over molecular weight using CPDB and 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid (DMP) as chain transfer agents.13 This study also characterized, for the first time, the dilute solution properties of PVDMA using static and dynamic light scattering experiments. Pascual et al. used experimental and computational studies to characterize the efficiency of block copolymerization of VDMA with methyl acrylate, styrene, and MMA using RAFT.32 The results of these experiments and DFT calculations provide a fundamental understanding of several factors governing the polymerization of VDMA that should prove useful for the preparation of a broader range of azlactone-functionalized materials.
Scheme 4.
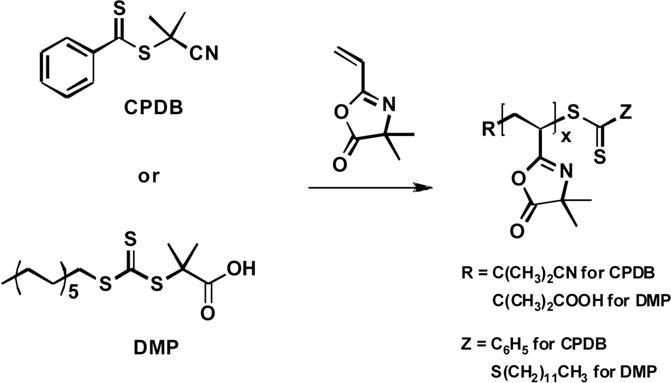
Synthesis of PVDMA by RAFT. Images re-drawn from reference [13].
Finally, we note that the use of living/controlled methods of polymerization offers the ability to not only define or control the molecular weights and PDIs of a polymer, but also to install well-defined functionality at polymer end groups. Ho et al. recently demonstrated that polymers synthesized by RAFT can be end-functionalized with azlactone groups by thiol-Michael addition of a thiol-terminated polymer to VDMA monomer.33 This approach provided access to polymers with azlactone-functionalized end groups that could be used to subsequently install other functionality (e.g., through bio-conjugation) with many of the reactivity-based benefits of azlactone groups outlined above.
Ring-opening metathesis polymerization (ROMP)
In addition to developing methods for the controlled radical polymerization of azlactone-functionalized vinyl monomers, Fontaine and coworkers have investigated the synthesis of well-defined, azlactone-functionalized polymers by ROMP (Scheme 5). Lapinte et al. reported the synthesis and subsequent polymerization of azlactone-functionalized norbornene monomers by ROMP using various Grubbs-type ruthenium alkylidenes and reported detailed characterization of the reactivity of these monomers and polymers.25,29 The azlactone functional groups were well tolerated by the ruthenium catalysts and remained intact during the polymerization, thus providing a reactive handle for further modification. The resulting polymers could be functionalized by treatment with nucleophiles such as sodium hydroxide, diethylamine, and glycine methyl ester hydrochloride, thereby providing additional routes to the post-polymerization synthesis of well-defined ROMP-based materials.
Scheme 5.
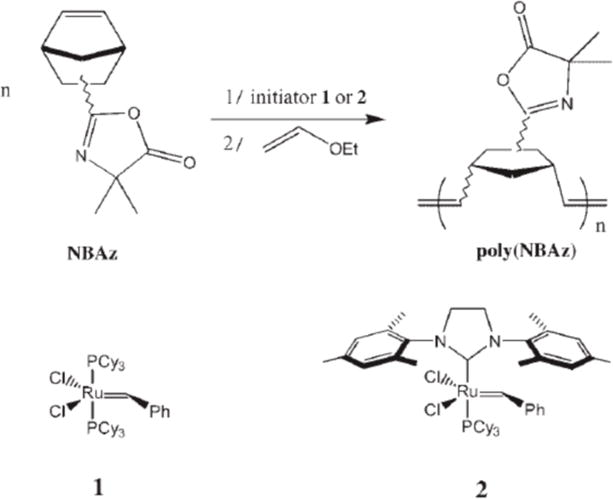
ROMP of norbornylazlactone (NBAz) using ruthenium alkylidenes. Reproduced with permission from reference [29].
Post-Polymerization Modification of Azlactone-Functionalized Polymers
As described in several of the examples in the preceding sections, reactive azlactone-functionalized polymers can be readily modified post-polymerization by treatment with a variety of different types of nucleophilic species. Several groups have continued to investigate PVDMA synthesized by conventional methods as a reactive template for the synthesis of functional materials.
In an initial report, Messman et al. used spectroscopic and chromatographic techniques to characterize ring-opening reactions that occur upon treatment of VDMA, PVDMA, and copolymers of VDMA and vinylpyrrolidone (VP) with amine- and hydroxyl-functionalized nucleophiles (including hexylamine, hexaethylene glycol monomethyl ether, dansyl cadaverine, Nα,Nα-bis(carboxymethyl)-L-lysine hydrate, and sodium hydroxide).34 This study also reported physicochemical characterization of the resulting functionalized polymers and provides further fundamental insight into the reactivity of azlactone-functionalized materials.
Our group has reported on the post-polymerization modification of PVDMA using a variety of different small-molecule diamines (containing both tertiary and primary amines) as an approach to the rapid synthesis of libraries of amine-functionalized polymers (see polymers P1-P12, Figure 1).15 Because tertiary amines do not react with the azlactone ring (Figure 1A), this approach resulted in polymers containing tertiary amine-functionalized side chains (Figure 1B) that were water soluble, positively charged at physiological pH, and able to form electrostatic complexes with DNA in aqueous media. One potential advantage of this reactive ‘template-based’ approach to parallel synthesis (relative to methods based on parallel step growth polymerization or the parallel polymerization of different vinyl monomers) is that the use of a common, ‘universal’ reactive polymer backbone permitted the synthesis of functionalized polymers having (i) different side-chain functionality, but (ii) uniform (or nearly uniform) molecular weights and molecular weight distributions.15 This approach thus facilitated the identification of some basic structure-property relationships that influence the efficiency with which these new polymers promote (or do not promote) the delivery of DNA to cells. This approach to the parallel functionalization of azlactone-based polymer templates could also be used to design libraries of functional polymers of interest in a broad range of other fundamental and applied contexts. We note that while the PVDMA templates used in this past study were synthesized using conventional free radical polymerization, living/controlled polymerization methods described in the preceding section could also be used to vary parameter space further and design more diverse libraries of functional polymers.
Figure 1.
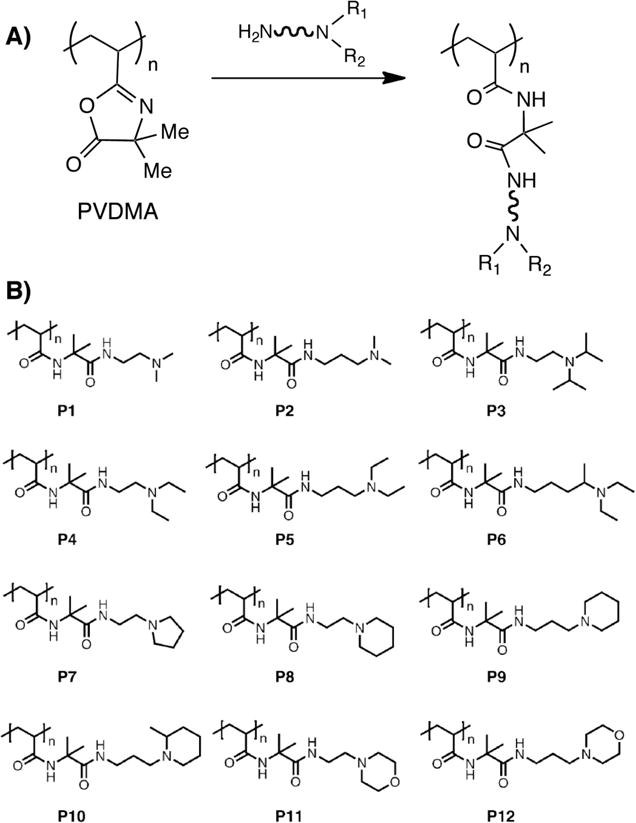
(A) Treatment of PVDMA with a small-molecule diamine containing both tertiary and primary amines results in a tertiary amine-functionalized polymer. (B) A library of amine-functionalized polymers generated by parallel synthesis using PVDMA and 12 different diamine molecules. Adapted with permission from reference [15].
We have also exploited reactions of PVDMA with hydroxyl-functionalized molecules to develop a new family of cationic “charge-shifting” polyelectrolytes.35–36 Scheme 6 shows an example of the reaction of PVDMA with a molecule possessing both hydroxyl and tertiary amine functionality (the amine base DBU is necessary in this case to catalyze the reaction between hydroxyl and azlactone functionality). This reaction results in a new polymer having protonatable tertiary amine side-chain functionality that is attached to the backbone of the polymer by an amide/ester functionalized linker. Hydrolysis of the ester bond in this linker leads to a loss of protonatable (cationic) side chain amine-functionality and the unmasking of a negatively-charged carboxylate group.35–36 Side chain hydrolysis thus results in a change (or a “shift”) in the net charge of the polymer and, as a result, the nature of its interactions with anionic polymers (such as DNA). We have used this azlactone-based synthetic approach and these new “charge-shifting” materials to design multilayered polyelectrolyte thin films (or polyelectrolyte multilayers) that promote and provide new levels of control over the release of DNA from surfaces.35–36
Scheme 6.
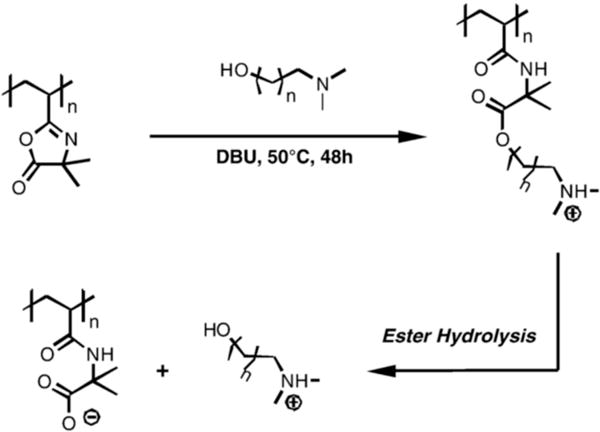
Synthesis and hydrolysis of ‘charge-shifting’ cationic polymers by reaction of pendant azlactone functionality of PVDMA with a tertiary amine containing hydroxyl functionality. The resulting amide/ester linker in the side chain of the polymer hydrolyzes in aqueous media to release amine functionality and unmask anionic carboxylate functionality. Adapted with permission from reference [36].
Finally, we have used post-polymerization-based approaches to the functionalization of PVDMA to synthesize amphiphilic copolymers having both hydrophobic and hydrophilic side chains (Figure 2).12,14 These polymers were synthesized by the sequential addition of controlled amounts of hydrophobic and hydrophilic primary amines to a solution of PVDMA. Similar to the approach to parallel synthesis described above, this post-polymerization approach to copolymer synthesis can provide access to several different copolymer compositions all having similar molecular weights and PDIs. This approach also facilitates the synthesis of random terpolymers and can be used to introduce low mole percentages of amine-functionalized fluorophores during synthesis.14 We have used this azlactone-based approach to design amphiphilic copolymers (such as those shown in Figure 2) that assemble at interfaces formed between aqueous solutions and thermotropic liquid crystals12,14 as a first step toward the development of new platforms for the design of liquid crystal-based sensors.
Figure 2.
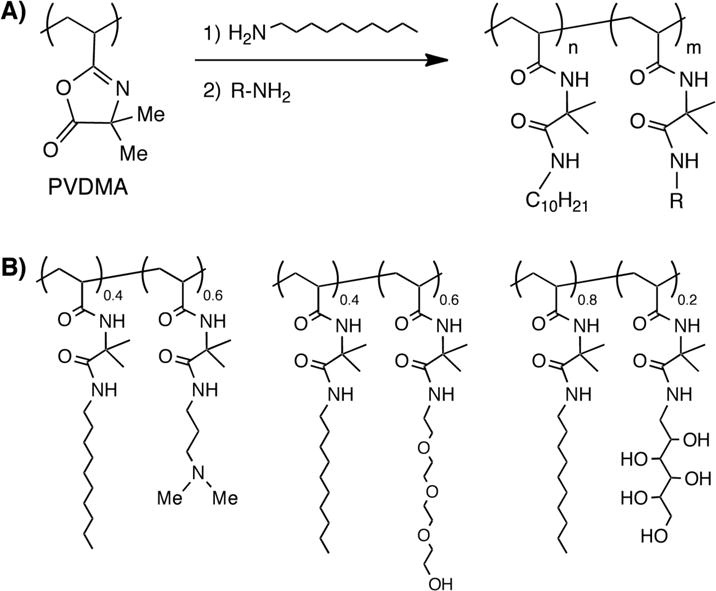
(A) Schematic showing synthesis of amphiphilic copolymers by sequential addition of hydrophobic and hydrophilic primary amines (designated as R) to PVDMA. (B) Structures of amphiphilic copolymers synthesized using this approach.
III. Porous Polymer Supports: Functional Monoliths and Polymer Beads
New Approaches to Reactive Filtration
Combinatorial chemistry-based approaches to small molecule synthesis often use solid phase synthesis methodologies to facilitate more rapid and efficient purification by simple removal of a polymer support from excess reagents and soluble side-products. However, solution phase reactions are often preferable because of the nearly unlimited number of chemical transformations that can be carried out in solution. Solid-phase approaches that facilitate the removal of unwanted soluble side-products from solution phase reactions (e.g., by simple filtration) are therefore of significant interest. A range of different solid-phase materials synthesized from, or functionalized with, azlactone-containing polymers have been developed to remove nucleophilic species from solution phase reactions (including the azlactone-functionalized supports reported by Fournier et al., as discussed above31).
Fréchet and coworkers have developed macroporous monolithic disks as alternatives to the porous polymer beads more commonly used as scavenger resins.37–38 Macroporous monoliths are readily amenable to flow-through processes that are useful for automated synthesis and, because solutions can be pumped through these monolithic materials, mass transport of reagents to the reactive sites on the pore surfaces is rapid. Tripp et al. reported the fabrication of reactive porous monoliths by first polymerizing the monomers chloromethylstyrene and divinylbenzene in the presence of a porogen (e.g., dodecanol) inside a polyethylene mold.37–38 VDMA was then grafted onto the surfaces of this insoluble polymer support by immobilization of an azo initiator on the surface of monolith (Scheme 7) and subsequent polymerization of VDMA (designated as VAZ in Scheme 7). The amount of grafted PVDMA could be controlled by the concentration of VDMA in the polymerization reaction solution, and these investigators demonstrated the ability to scavenge (or remove by “reactive filtration”) excess amine-functionalized reagents (e.g., benzylamine, butylamine, diethylamine, etc.) from model reactions of isocyanates and amines. These azlactone-functionalized monoliths were able to scavenge ~75% of primary amine nucleophiles and ~90% of secondary amines after just eight minutes in the presence of the monolith.37–38 Notably, because amine-based compounds react faster with azlactone groups than hydroxyl groups do,8,38 azlactone-functionalized monoliths can be used to scavenge amine-based nucleophiles from reactions carried out in water or in alcohol solvents. This contrasts with many commercially available resins that often exploit isocyanate functional groups that react readily with alcohols and water.
Scheme 7.
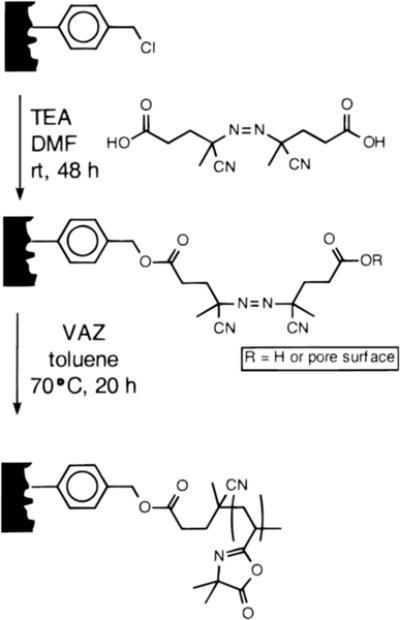
Preparation of polymeric monoliths grafted with PVDMA by surface-initiated polymerization of VDMA (designated as VAZ in this figure). Reproduced with permission from reference [37].
Fontaine and coworkers have developed azlactone-functionalized polymer supports useful for the solid phase synthesis of small molecules39 and for reactive filtration/nucleophile scavening.31,40–42 As described above in the discussion of ATRP-mediated synthesis, Fournier et al. reported reactive resins that scavenged benzylamine by polymerization of PVDMA from appropriately functionalized Wang resins.31 Although both immobilized PVDMA homopolymers and poly(VDMA-stat-S) effectively removed benzylamine from reaction solutions (~98% scavenged in 24 h), it was observed that the copolymer-functionalized supports reacted more rapidly and were more efficient at scavenging benzylamine.31 This improved behavior was attributed to the physical separation of azlactone moieties in the statistical copolymer, which was thought to improve the accessibility of azlactone groups on the grafted polymer chains.
Fontaine and coworkers have also reported the preparation of insoluble crosslinked polymer beads by direct polymerization of azlactone-functionalized monomers. In an initial report, polymer beads were synthesized using the vinyl monomer N-(p-vinylbenzoyl)-2-methylalanine (VBM) (a monomer that contains a hydrolyzed azlactone group). Guyomard et al. prepared a series of polymer beads by emulsion polymerization of VBM, styrene, and divinylbenzene.40 FT-IR and Raman spectroscopy, scanning electron microscopy (SEM), and elemental analysis revealed that VBM was incorporated homogenously throughout the beads. VBM-functionalized supports were then converted to reactive azlactone groups by refluxing the beads in acetic anhydride for 24 h. These azlactone-functionalized beads were able to scavenge approximately 91% of benzylamine dissolved in THF after six hours of mixing. Lucchesi et al. subsequently optimized emulsion polymerization conditions such that azlactone groups could be directly incorporated into the beads rather than requiring a reflux step to cyclize and re-form reactive azlactone groups.41
The usefulness of polymer beads is somewhat limited in certain applications because they can often only be used in batch format (i.e., mixing with a reaction solution) and typically do not perform as well in flow systems for which reaction solutions are pumped through the beads (e.g, to improve mass transport of reagents to the support surface).42 Fontaine and coworkers recently reported the fabrication of azlactone-functionalized polymer supports in the form of a poly(HIPE)s (or high internal phase emulsions) that can be used under flow conditions.42–43 Although the materials were highly porous and reactive, initial experiments revealed that these materials were more efficient at scavenging 4-fluorobenzylamine in batch processes (~60% after 24 h) than in flow-through processes (~20% after one week of flow).
Bioreactors: Immobilization of Proteins and Other Catalysts on Porous Polymer Supports
Several academic and industrial groups have investigated the utility of azlactone-functionalized solid supports for the immobilization of enzymes and the development of enzymatic bioreactors. Azlactone-functionalized supports are particularly well-suited for the immobilization of enzymes because azlactone groups react rapidly and selectively with lysine residues present on the surfaces of many proteins.8 3M has devoted significant effort over the last several decades to the development of azlactone-functionalized porous polymer supports for the immobilization of a range of different enzymes,8 and azlactone-functionalized porous polymer beads have been commercialized as 3M’s EMPHAZE™ Biosupport Medium. In addition to ongoing work at 3M, the last decade has seen an increase in literature reports describing azlactone-based supports for the immobilization of enzymes.
Porous Polymer Monoliths
In their initial work using azlactone-functionalized polymer monoliths (described above), Fréchet, Svec, and coworkers reported the fabrication of porous poly(2-vinyl-4,4-dimethylazlactone-co-acrylamide-co-ethylene dimethacrylate) monoliths and the subsequent immobilization of trypsin.21 Aqueous solutions containing trypsin were pumped through azlactone-containing monoliths, resulting in immobilization of trypsin through reaction of lysine residues on the protein with the azlactone-functionalized support (e.g., Figure 3C). Optimization of the pore sizes and hydrophilicity/hydrophobicity of the monoliths resulted in high activities of immobilized trypsin for both low molecular weight (L-benzoyl arginine ethyl ester, BAEE) and high molecular weight (casein) substrates. Notably, the catalytic activity of these trypsin-containing monoliths was high even at flow rates (i.e., 180 cm/min) that far exceeded those reported for packed bed reactors of immobilized enzymes.21
Figure 3.
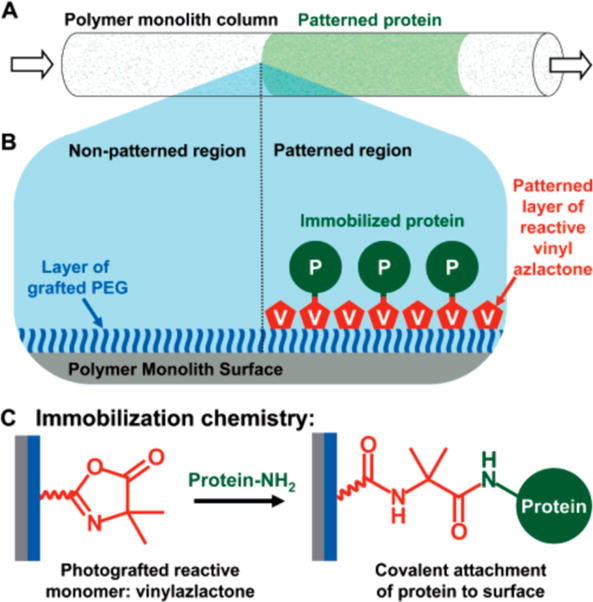
Schematic illustration of the process used to spatially pattern proteins within an azlactone-functionalized monolith. (A) Illustration of a monolith patterned in well-defined areas with protein (green). (B) The proteins are patterned by first immobilizing a layer of PEG on the monolith to prevent non-specific adsorption. VDMA is then photografted in defined areas of the monolith using a photomask. Proteins can then react selectively with regions patterned with VDMA through nucleophilic ring opening reactions (reaction shown in C) between lysines on the proteins and available azlactone groups on the monolith. Reproduced with permission from reference [47].
The Fréchet group has subsequently miniaturized their azlactone-functionalized monoliths to microfluidic formats for the development of enzymatic microreactors and proteomics tools.44–46 In these systems, monoliths were formed directly inside microfluidic chambers or capillary tubes using photoinitiated polymerization.44–47 Photoinitiation permitted in situ construction of the monolith at precise locations within microfluidic devices (e.g., fused silica capillaries and microfluidic channels). Characterization of digested myoglobin using mass spectrometry revealed sequence coverage to be ~67% at short residence times,44 which was comparable to coverage obtained using other approaches that require longer reaction times.48–49 Subsequent modifications of this basic system improved the sequence coverage and reusability of the microreactors by incorporating non-reactive monolith segments that concentrated45 and separated46 the digested peptides. Finally, using these azlactone-containing microreactors, this group developed approaches for patterning the monoliths with multiple different enzymes that can be used to catalyze spatially separated multienzymatic reactions.47 By controlling the spatial placement of PVDMA-grafted regions of the monolith using photoinitiated grafting reactions and appropriate photomasks (Figure 3), the immobilization of multiple enzymes could be spatially controlled. Directional flow through the monolith such that the product of the first enzymatic reaction became the substrate for a second enzyme immobilized downstream of the first enzyme resulted in multi-transformation chemical reactions. When combined, these reports demonstrate the potential of azlactone-functionalized microreactors as new tools for proteomic analysis and chemical synthesis.
Several other groups have investigated azlactone-functionalized monoliths for the immobilization of functional proteins. Krenkova et al. photopolymerized VDMA onto poly(glycidyl methacrylate-co-ethylene dimethacrylate) and poly(butyl methacrylate-co-ethylene dimethacrylate) monoliths for the immobilization of peptide-N-glycosidase F.50 Using an optimized monolithic reactor, several proteins were demonstrated to be de-glycosylated at room temperature in 5.5 min to an extent similar to that achieved by soluble protein in 24 h at 37 °C. Chen et al. developed an immunoaffinity capillary column by fabricating poly(VDMA-co-2-hydroxyethyl methacrylate-co-ethylene dimethacrylate) monoliths inside capillary tubes followed by immobilizing an anti-testosterone polyclonal antibody.51 Testosterone could be captured efficiently and subsequently eluted from the monolith to enrich the testosterone concentration in solution. Connolly et al. reported the use of capacitively coupled contactless conductivity detection as a means to probe the presence of charged species on the surfaces of polymer-based monoliths.52 This approach could be used to detect the presence of proteins (e.g., bovine serum albumin) immobilized on azlactone-functionalized polymer supports. Finally, we note that Paull and coworkers have also used azlactone-functionalized monoliths to immobilize gold nanoparticles.53
Ongoing Work at 3M
Heilmann and coworkers at 3M have continued to develop industrial-scale enzymatic bioreactors based on azlactone-functionalized polymer supports.54–56 Using crosslinked dispersion polymer supports, the enzymes pig liver esterase (PLE) and penicillin G acylase (PGA) were efficiently coupled to the supports and used to catalyze enzymatic reactions.54–55 At a catalyst loading of 4 wt %, immobilized PLE exhibited 68% of the activity of free enzyme in solution. Using supports containing 20 wt % immobilized PGA, enzyme activities of 80-90% could be readily achieved.55 Furthermore, immobilization of PGA on azlactone-functionalized supports resulted in shorter coupling times and higher activities relative to an oxirane-functional commercial standard.
This group subsequently developed a cartridge filter system containing azlactone-functionalized polymer supports and recently described important process parameters for conducting preparative organic reactions using both PGA and PLE using this system.56 The cartridge filter system was configured such that enzymes immobilized on azlactone-functionalized polymer particulates were positioned on the upstream surface of a cartridge filter. This configuration offers several advantages: (i) the biocatalyst is separated from the bulk of the reactant and product solutions and, thus, repetition of the reaction process is straightforward, (ii) the lifetime of the biocatalysts is significantly improved by isolating the catalyst from mechanical and chemical stresses normally associated with batch operations, and (iii) the use of high recirculation flow rates provides conversion rates comparable to slurry batch processes.56 Further development of azlactone-based catalytic systems could thus provide enhanced catalyst lifetimes and high turnovers necessary for industrial scale biocatalytic systems.
IV. Other Approaches to the Modification of Surfaces with Azlactone Functionality
Fabrication of Polymer Brushes
To date, the use of azlactone-functionalized monomers and polymers for the fabrication of polymer brushes on surfaces has not been extensively reported. In one study, Fontaine et al. reported the polymerization of VDMA from the surfaces of poly(propylene) films and fabrics activated using electron-beam irradiation.57 Characterization of the modified fibers and fabrics using several spectroscopic methods confirmed that PVDMA was grafted from the surfaces. These investigators subsequently demonstrated the ability to use the azlactone groups of grafted PVDMA to immobilize a broad range of amine-functionalized chemical and biological species on the surfaces of films and fabrics (including benzylamine, the polyether amine Jeffamine® M-600, the peptide poly(benzyl aspartate) omega-benzylamide, and the protein sericin).
Gopalan and coworkers also recently reported the surface-initiated ATRP of VDMA from silicon surfaces functionalized with a silane initiator (Figure 4).58 Several proteins, including RNase A, glucose oxidase, DNase I, glucoamylase, and trypsin were subsequently immobilized on these brushes via reaction of available lysine resides on the proteins with accessible azlactone functionality on the polymer brushes. Immobilized RNase A showed activities similar to free protein in solution and immobilized DNase I, glucose oxidase, glucoamylase, and trypsin showed activities similar to or higher than values reported using other immobilization strategies.
Figure 4.
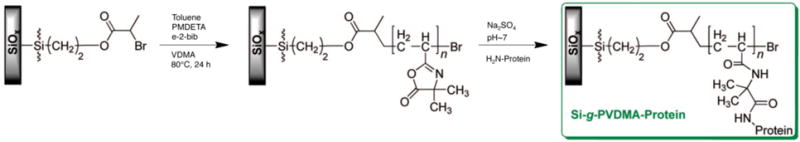
Synthesis of reactive polymer brushes by surface-initiated ATRP of VDMA and subsequent immobilization of proteins. Adapted with permission from reference [58].
Stamping of Nano- and Micrometer-Scale Topographic Patterns into Reactive Thin Films
Our laboratory has reported methods to transfer well-defined, nanometer- and micrometer-scale topographic patterns to thin films of PVDMA and azlactone-containing copolymers.59 We used nano-imprint lithography (NIL) to stamp topographic patterns (e.g., using silicon masters having patterns of ridges and troughs) into spin-coated thin films of PVDMA or random copolymers of VDMA/MMA heated above Tg (Figure 5A). Fredin et al. demonstrated proof-of-concept of this approach using silicon masters having patterns of lines with widths ranging from 400 nm to 2 μm, demonstrating that this method can be used to transfer patterns into these reactive films over a broad range of feature sizes. Further characterization by FTIR revealed that the azlactone groups were not substantially degraded at the temperatures and pressures used in the NIL process, providing opportunities to chemically functionalize the surfaces of these topographically patterned films by treatment with primary-amine-containing nucleophiles (e.g., amine-functionalized fluorophores or other agents) (Figure 5B–5C).59
Figure 5.
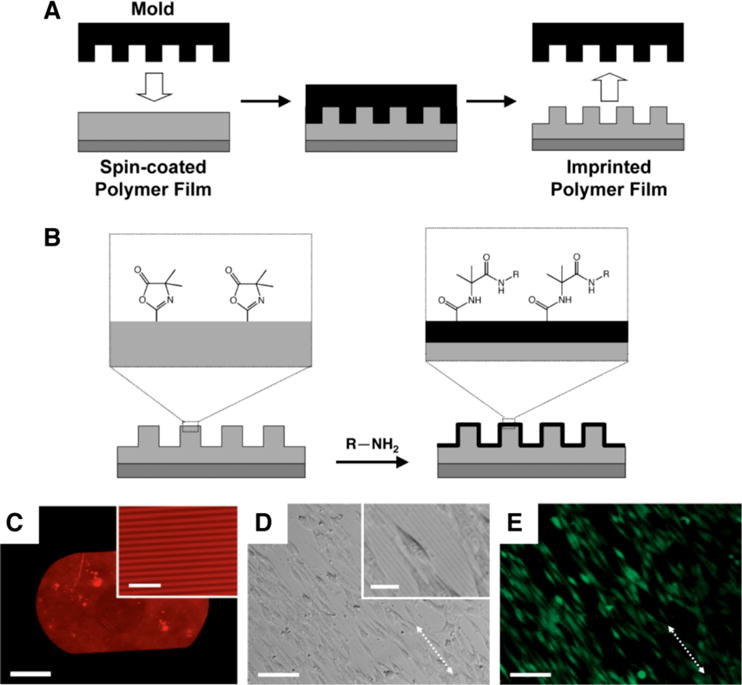
(A) Schematic illustration the nano-imprinting process used to transfer nanometer- or micrometer-scale topographic features to a thin film of a reactive, azlactone-containing polymer. (B) Schematic illustration of post-fabrication modification of the surfaces of imprinted films by reaction of azlactone functional groups with amine-functionalized nucleophiles. (C) Fluorescence microscopy image of a nano-imprinted thin film of PVDMA (pitch of patterned lines = 4 μm) functionalized by treatment with the amine-functionalized fluorophore tetramethylrhodamine cadaverine (TMR-cadaverine). (D, E) Phase contrast (D) and fluorescence microscopy (E) images of NIH 3T3 fibroblasts (stained with Calcein AM) seeded on a thin film of poly(VDMA-co-MMA) imprinted with a topographic pattern of lines similar to that shown in (C). Scale bars: (C) 500 μm (20 μm for inset), (D-E) 100 μm (25 μm for inset in D). Adapted with permission from reference [59].
The ability to combine methods for the topographic patterning of polymer films with materials that permit facile post-fabrication chemical functionalization could contribute significantly to advances in biotechnology and a range of other areas. For example, the introduction of topographic patterns has been used to design surfaces that can influence or guide the behavior of cells,60–61 but conventional materials used for the design of such surfaces (e.g., silicon, PMMA, etc.) are often not readily amenable to the introduction of the range of chemical or biological functionality that could be used to improve biocompatibility or guide other aspects of cell behavior. The reactivity of surface-accessible azlactone groups in imprinted films of VDMA-containing polymers could thus facilitate investigations of the combinatorial effects of changes in topographic and chemical patterning on cell behavior. As a first step toward this broader goal, we demonstrated (i) that PVDMA and copoly(VDMA/MMA) films imprinted with lined patterns having a pitch of 4 μm promoted the alignment of mammalian fibroblast cells in the direction of the topographically imprinted pattern (Figure 5D–5E), and (ii) that treatment of these reactive films with amine-functionalized poly(ethylene glycol) could be used to chemically pattern regions that prevented the attachment and growth of cells.59 It should prove possible in future studies to exploit the reactivity of surface-accessible azlactone groups to functionalize the surfaces of these films with a broader range of other chemical or biological functionality (e.g., peptides, proteins, carbohydrates, etc.). More generally, as noted above, the ability to define both the physical topography and the chemical functionality of these reactive thin films could also prove useful in a range of other applications.
Fabrication of Multicomponent Thin Films
Barringer et al. developed a multilayered polymer scaffold to immobilize small-molecule amines on reactive, azlactone-functionalized surfaces.62 In this approach, poly(glycidyl methacrylate) was first tethered to clean silicon substrates through reaction with free hydroxyl groups on the surface. Reaction of this base layer with 1,6-diaminohexane created amine-functionalized surfaces that could be used to immobilize the copolymer PVDMA-co-PVP. Residual unreacted azlactone functionality in these multilayer scaffolds could then be used to covalently immobilize the fluorescent small molecule dansyl cadaverine on the surfaces either through solution-based reactions or by drop-on-demand printing processes.
Reactive Layer-by-Layer Assembly of Polymer Multilayers
Our group has developed methods for the ‘reactive’ layer-by-layer assembly63 of thin polymer multilayers using poly(2-alkenyl azlactone)s. This approach builds conceptually upon methods developed for the layer-by-layer assembly of charged polyelectrolytes (i.e., to fabricate ‘polyelectrolyte multilayers’, or PEMs)64–65 on a variety of different types of surfaces. However, rather than the stepwise, layer-by-layer assembly of films mediated by ionic interactions between oppositely charged polyelectrolytes, our ‘reactive’ approach to assembly is mediated by fast, interfacial chemical reactions between azlactone-functionalized polymers and polymers containing primary amines.66 Figure 6 shows a schematic illustration of this approach. This approach preserves many of the advantages of conventional layer-by-layer assembly,64,67–70 but the ‘reactive’ nature of this process leads to the formation of covalently-crosslinked multilayers (as opposed to ionically-crosslinked multilayers, or films held together by other weak intramolecular reactions). In addition, this approach can be used to fabricate films using organic solvents rather than the aqueous solvents that are generally required for the layer-by-layer assembly of water-soluble polyelectrolytes. The majority of work in our group has focused on films fabricated using PVDMA and branched poly(ethylene imine) (PEI), but we have demonstrated that it is also possible to use VDMA-containing copolymers,71 and it should, in principle, be possible to expand the scope of this general approach using a range of other primary amine-containing polymers as well.
Figure 6.
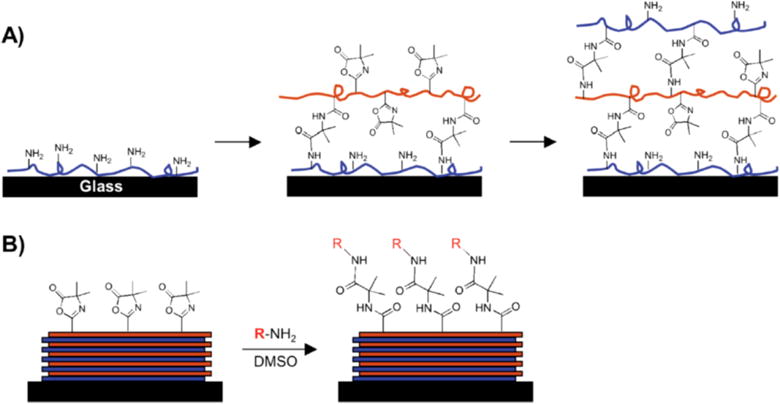
Schematic illustration showing the ‘reactive’ layer-by-layer assembly (A) and subsequent chemical functionalization (B) of polymer multilayers fabricated using azlactone-functionalized polymers. A) Branched poly(ethylene imine) (PEI) is first adsorbed onto a substrate followed by treatment with a solution of an azlactone-containing polymer. Repetition of this process results in layer-by-layer buildup of covalently crosslinked multilayers containing residual azlactone functionality. B) A broad range of surface functionality can be imparted to the films post-fabrication by treatment of residual azlactone functionality with primary amine-containing nucleophiles. Reproduced with permission from reference [72].
We demonstrated in an initial report that the alternate and repetitive immersion of planar silicon substrates into solutions of PEI and PVDMA results in the stepwise growth of polymer multilayers (e.g., ~100 nm thick), and that these films contain residual, unreacted azlactone functionality (as revealed by reflective IR spectroscopy experiments).66 Additional experiments demonstrated that these residual azlactone groups could be used to functionalize these films and, in some cases, modify their surface properties (e.g., water contact angles) by treatment with appropriately-selected primary amines.
In a series of subsequent reports,66,71–79 we have reported on the fabrication and characterization of these reactive multilayers on the surfaces of a variety of different microscopic and macroscopic substrates, and we have investigated the extent to which these materials could serve as a general platform for the chemical functionalization of film-coated surfaces. For example, these films can be fabricated on the surfaces of planar silicon66 and glass72 substrates, as well as on the more chemically varied and topographically complex surfaces of hair, paper, cotton thread (Figure 7A) and commercial wound dressings (Figure 7B).74 The organic solvent-based nature of this assembly process also permits the assembly of films at liquid/liquid interfaces created between organic phases and aqueous phases ([e.g., enabling the straightforward fabrication of suspended thin films and porous membranes covering the ends of water-filled capillaries (e.g., Figures 7C–7D), etc.)].73 In addition to fabrication of reactive films on macroscopic substrates, this layer-by-layer approach is also compatible with the assembly of films on the surfaces of microparticles (Figure 7E), and can be used to fabricate reactive hollow microcapsules (e.g., by assembly of films on, and then subsequent removal of, a sacrificial microparticle core, Figure 7F).79 Finally, we have reported methods for the fabrication of amine-reactive free-standing thin films by the delamination of PEI/PVDMA multilayers from the surfaces of curved or planar substrates on which they were deposited.75 These delamination processes can be conducted under conditions that do not degrade substantially the azlactone functionality in the films, and yield flexible, free-standing films (Figure 7G) that can be manipulated and transferred to the surfaces of other substrates (e.g., onto substrates containing holes or pores, to create arrays of suspended membranes; Figure 7H).
Figure 7.
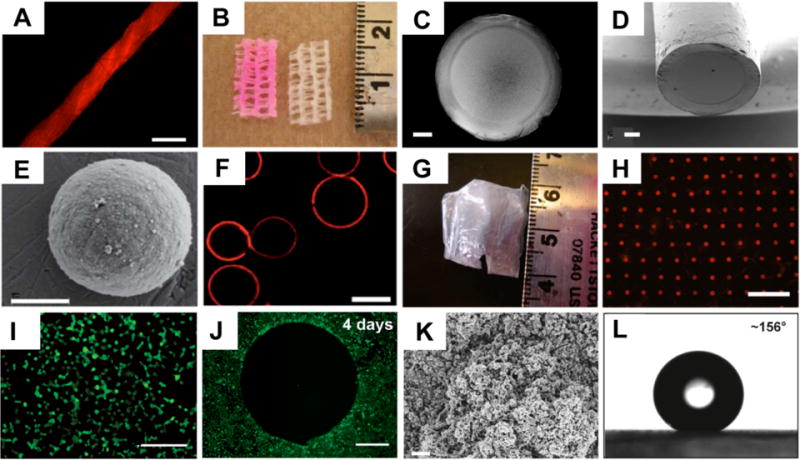
(A) Fluorescence microscopy image of a cotton thread coated by ‘reactive’ layer-by-layer assembly of PEI and PVDMA, and subsequently treated with a primary amine-functionalized fluorescent dye (TMR-cadaverine). (B) Digital picture of a strip of commercial gauze coated with PEI/PVDMA (followed by treatment with TMR-cadaverine; left) and an uncoated/untreated strip of gauze (right) used as a control. (C-E) SEM images of (C, D) the film-coated end of a glass capillary and (E) a CaCO3 microparticle coated with a reactive PEI/PVDMA film. (F) Confocal microscopy image of hollow microcapsules fabricated by fabrication of PEI/PVDMA films on (and subsequent etching of) sacrificial glass microspheres (films in this example were reacted with TMR-cadaverine). (G) An image of a free-standing PEI/PVDMA film obtained by removal from an underlying planar silicon substrate. (H) Fluorescence micrograph of a TMR-functionalized free-standing PEI/PVDMA film film similar to that shown in (G) transferred onto a planar glass substrate patterned with an array of microholes. (I-J) Fluorescence microscopy images of COS-7 cells either (I) 24 h or (J) 4 days after seeding on planar glass substrates coated with (I) an unmodified (i.e., azlactone-containing) PEI/PVDMA film or (J) a PEI/PVDMA film patterned by treatment with a small spot of D-glucamine (in the center of the image; regions surrounding this spot were subsequently treated with the hydrophobic alkylamine n-decylamine). Cells were stained with the live cell stain Calcein AM prior to imaging to aid in visualization of cell locations and morphologies. (K) SEM image of a 100-bilayer PEI/PVDMA film (fabricated in the presence of cyclic azlactone-functionalized oligomers, see text) treated with the semi-fluorinated amine heptadecafluoroundecylamine. The image was acquired after soaking the film in water for 6 weeks. (L) Image of a water droplet (4 μL) on the surface of a heptadecafluoroundecylamine-treated film similar to that shown in (K) showing a contact angle of >150° after immersion in water for 6 weeks. Scale bars correspond to (A) 500 μm, (B) scale in cm, (C-D) 200 μm, (E) 2 μm, (F) 30 μm, (G) scale in cm, (H) 100 μm, (I) 300 μm, (J) 500 μm, and (K) 2 μm. Adapted with permission from references [72–76, 79].
In each of the studies described above, the presence of residual azlactone functionality after film fabrication enabled straightforward functionalization of the films by treatment with a range of functional amine-based nucleophiles, including hydrophobic and hydrophilic small molecules, proteins, and other macromolecules, as well as amine-functionalized micrometer-scale droplets of thermotropic liquid crystal.66,71–77 Treatment with increasingly hydrophobic primary amines, for example, provides control over water contact angles on film-coated silicon substrates66 and the interactions of water with samples of paper coated with PEI/PVDMA films.74 It is also possible to functionalize and pattern the surfaces of these azlactone-containing films with motifs that promote or prevent the adsorption of protein or the attachment of cells (Figure 7I–7J).72 For example, treatment of films with the hydrophobic amine n-decylamine resulted in films that promoted the adsorption of bovine serum albumin and supported the adhesion and proliferation of mammalian fibroblast cells. However, treatment of films with the small molecule D-glucamine (a hydrophilic, carbohydrate-based antifouling motif used in past studies on self-assembled monolayers80) results in film-coated surfaces that prevent almost completely the adhesion or migration of mammalian cells for periods of at least one month (Figure 7J).72 The results of more recent studies demonstrate that it is also possible to modulate and tune further the behaviors of cells using films assembled from PEI and copolymers containing various ratios of VDMA and MMA (followed by treatment with D-glucamine).71
The versatility of the layer-by-layer assembly process used to fabricate these films also renders this approach particularly well suited for the deposition of amine-reactive films on the surfaces of topographically and topologically complex objects. We recently demonstrated, for example, that PEI/PVDMA films can be deposited on the surfaces of 3D polyurethane-based microwell cell culture arrays (Figures 8A–8C).78 Additional experiments demonstrated that this approach provides a convenient route to the chemical functionalization of different structural features of these arrays (e.g., the interior walls of the wells vs. the areas located between the wells) to create dual-functionalized microwell arrays that provide spatial control over the attachment and growth of cells (Figure 8D–8E). This azlactone-based approach could thus provide new opportunities for the design of substrates for the long-term culture of cell types for which chemically well-defined 3D culture environments are thought to be useful for control over cell growth, differentiation, or other important cell behaviors. More generally, the ability to coat and then functionalize the surfaces of otherwise unreactive topographically patterned substrates could also be useful in a range of other areas.
Figure 8.
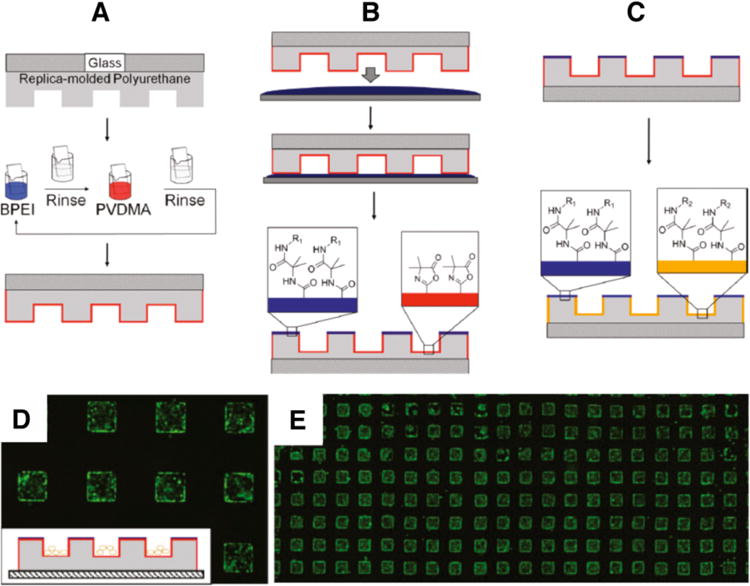
Schematic showing: (A) Replica-molded polyurethane microwell array (gray) on a glass slide (cross-hatched), coated with PEI/PVDMA films using reactive layer-by-layer assembly. (B) Process for selective chemical functionalization of the regions on the top surfaces of microwells (i.e., the areas between the wells) by inverting and exposing the film-coated arrays to a thin film of an aqueous solution of an amine-functionalized molecule. (C) Subsequent functionalization of remaining, unreacted azlactone groups inside the wells by immersion of film-coated arrays in a second aqueous solution containing a different amine-functionalized molecule; this process yields dual-functionalized arrays. (D) The growth of cells seeded on film-coated arrays patterned with glucamine in areas outside of the wells was confined to the microwells, as shown in the fluorescence microscopy image. (E) A lower magnification image of the same cell-seeded array shown in (D), demonstrating that the patterning of cells within wells is maintained with fidelity over large areas of the substrate. Cells were stained with Calcein AM prior to imaging. Microwell dimensions in (D) and (E) are 300 μm on each side. Adapted with permission from reference [78].
Finally, we have demonstrated that this reactive layer-by-layer process can be used to fabricate reactive PEI/PVDMA-based multilayer coatings that are superhydrophobic.76 This work originated from basic observations that thicker PEI/PVDMA films (e.g., consisting of ~50-100 layer pairs) often resulted in optically opaque films,75–76 and that this opacity correlated with an increase in the micro- and nanoscale surface roughness (as characterized by SEM; Figure 7K).76 Subsequent characterization of the water contact angles of these films, as-fabricated, revealed them to exhibit advancing water contact angles in excess of 150°. The results of several experiments revealed that this surface roughness was dependent upon both film thickness and the presence of cyclic azlactone-functionalized oligomers that have been reported to form, under certain conditions,81 upon storage of VDMA (films fabricated using PVDMA synthesized using freshly distilled monomer were smooth and exhibited contact angles of ~75°).
The addition of controlled amounts of independently synthesized oligomer81 to samples of distilled VDMA provided samples of PVDMA that could be used to fabricate superhydrophobic thin films reproducibly on a variety of different surfaces. Although the surfaces of these films were superhydrophobic immediately after fabrication (i.e., in the absence of any other processing or treatment), they became hydrophilic upon exposure to aqueous media for extended periods of time (i.e., after two days in water or prolonged exposure to humid environments). This behavior is presumably a result of ring-opening of residual azlactone functionality and the subsequent introduction of hydrophilic, carboxylate groups on the surfaces of the films.76 Additional experiments, however, demonstrated that this superhydrophobic character could be stabilized and preserved for long periods of time simply by treatment of the residual azlactone groups in these materials with hydrophobic amines (e.g., aliphatic or semifluorinated primary amines). Films treated in this manner exhibited minimal changes in contact angles and remained superhydrophobic even after complete submersion in water for at least six weeks (Figure 7L).
IV. Summary and Outlook
Azlactone-functionalized polymers and other macromolecular structures fabricated from these reactive materials provide facile routes to the synthesis of a broad range of functional materials and interfaces. In the sections above, we have highlighted work over the last ten years demonstrating new methods for (i) the synthesis of well-defined, azlactone-functionalized polymers, (ii) new synthetic approaches to the post-polymerization modification of these reactive materials, and (iii) methods for the fabrication of azlactone-functionalized reactive polymer supports, surface, and interfaces. These reports demonstrate both the versatility of the azlactone functional group in the context of macromolecular science and the increasing awareness of the ways that the unique reactivity of this functionality can be exploited in a variety of both fundamental and applied contexts.
Although the number of literature reports on azlactone-functionalized materials has increased over the last ten years, azlactone-functionalized polymers have not been investigated or characterized to the same extent as several other classes of reactive polymers (e.g., polymers bearing activated ester N-hydroxysuccinimide or pentafluorophenyl groups, etc.).1–2 It is clear from the work highlighted above, however, that the range of applications for which azlactone-functionalized materials can be tailored is both broad and diverse (extending, for example, from the design of polymer-based drug delivery agents to the fabrication of insoluble supports for the immobilization of biocatalysts). The ability to copolymerize VDMA and other azlactone-containing structures with a variety of different co-monomers should also permit the design and synthesis of statistical and block copolymers with interesting and useful physicochemical properties. The potential to incorporate orthogonally reactive co-monomers also opens additional opportunities to create complex, multifunctional materials with well-defined compositions. It is our hope that this review, and the work described herein, will contribute to the growing awareness of the properties of azlactone-functionalized monomers and polymers and help stimulate creative new directions and applications for these reactive materials.
Acknowledgments
Work from the author’s laboratory was supported in part by the National Science Foundation (DMR-0520527) through a grant to the Materials Research Science and Engineering Center (MRSEC) at the University of Wisconsin. The authors are grateful to Dr. Steven M. Heilmann and Dr. Jerald K. Rasmussen (3M) for providing generous samples of VDMA monomer and for numerous insightful discussions related to the chemistry and characterization of azlactones and azlactone-functionalized polymers. M.E.B. was funded in part by an NIH Chemistry Biology Interface Training Grant (NIGMS T32 GM008505).
Biographies

Maren E. Buck obtained a B.S. in Chemistry in 2005 from the University of Puget Sound in Tacoma, WA. She graduated from the University of Wisconsin-Madison in 2010 with a Ph.D. in Chemistry. While at UW-Madison, she worked under the supervision of Professor David M. Lynn investigating the layer-by-layer assembly and post-fabrication modification of reactive thin films and membranes assembled using azlactone-functionalized polymers. She then joined Professor David A. Tirrell’s lab at the California Institute of Technology as a postdoctoral scholar where she is currently developing artificial protein polymers for the assembly of physical hydrogels of interest in the contexts of cell encapsulation and tissue regeneration and repair.

David M. Lynn was born in York, Pennsylvania, in 1972. He received a B.S. in Chemistry from the University of South Carolina in 1994 and a Ph.D. in Chemistry from Caltech in 1999, where he worked under the supervision of Professor Robert H. Grubbs. After a postdoctoral stint at the Massachusetts Institute of Technology with Professor Robert Langer, he joined the faculty in the Department of Chemical and Biological Engineering at the University of Wisconsin – Madison in 2002. His current interests include the design of functional polymers, macromolecular assemblies, and surfaces/interfaces, with a particular focus on the development of new materials platforms that address problems of biomedical and biotechnological importance.
References
- 1.Gauthier MA, Gibson MI, Klok HA. Angew Chem Int Ed. 2009;48:48–58. doi: 10.1002/anie.200801951. [DOI] [PubMed] [Google Scholar]
- 2.Theato P. J Polym Sci Part A. 2008;46:6677–6687. [Google Scholar]
- 3.Boaen NK, Hillmyer MA. Chem Soc Rev. 2005;34:267–275. doi: 10.1039/b311405h. [DOI] [PubMed] [Google Scholar]
- 4.Evans RA. Aust J Chem. 2007;60:384–395. [Google Scholar]
- 5.Pedone E, Li XW, Koseva N, Alpar O, Brocchini S. J Mater Chem. 2003;13:2825–2837. [Google Scholar]
- 6.Gibson MI, Frohlich E, Klok HA. J Polym Sci Part A. 2009;47:4332–4345. [Google Scholar]
- 7.Wong SY, Sood N, Putnam D. Mol Ther. 2009;17:480–490. doi: 10.1038/mt.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heilmann SM, Rasmussen JK, Krepski LR. J Polym Sci Part A. 2001;39:3655–3677. [Google Scholar]
- 9.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Barner-Kowollik C, Du Prez FE, Espeel P, Hawker C, Junkers T, Schlaad H, Van Camp W. Angew Chem Int Ed. 2011;50:60–62. doi: 10.1002/anie.201003707. [DOI] [PubMed] [Google Scholar]
- 11.Guichard B, Noel C, Reyx D, Thomas M, Chevalier S, Senet JP. Macromol Chem Phys. 1998;199:1657–1674. [Google Scholar]
- 12.Kinsinger MI, Buck ME, Campos F, Lynn DM, Abbott NL. Langmuir. 2008;24:13231–13236. doi: 10.1021/la803376u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lokitz BS, Messman JM, Hinestrosa JP, Alonzo J, Verduzco R, Brown RH, Osa M, Ankner JF, Kilbey SM. Macromolecules. 2009;42:9018–9026. [Google Scholar]
- 14.Kinsinger MI, Buck ME, Meli MV, Abbott NL, Lynn DM. J Colloid Interface Sci. 2010;341:124–135. doi: 10.1016/j.jcis.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun B, Liu XH, Buck ME, Lynn DM. Chem Commun. 2010;46:2016–2018. doi: 10.1039/b921664b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen JK, Heilmann SM, Krepski LR, Jensen KM, Mickelson J, Johnson KZ, Coleman PL, Milbrath DS, Walker MM. React Polym. 1992;16:199–212. [Google Scholar]
- 17.Coleman PL, Walker MM, Heilmann SM, Rasmussen JK, Krepski LR, Jensen KM. Faseb J. 1988;2:A1770–A1770. [Google Scholar]
- 18.Coleman PL, Walker MM, Milbrath DS, Stauffer DM, Rasmussen JK, Krepski LR, Heilmann SM. J Chromatogr A. 1990;512:345–363. [Google Scholar]
- 19.Rasmussen JK, Heilmann SM, Krepski LR, Jensen KM, Mickelson J, Johnson KZ, Coleman PL, Milbrath DS, Walker MM. React Polym. 1992;16:199–212. [Google Scholar]
- 20.Drtina GJ, Heilmann SM, Moren DM, Rasmussen JK, Krepski LR, Smith HK, Pranis RA, Turek TC. Macromolecules. 1996;29:4486–4489. [Google Scholar]
- 21.Xie SF, Svec F, Frechet JMJ. Biotechnol Bioeng. 1999;62:30–35. doi: 10.1002/(sici)1097-0290(19990105)62:1<30::aid-bit4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 22.Rasmussen JK, Heilmann SM, Krepski LR. In: Encyclopedia of Polymer Science and Engineering. 2nd. Mark HF, Bikales N, Overberger CG, Menges G, editors. Vol. 11. Wiley-Interscience; New York: 1988. p. 558. [Google Scholar]
- 23.Stanek LG, Heilmann SM, Gleason WB. J Polym Sci Part A. 2003;41:3027–3037. [Google Scholar]
- 24.Matyjaszewski K. Advances in Controlled/Living Radical Polymerization. American Chemical Society; Washington, D.C: 2003. p. 688. (ACS Symposium Series 854). [Google Scholar]
- 25.Lapinte V, Fontaine L, Montembault V, Campistron L, Reyx D. J Mol Catal A: Chem. 2002;190:117–129. [Google Scholar]
- 26.Schilli CM, Muller AHE, Rizzardo E, Thang SH, Chong YK. RAFT polymers: Novel precursors for polymer-protein conjugates. In: Matyjaszewski K, editor. Advances in Controlled/Living Radical Polymerization. Vol. 854. American Chemical Society; Washington: 2003. pp. 603–618. [Google Scholar]
- 27.Tully DC, Roberts MJ, Geierstanger BH, Grubbs RB. Macromolecules. 2003;36:4302–4308. [Google Scholar]
- 28.Fournier D, Pascual S, Fontaine L. Macromolecules. 2004;37:330–335. [Google Scholar]
- 29.Lapinte V, Brosse JC, Fontaine L. Macromol Chem Phys. 2004;205:824–833. [Google Scholar]
- 30.Fournier D, Pascual S, Montembault V, Fontaine L. J Polym Sci Part A. 2006;44:5316–5328. [Google Scholar]
- 31.Fournier D, Pascual S, Montembault V, Haddleton DM, Fontaine L. J Comb Chem. 2006;8:522–530. doi: 10.1021/cc0600122. [DOI] [PubMed] [Google Scholar]
- 32.Pascual S, Blin T, Saikia PJ, Thomas M, Gosselin P, Fontaine L. J Polym Sci Part A. 2010;48:5053–5062. [Google Scholar]
- 33.Ho H, Levere M, Soutif JC, Montembault V, Pascual S, Fontaine L. Polym Chem. 2011;2:1258–1260. [Google Scholar]
- 34.Messman JM, Lokitz BS, Pickel JM, Kilbey SM. Macromolecules. 2009;42:3933–3941. [Google Scholar]
- 35.Zhang JT, Lynn DM. Adv Mater. 2007;19:4218–4223. [Google Scholar]
- 36.Sun B, Lynn DM. J Controlled Release. 2010;148:91–100. doi: 10.1016/j.jconrel.2010.07.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tripp JA, Stein JA, Svec F, Frechet JMJ. Org Lett. 2000;2:195–198. doi: 10.1021/ol9912837. [DOI] [PubMed] [Google Scholar]
- 38.Tripp JA, Svec F, Frechet JMJ. J Comb Chem. 2001;3:216–223. doi: 10.1021/cc000092o. [DOI] [PubMed] [Google Scholar]
- 39.Lucchesi C, Arbore A, Pascual S, Fontaine L, Maignan C, Dujardin G. Carbohydrate Res. 2010;345:844–849. doi: 10.1016/j.carres.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 40.Guyomard A, Fournier D, Pascual S, Fontaine L, Bardeau JF. Eur Polym J. 2004;40:2343–2348. [Google Scholar]
- 41.Lucchesi C, Pascual S, Jouanneaux A, Dujardin G, Fontaine L. J Polym Sci Part A. 2007;45:3677–3686. [Google Scholar]
- 42.Lucchesi C, Pascual S, Dujardin G, Fontaine L. React Funct Polym. 2008;68:97–102. [Google Scholar]
- 43.Brown JF, Krajnc P, Cameron NR. Ind Eng Chem Res. 2005;44:8565–8572. [Google Scholar]
- 44.Peterson DS, Rohr T, Svec F, Frechet JMJ. Anal Chem. 2002;74:4081–4088. doi: 10.1021/ac020180q. [DOI] [PubMed] [Google Scholar]
- 45.Peterson DS, Rohr T, Svec F, Frechet JMJ. Anal Chem. 2003;75:5328–5335. doi: 10.1021/ac034108j. [DOI] [PubMed] [Google Scholar]
- 46.Geiser L, Eeltink S, Svec F, Frechet JMJ. J Chromatogr A. 2008;1188:88–96. doi: 10.1016/j.chroma.2008.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Logan TC, Clark DS, Stachowiak TB, Svec F, Frechet JMJ. Anal Chem. 2007;79:6592–6598. doi: 10.1021/ac070705k. [DOI] [PubMed] [Google Scholar]
- 48.Ekstrom S, Onnerfjord P, Nilsson J, Bengtsson M, Laurell T, Marko-Varga G. Anal Chem. 2000;72:286–293. doi: 10.1021/ac990731l. [DOI] [PubMed] [Google Scholar]
- 49.Lazar IM, Ramsey RS, Ramsey JM. Anal Chem. 2001;73:1733–1739. doi: 10.1021/ac001420+. [DOI] [PubMed] [Google Scholar]
- 50.Krenkova J, Svec F. J Sep Sci. 2009;32:706–718. doi: 10.1002/jssc.200800641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen HX, Huang T, Zhang XX. Talanta. 2009;78:259–264. doi: 10.1016/j.talanta.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 52.Connolly D, O’Shea V, Clark P, O’Connor B, Paull B. J Sep Sci. 2007;30:3060–3068. doi: 10.1002/jssc.200700365. [DOI] [PubMed] [Google Scholar]
- 53.Connolly D, Twamley B, Paull B. Chem Commun. 2010;46:2109–2111. doi: 10.1039/b924152c. [DOI] [PubMed] [Google Scholar]
- 54.Heilmann SM, Drtina GJ, Haddad LC, Rasmussen JK, Gaddam BN, Liu JJ, Fitzsimons RT, Fansler DD, Vyvyan JR, Yang YN, Beauchamp TJ. J Mol Catal B: Enzym. 2004;30:33–42. [Google Scholar]
- 55.Drtina GJ, Haddad LC, Rasmussen JK, Gaddam BN, Williams MG, Moeller SJ, Fitzsimons RT, Fansler DD, Buhl TL, Yang YN, Weller VA, Lee JM, Beauchamp TJ, Heilmann SM. React Funct Polym. 2005;64:13–24. [Google Scholar]
- 56.Heilmann SM, Drtina GJ, Eitzman PD, Haddad LC, Coleman PL, Hyde FW, Johnson TW, Rasmussen JK, Smith HK, Liu RJ, Fitzsimons RT, Williams MG, Moeller SJ, Nakamura MM, Gibbens KJ, Buhl TL. J Mol Catal B: Enzym. 2007;45:1–9. [Google Scholar]
- 57.Fontaine L, Lemele T, Brosse JC, Sennyey G, Senet JP, Wattiez D. Macromol Chem Phys. 2002;203:1377–1384. [Google Scholar]
- 58.Cullen SP, Mandel IC, Gopalan P. Langmuir. 2008;24:13701–13709. doi: 10.1021/la8024952. [DOI] [PubMed] [Google Scholar]
- 59.Fredin NJ, Broderick AH, Buck ME, Lynn DM. Biomacromolecules. 2009;10:994–1003. doi: 10.1021/bm900045c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Curtis A, Wilkinson C. Biomaterials. 1997;18:1573–1583. doi: 10.1016/s0142-9612(97)00144-0. [DOI] [PubMed] [Google Scholar]
- 61.Jung DR, Kapur R, Adams T, Giuliano KA, Mrksich M, Craighead HG, Taylor DL. Crit Rev Biotechnol. 2001;21:111–154. doi: 10.1080/20013891081700. [DOI] [PubMed] [Google Scholar]
- 62.Barringer JE, Messman JM, Banaszek AL, Meyer HM, Kilbey SM. Langmuir. 2009;25:262–268. doi: 10.1021/la802925g. [DOI] [PubMed] [Google Scholar]
- 63.Bergbreiter DE, Liao KS. Soft Matter. 2009;5:23–28. [Google Scholar]
- 64.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 65.Bertrand P, Jonas A, Laschewsky A, Legras R. Macromol Rapid Comm. 2000;21:319–348. [Google Scholar]
- 66.Buck ME, Zhang J, Lynn DM. Adv Mater. 2007;19:3951–3955. [Google Scholar]
- 67.Hammond PT. Adv Mater. 2004;16:1271–1293. [Google Scholar]
- 68.Peyratout CS, Dahne L. Angew Chem Int Ed. 2004;43:3762–3783. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]
- 69.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]
- 70.Boudou T, Crouzier T, Ren K, Blin G, Picart C. Adv Mater. 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 71.Buck ME, Lynn DM. Adv Biomater. 2011 doi: 10.1002/adem.201080085. [DOI] [Google Scholar]
- 72.Buck ME, Breitbach AS, Belgrade SK, Blackwell HE, Lynn DM. Biomacromolecules. 2009;10:1564–1574. doi: 10.1021/bm9001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buck ME, Lynn DM. Adv Mater. 2010;22:994–998. doi: 10.1002/adma.200903054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buck ME, Lynn DM. ACS Appl Mater Interfaces. 2010;2:1421–1429. doi: 10.1021/am1000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buck ME, Lynn DM. Langmuir. 2010;26:16134–16140. doi: 10.1021/la103009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buck ME, Schwartz SC, Lynn DM. Chem Mater. 2010;22:6319–6327. doi: 10.1021/cm102115e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kinsinger MI, Buck ME, Abbott NL, Lynn DM. Langmuir. 2010;26:10234–10242. doi: 10.1021/la100376u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Broderick AH, Azarin SM, Buck ME, Palecek SP, Lynn DM. Biomacromolecules. 2011;12:1998–2007. doi: 10.1021/bm200296a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saurer EM, Flessner RM, Buck ME, Lynn DM. J Mater Chem. 2011;21:1736–1745. doi: 10.1039/C0JM02633F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luk YY, Kato M, Mrksich M. Langmuir. 2000;16:9604–9608. [Google Scholar]
- 81.Heilmann SM, Moren DM, Krepski LR, Rasmussen JK, Gaddam BN, Roscoe SB, Lewandowski KM, McIntosh LH, Roberts RR, Fansler DD, Szekely GG, Weil DA, Thakur KA, Pathre SV, Battiste JL, Hanggi DA. J Macromol Sci-Pure Appl Chem. 2003;A40:755–790. [Google Scholar]