

SUMMARY
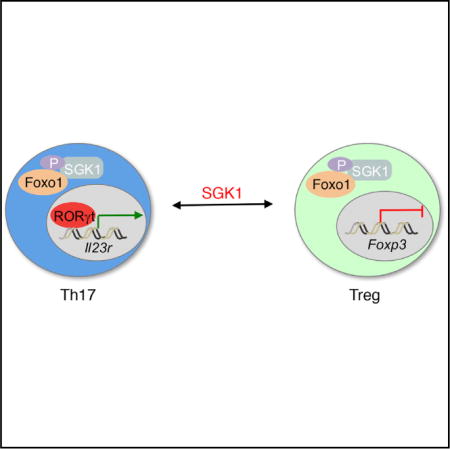
A balance between Th17 and regulatory T (Treg) cells is critical for immune homeostasis and tolerance. Our previous work has shown Serum- and glucocorticoid-induced kinase 1 (SGK1) is critical for the development and function of Th17 cells. Here, we show that SGK1 restrains the function of Treg cells and reciprocally regulates development of Th17/Treg balance. SGK1 deficiency leads to protection against autoimmunity and enhances self-tolerance by promoting Treg cell development and disarming Th17 cells. Treg cell-specific deletion of SGK1 results in enhanced Treg cell-suppressive function through preventing Foxo1 out of the nucleus, thereby promoting Foxp3 expression by binding to Foxp3 CNS1 region. Furthermore, our data suggest that SGK1 also plays a critical role in IL-23R-mediated inhibition of Treg and development of Th17 cells. Therefore, we demonstrate that SGK1 functions as a pivotal node in regulating the reciprocal development of pro-inflammatory Th17 and Foxp3+ Treg cells during autoimmune tissue inflammation.
Graphical Abstract
INTRODUCTION
Foxp3+ T (Treg) cells, a distinct subset of CD4+ T cells, play an essential role in regulating immune tolerance and homeostasis (Sakaguchi et al., 2008). As a master transcription factor, Foxp3 plays a critical role in regulating Treg cell development and function. Foxp3-deficient mice (e.g., Scurfy mice) exhibit hyperactivated effector T cells and dramatically enhanced pro-inflammatory cytokine production and are characterized by early lethality at around 16–25 days after birth due to lymphadenopathy, splenomegaly, and inflammation within multiple organs (Brunkow et al., 2001; Kim et al., 2007). A similar phenotype has also been observed in humans with a loss of function in FOXP3. These patients develop IPEX syndrome (immunodysregulation, polyendocrinopathy, and enteropathy, X-linked syndrome) with multi-organ inflammation and autoimmunity, and a considerable reduction in the lifespan of the individual (d’Hennezel et al., 2009; Le Bras and Geha, 2006).
Two distinct Treg cell populations express Foxp3, thymus-derived Treg (tTreg) cells, and peripherally induced Treg (pTreg) cells (Gavin et al., 2007; Josefowicz et al., 2012; Lin et al., 2007; Williams and Rudensky, 2007). For both tTreg and pTreg cells, Foxp3 is known to be induced through T cell receptor (TCR) stimulation together with various cytokine signals, including interleukin-2 (IL-2) and transforming growth factor β (TGF-β) (Fu et al., 2004; Horwitz et al., 2008; Hsieh et al., 2004; Zheng et al., 2007). TCR and CD28 stimulation activates the NF-κB signaling pathway (Rudd et al., 2009; Vang et al., 2010), while IL-2 and TGF-β signals initiate the activation of STAT5 and SMADs (Zheng et al., 2007), respectfully. All these transcription factors are able to bind to the Foxp3 locus and coordinately regulate the expression of Foxp3 and development of Treg cells (Zheng et al., 2010). In contrast, IL-23, a cytokine that promotes development of pathogenic Th17 cells, has been shown to suppress development of Tregs (Izcue et al., 2008), but the mechanism by which IL-23 mediates this effect on Tregs has not been studied.
Full commitment of the Treg cell lineage also requires integration of additional signaling pathways with the Foxp3 program, including some of the members of AGC family of kinases that are induced downstream of the TCR. It has been reported that phosphoinositide-3-kinase (PI3K) and Akt pathways play an essential role in regulating Treg cell differentiation (Sauer et al., 2008).
Our regulatory network analysis of Th17 cells predicted that Serum- and glucocorticoid-induced kinase 1 (SGK1), a member of the AGC family of kinases that is downstream of IL-23R, may also play a critical role in regulating the balance between Foxp3+ Tregs and pro-inflammatory Th17 cells. Its expression is modulated by glucocorticoids and serum (Leong et al., 2003; Mikosz et al., 2001), and it plays an important part in activating the epithelial sodium channel, ENaC (Canessa et al., 1993), in the aldosterone-sensitive distal nephron (ASDN) (Wade et al., 2001). So far, the role of SGK1 in different processes, such as the regulation of ion transport, enzymatic activities, transcriptional regulation, release of hormones, cell volume, and cell proliferation, and apoptosis, has been extensively studied (Fejes-Tóth et al., 2008; Loffing et al., 2006; Loffing et al., 2001). However, to date, there are limited studies exploring the role of SGK1 in the immune system, particularly during T cell differentiation. Our previous work first demonstrated that SGK1 plays a critical role in the differentiation of Th17 cells by regulating IL-23R expression (Wu et al., 2013). Loss of SGK1 reduces the expression of IL-23R and limits development of pathogenic Th17 cells. A recent study also indicates that activation of SGK1 by mTORC2 can regulate the development of Th1 and Th2 cells (Heikamp et al., 2014).
In the present study, we show that SGK1 functions as a negative regulator for Treg cell development. Loss of SGK1 in Treg cells results in increased Treg-suppressive function. Mice deficient in SGK1 specifically in Treg cells exhibit improved immune homeostasis and are protected from the development of multiple autoimmune diseases. Moreover, the activity of SGK1 in CD4+ T cells that is required for promoting a pathogenic Th17 cell response is also required for disarming the protective Treg cell response in an IL-23-dependent manner. Deficiency of SGK1 specifically in Treg cells leads to an increased frequency of Treg cells with improved function in vivo in the context of gut inflammation. Mechanistically, the loss of SGK1 limits phosphorylation of Foxo1, resulting in nuclear retention of Foxo1, which directly promotes the transactivation and maintenance of Foxp3. Therefore, we demonstrate that SGK1 functions as a pivotal node in regulating the balance of pro- and anti-inflammatory T cells development during autoimmunity.
RESULTS
SGK1 Signaling Is Active in Treg Cells
We have previously shown that Sgk1 mRNA is highly expressed under Th17 differentiation conditions and that exposure of naive T cells to TGF-β can also induce substantial transcription of Sgk1 (Wu et al., 2013). We confirmed here that in addition to Th17 cells, Sgk1 mRNA is expressed in both pTreg and tTreg cells with tTregs expressing the highest level, as compared with Th17 cells (Figure 1A). However, after IL-2 stimulation either in vitro or in vivo, which promotes Treg cell expansion and presumably function, Sgk1 expression was reduced in tTreg cells (Figures S1A and S1B), suggesting IL-2 exerts as a negative signal for Sgk1 expression. To investigate the function of SGK1 in tTreg cells, we first compared SGK1 activity between tTreg and naive T cells at steady state. We observed that tTreg cells exhibited enhanced phosphorylation of p27, one downstream target of SGK1 (Hong et al., 2008). Moreover, we found tTreg cells also contain higher levels of p27, phospho-p27, Foxo1, and raptor compared with naive T cells, whose expression all depend on SGK1 activity (Brown et al., 2011; Brunet et al., 2001) (Figures 1B and 1C). It is known that TCR signaling is crucial for Treg cell function (Sauer et al., 2008). Using anti-CD3/CD28 stimulation, we observed increased phosphorylation of p27 and Foxo1 in tTreg cells (Figures 1D and 1E), suggesting that SGK1 activity is likely promoted by TCR signaling in tTreg cells and regulates tTreg function. To examine the role of SGK1 in Treg cells, we generated mice with an SGK1 deficiency specifically in Treg cells (Foxp3CreSgk1fl/fl). Under steady state, the Foxp3CreSgk1fl/fl mice had similar frequencies of Foxp3+ Treg cells from spleen, draining lymph nodes (dLN), and lamina propria (LP), as compared to wild-type (WT) mice (Figures 1F and 1G). Interestingly, upon TCR stimulation, tTreg cells from the Foxp3CreSgk1fl/fl mice displayed a higher proliferative rate as indicated by their expression of Ki-67, a cellular marker for proliferation (Figure 1H).
Figure 1. SGK1 Signaling Is Active in Treg Cells and Inhibits Treg Cell Frequency.
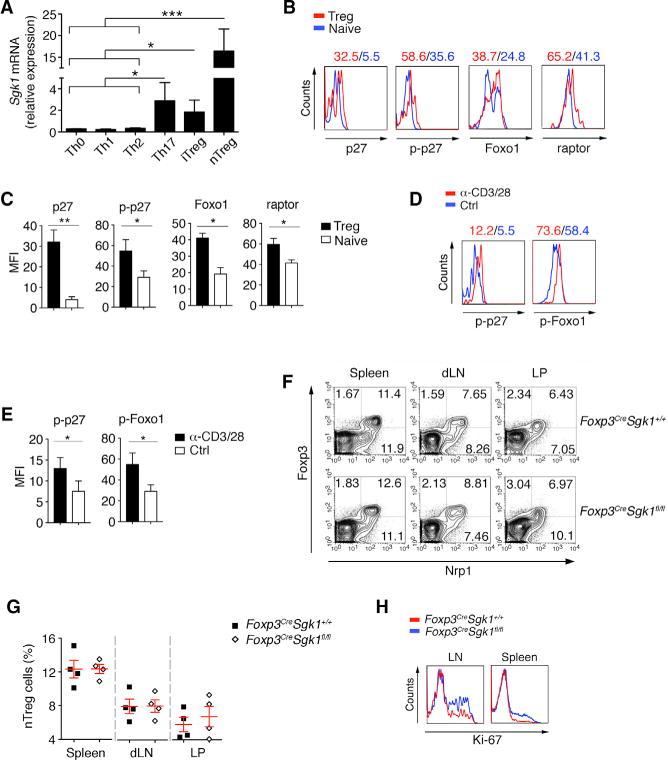
(A) In-vitro-differentiated Th0, Th1, Th2, Th17, and pTreg cells and tTreg cells were harvested for qPCR. mRNA levels of Sgk1 genes for different subsets are shown.
(B) The expression of indicated molecules on naive T cells and tTreg cells was determined by flow cytometry.
(C) Quantification of the mean fluorescence intensity (MFI) of the molecules indicated in (B).
(D) Comparison of phosphorylation of p27 and Foxo1 between freshly isolated and pre-activated (anit-CD3/28) Treg cells by flow cytometry.
(E) Quantification of the MFI of the molecules indicated in (D).
(F) The percentage of Foxp3+ Treg cells in spleen, drain lymphoid nodes (dLN), mesenteric LN (mLN), lamina propria (LP), liver, and lung from WT and Foxp3CreSgk1fl/fl mice was determined by flow cytometry.
(G) Quantification of the frequency of Foxp3+ Npr1+ in CD4+ tTreg cells within indicated mice as in (G).
(H) Comparison of Ki67 between activated (anit-CD3/28) tTreg cells from spleen and LNs of WT and Foxp3CreSgk1fl/fl mice.
Data are pooled from three independent experiments (A, C, and E) or representative of three independent experiments (B, D, and F–H) with n R 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test, error bars, SD). See also Figure S1.
SGK1 Limits tTreg Cell Function
It has been reported that Treg cells are crucial for immune self-tolerance and controlling inflammation (Fontenot et al., 2003; Williams and Rudensky, 2007; Wu et al., 2014). We next assessed the function of the tTreg cells from Foxp3CreSgk1fl/fl mice by in vitro suppression assay. CD4+Foxp3+ Treg cells were isolated from spleen and LNs by using Cre-YFP as a sorting marker, and co-cultured with naive WT T cells. Compared with WT tTreg cells, Foxp3CreSgk1fl/fl tTreg cells showed an enhanced ability to inhibit the TCR-stimulated proliferation of WT naive CD4+ T cells (Figure 2A). The signature markers and co-stimulatory molecules, such as CTLA-4, Lag-3, GITR, CD25, and ICOS, on activated tTreg cells from Foxp3CreSgk1fl/fl mice were increased, compared with those from WT mice (Figure 2B).
Figure 2. SGK1 Limits tTreg Cell function.
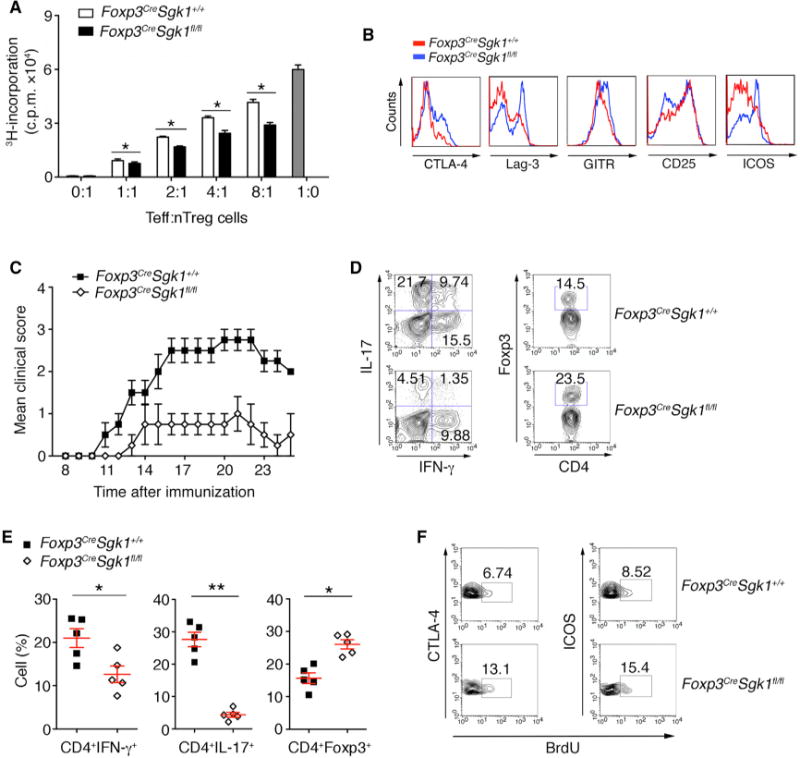
(A) Proliferation of WT naive CD4+ T cells in the presence of anti-CD3 and irritated antigen presentation cells (APCs) and tTreg cells from WT and Foxp3CreSgk1fl/fl mice. Data shown are presented as mean [3H]-thymidine incorporation.
(B) The expression of indicated molecules on tTreg cells from WT and Foxp3CreSgk1fl/fl mice was determined by flow cytometry.
(C) Disease score of EAE development in Foxp3CreSgk1+/+ and Foxp3CreSgk1fl/fl mice by MOG immunization.
(D) IL-17 and IFN-γ secretion and Foxp3 expression by CD4+ T cells within the CNS was determined by flow cytometry.
(E) Quantification of the frequency of IFN-γ+, IL-17+, and Foxp3+ in CD4+ cells within indicated mice CNS as in Figure 2D.
(F) BrdU incorporation in proliferating CTLA-4+ and ICOS+ Treg cells in the CNS of indicated mice was determined by flow cytometry.
Data are pooled from three independent experiments (A) or representative of three independent experiments (B–F) with n ≥ 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01 (Student’s t test, error bars, SD).
We next examined tTreg cell function in vivo in Foxp3Cre Sgk1fl/fl mice by induction of experimental autoimmune encephalomyelitis (EAE), a murine model of human disease multiple sclerosis. After immunization with myelin oligodendrocyte glycoprotein (MOG) peptide, Foxp3CreSgk1fl/fl mice developed much less severe EAE than control mice (Figure 2C). Analysis of the infiltrating T cells within the CNS showed that Foxp3CreSgk1fl/fl mice had fewer Th1 and Th17 cells, but more Treg cells (Figures 2D and 2E). Consistent with the in vitro assay (Figure 1F), in vivo bromodeoxyuridine (BrdU) incorporation showed that Treg cells from Foxp3CreSgk1fl/fl mice with EAE exhibited higher proliferative rates for Foxp3+ Treg cells than WT mice (Figure 2F). Given that tTreg cells are the main regulators during active EAE development (Korn et al., 2007), these results demonstrate a critical role for SGK1 in restraining tTreg cell expansion and function, both in vitro and in vivo.
SGK1 Represses Treg Cell Homeostasis
We then asked whether SGK1 deletion in Treg cells altered Treg function in systemic immune tolerance and homeostasis. While the Foxp3CreSgk1fl/fl mice at 6–8 weeks old exhibited normal T cell composition in the peripheral lymph nodes (LNs) and spleens (data not shown), the mice at 24 weeks old showed reduced proportions of activated CD4+ (CD62LloCD44+) T cells (Figures 3A and 3B). Their CD4+CD44hi memory T cells showed decreased interferon (IFN)-γ expression (Figures 3C and 3D), suggesting that a reduction in activated/memory cells may be because of increase in Treg activity in Foxp3CreSgk1fl/fl mice. To further test the ability of Treg cells in the absence of SGK1 to control spontaneous inflammation and regulate immune tolerance in vivo, we performed a Treg rescue assay in Foxp3-deficient Scurfy mice, which develop a spontaneous Th1 inflammatory response associated with severe multiorgan tissue inflammation (Clark et al., 1999). Transfer of purified tTreg cells into neonatal Scurfy mice rescues these pathological phenotypes (Kasprowicz et al., 2003). Although both WT and SGK1-deficient tTreg cell transfer prevented development of tissue inflammation in Scurfy mice, the frequency of CD4+Foxp3+ cells in the SGK1-deficient tTreg cells recipients was higher than that of WT Treg cells (Figures 3E and 3F). Additionally, as compared to WT tTreg cells, the CD4+CD44hi effector T cells from the recipient Scurfy mice that received SGK1-deficient tTreg cells displayed a reduced Th1 response (Figures 3G and 3H), consistent with the better repressive ability of SGK1-deficient tTreg cells in vitro (Figure 2A). These data illustrate that loss of SGK1 in tTreg cells facilitates tTreg cell function and SGK-deficient Tregs are therefore are better able to control autoimmune and inflammatory responses in vivo.
Figure 3. SGK1 Represses Treg Cell Homeostasis.
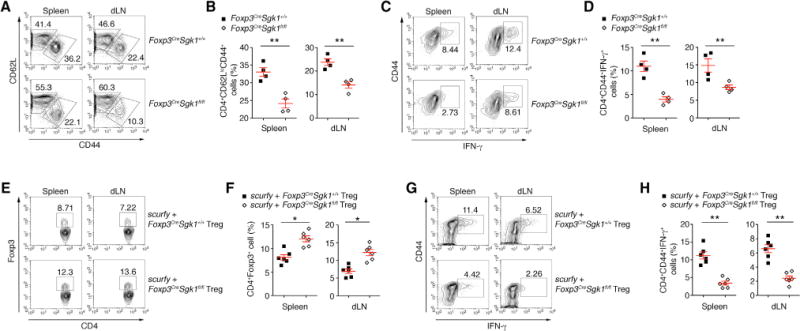
(A and B) Flow cytometry (A) and quantitative analysis (B) of CD44 and CD62L expression on CD4+ T cells from spleen and draining lymph nodes (dLNs) of WT and Foxp3CreSgk1fl/fl mice at 30 weeks of age.
(C and D) Flow cytometry (C) and quantitative analysis (D) of the IFN-γ expression in isolated CD4+CD44hi cells from WT and Foxp3CreSgk1fl/fl mice at 30 weeks of age.
(E and F) Flow cytometry (E) and quantitative analysis (F) of Foxp3 expression by cells isolated from the spleens and dLNs of scurfy recipients of WT or Foxp3CreSgk1fl/fl Treg cells.
(G and H) Flow cytometry (G) and quantitative analysis (H) of Foxp3 expression by cells isolated from the spleens and dLNs of scurfy recipients of WT or Foxp3CreSgk1fl/fl Treg cells.
Data are representative of three independent experiments (A–F) with n ≥ 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01 (Student’s t test, error bars, SD).
SGK1 Represses Foxp3 Expression via Regulating IL-23R
Our network analysis predicted that SGK1 is downstream of IL-23R signaling, where it has been reported to regulate Treg cells (Izcue et al., 2008). Activation of SGK1 is also implicated in repression of the Treg lineage in response to high salt exposure (Hernandez et al., 2015). To understand the relationship between IL-23R signaling and development of Foxp3+ Tregs in vivo, we transferred CD4+CD25−CD45RBhi T cells from IL-23R-GFP reporter mice into Rag2−/− recipients to induce colitis. Six weeks after transfer, CD4+GFP+ and CD4+GFP− T cells from different organs were sorted and gene expression analysis by real-time PCR was performed. IL-23R-expressing (IL-23R-GFP+) T cells expressed high levels of Sgk1 when multiple organs were interrogated, suggesting that IL-23R signaling may promote SGK1 expression in T cells during tissue inflammation (Figure 4A). Given that γδ T cells were reported to express IL-23R (Petermann et al., 2010), we also found upregulated Sgk1 expression in γδ T cells after IL-23 treatment (Figure S2).
Figure 4. IL-23R and SGK1 Form Positive Signaling Feedback Loop.
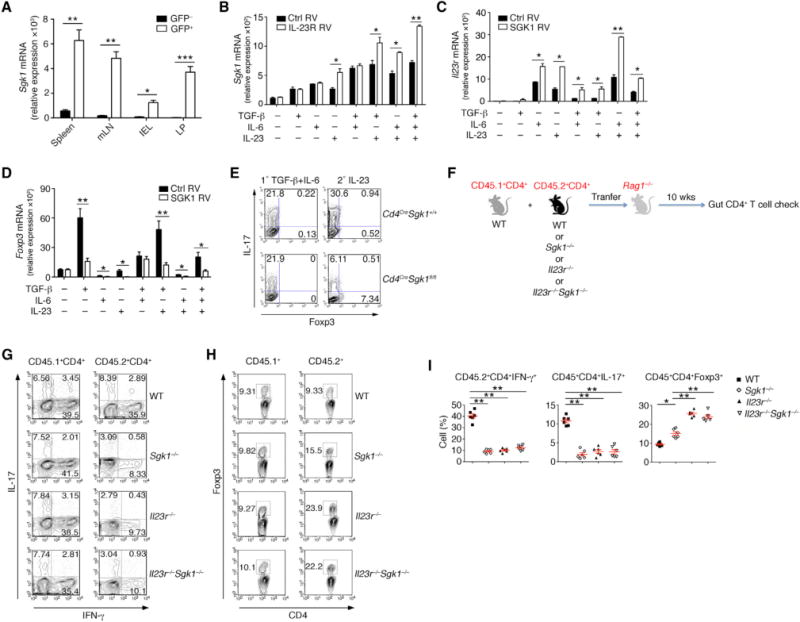
(A) T cell-induced colitis was performed on Rag2−/− mice by using Il23rgfp donor CD4+CD25−CD45RBhi GFP− T cells. 6 weeks after transfer, qPCR analysis of the indicated genes in CD4+GFP+ or CD4+GFP− cells sorted from the colon Rag2−/− mice.
(B) qPCR of Sgk1 mRNA expression in naive CD4+ T cells transduced with retrovirus expressing control vector (Ctrl RV) or IL-23R and treated with indicated cytokines; on day 3, RNA was isolated from GFP+ cells for analysis.
(C and D) qPCR of (C) Il23r or (D) Foxp3 mRNA expression in naive CD4+ T cells transduced with retrovirus expressing control vector (Ctrl RV) or SGK1 and treated with indicated cytokines; on day 3, RNA was isolated from GFP+ cells for analysis.
(E) Naive CD4+ T cells from Cd4CreSgk1fl/fl and WT mice were differentiated into Th17 cells with TGF-β and IL-6 (left) or restimulated with IL-23 (right). IL-17 and Foxp3 expression were assessed by flow cytometry.
(F) Schematic illustration of adoptive transfer experiments shown in (G).
(G) CD45.2+CD4+CD45RBhiCD25− cells were transferred into Rag2−/− mice from WT, Il23r−/−, Sgk1−/−, or Il23r−/−Sgk1−/− donor mice, together with congenic CD45.1+CD4+CD45RBhiCD25− cells as internal control at a ratio of 1:1. 10 weeks after transfer, IFN-γ and IL-17 production from CD4+ T cells within the LP were examined by flow cytometry.
(H) Foxp3 expression from CD4+ T cells in the samples as shown in (G) was examined by flow cytometry.
(I) Quantification of the frequency of IFN-γ, IL-17, or Foxp3 among CD4+ T cells from CD45.2+ donor mice in the LP.
Data are pooled from three independent experiments (A–D) or representative of three independent experiments (E–I) with n ≥ 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test, error bars, SD). See also Figures S2 and S3.
To further study the relationship between SGK1 and IL-23R signaling, we determined the effects of overexpressing SGK1 and IL-23R in activated naive CD4+ T cells in the presence of various cytokines. Overexpression of IL-23R significantly enhanced SGK1 expression (Figure 4B) and kinase activity (Figure S3) specifically in the presence of IL-23, suggesting that IL-23 signaling can induce the expression of SGK1 during Th17 cell differentiation. It has been reported that IL-23R cannot be induced prior to the initiation of the Th17 program (Stritesky et al., 2008). Interestingly, forced expression of SGK1 increased the expression of IL-23R in T cells (Figure 4C). IL-23R expression is suppressed in the presence of TGF-β. However, forced over-expression of SGK1 overcame this inhibition and enhanced Il23r even under TGF-β conditions (Figure 4C). Thus, IL-23R and SGK1 function as a positive feedforward loop to enhance each other’s expression. We also analyzed Foxp3 expression under these same conditions to study the effect of SGK1 and IL-23 stimulation on Foxp3 expression. We observed that SGK1 over-expression inhibited expression of Foxp3, even when Foxp3 expression was further enhanced by activation with TGF-β or IL-23 (Figure 4D). Given that Th17 and pTreg cells can be reciprocally regulated (Bettelli et al., 2006; Xu et al., 2007), we examined the expression of Foxp3 following IL-23 stimulation of WT and Cd4CreSgk1fl/fl T cells in vitro. We found that not only was the production of IL-17 compromised, but also Foxp3 expression was enhanced simultaneously in the SGK1-deficient T cells (Figure 4E). These data therefore suggest that the activation of SGK1 during Th17 cell differentiation initiates a feedback inhibitory loop to suppress induction of Foxp3 and promote IL-23R expression in an IL-23-dependent manner.
IL-23 Restrains Treg Cell Development In Vivo Partly via SGK1
IL-23 signaling has been shown to affect the development of intestinal Treg cells (Izcue et al., 2008). We tested the role of the IL-23R-SGK1 axis during in vivo pTreg generation. We transferred CD45.2+CD4+CD45RBhiCD25− cells into Rag2−/− mice from WT, Il23r−/−, Sgk1−/− or Il23r−/−Sgk1−/− donor mice, together with CD45.1+CD4+CD45RBhiCD25− cells as an internal control at a ratio of 1:1. Ten weeks after transfer, we isolated the T cells from the gut and analyzed their cytokine profile (Figure 4F). As expected, CD45.1+ and CD45.2+ WT donor cells exhibited identical IL-17 and IFN-γ production in this transfer model (Figure 4G). Moreover, consistent with previous data (Hue et al., 2006), we observed a reduction in both IFN-g and IL-17-producing T cells in the LP from Il23r−/− donor mice (Figure 4G). Similar to Il23r−/− T cells, reduced Th1 and Th17 responses were also found in either Sgk1−/− or Il23r−/−Sgk1−/− T cells (Figures 4G and 4I). In contrast, we found an increased frequency of Foxp3+ T cells from the Sgk1−/− T cells compared with WT T cells. Interestingly, Il23r−/− cells donor cells showed an even greater increase in Foxp3 expression compared to the Sgk1−/− transferred T cells, suggesting that SGK1 may only partially account for restraint of Foxp3 expression in Treg cells. However, the increase in Foxp3 expression was similar in both Il23r−/−Sgk1−/− and Il23r−/− T cells compared to Sgk1−/− T cells (Figures 4H and 4I). These results reveal that both IL-23R and its downstream kinase SGK1 are able to repress expression of Foxp3 within pTregs.
SGK1 Regulates Reciprocal Development of Th17 and pTreg Cells during Intestinal Inflammation
We then asked whether such SGK1 could regulate development of Th17 cells and Tregs in vivo, without using very strong immunization regimens that would bias T cell fates. We transferred WT or SGK1-deficient CD4+CD25−CD45RBhi T cells into Rag2−/− recipients and observed the mice for the development of intestinal inflammation as measured by weight loss and histopathology. We observed that WT T cells when transferred to the Rag2−/− mice induced weight loss and clinical histopathological signs of colitis whereas T cells from the Sgk1−/− mice were not able to transfer colitis and appear to be protected from development of colitis (Figures 5A and 5B). SGK1-deficient CD4+ T cells isolated from Rag2−/− recipients exhibited reduced expression of IL-23R and production of pro-inflammatory cytokines IFN-γ and IL-17. The transferred T cells from the Sgk1−/− mice showed an increase in the frequency of Foxp3+ T cells in the gut compared with WT T cells (Figures 5C–5F). These data suggest that SGK1 is also involved in the regulation of reciprocal differentiation of Th17 and FoxP3+Treg cells in vivo.
Figure 5. SGK1 Regulates IL-23R Expression and Reciprocal Differentiation of Th17 and Treg Cells during Intestinal Inflammation.
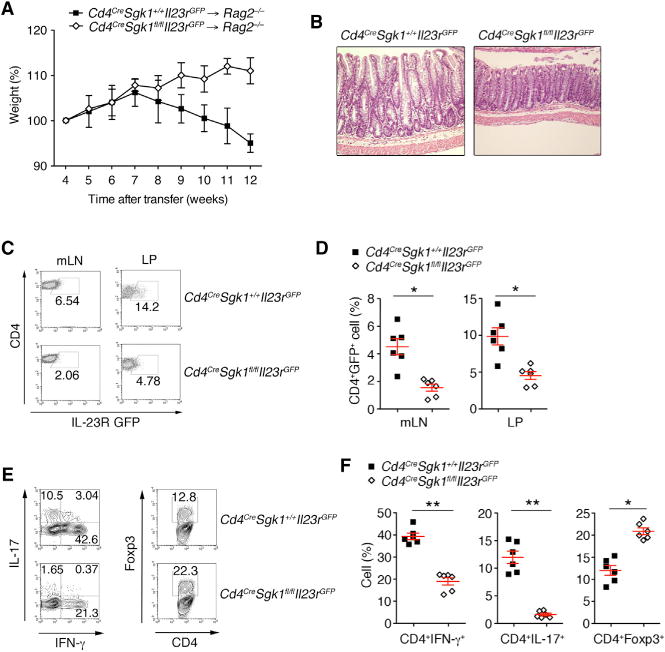
(A) Body weight of Rag2−/− mice transferred with CD4+CD25−CD45RBhi T cells from WT or Cd4CreSgk1fl/fl mice.
(B) H&E staining of colon samples from the different groups as in (A) (original magnification, ×20).
(C and D) Flow cytometry (C) and quantitative analysis (D) of IL-23R expression from CD4+ T cells isolated from mLN and LP from indicated groups as in (A) 10 weeks after colitis induction.
(E and F) Flow cytometry (E) and quantitative analysis (F) of IL-17 and IFN-γ, Foxp3 expression by CD4+ T cells isolated from LP from indicated groups as in (A) 10 weeks after colitis induction. Data are representative of three independent experiments. Error bars represent SEM. *p < 0.05, **p < 0.01 (Student’s t test, error bars, SD).
Cell-Intrinsic Role of SGK1 in Regulating pTreg Differentiation and Function
In order to examine whether SGK1 plays an intrinsic role within pTreg cells for Foxp3 expression, we differentiated naive T cells from Foxp3CreSgk1fl/fl in presence of TGF-β to induce expression of Foxp3 in vitro. Under these in vitro differentiation conditions, Foxp3CreSgk1fl/fl T cells displayed increased Foxp3 compared with WT T cells (Figure 6A). Similar to tTreg cells, SGK1-deficient Foxp3+ pTreg cells exhibited an increased suppressive capacity in inhibiting the proliferation of WT T effector cells (Figure 6B). We next examined the functional ability of pTregs differentiated in vitro from SGK1-deficient T cells to induce colitis upon adoptive transfer model. We transferred CD4+CD45RBhiYFP− cells from WT and Foxp3CreSgk1fl/fl mice into Rag2−/− mice to induce colitis. We observed that, whereas WT T cells could induce both clinical and histopathological disease, Foxp3CreSgk1fl/fl T cells protected mice from development of intestinal inflammation in T cell-induced colitis (Figures 6C and 6D). We observed reduced Th1 and Th17 development in the gut in the mice that received Foxp3Cre Sgk1fl/fl T cells 10 weeks after transfer, concomitant with an increase in the frequency of pTreg cells (Figures 6E and 6F). Thus, deficiency in SGK1 resulted in both in vitro and in vivo elevated pTreg cells and protected mice from intestinal inflammation.
Figure 6. SGK1 Reduces pTreg Differentiation and Function.
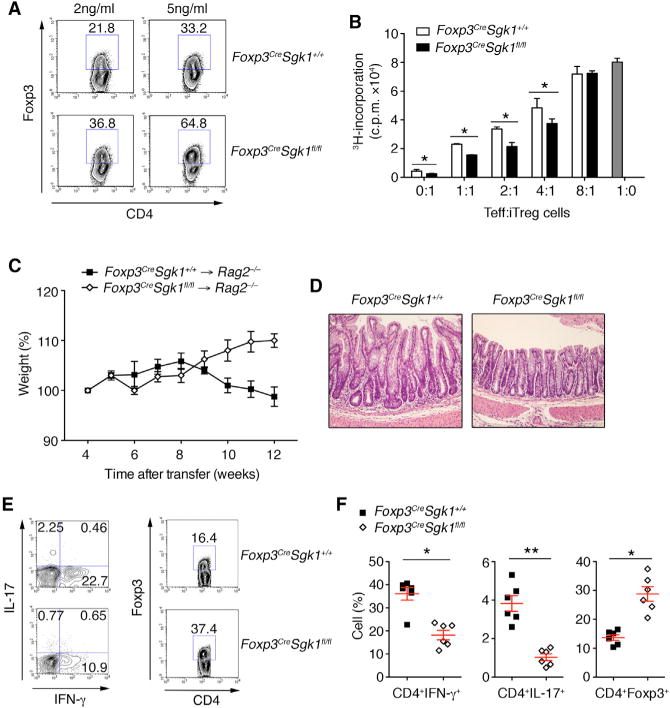
(A) Naive CD4+ T cells from WT and Foxp3Cre Sgk1fl/fl mice were differentiated into Treg cells with indicated dose of TGF-β. Foxp3 expression was assessed by flow cytometry.
(B) Proliferation of WT naive CD4+ T cells in the presence of anti-CD3 and irritated APCs and GFP+ (Foxp3+) pTreg cells from Foxp3CreSgk1+/+ and Foxp3CreSgk1fl/fl mice.
(C) Body weight of Rag2−/− mice after transferred i.p. with CD4+CD25−CD45RBhiFoxp3− T cells from WT or Foxp3CreSgk1fl/fl mice.
(D) H&E staining of colon samples from the different groups as in (C) (original magnification, 320).
(E and F) Flow cytometry (E) and quantitative analysis (F) of IL-17, IFN-γ, and Foxp3 expression by CD4+ T cells isolated from LP from indicated groups as in (C) 10 weeks after colitis induction. Data are representative of three independent experiments with n ≥ 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01 (Student’s t test, error bars, SD).
SGK1 Restrains Foxp3 Expression via Reduced Foxo1 Nuclear Exclusion
To gain more molecular insight into how SGK1 regulates Treg cell development and function, we used activated Foxp3+ tTreg cells from spleens of WT and Foxp3CreSgk1fl/fl mice and performed a multiplex expression profiling experiment using nanostring nCounter analysis. Nanostring multiplex expression analysis of WT and Foxp3CreSgk1fl/fl Foxp3+ tTreg cells revealed an increase in the expression of Treg related genes and decreased expression of Th17 genes in SKG1-deficient Treg cells (Figure 7A). Expression of selected genes was further confirmed by RT-PCR, including Foxo1 and Il23r, showing a decreased Il23r expression and an increased Foxo1 expression in Foxp3CreSgk1fl/fl tTreg (Figure 7B). SGK1 has been shown to directly phosphorylate Foxo1, resulting in nuclear exclusion of Foxo1 (Ouyang et al., 2012). Other studies have shown that Foxo1 not only has a role in affecting the genomic landscape for Treg cell development but also directly transactivates Foxp3 expression (Merkenschlager and von Boehmer, 2010; Ouyang et al., 2012). We isolated Foxp3+ Treg cells from either WT or Foxp3CreSgk1fl/fl mice and then activated them with anti-CD3 and anti-CD28. Immunostaining showed attenuated Foxo1 nuclear clearance and increased accumulation of Foxo1 in the nucleus of SGK1-deficient compared with WT Treg cells upon activation (Figures 7C and 7D), consistent with the increase in Foxp3 activity of the SGK1-deficient Treg cells. It has been reported that Foxo1 could directly bind to the Foxp3 CNS1 region and promote Foxp3 expression (Ouyang et al., 2010). To determine how SGK1 regulates Foxp3 expression, we transfected WT or Foxp3CreSgk1fl/fl naive T cells with Foxp3 luciferase reporter constructs with the CNS1 region and tested luciferase activity under Th0 or pTreg conditions. We observed enhanced Foxp3 transcriptional activity in Foxp3CreSgk1fl/fl as compared to WT T cells. To further determine the specificity of binding and transactivation, we observed that the transactivation of Foxp3 was inhibited when the Foxo-binding site in CNS1 was mutated (Figure 7E). In addition, given that Foxo1 phosphorylation by SGK1 directly affects Foxp3 transactivation, we next tested how activation status of SGK1 affects Foxp3 expression. When retroviral constructs of WT, the constitutively active (S422DSGK1) or inactive (K127NSGK1) form of SGK1 were transfected into WT naive T cells that were then activated in the presence of TGF-β, we observed that Foxp3 expression was inhibited when WT SGK1 was overexpressed and further inhibited by expression of constitutively active SGK-1 (S422DSGK1) (Figure 7F). We also confirmed that the control group and K127NSGK1-infected group exhibited more nuclear expression of Foxo1 than WT SGK1 or S422DSGK1-infected groups (Figure 7G), suggesting the kinase activity of SGK1 determines Foxo1 location. Foxo1 acts as a transcription factor in a positive feedback manner (Essaghir et al., 2009; Wu et al., 2013); therefore, nuclear Foxo1 can enforce its own transactivation and expression. We examined the different SGK1-mutant-infected groups and found higher SGK1 kinase activity correlated with lower Foxo1 expression (Figure 7H). These findings support the idea that as phosphorylation target of Sgk-1, Foxo1, which regulates Foxp3 expression is limited by both expression and activation of SGK1. Therefore, SGK1 restrains regulatory T cell function by controlling Foxo1 activity.
Figure 7. SGK1 Deficiency Results in Compromised Foxo1 Nuclear Exclusion, Promoting Foxp3 Expression.
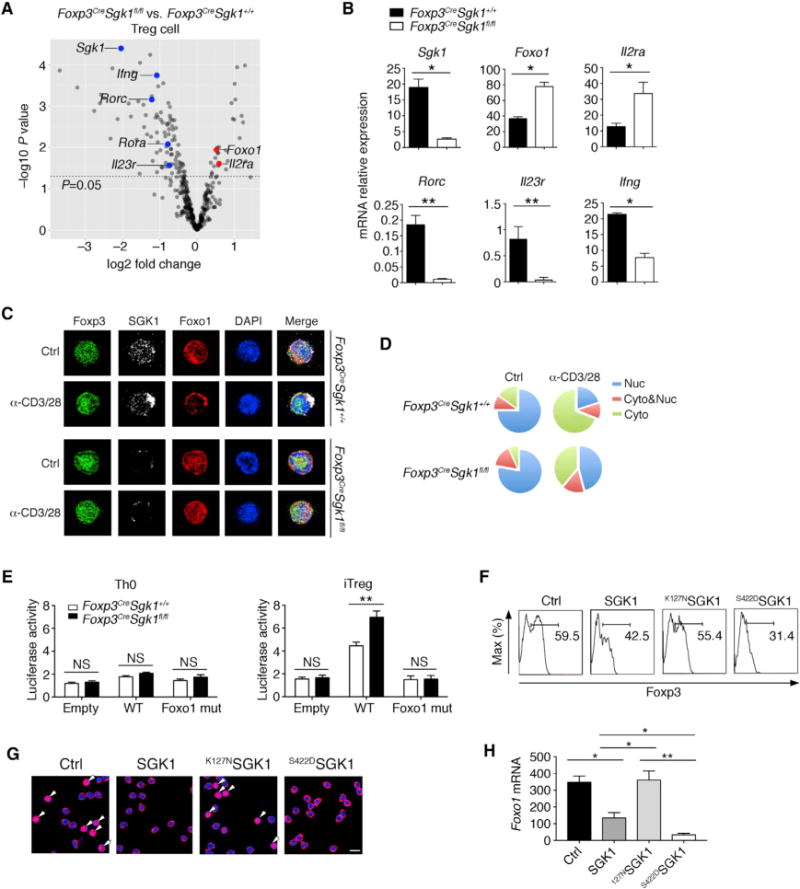
(A) Volcano plot comparing the p value versus fold change for probes of nanostring data from Foxp3CreSgk1fl/fl versus WT tTreg cells. The key genes of Treg and Th17 cells are highlighted in red and blue, respectively (transcripts upregulated in Treg cells) and green (transcripts downregulated in Th17 cells).
(B) qPCR validation of signature genes in (A) from WT and Foxp3CreSgk1fl/fl tTreg cells.
(C) Representative immunostaining images of Foxp3, Foxo1, and SGK1. CD4+Foxp3+ tTreg cells isolated from WT or Foxp3CreSgk1fl/fl mice were left untreated or stimulated with anti-CD3/CD28 on chamber slides at 37° C for 20 min.
(D) Quantitative analysis of Foxo1 localization in Treg cells between WT or Foxp3CreSgk1fl/fl mice. Cyto, Foxo1 localized in the cytosol; Cyto/Nuc, Foxo1 localized in both the cytosol and the nucleus; Nuc, Foxo1 localized in the nucleus.
(E) WT or Foxp3CreSgk1fl/fl CD4+ naive T cells were transfected with a luciferase reporter containing Foxp3 CNS1 region construct with or without Foxo1 binding site mutation and stimulated with Th0 or pTreg conditions. Luciferase activity was measured 24 hr later.
(F–H) Naive CD4+ T cells were transduced with retrovirus expressing control vector (Ctrl), WT SGK1, the constitutively active (S422DSGK1) or the inactive forms of SGK1 (K127NSGK1) under pTreg conditions. (F) Foxp3 expression in transfected cells was determined by flow cytometry. (G) Foxo1 localization within indicated groups was examined. Arrows indicate nuclear Foxo1. DAPI (blue), Foxo1 (red). Scale, 500 μm. (H) Foxo1 mRNA expression within different groups was determined by qPCR.
Data are representative of three independent experiments (A, C, D, F, and G) or pooled from three independent experiments (B, E, and H) with n ≥ 4 mice each group. Error bars represent SEM. *p < 0.05, **p < 0.01 (Student’s t test, error bars, SD).
DISCUSSION
We previously developed a high-density temporal transcriptional network for differentiation of Th17 cells and connected this transcriptional network to known protein interactions associated with IL-23R signaling (Wu et al., 2013; Yosef et al., 2013). This network analysis identified SGK1, a member of the AGC family of serine/threonine protein kinases, as a pivotal node that regulates the differentiation of pathogenic Th17 cells and the resulting balance between Th17 cells and Foxp3+ Tregs. In this paper, we validated this network analysis, by showing that loss of SGK1 not only inhibits the development of Th17 cells but also found that it induces stable Tregs such that deletion of SGK1 in Tregs makes mice highly resistant to the development of autoimmunity.
Loss of SGK1 within Treg cells makes them highly potent and stable, promoting their ability to suppress pro-inflammatory responses and maintain tissue homeostasis and tolerance. Phosphorylation of Foxo1 by SGK1 deactivates the protein by exporting it out of the nucleus, thereby preventing it from binding to the Foxp3 locus, and limiting its ability to prepare the genomic landscape for expression of Foxp3 gene (Brunet et al., 2001; Di Pietro et al., 2010). This is consistent with our data showing an enhancement of Treg cell generation and function by conditional deletion of SGK1 within Tregs. As a critical transcription factor controlling Foxp3 expression, loss of Foxo1 has been shown to disrupt Treg function, resulting in hyperactivation of Th1-like immune responses (Ouyang et al., 2012). Consistent with this observation, our studies demonstrate that conditional deletion of SGK1 in Treg cells stabilizes Foxo1 in the nucleus and promotes Treg-suppressive function. It has previously been reported that Foxo1 serves as a ‘‘placeholder’’ for Foxp3 during Treg cell development (Samstein et al., 2012). Thus, nuclear exclusion of Foxo1 by SGK1 could not only inhibit Foxp3 expression, but also alter the genomic landscape during Treg cell development. Our expression profiling showed that besides enhanced Foxo1 expression, there was a decrease in Th1- and Th17-associated genes in SGK1-deficient Treg cells, including Ifng, Rorc, Rora, and Il23r. We also found that SGK1-deficient Treg cells suppressed Th1 and Th17 responses more efficiently at both steady and inflammatory states. Consistent with this observation, Foxo1-deficient Treg cells display the opposite phenotype of SGK1-deficient Treg cells, producing large amounts of IFN-γ and IL-17 at a quiescent state (Ouyang et al., 2010). These data implicate the SGK1-Foxo1 axis in specification of Treg cell identity and stability. Activation of Foxo1 results in destabilization of Treg cell function and development. Therefore, SGK1 is a pivotal kinase that regulates the balance between Tregs and Th17 cells through activation of Foxo1, which reciprocally drives the expression of Foxp3 and IL-23R.
IL-23R signaling has been previously shown to suppress development and accumulation of Foxp3+ Treg cells by a direct cell-intrinsic mechanism during intestinal inflammation (Izcue et al., 2008). It has also been reported that, as opposed to in-vitro-derived pTreg cells, intestinal Foxp3+ Treg cells also co-express RORγt, which potentially induces expression of IL-23R (Sefik et al., 2015; Yang et al., 2016). This intermediate phase of T cell development can be observed in the gut environment. The fact that IL-23 regulates Treg cells in the gut indicates that IL-23 potentially functions synergistically with environmental cues to regulate the development of Tregs and Th17 cells. As one of the downstream targets of IL-23R, we showed that SGK1 expression correlates with IL-23R induction during intestinal inflammation. It is known that SGK1 plays a critical role on the epithelia cells for Na+ transport and could lead to an imbalance of sodium homeostasis in the intestinal tract. We and others also found that Na+ concentration is not only critical for T cell differentiation and function (Hernandez et al., 2015; Wu et al., 2013) but also affects gut commensalism. The loss of SGK1 in Tregs could therefore affect immune regulation via both intrinsic molecular signaling mechanisms by regulating by impacting Treg stability and function but also by cell-extrinsic mechanisms such as by affecting microbiota. Therefore, regulation of pTreg development by SGK1 following IL-23 signaling must be context dependent. Our gain-of-function experiments demonstrate that IL-23R and SGK1 form a positive feedforward loop, indicating that IL-23 represses Foxp3 expression in a SGK1-dependent manner. Consistent with previous observations (Izcue et al., 2008), our studies thus provide a mechanism by which IL-23R signaling regulates Treg function in the gut.
Although superficially, the phenotype of Il23r−/− mice looks very similar to Sgk1−/− mice in terms of elevated Treg function, and, upon closer observation, the phenotype of Il23r−/− or Il23r−/−Sgk1−/− mice appears to be enhanced as compared with Sgk1−/− mice, suggesting that IL-23R signaling must employ additional mechanisms to suppress Treg function in addition to SGK1. STAT3, which is downstream of IL-23R signaling has been shown to be a negative regulator of Foxp3 expression (Bettelli et al., 2006; Nurieva et al., 2007). Therefore, SGK1 and STAT3 downstream of IL-23R could work together to induce stronger inhibition of Treg development and function.
A balance between effector and Treg cells is crucial for maintaining immune homeostasis and tolerance during various physiological or pathological processes. SGK1 appears to be a pivotal kinase that regulates the balance between Th17 and Treg cells by activation of the transcription factor Foxo1. Although the role of SGK1 in Th17 development has been studied extensively, this current study highlights a cell-intrinsic role for SGK1 in Treg development and function. Based on our studies, blocking SGK1 activation using a small-molecular-weight inhibitor could be a potential strategy to ameliorate Th17-mediated inflammation and enhance development of Treg cells, shifting the balance away from a pro-inflammatory Th17 response to a more tissue protective Treg response.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6 (B6), Rag2−/−, Foxp3Cre, and Scurfy mice were purchased from Jackson Laboratory; Cd4Cre mice were purchased from Taconic; Il23rgfp, Il23r−/−, and Sgk1fl/fl mice were described previously (Croxford et al., 2009; Fejes-Tóth et al., 2008; Petermann et al., 2010). All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School.
Antibodies
The following antibodies were used in fluorescence-activated cell sorting (FACS) and cell sorting: CD45.1 (A20, BioLegend), CD45.2 (104, BioLegend), CD4 (H129.19, BD Pharmingen), CD25 (7D4, BD Pharmingen), IL-17A (TC11-18H10.1, BD Pharmingen), IFN-g (XMG1.2, BD Pharmingen), CD44 (IM7, Bio-Legend), CD62L (MEL-14, BioLegend), CD25 (7D4, BD Pharmingen), Lag3 (46-2231-80, eBioscience), Nrp1 (FAB566A, R&D Systems), Ki67 (SolA15, eBioscience), CTLA4 (14D3, eBioscience), GITR (DTA-1, eBioscience), ICOS (C398.4A, BioLegend), Foxp3 (FJK-16 s, eBioscience), p27 (sc-776, Santa Cruz Biotechnology), p-p27 (ab85047, Abcam), Foxo1 (ab39670, Abcam), p-Foxo1 (9461S, Cell Signaling Technology), Raptor (ab5454, Abcam), Brdu (B44, BD Pharmingen).
In Vitro T Cell Differentiation
Naive (CD44loCD62L+CD25−) or memory (CD44hiCD62LloCD25−) CD4+ T cells was obtained from spleens and LNs of indicated mice by flow cytometry accordingly. The purity of isolated T cell populations routinely exceeded 98%. Naive T cells were stimulated with plate-bound anti-CD3 (145-2C11, 1 μg/mL) and anti-CD28 (PV-1, 1 μg/mL) and polarizing cytokines (R&D Systems).
Flow Cytometry
For intracellular cytokine staining, cells were cultured as described above and stimulated for 4 hr at 37° C in culture medium containing phorbol 12-myristate 13-acetate (PMA) (50 ng/mL; Sigma), ionomycin (1 μg/mL; Sigma), and monensin (GolgiStop; 1 μg/mL; BD Bioscience). Surface markers were stained in PBS with 1% FCS for 20 min at room temperature and then subsequently fixed in Cytoperm/Cytofix (BD Bioscience), permeabilized with Perm/Wash Buffer (BD Bioscience), and stained with cytokine antibodies diluted in Perm/Wash. For Foxp3 stating, cells were fixed and permeabilized with the Foxp3 Staining Buffer Set, according to the manufacturer’s instructions (eBioscience). All flow cytometry data were acquired on a FACS Calibur (Becton Dickinson) and analyzed with FlowJo software (Tree Star).
Suppression Assay
Naive CD4+ T effectors (5 × 105) were cultured with iTregs (5 × 105, titrated down as described) in 96-well round-bottom plates (Corning). Cells were stimulated with anti-CD3/28 and left in culture for 5 days. Cells were pulsed with 1 μCi 3H thymidine for the last 16 hr of incubation. Mean thymidine incorporation in triplicate wells was measured using a β counter (LS 5000; Beckman Coulter).
qRT-PCR
For gene expression detection, total RNA was isolated from whole cells using the QIAGEN miniRNA extraction kit following the manufacturer’s instructions. RNA was quantified and complementary DNA was reverse-transcribed using the cDNA archival kit (Applied Biosystems) following the manufacturer’s instructions. The cDNA samples were used at 20 ng/well in a 384-well plate and run in triplicate. PCRs were set up using TaqMan Universal PCR Master Mix (Applied Biosystems) on an ABI Prism 7500 Sequence Detection System. Quantification of relative mRNA expression was normalized to the expression of GAPDH. Primer-probe mixtures were purchased from Applied Biosystems: Foxp3 (Mm00475162); Foxo1 (Mm00490672-m1); Il23r (Mm00519943-m1); Rorc (Mm01261022-m1); Il2r (Mm01340213_m1); Sgk1 (Mm00441380-m1); Ifng (Mm01168134_m1); GAPDH (4352339E).
Plasmids
Murine SGK1 or IL-23R (IMAGE cDNA clone) was cloned into the MSCV-IRES-GFP and MSCV-IRES-Thy1.1 vectors at EcoRI/XhoI and BglII/XhoI sites, respectively.
Retroviral Transduction
Retroviral particles were produced by transiently transfecting HEK293T cells with retroviral packaging constructs (PCL, Gag/pol) and expression plasmids MSCV-IRES-GFP or MSCV-IRES-Thy1.1 using Fugene HD (Roche). 72 hr after transfection, viral culture supernatants were harvested, supplemented with polybrene (8 mg/mL) and added to previously stimulated T cells (5 × 105/well, plate-bound anti-CD3/CD28 and different combination of cytokines for 24 hr). Cultures were centrifuged at 800 × g for 45 min at 25° C.
Reporter Assays
HEK293T cells (1 × 105 cells/well, 48-well plate) were transiently transfected with the indicated expression vectors, empty vector controls as well as the promoter firefly luciferase-reporter constructs and Renilla luciferase reporter vector (Promega) with Fugene HD (Promega). 48 hr after transfection, luciferase expression was determined by measuring luminescence with the Dual-Luciferase Reporter Assay System (Promega). The firefly luciferase activity was normalized to Renilla luciferase activity. Data are representative of at least two independent experiments; each data point represents duplicate values.
Immunofluorescence Microscopy
Treg cells were isolated from WT and SGK1-deficient were stimulated with plate-bound anti-CD3 (1 μg/mL) and anti-CD28 (1 μg/mL) on chamber slides at 37° C for 20 min. After fixation with 4% paraformaldehyde, cells were permeabilized with Foxp3 Fixation/Permeabilization buffer (eBioscience) according to the manufacturer’s instructions. After blocking with Permeabilization buffer and 3% BSA, cells were incubated with 1:250 diluted mouse anti-Foxo1 (ab1102, Abcam), rat anti-Foxp3 (FJK-16 s, eBioscience), and rabbit anti-SGK1 (ab43606, Abcam), followed by Alexa555-anti-mouse, allophyco-cyanin (APC)-anti-rabbit and fluorescein isothiocyanate (FITC)-anti-rat secondary antibodies in Permeabilization buffer and 1% BSA. Slides were mounted with gold anti-fading mounting buffer (Invitrogen). Images were acquired with a Leica TCS SP5-II confocal microscope. For quantitative analysis, five fields were selected randomly and total cells in the field were manually counted and grouped with Volocity software (PerkinElmer), on the basis of their Foxo1 nuclear or cytosolic localization.
T Cell-Induced Colitis
CD4+ T cells were purified from the spleen and LNs of WT and SGK1-deficient mice by positive selection with magnetic beads (Miltenyi Biotec), and naive CD4+CD25− CD45RBhi T cells were purified by sorting with flow cytometry. 7 × 105 naive T cells were injected intraperitoneally (i.p.) into age- and sex-matched Rag2 −/− mice and weight loss was monitored.
Assessment of Intestinal Inflammation
Tissues were graded semiquantitatively from 0 to 5 in a blinded fashion: score 0, no changes observed; score 1, minimal scattered mucosal inflammatory cell infiltrates, with or without minimal epithelial hyperplasia; score 2, mild scattered to diffuse inflammatory cell infiltrates, sometimes extending into the sub-mucosa and associated with erosions, with minimal to mild epithelial hyperplasia and minimal to mild mucin depletion from goblet cells; score 3, mild to moderate inflammatory cell infiltrates that were sometimes transmural, often associated with ulceration, with moderate epithelial hyperplasia and mucin depletion; score 4, marked inflammatory cell infiltrates that were often transmural and associated with ulceration, with marked epithelial hyperplasia and mucin depletion; score 5, marked transmural inflammation with severe ulceration and loss of intestinal glands.
EAE Induction
Mice were immunized subcutaneously (s.c.) in the flanks with an emulsion containing MOG35–55 (100 μg/mouse) and M. tuberculosis H37Ra extract (3 mg/mL, Difco Laboratories) in CFA (100 μL/mouse). Pertussis toxin (100 μg/mouse, List Biological Laboratories) was administered i.p. on days 0 and 2. Mice were monitored and assigned grades for clinical signs of EAE using the following scoring system: 0, healthy; 1, limp tail; 2, impaired righting reflex or waddled gait; 3, hind limb paralysis; 4, total limb paralysis; 5, moribund or death.
Adoptive Transfers
CD45.2+CD4+CD45RBhiCD25− cells from WT, Il23r−/−, Sgk1−/−, or Il23r−/− Sgk1−/− donor mice, together with CD45.1+CD4+CD45RBhiCD25− cells as internal control at a ratio of 1:1, were injected intravenously into Rag2−/− mice. 10 weeks after transfer, mice were sacrificed for analysis. For neonatal transfer, GFP+ Treg cells were isolated from WT or SGK1-deficient mice and were resuspended in 1 × 106 cells/25 mL PBS. The cells were injected intraperitoneally into 1- to 2-day-old scurfy neonates. Mice were monitored for external signs of disease and were killed after 25–60 days for analysis.
NanoString Gene Expression Analysis
Purified RNA was hybridized with a custom-made CodeSet provided by NanoString Technologies. Bar codes were counted (1,150 fields of view per sample) on an nCounter Digital Analyzer. Data were processed using nSolver Analysis Software by normalization to the geometric mean of the positive control spike count in addition to the 4 reference genes (Actv, Gapdh, Hprt, and Tubb5).
Statistical Analysis
Statistical analysis was performed using Prism software (GraphPad, La Jolla, CA, USA). p values <0.05 were considered significant.
Supplementary Material
Highlights.
SGK1 restrains Treg cell expansion and function
SGK1 represses Foxp3 expression via regulating IL-23R
SGK1 reciprocally regulates development of Th17/Treg balance
SGK1 restrains Foxp3 expression via reduced Foxo1 nuclear exclusion
Acknowledgments
We thank Deneen Kozoriz for cell sorting, M. Collins for advice and editing the manuscript, and L.G. Palmer (Weill-Cornell Medical College) for Sgk1fl/fl mice. This work is supported by the National Multiple Sclerosis Society (Career Transition Award TA 3059-A-2 to C.W.) and the US NIH (R00 NIH Pathway to Independence Award R00AI110649A to C.W., K01DK090105 to S.X., and P01AI073748, P01AI056299, P01AI039671, R01NS045937, and R01 NS030843 to V.K.). T.T. was supported by the Austrian Science Fund (FWF, J 3091-B12).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.068.
AUTHOR CONTRIBUTIONS
C.W. designed and performed experiments and wrote the manuscript; Z.C. performed experiments and wrote the manuscript; S.X., T.T., and T.H. performed experiments; A.M. analyzed the data; and V.K. supervised the study and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem. 2011;286:44295–44305. doi: 10.1074/jbc.M111.258053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–2554. [PubMed] [Google Scholar]
- Croxford AL, Kurschus FC, Waisman A. Cutting edge: An IL-17F-CreEYFP reporter mouse allows fate mapping of Th17 cells. J Immunol. 2009;182:1237–1241. doi: 10.4049/jimmunol.182.3.1237. [DOI] [PubMed] [Google Scholar]
- d’Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, Noya FJ, Jabado N, Mazer B, Piccirillo CA. FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N Engl J Med. 2009;361:1710–1713. doi: 10.1056/NEJMc0907093. [DOI] [PubMed] [Google Scholar]
- Di Pietro N, Panel V, Hayes S, Bagattin A, Meruvu S, Pandolfi A, Hugen-dubler L, Fejes-Tóth G, Naray-Fejes-Tóth A, Mueller E. Serum-and glucocorticoid-inducible kinase 1 (SGK1) regulates adipocyte differentiation via forkhead box O1. Mol Endocrinol. 2010;24:370–380. doi: 10.1210/me.2009-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284:10334–10342. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes-Tóth G, Frindt G, Náray-Fejes-Tóth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol. 2008;294:F1298–F1305. doi: 10.1152/ajprenal.00579.2007. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, Zhang H, Ding Y, Bromberg JS. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant. 2004;4:1614–1627. doi: 10.1111/j.1600-6143.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Heikamp EB, Patel CH, Collins S, Waickman A, Oh MH, Sun IH, Illei P, Sharma A, Naray-Fejes-Toth A, Fejes-Toth G, et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol. 2014;15:457–464. doi: 10.1038/ni.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, Hafler DA. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125:4212–4222. doi: 10.1172/JCI81151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Horwitz DA, Zheng SG, Wang J, Gray JD. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp3+CD4+ Treg. Eur J Immunol. 2008;38:912–915. doi: 10.1002/eji.200738109. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwa-kura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: Mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprowicz DJ, Smallwood PS, Tyznik AJ, Ziegler SF. Scurfin (FoxP3) controls T-dependent immune responses in vivo through regulation of CD4+ T cell effector function. J Immunol. 2003;171:1216–1223. doi: 10.4049/jimmunol.171.3.1216. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäck-ström BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest. 2006;116:1473–1475. doi: 10.1172/JCI28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–5882. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- Loffing J, Zecevic M, Féraille E, Kaissling B, Asher C, Rossier BC, Fire-stone GL, Pearce D, Verrey F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: Possible role of SGK. Am J Physiol Renal Physiol. 2001;280:F675–F682. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- Loffing J, Flores SY, Staub O. Sgk kinases and their role in epithelial transport. Annu Rev Physiol. 2006;68:461–490. doi: 10.1146/annurev.physiol.68.040104.131654. [DOI] [PubMed] [Google Scholar]
- Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649–16654. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, Prinz I, Hemmer B, Kuchroo VK, Oukka M, Korn T. γδ T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33:351–363. doi: 10.1016/j.immuni.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. 2015;349:993–997. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang KB, Yang J, Pagán AJ, Li LX, Wang J, Green JM, Beg AA, Farrar MA. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol. 2010;184:4074–4077. doi: 10.4049/jimmunol.0903933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JB, Welling PA, Donowitz M, Shenolikar S, Weinman EJ. Differential renal distribution of NHERF isoforms and their colocalization with NHE3, ezrin, and ROMK. Am J Physiol Cell Physiol. 2001;280:C192–C198. doi: 10.1152/ajpcell.2001.280.1.C192. [DOI] [PubMed] [Google Scholar]
- Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C, Zhu C, Hirashima M, Anderson AC, Kuchroo VK. Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity. 2014;41:270–282. doi: 10.1016/j.immuni.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Fuss I, Strober W. Cutting edge: Regulatory T cells induce CD4+CD25-Foxp3-T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, Fohse L, Prinz I, Pezoldt J, Suerbaum S, et al. Foxp3 T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 2016;9:444–457. doi: 10.1038/mi.2015.74. [DOI] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–468. doi: 10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25−cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–2027. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.