

Abstract
An improved and enantioselective total synthesis of antibiotic CJ-16,264 through a practical kinetic resolution and an iodolactonization reaction to form the iodo pyrrolizidinone fragment of the molecule is described. A series of racemic and enantiopure analogues of CJ-16,264 was designed and synthesized through the developed synthetic technologies and tested against drug-resistant bacterial strains. These studies led to interesting structure–activity relationships and the identification of a number of simpler, and yet equipotent, or even more potent, antibacterial agents than the natural product, thereby setting the foundation for further investigations in the quest for new anti-infective drugs.
Graphical Abstract
1. INTRODUCTION
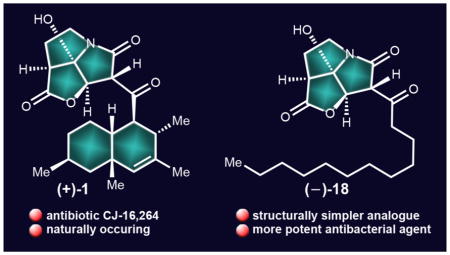
The continued proliferation of drug-resistant bacterial strains is currently viewed as one of the most challenging problems facing society. The search for new antibiotics, therefore, is considered as an urgent need and a priority for biomedical research. Naturally occurring antibiotics and their structural variations have been the major source of clinical agents to treat human infections caused by bacterial pathogens.1 The ability of pathogenic bacteria to evolve beyond our latest and most powerful antibacterial agents made the fight against them particularly challenging and, thus far, continuous.1 Antibiotic CJ-16,264 [(+)-1: revised structure;2 (+)-1a: originally assigned structure,3 Figure 1] is one of the latest antibacterial agents to be discovered from Nature.3 Isolated from fungus CL39457, antibiotic CJ-16,264 exhibits potent antibacterial properties (MIC = 0.78 μg mL−1 against Staphylococcus aureus 01A1120; MIC = 0.39 μg mL−1 against Moraxella catarrhalis 87A1055, and MIC = 6.25 μg mL−1 against Escherichia coli 51A1051 with altered permeability) and cytotoxic properties (IC90 = 8.0 μg mL−1 against HeLa cells).3 Other members of this class of antibiotics include CJ-16,367 (A),3 pyrrolizilactone (B),4 UCS-1025A (C),5 and UCS-1025B (D)5a,b (Figure 1). The first total synthesis of antibiotic CJ-16,264 has recently been reported from our laboratories,2 an accomplishment that also led to its structural revision. The chemical detective work that led to the identification of the correct structure of antibiotic CJ-16,264 [(+)-1] was accompanied by the synthesis of a number of isomers of the natural product [(−)-1, (−)-1b, (−)-1c, (+)-1c, and (+)-1d, Figure 2B]. In this article we report an improved synthesis of CJ-16,264 [(+)-1], which includes an enantioselective synthesis of the pyrrolizidine structural domain of the molecule (as opposed to its racemic form employed in our first synthesis).2 In addition, herein we describe the design, synthesis, and biological evaluation of an array of racemic analogues [i.e., (±)-2–(±)-6, 7, and (±)-8–(±)-12, Figure 2A] and a series of enantiopure analogues [i.e., (−)-13, (+)-7, (+)-8, and (−)-14–(+)-20, Figure 2B] for bioactivity comparison purposes. These investigations resulted in improvements of our original synthesis2 and enabled the identification of a number of equipotent, or even more potent, and yet significantly less complex than the natural product, antibacterial agents.
Figure 1.

Molecular structures of antibiotic CJ-16,264 [(+)-1], its originally assigned structure [(+)-1a], and other related pyrrolizidine natural products CJ-16,367 (A), pyrrolizilactone (B), UCS-1025A (C), and UCS-1025B (D).
Figure 2.

Synthesized analogues of antibiotic CJ-16,264 [(±)-2–(±)-6, 7, (±)-8–(±)-12, (−)-1, (−)-1b, (−)-1c, (+)-1c, (+)-1d, (−)-13–(−)-15, (+)-16, (+)-7, (+)-17, (−)-18, (+)-19, (+)-8, and (+)-20].
2. RESULTS AND DISCUSSION
Summarized in retrosynthetic format, and shown in Figure 3, our original total synthesis of antibiotic CJ-16,264 [(+)-1]2 utilized racemic iodo pyrrolizidinone fragment (±)-21 and enantiopure aldehyde (−)-22, the latter obtained from (S)-citronellal (25) via intermediates 23 and 24. Racemic pyrrolizidinone (±)-21 was prepared following the procedure reported by Danishefsky et al.5c and summarized in Figure 4A [26 + 27 → 28 → (±)-29 → (±)-21, 38% overall yield from 27]. Due to the racemic nature of fragment (±)-21, that synthesis suffered from the loss of half of the material at the coupling stage of the two advanced intermediates [i.e., (±)-21 and (−)-22, Figure 3], which ended up in another diastereoisomer of the natural product. In contemplating an enantioselective synthesis of the required pyrrolizidinone fragment (±)-21, we were cognizant of the elegant enantioselective approaches to its antipode [(−)-21] developed by the Danishefsky5c and Christmann groups5e and highlighted in Figures 4B [30+(+)-31 → (−)-29 → (−)-21, 9% overall yield from 31]5c and 4C [32 → (±)-29 → (−)-29 → (−)-21, < 13% overall yield from 32], respectively.5e Since neither of those approaches was suitable to the synthesis of the desired (+)-antipode of fragment 21, we sought to develop our own enantioselective synthesis of this key building block and then utilize it to achieve a completely enantioselective total synthesis of antibiotic CJ-16,264 [(+)-1].
Figure 3.

Previous strategy employed in the first total synthesis of CJ-16,264 [(+)-1] presented in retrosynthetic format; TBS = tert-butyldimethylsilyl.
Figure 4.

Previous approaches to the racemic iodo pyrrolizidinone core [(±)-21, panel A], enantiopure iodo pyrrolizidinone core [(−)-21, panel B] and [(−)-21, panel C].
2.1. Enantioselective Total Synthesis of Antibiotic CJ-16,264
In our efforts to develop a selective synthesis of the required enantiomer of iodo pyrrolizidinone (+)-21, we opted to employ the diastereofacially selective iodolactonization approach, which has been used previously with simple olefinic substrates and chiral auxiliaries.6 The challenge for our strategy (outlined in Figure 5) to synthesize (+)-21 enantioselectively and efficiently was based on the premise of finding a chiral auxiliary that would (a) achieve kinetic resolution during its attachment on our substrate [(±)-29 → 33, Figure 5] and (b) facilitate iodolactonization of the resulting derivative to the desired product [33 → 33a → 33b → (+)-21, Figure 5].
Figure 5.

Proposed enantioselective synthesis of iodo pyrrolizidinone (+)-21 based on diastereofacial iodolactonisation strategy.
Initial attempts to stereoselectively synthesize carboxylic acid 29 through chiral esters failed due to difficulties upon removal of the chiral auxiliaries. Thus, we opted to prepare chiral amide substrates that could undergo iodolactonization directly without further hydrolysis or cleavage of the auxiliaries. We, therefore, undertook a systematic study in order to test this hypothesis. The results are shown in Scheme 1. Carboxylic acid (±)-295c,e was reacted with commercially available sulfonamide 34a in the presence of PivCl, LiCl, and Et3N7 to afford a ca. 1:1 diastereomeric mixture of sulfonamide derivatives 35a and 36a, which were chromatographically separated. To our disappointment, however, neither 35a nor 36a underwent iodolactonization under a variety of conditions, including a number of different iodide reagents [e.g., I(sym-collidine)2ClO4, I2, KI/I2, ICl, NIS/Ag2CO3, N-iodosaccharin]. Reasoning that the electron-withdrawing effect of the sulfonamide group lessens the ability of the lone pair of electrons on the adjacent nitrogen to participate in the expected iodolactonization, we prepared the corresponding 2-oxo-1,3-oxazolidin-3-yl derivatives from carboxylic acid (±)-29 and oxazolidinone 34b.
Scheme 1. Enantioselective Iodolactonization of Amide Derivatives to Iodo Pyrrolizidinone (+)-21a.

aReagents and conditions: (a) (±)-29 (1.0 equiv), 34a (1.2 equiv), PivCl (1.05 equiv), LiCl (1.7 equiv), Et3N (2.5 equiv), CH2Cl2, −20 °C, 6 h, 35a+36a (82%, 1:1 dr); (b) (±)-29 (1.0 equiv), 34b (1.2 equiv), PivCl (1.05 equiv), LiCl (1.7 equiv), Et3N (2.5 equiv), CH2Cl2, −20 °C, 6 h, 35b+36b (87%, 1:1 dr); (c) (±)-29 (1.0 equiv), EDCI (1.5 equiv), HOAt (1.0 equiv), 34c (2.0 equiv), i-Pr2NEt (3.0 equiv), CH2Cl2, 0 → 25 °C, 16 h, 35c+36c (68%, 1:1 dr); (d) (±)-29 (1.0 equiv), EDCI (1.5 equiv), HOAt (1.0 equiv), 34d (1.0 equiv), i-Pr2NEt (3.0 equiv), CH2Cl2, 0 → 25 °C, 24 h, 35d (42%); (e) I(sym-collidine)2ClO4 (5.0 equiv), CH2Cl2:MeOH:H2O 1:1:0.05 (v/v/v), 25°C, 7 d, 3%, (98:2 er) from 35b; 72 h, 28% (24:76 er) from 35c+36c; 47%, 73% brsm (>99:1 er) from 35d. PivCl = pivaloyl chloride; EDCI = 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine, HOAt = 3-hydroxytriazolo[4,5-b]pyridine.
Reaction of (±)-29 with 34b under the influence of PivCl, LiCl, and Et3N furnished derivatives 35b and 36b as a 1:1 mixture of diastereoisomers, which were chromatographically separated and individually subjected to iodolactonization with various reagents and conditions as for 35a and 36a. It was after considerable experimentation that we found that isomer 35b, upon treatment with I(sym-collidine)2ClO4,8 led to trace amounts of iodolactonization product (+)-21 (3% yield, 98:2 er). The absolute configuration of this product was evident from its optical rotation and confirmed by HPLC analysis and comparison with (±)-21.5c Encouraged by this observation we proceeded to prepare the amide of carboxylic acid (±)-29 and (S)-prolinol (34c), expecting that the enhanced electron density on the oxygen of the amide (as compared to its sulfonamide and 2-oxo-1,3-oxazolidin-3-yl substrates that failed before it) would be sufficient to promote the coveted iodolactonization. Thus, treatment of a mixture of (±)-29 and 34c with EDCI, HOAt, and i-Pr2NEt led to an inseparable mixture (ca. 1:1) of diastereoisomeric amides 35c and 36c. The latter mixture was treated with I(sym-collidine)2ClO4, leading to the formation of the antipodal iodolactone (−)-21 (for structure see Figure 4B) [28% yield, 23:77 er, enriched in (−)-21], as determined by HPLC comparison with (±)-215c and (+)-21. Through additional experimentation, we discovered that reaction of (±)-29 with 1 equiv of (R,R)-dimethoxyamine (−)-34d9 in the presence of EDCI, HOAt, and i-Pr2NEt furnished (exclusively) only one of the two diastereoisomers of the corresponding amides (i.e., 35d, 42% yield). Pleasantly, iodolactonization of 35d under the influence of I(sym-collidine)2ClO4 furnished the coveted enantiomer (+)-21 in 47% yield (73% brsm) and >99:1 enantiomeric ratio as determined by HPLC comparison with (±)-21.
Further refinement of this process led to a practical enantioselective synthesis of iodo pyrrolizidinone (+)-21 as shown in Scheme 2. Thus, amide formation between excess of the readily available (±)-295c,e (1.0 equiv) and commercially available dimethoxyamine (−)-34d (0.6 equiv) in the presence of EDCI and HOAt resulted in kinetic resolution of the former, furnishing exclusively amide (+)-35d [45% yield based on (±)-29, 75% yield based on 34d; see Supporting Information for the crude 1H NMR spectra] and recovered unreacted enantiomerically enriched carboxylic acid (−)-29 [42% yield based on (±)-29]. The coveted iodo pyrrolizidinone (+)-21 was obtained in two steps from the readily available (±)-295c,e in 33% overall yield (and on gram scale). Furthermore, (−)-29 was converted to the chiral antipode of the iodo pyrrolizidinone building block [i.e., (−)-29; 91:9 er], as shown in Scheme 2, following a previously reported procedure.5c It should be noted (±)-29 is the less costly of the two starting materials used in this process.
Scheme 2. Enantioselective Synthesis of Iodo Pyrrolizidinone (+)-21a.

aReagents and conditions: (a) (±)-29 (1.0 equiv), EDCI (1.5 equiv), HOAt (1.0 equiv), (−)-34d (0.6 equiv), i-Pr2NEt (3.0 equiv), CH2Cl2, 0°C, 48 h, 75% based on 34d, 45% based on (±)-29; (b) I(sym-collidine)2ClO4 (5.0 equiv), CH2Cl2:MeOH:H2O 1:1:0.05 (v/v/v), 25 °C, 72 h, 47% (73% brsm); (c) I2 (3.0 equiv), NaHCO3 aq:Et2O 1:1 (v/v), 25 °C, 24 h, 74%.
With both fragments (+)-21 and (−)-22 in hand, the latter obtained from (S)-citronellal as previously described,2 their coupling proceeded well under the influence of BEt3 to afford alcohol (+)-37 as a single diastereoisomer and in 82% yield as shown in Scheme 3. Comparison of the spectral data and optical rotation of (+)-37 matched those previously obtained for the same compound,2 confirming its absolute stereochemistry, including that at C8′. The latter could be deduced from mechanistic considerations [boron-induced (Z)-enolate → syn-product]10 and additionally was unambiguously confirmed by X-ray crystallographic analysis during our original total synthesis of CJ-16,264.2 Following the same rationale, all synthesized coupling products with a hydroxy group at C8′ to be discussed below are assumed to have the same syn-configuration. Removal of the silyl group from (+)-37 required buffered TBAF conditions (AcOH) (in order to avoid rupture of the γ-lactone) and yielded dihydroxy compound (−)-14. Oxidation of the latter with IBX furnished, in 68% yield, CJ-16,264 [(+)-1] in its naturally occurring enantiomeric form. The same transformation [(−)-14 → (+)-1] could be achieved through the use of DMP (83% yield) as shown in Scheme 3. It should be noted that IBX11 was found to be more reliable in giving consistent yields of the oxidation product [i.e., (+)-1] as opposed to DMP, whose performance was dependent on its varying quality. Certain complications arising from the use of DMP in such oxidations that we encountered will be discussed below.
Scheme 3. Total Synthesis of CJ-16,264 [(+)-1]a.

aReagents and conditions: (a) (+)-21 (1.0 equiv), (−)-22 (2.0 equiv), BEt3 (1.1 equiv), toluene, −78 °C, 6 h, 82%; (b) TBAF (1.0 equiv), AcOH (1.05 equiv), THF, 0 °C, 30 min, 88%; (c) IBX (5.0 equiv), EtOAc, 70 °C, 9 h, 68%; (d) DMP (1.5 equiv), CH2Cl2, 0 → 25 °C, 2 h, 83%; BEt3 = triethyl borane, IBX = 2-iodoxybenzoic acid, DMP = Dess–Martin periodinane.
2.2. Design and Synthesis of Racemic CJ-16,264 Analogues
Prior to the development of our enantioselective synthesis of iodo pyrrozilidinone (+)-21, we undertook the design and synthesis of a series of racemic analogues [(±)-2–(±)-6, 7, and (±)-8–(±)-12, Figure 2A] by utilizing racemic iodo pyrrozilidinone (±)-215c in order to develop structure–activity relationships (SARs) within the CJ-16,264 structural type. Scheme 4 summarizes the synthesis of compounds (±)-2–(±)-7 from (±)-21 and a variety of rather simple and readily available aldehydes. Removal of the TBS group from (±)-21 (TASF, 35% yield) led to hydroxy iodo pyrrozilidinone (±)-2. Reaction of (±)-21 with acetaldehyde (38) under the influence of BEt3 resulted in the formation of alcohol 39 (83% yield) which, on sequential exposure to TASF (88% yield) and IBX (57% yield), furnished methyl keto analogue (±)-3 via dihydroxy intermediate 40. Following the same protocol, analogue (±)-4 was synthesized from (±)-21 and crotyl aldehyde (41), via 42 and 43, in comparable yields as shown in Scheme 4. Cyclohexyl [(±)-5] and phenyl [(±)-6] CJ-16,264 analogues were similarly, and in similar yields, prepared from (±)-21 employing cyclohexyl carboxaldehyde (44) and benzaldehyde (47), respectively, as depicted in Scheme 4, with the only difference being the use of DMP for the final oxidation step. Analogue 7 (ca. 1:1 dr) was synthesized from (±)-21 and aldehyde 50 (racemic mixture) through the use of BEt3 as coupling inducer (81% yield) and DMP as the oxidant (63% yield), as shown in Scheme 4.
Scheme 4. Synthesis of CJ-16,264 Analogues (±)-2–7a.

aReagents and conditions: (a) TASF (1.8 equiv), THF, 0 °C, 5 min, 35%; (b) (±)-21 (1.0 equiv), 38 (5.0 equiv), BEt3 (1.0 equiv), toluene, −78 °C, 6 h, 83%; (c) TASF (1.5 equiv), THF, 0 °C, 5 min, 88%; (d) IBX (5.0 equiv), EtOAc, 70 °C, 6 h, 57%; (e) (±)-21 (1.0 equiv), 41 (3.0 equiv), BEt3 (1.1 equiv), toluene, −78 °C, 6 h, 72%; (f) TBAF (1.0 equiv), THF, 0 °C, 5 min, 90%; (g) IBX (6.0 equiv), EtOAc, 70 °C, 6 h, 54%; (h) (±)-21 (1.0 equiv), 44 (5.0 equiv), BEt3 (1.0 equiv), toluene, −78 °C, 6 h, quant; (i) TASF (1.5 equiv), THF, 0 °C, 5 min, 66%; (j) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 63%; (k) (±)-21 (1.0 equiv), 47 (3.0 equiv), BEt3 (1.1 equiv), toluene, −78 °C, 6 h, quant; (l) TASF (1.5 equiv), THF, 0 °C, 5 min, 56%; (m) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 61%; (n) (±)-21 (1.0 equiv), 50 (3.0 equiv), BEt3 (1.0 equiv), toluene, −78 °C, 6 h, 81% (1:1 dr); (o) TASF (1.5 equiv), THF, 0 °C, 5 min, 68% (1:1 dr); (p) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 63% (1:1 dr); TASF = tris(dimethylamino)sulfonium difluorotrimethylsilicate.
Analogue (±)-8 (Scheme 5A) was designed as an isomer of antibiotic CJ-16,264, containing in its structure a much simpler lipophilic domain (C14H23) and without chiral centers as a replacement of the more complex decalin domain (also C14H23) of the natural product. Its synthesis is summarized in Scheme 5. Embedded within the molecule of farnesal (53), this side chain was introduced into the desired structure through the standard three-step sequence from (±)-21 and 53 {BEt3-induced coupling (54, 84% yield); TBAF promoted desilylation (55, 72% yield); and DMP oxidation [(±)-8, 24% yield]}. Interestingly, the last reaction (i.e., DMP oxidation of 55) yielded, in addition to the desired product (±)-8, chloride (±)-9 (11% yield) as a byproduct. The tentative assignment of the designated configuration for the chloride residue in the latter compound was based on steric hindrance considerations. One speculative mechanism for this rare reaction is shown in Scheme 5B, whereby a chloride anion (presumably from trace amounts of HCl present in the CH2Cl2 used as a solvent in this reaction) generates chloro DMP derivative II from DMP (with loss of one acetate). The latter species may then suffer attack from the enol form III of product (±)-8, either at the chlorine center (pathway A, red) with loss of one acetate to form product (±)-9 and iodine(III) species V or at the iodine residue (pathway B, green) with loss of chloride to afford species IV. The latter iodine(V) species can then undergo attack from the generated chloride (with concomitant loss of one acetate) to furnish the observed product [i.e., chloride (±)-9] and iodine(III) species V. A third pathway (pathway C, blue) includes the attack of DMP (I) by enol III causing the displacement of one acetate group and formation of intermediate VII. The latter can then suffer allylic attack from acetoxyiodinane oxide VI (see caption of Scheme 5) at C7′, leading to adduct VIII and acetoxybenziodoxolone V. Finally, product (±)-9 can be generated through a nucleophilic attack of a chloride anion at C7′ with concomitant release of VI. This hypothesis is supported by previously reported studies on DMP-mediated oxidations.12
Scheme 5. Synthesis of CJ-16,264 Analogues (±)-8 and (±)-9 (A) and Tentative Mechanism for the Formation of (±)-9 (B)a.

aReagents and conditions: (a) (±)-21 (1.0 equiv), 53 (3.0 equiv), BEt3 (1.0 equiv), toluene, −78 °C, 6 h, 84%; (b) TBAF (1.0 equiv), THF, 0 °C, 5 min, 72%; (c) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, (±)-8 (24%), (±)-9 (11%); (d) IBX (5.0 equiv), EtOAc, 70 °C, 5 h, 58%; compound VI is a common contaminant of Dess–Martin reagent, either arising from incomplete acetylation of IBX during its preparation or due to partial hydrolysis of DMP; TBAF = tetra-n-butylammonium fluoride.
In order to test the effect of a basic nitrogen on the antibacterial properties of the molecule, we designed morpholine analogue (±)-10. Scheme 6 summarizes the synthesis of this compound starting from allylic alcohol (±)-42 (see Scheme 4 for preparation) and olefinic carboxylic acid 57. Thus, 57 was converted to its morpholine amide [58, (COCl)2, Et3N, morpholine, 75% yield] and thence to olefinic morpholine derivative 5913 through reduction (LiAlH4, 85 °C, 80% yield). Cross metathesis between (±)-42 and 59 was effected with Grubbs II cat. 56, affording product 60 (48% yield, 86% based on recovered starting material). Removal of the silyl group (TBAF, 61, 62% yield) followed by IBX oxidation led to desired analogue (±)-10 (24% yield) as shown in Scheme 6.
Scheme 6. Synthesis of CJ-16,264 Analogue (±)-10a.

aReagents and conditions: (a) (±)-42 (1.0 equiv), 59 (6.0 equiv), Grubbs II catalyst 56 (0.10 equiv), CH2Cl2, 40 °C, 24 h, 48%, 86% brsm; (b) TBAF (1.0 equiv), THF, 0 °C, 5 min, 62%; (c) IBX (5.0 equiv), EtOAc, 70 °C, 2 h, 24%; d) 57 (1.0 equiv), (COCl)2 (2.0 equiv), CH2Cl2, 0 → 25 °C, 3h; Et3N (3.0 equiv), morpholine (2.0 equiv), CH2Cl2, 0 → 25 °C, 9 h, 75%; (e) LiAlH4 (2.0 equiv), THF, 85°C, 12 h, 80%.
Next, we designed macrocyclic CJ-16,264 analogue (±)-11 (Scheme 7) with a lipophilic chain stretching between the carbonyl and hydroxy groups attached onto the pyrrozilidinone core of the molecule. Its synthesis is shown in Scheme 7. Cross metathesis between pyrrozilidinone intermediate (±)-42 (see Scheme 4 for its synthesis) and olefinic silyl ester 62 (see Supporting Information for preparation) as promoted by Grubbs II catalyst 56 furnished olefinic TBS-ether carboxylic acid 63 possessing the (E)-geometry (20:1, by 1H NMR spectroscopic analysis) and in good yield (63% yield). Desilylation of the latter (TBAF, AcOH) then furnished hydroxy acid 64 in 82% yield. Shiina macrolactonization (MNBA, DMAP)14 of seco acid 64 gave macrolactone 65 in 84% yield. Given the presence of the secondary allylic alcohol in the lactonization precursor 64, this result is impressive. An explanation for this observation may be the strained nature of the 13-membered ring expected from the latter mode of cyclization as opposed to the 16-membered ring formed, the latter being one of the less strained and favored macrocycles in such macrolactonizations.15 Furthermore, the allylic alcohol may be sufficiently deactivated by hydrogen bonding to the lactam carbonyl oxygen so as to render it noncompetitive with the tertiary hydroxyl group in this reaction. Finally, oxidation of 65 with IBX provided macrocyclic analogue (±)-11, which existed in CDCl3 solution as a mixture of its keto and enol [(±)-11a] tautomers (ca. 60:40 by 1H NMR spectroscopic analysis, 600 MHz, CDCl3).
Scheme 7. Synthesis of CJ-16,264 Analogue (±)-11a.

aReagents and conditions: (a) 62 (3.0 equiv), Grubbs II catalyst 56 (0.2 equiv), CH2Cl2, 25 °C, 24 h, 63%; (b) TBAF (1.0 equiv), AcOH (2.0 equiv), THF, 0 °C, 5 min, 82%; (c) MNBA (1.4 equiv), DMAP (3.0 equiv), CH2Cl2, 25 °C, 12 h, 84%; (d) IBX (5.0 equiv), EtOAc, 70°C, 7 h, 48%; MNBA = 2-methyl-6-nitrobenzoic anhydride, DMAP = N,N-dimethylpyridin-4-amine.
CJ-16,264 analogue (±)-12 was designed to test the effect of shifting the lipophilic domain of the molecule from the original side chain position to the tertiary alcohol site of the pyrrozilidinone domain on its biological properties. Its synthesis (Scheme 8) required selective esterification of diol 40 (for preparation see Scheme 4) with farnesoic acid (66),16 a task accomplished under Yamaguchi conditions (2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, 68), albeit in modest yield (39%), presumably due to interference from the allylic hydroxy group and/or other side reactions. Oxidation of the resulting product (68) with IBX then furnished targeted analogue (±)-12 (47% yield). This compound existed in equilibrium with its enol form (±)-12a (as shown in Scheme 8) as revealed by 1H NMR spectroscopic analysis [(±)-12:(±)-12a ca. 45:55, 600 MHz, CDCl3].
Scheme 8. Synthesis of CJ-16,264 Analogue (±)-12a.

aReagents and conditions: (a) 40 (1.0 equiv), 66 (1.0 equiv), 67 (1.0 equiv), Et3N (3.0 equiv), DMAP (2.0 equiv), CH2Cl2, 0 °C, 3 h, 39%; (b) IBX (5.0 equiv), EtOAc, 70 °C, 6 h, 47%.
2.3. Design and Synthesis of Enantiopure CJ-16,264 Analogues
As mentioned above, our total synthesis of antibiotic CJ-16,264 [(+)-1] endeavor led to, in addition to the revised and true structure of the natural product, a number of its enantiopure isomers [i.e., (−)-1, (−)-1b, (−)-1c, (+)-1c, and (+)-1d, Figure 2] for biological evaluation.
In order to test the importance of enolization of the 1,3-dicarbonyl system present in the CJ-16,264 molecule, we decided to replace the C-7 hydrogen with a hydroxy moiety as a means to block this enolization. Such a compound was previously reported as an isolation product5a and synthesized,5b but it is not clear whether this compound is a natural product or an artifact. Inspired by observations of hydroxy byproducts in the oxidation of the hydroxy precursor of (+)-1 (and other similar substrates) with DMP, we explored the use of this reagent as a means to synthesize the targeted C7′ hydroxy CJ-16,264 analogue (−)-13. Thus, and as shown in Scheme 9, we found that treatment of the hydroxy TBS-ether derivative 692 with DMP in CH2Cl2 at 0 °C led to the formation of TBS-ether hydroxy ketone 70 in 61% yield (ca. 1:1 dr), from which the C7′-hydroxylated analogue (−)-13 was generated through TASF induced desilylation (33% yield). Interestingly, the latter compound was obtained as a single diastereoisomer, presumably the other undergoing decomposition to other unidentified product(s). The susceptibility of substrate 692 toward C7′ hydroxylation in the presence of DMP stands in contrast to the resistance of its desilylated counterpart [i.e., (−)-14, Scheme 3] to undergo this step under the same conditions, presumably due to the negative effect of the hydrogen bonding of the free tertiary hydroxy group with the exocyclic carbonyl moiety within the latter.
Scheme 9. Synthesis of CJ-16,264 Analogue (−)-13a.

aReagents and conditions: (a) DMP (3.0 equiv), CH2Cl2, 0 → 25 °C, 45 min, 61% (ca. 1:1 dr); (b) TASF (10 equiv), THF, 0 °C, 5 min, 33%.
The development of our enantioselective route to iodo pyrrozilidinone (+)-21 opened the possibility of synthesizing simpler designed analogues of antibiotic CJ-16,264 in their enantiopure form. To this end, and as shown in Scheme 10, we reacted (+)-21 with readily available aldehydes 71,17 72, and 73 (the latter two being commercially available) in the presence of BEt3 to obtain coupling products 74 (66% yield), 76 (81% yield), and 78 (70% yield), respectively. The TBS group was removed from these products with TBAF to afford diols 75 (67% yield), 77 (64% yield), and 79 (77% yield), respectively. Exposure of the latter compounds to DMP provided the targeted analogues (−)-15 (55% yield), (+)-16 (60% yield), and (+)-7 (60% yield), respectively.
Scheme 10. Synthesis of CJ-16,264 Analogues (−)-15, (+)-17, and (+)-7a.

aReagents and conditions: (a) (+)-21 (1.0 equiv), 71 (3.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 66%; (b) TBAF (1.0 equiv), THF, 0°C, 5 min, 67%; (c) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 55%; (d) (+)-21 (1.0 equiv), 72 (3.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 81%; (e) TBAF (1.0 equiv), THF, 0 °C, 5 min, 64%; (f) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 60%; (g) (+)-21 (1.0 equiv), 73 (3.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 70%; (h) TBAF (1.0 equiv), THF, 0 °C, 5 min, 77%; (i) DMP (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 60%.
Analogue (+)-17 carrying methyl groups at C1, C4, and C8 was synthesized from fragment (+)-21 and aldehyde 84 as summarized in Scheme 11. The latter was obtained from known iodide 8018 through a stereoselective process involving ephedrine propionate 81.19 Thus, alkylation of 81 (LDA, LiCl) with citronellyl iodide (80)18 furnished amide 82 (85% yield), which was reductively cleaved with H3N·BH3 in the presence of LDA to afford primary alcohol 83 in excellent diastereoselectivity (72% yield). DMP oxidation of the latter furnished aldehyde 84 (82% yield). Coupling of 84 with (+)-21 under the standard BEt3 conditions gave alcohol 85. Desilylation of the latter (TBAF, 85% yield) then led to precursor diol 86, whose oxidation with DMP resulted in the desired analogue (+)-17 (61% yield).
Scheme 11. Synthesis of CJ-16,264 Analogue (+)-17a.

aReagents and conditions: (a) 81 (2.1 equiv), 80 (1.0 equiv), LDA (4.0 equiv), LiCl (12.7 equiv), THF, 0 °C, 24 h, 85%; (b) LDA (3.9 equiv), H3N·BH3 (4.0 equiv), THF, 0 → 25 °C, 6 h, 72%; (c) DMP (1.5 equiv), CH2Cl2, 0 → 25 °C, 1 h, 82%; (d) (+)-21 (1.0 equiv), 84 (3.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 63%; (e) TBAF (1.3 equiv), THF, 0 °C, 5 min, 85%; (f) DMP (1.5 equiv), CH2Cl2, 0 °C, 2 h, 61%; LDA = lithium diisopropylazanide.
CJ-16,264 analogue (+)-18 possessing a straight C12 aliphatic chain was synthesized as summarized in Scheme 12. Iodo pyrrolizidinone (+)-21 was coupled with commercially available aldehyde 87 in the presence of BEt3 to afford product 88 (83% yield), whose TBS group was cleaved with TBAF/ AcOH to give dihydroxy intermediate 89 (87% yield). On exposure to DMP, 89 gave analogue (−)-18 (46% yield) plus its hydroxylated counterpart analogue (+)-19 (11% yield). A cleaner and more efficient conversion of dihydroxy compound 89 to analogue (−)-18 (83% yield) was achieved through the use of IBX as shown in Scheme 12. Interestingly, use of excess DMP (6.0 equiv) resulted in the formation of hydroxylated product (+)-19 in 58% yield.
Scheme 12. Synthesis of CJ-16,264 Analogue (+)-18 and (+)-19a.

aReagents and conditions: (a) (+)-21 (1.0 equiv), 87 (3.0 equiv), BEt3 (1.0 equiv), toluene, −78 °C, 6 h, 83%; (b) TBAF (1.1 equiv), AcOH (1.1 equiv), THF, 0 °C, 15 min, 87%; (c) DMP (2.0 equiv), CH2Cl2, 0 → 25 °C, 3 h, (−)-18 (46%), (+)-19 (11%, one C7′ epimer); (d) IBX (5.0 equiv), EtOAc, 70 °C, 7 h, 83%; (e) DMP (6.0 equiv), CH2Cl2, 25°C, 6 h, 58%.
Scheme 13 depicts the synthesis of analogues (+)-8 and (+)-20 from iodo pyrrozilidinone (+)-21 and readily available aldehydes 5320 and 9021 through intermediates 91 and 92 [for (+)-8], and 93 and 94 [for (+)-20], respectively, in good overall yields though the standard reagents and conditions as shown in Scheme 13.
Scheme 13. Synthesis of CJ-16,264 Analogues (+)-8 and (+)-20a.

aReagents and conditions: (a) (+)-21 (1.0 equiv), 53 (2.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 77%; (b) TBAF (1.0 equiv), THF, 0 °C, 5 min, 66%; (c) MnO2 (40 equiv), CH2Cl2, 25 °C, 10 h, 39%, 72% brsm; (d) (+)-21 (1.0 equiv), 90 (2.0 equiv), BEt3 (1.3 equiv), toluene, −78 °C, 6 h, 69%; (e) TBAF (1.0 equiv), THF, 0 °C, 5 min, 67%; (f) MnO2 (40 equiv), CH2Cl2, 25 °C, 10 h, 44%, 66% brsm.
2.4. Biological Evaluation of Synthesized Compounds
The synthesized CJ-16,264 analogues (±)-2–(±)-6, 7, (±)-8–(±)-12, (−)-1, (−)-1b, (−)-1c, (+)-1c, (+)-1d, (−)-13–(−)-15, (+)-16, (+)-7, (+)-17, (−)-18, (+)-19, (+)-8, and (+)-20 (Figure 2) together with synthetic CJ-16,264 [(+)-1], minocycline (Minomycin), and tigecycline (Tygacil) were tested against the Gram-positive drug-resistant strains Bacillus subtilis 168, Enterococcus faecalis S613, Enterococcus faecium 105, and methicillin-resistant Staphylococcus aureus 371. The results are shown in Table 1. As seen in this table, a number of these newly synthesized compounds, including the enantiomer and diastereoisomers of the natural product [(+)-1], compare well with minocycline and tigecycline (see highlighted entries in Table 1). Thus, synthetic (+)-1 and (+)-1d show similar potencies against all four bacterial strains [(+)-1: MIC = 0.5 μg/mL (B. subtilis 168); 8 μg/mL (E. faecalis S613); 2 μg/mL (E. faecium 105); 1 μg/mL (MRSA 371); (+)-1d: MIC = 1 μg/ mL (B. subtilis 168); 8 μg/mL (E. faecalis S613); 4 μg/mL (E. faecium 105); 2 μg/mL (MRSA 371)], while racemic analogue (±)-8 (whose lipophilic domain carries the same number of carbon and hydrogen atoms as that of CJ-16,264) exhibited approximately the same potency as its enantiomerically pure counterpart [(±)-8: MIC = 0.5 μg/mL (B. subtilis 168); 2 μg/ mL (E. faecalis S613); 4 μg/mL (E. faecium 105); 1 μg/mL (MRSA 371); (+)-8: MIC = 1 μg/mL (B. subtilis 168); 4 μg/ mL (E. faecalis S613); 4 μg/mL (E. faecium 105); 2 μg/mL (MRSA 371)]. These results indicate that the pyrrolizidinone domain of the molecule carries its pharmacophore, while its decalin system is simply a lipophilic domain that may facilitate its delivery to the active site of its biological target. The validity of this hypothesis was supported further by the high potencies of analogues (+)-17 [MIC = 0.5 μg/mL (B. subtilis 168); 1 μg/ mL (E. faecalis S613); 2 μg/mL (E. faecium 105); 1 μg/mL (MRSA 371)], (−)-18 [MIC = 0.25 μg/mL (B. subtilis 168); 1 μg/mL (E. faecalis S613); 1 μg/mL (E. faecium 105); 1 μg/mL (MRSA 371)] and (+)-20 [MIC = 0.5 μg/mL (B. subtilis 168); 4 μg/mL (E. faecalis S613); 1 μg/mL (E. faecium 105); 1 μg/ mL (MRSA 371)], which incorporate in their structures simple straight chain lipophilic residues of comparable carbon content to the natural product’s lipophilic domain.
Table 1.
MIC Data of Compounds Against Gram-Positive Bacteria and Comparison with Standard Antibiotics
| Compound | Gram-(+)a | |||
|---|---|---|---|---|
|
| ||||
| B. subtilis 168 | E. faecalis S613 | E. faecium 105 | MRSA371 | |
| minocycline | <0.125 | 8 | 8 | 1 |
| tigecycline | — | 0.5 | 0.5 | 1 |
|
| ||||
| (+)-1 | 0.5 | 8 | 2 | 1 |
|
| ||||
| (±)-2 | >64 | >64 | >64 | >64 |
| (±)-3 | >64 | >64 | >64 | >64 |
| (±)-4 | >64 | >64 | >64 | >64 |
| (±)-5 | 16 | 32 | 32 | 8 |
| (±)-6 | 16 | 32 | 64 | 16 |
| 7 | 2 | 16 | 16 | 4 |
|
| ||||
| (±)-8 | 0.5 | 2 | 4 | 1 |
|
| ||||
| (±)-9 | 2 | 64 | 8 | 8 |
| (±)-10 | >64 | >64 | >64 | >64 |
| (±)-11 | >64 | >64 | >64 | >64 |
| (±)-12 | >64 | >64 | >64 | >64 |
| (−)-1 | 2 | 16 | 4 | 4 |
| (−)-1b | 1 | 16 | 4 | 4 |
| (−)-1c | 1 | 16 | 4 | 2 |
| (+)-1c | 1 | 16 | 4 | 2 |
|
| ||||
| (+)-1d | 1 | 8 | 4 | 2 |
|
| ||||
| (−)-13 | >64 | >64 | >64 | >64 |
| (−)-14 | >64 | >64 | >64 | >64 |
| (−)-15 | 32 | 32 | 32 | 32 |
| (+)-16 | 32 | 32 | 32 | 32 |
| (+)-7 | 2 | 8 | 8 | 8 |
|
| ||||
| (+)-17 | 0.5 | 1 | 2 | 1 |
|
| ||||
| (−)-18 | 0.25 | 1 | 1 | 1 |
|
| ||||
| (+)-19 | 32 | 32 | 32 | >64 |
|
| ||||
| (+)-8 | 1 | 4 | 4 | 2 |
|
| ||||
| (+)-20 | 0.5 | 4 | 1 | 1 |
MIC assays were run in triplicate; data are given in units of μg/mL.
Interestingly, the natural product [(+)-1] and its equipotent diastereoisomer (+)-1d include in their structure the same enantiomeric structural motif of the pyrrolizidinone domain (i.e., its presumed pharmacophore) but the opposite enantiomeric form of the decalin system (i.e., lipophilic domain), providing additional support for the roles of the two domains of the molecule as the pharmacophore and navigational facilitator, respectively.
Considering the lack of activity for iodo pyrrolizidinone (±)-2 and the low potencies of analogues (±)-3–(±)-6 in which the side chain contains no carbonyl functionality, or includes relatively short lipophilic chains or rings on the carbonyl moiety, it is reasonable to conclude that both the exocyclic carbonyl group and a relatively highly lipophilic moiety are essential for high potency. It is also suggested from the results with the tertiary amine containing analogue (±)-10 that a basic nitrogen within the side chain deactivates the molecule toward the bacterial strains tested and so does the blocking of the tertiary hydroxyl group on the pyrrolizidinone moiety as in analogues (±)-11 and (±)-12.
Replacing the C7′ hydrogen with another substituent [i.e., OH or Cl as in analogues (±)-9, (−)-13 and (+)-19; see Figure 2 and Table 1] also prevents the molecule from exerting its antibacterial action,5a,b suggesting the involvement of an enolic intermediate in the mechanism of action of these molecules and pointing to the importance of their 1,3-dicarbonyl system for biological activity. It is also interesting to note that, as revealed by these data, the isomeric forms of CJ-16,264 and its analogues exhibit comparable antibacterial properties [cf. (+)-1 with (−)-1, (−)-1b, (−)-1c, (+)-1c, and (+)-1d; see Table 1].
3. CONCLUSION
An efficient, enantioselective, and practical total synthesis of antibiotic CJ-16,264 [(+)-1] has been achieved. The synthetic technologies developed were applied to the synthesis of an array of racemic and a series of enantiopure analogues of antibiotic CJ-16,264, biological evaluation of which revealed interesting SARs and identified a number of highly potent and yet structurally simpler analogues as compared to the natural product. From the latter, the most impressive were (±)-8 and (+)-8, (+)-17, (−)-18, and (+)-20. Due to their relative structural simplicities and high potencies, the latter deserve further optimization and pharmacological investigation as potential antibacterial agents. The present synthetic studies also led to a number of interesting chemical reactions and revealed useful SARs inherent in the pyrrolizidinone–exocyclic carbonyl system and the two main domains of antibiotic CJ-16,264. They also uncovered the unusual DMP-induced chlorination and hydroxylation reactions at the C7′ position of the molecule and the loss of antibacterial activity upon substitution of the C7′-H or the H of the tertiary hydroxy group. These investigations also led to the recognition that the elaborate lipophilic domain of the molecule does not necessarily need its polycyclic framework or stereochemical intricacies for biological activity. Rather a simple aliphatic chain of similar lipophilicity suffices for its biological action and potency. These phenomena may be linked to the mechanism of action of these novel antibiotics.22
Supplementary Material
Acknowledgments
We thank Drs. Lawrence B. Alemany and Quinn Kleerekoper (Rice University) for NMR-spectroscopic assistance and Drs. Christopher L. Pennington (Rice University) and Ian Riddington (University of Texas at Austin) for mass-spectrometric assistance. This work was supported by the National Institutes of Health (USA) (grant AI055475) and The Welch Foundation (grant C-1819).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08749.
Experimental procedures and characterization data for all compounds. MIC data against B. subtilis 168, E. faecalis S613, E. faecium 105, and MRSA 371 (PDF)
References
- 1.Nicolaou KC, Rigol S. J Antibiot. 2018 doi: 10.1038/ja.2017.62. [DOI] [PubMed] [Google Scholar]
- 2.Nicolaou KC, Shah AA, Korman H, Khan T, Shi L, Worawalai W, Theodorakis EA. Angew Chem, Int Ed. 2015;54:9203–9208. doi: 10.1002/anie.201504337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugie Y, Hirai H, Kachi-Tonai H, Kim YJ, Kojima Y, Shiomi Y, Sugiura A, Sugiura A, Suzuki Y, Yoshikawa N, Brennan L, Duignan J, Huang LH, Sutcliffe J, Kojima N. J Antibiot. 2001;54:917–925. doi: 10.7164/antibiotics.54.917. [DOI] [PubMed] [Google Scholar]
- 4.Nogawa T, Kawatani M, Uramoto M, Okano A, Aono H, Futamura Y, Koshino H, Takahashi S, Osada H. J Antibiot. 2013;66:621–623. doi: 10.1038/ja.2013.55. [DOI] [PubMed] [Google Scholar]
- 5.(a) Nakai R, Ogawa H, Asai A, Ando K, Agatsuma T, Matsumiya S, Akinaga S, Yamashita Y, Mizukami T. J Antibiot. 2000;53:294–296. doi: 10.7164/antibiotics.53.294. [DOI] [PubMed] [Google Scholar]; (b) Agatsuma T, Akama T, Nara S, Matsumiya S, Nakai R, Ogawa H, Otaki S, Ikeda S-i, Saitoh Y, Kanda Y. Org Lett. 2002;4:4387–4390. doi: 10.1021/ol026923b. [DOI] [PubMed] [Google Scholar]; (c) Lambert TH, Danishefsky SJ. J Am Chem Soc. 2006;128:426–427. doi: 10.1021/ja0574567. [DOI] [PubMed] [Google Scholar]; (d) Hoye TR, Dvornikovs V. J Am Chem Soc. 2006;128:2550–2551. doi: 10.1021/ja0581999. [DOI] [PubMed] [Google Scholar]; (e) de Figueiredo RM, Fröhlich R, Christmann M. Angew Chem, Int Ed. 2007;46:2883–2886. doi: 10.1002/anie.200605035. [DOI] [PubMed] [Google Scholar]; (f) Sizova EP. PhD Dissertation. University of Minnesota; Minneapolis, MN: 2009. Second Generation Synthesis of UCS1025A. Synthetic Efforts Toward Total Syntheses of CJ-16,264 and Phomopsichalasin. [Google Scholar]; (g) Uchida K, Ogawa T, Yasuda Y, Mimura H, Fujimoto T, Fukuyama T, Wakimoto T, Asakawa T, Hamashima Y, Kan T. Angew Chem, Int Ed. 2012;51:12850–12853. doi: 10.1002/anie.201207800. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kaoru F, Manabu N, Yoshimitsu N, Takeo K. Tetrahedron Lett. 1990;31:3175–3178. [Google Scholar]; (b) Yokomatsu T, Iwasawa H, Shibuya S. J Chem Soc, Chem Commun. 1992:728–729. [Google Scholar]; (c) Kitagawa O, Hanano T, Kikuchi N, Taguchi T. Tetrahedron Lett. 1993;34:2165–2168. [Google Scholar]; (d) Moon H, Eisenberg SWE, Wilson ME, Schore NE, Kurth MJ. J Org Chem. 1994;59:6504–6505. [Google Scholar]; (e) Nolsøe JMJ, Hansen TV. Eur J Org Chem. 2014;2014:3051–3065. [Google Scholar]
- 7.Chakraborty TK, Suresh VR. Tetrahedron Lett. 1998;39:7775–7778. [Google Scholar]
- 8.Lemieux RU, Morgan AR. Can J Chem. 1965;43:2190–2197. [Google Scholar]
- 9.Yamamoto Y, Hoshino Ji, Fujimoto Y, Ohmoto J, Sawada S. Synthesis. 1993;1993:298–302. [Google Scholar]
- 10.Mahrwald R. Aldol Reactions. Springer Netherlands; Dordrecht: 2009. pp. 23–38. [Google Scholar]
- 11.Bartlett SL, Beaudry CM. J Org Chem. 2011;76:9852–9855. doi: 10.1021/jo201810c. [DOI] [PubMed] [Google Scholar]
- 12.Meyer SD, Schreiber SL. J Org Chem. 1994;59:7549–7552. [Google Scholar]
- 13.Coan SB, Papa D. J Am Chem Soc. 1955;77:2402–2404. [Google Scholar]
- 14.(a) Shiina I, Kubota M, Oshiumi H, Hashizume M. J Org Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]; (b) Shiina I. Bull Chem Soc Jpn. 2014;87:196–233. [Google Scholar]
- 15.(a) Corey EJ, Nicolaou KC. J Am Chem Soc. 1974;96:5614–5616. [Google Scholar]; (b) Nicolaou KC. Tetrahedron. 1977;33:683–710. [Google Scholar]
- 16.Kim S, Kim E, Shin D-S, Kang H, Oh K-B. Bioorg Med Chem Lett. 2002;12:895–898. doi: 10.1016/s0960-894x(02)00038-0. [DOI] [PubMed] [Google Scholar]
- 17.(a) Nicolaou KC, Reingruber R, Sarlah D, Bräse S. J Am Chem Soc. 2009;131:2086–2087. doi: 10.1021/ja809405c. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Reingruber R, Sarlah D, Bräse S. J Am Chem Soc. 2009;131:6640. doi: 10.1021/ja809405c. [DOI] [PubMed] [Google Scholar]
- 18.Basar N, Damodaran K, Liu H, Morris GA, Sirat HM, Thomas EJ, Curran DP. J Org Chem. 2014;79:7477–7490. doi: 10.1021/jo5012027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496–6511. [Google Scholar]
- 20.Ishihara K, Ishibashi H, Yamamoto H. J Am Chem Soc. 2002;124:3647–3655. doi: 10.1021/ja0124865. [DOI] [PubMed] [Google Scholar]
- 21.Pattenden G, Smithies AJ. J Chem Soc, Perkin Trans 1. 1996:57–61. [Google Scholar]
- 22.Beabout K, McCurry MD, Mehta H, Shah AA, Pulukuri KK, Rigol S, Wang Y, Nicolaou KC, Shamoo Y. ACS Infect Dis. 2017 doi: 10.1021/acsinfecdis.7b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.