

SUMMARY
T cells coordinate multiple aspects of adaptive immunity throughout life, including responses to pathogens, allergens, and tumors. In mouse models, the role of T cells is studied in the context of a specific type of pathogen, antigen, or disease condition over a limited timeframe, while in humans, T cells control multiple insults simultaneously throughout the body and maintain immune homeostasis over decades. In this review, we discuss how human T cells develop and provide essential immune protection at different life stages and highlight that tissue localization and subset delineation are key determinants of the T cell functional role in immune responses. We also discuss how anatomic compartments undergo distinct age-associated changes in T cell subset composition and function over a lifetime. Considering age and tissue influences on human T cells is important for developing targeted strategies to modulate T cell-mediated immunity in vaccines and immunotherapies.
Keywords: Human Immunology, memory T cells
ETOC BLURB
Recent studies on human T cells in diverse tissue sites have revealed that the functional role of T cells is closely linked to the anatomical location, subset and developmental stage. Kumar et al. review these advances and highlight aspects of T cell immunity that are specific to humans.
Introduction
The establishment and maintenance of immune responses, homeostasis, and memory depends on T cells. T cells express a receptor with the potential to recognize diverse antigens from pathogens, tumors, and the environment, and also maintain immunological memory and self-tolerance. T cells are also implicated as major drivers of many inflammatory and autoimmune diseases. The in vivo functional role of T cells in immunity and immunopathology and the underlying mechanisms involved have been largely elucidated from mouse models, and have led to the development and advancement of immune-based cures and immunotherapies in humans (Cohen, 2014; Rosenberg, 2014). However, the power and utility of mouse models to test hypotheses depends on reducing the scope of inquiry to one type of infection or disease perturbation over a defined time period in sterile, pathogen-free conditions. By contrast, humans are continuously exposed to multiple benign and pathogenic microorganisms, harbor chronic pathogens, yet can survive for many decades free of major infections even in advanced years (Evans et al., 2014). In order to elucidate mechanisms for the unique longevity and stability of human immunity, it is necessary to study T cells within the complex environment of the human body—in multiple sites, at all ages, and across many individuals.
T lymphocytes originate from bone marrow progenitors that migrate to the thymus for maturation, selection, and subsequent export to the periphery. Peripheral T cells comprise different subsets including naïve T cells, which have the capacity to respond to new antigens, memory T cells that derive from previous antigen activation and maintain long-term immunity, and regulatory T (Treg) cells which keep immune responses in check. Immune responses commence when naïve T cells encounter antigen and costimulatory ligands presented by dendritic cells (DC), resulting in interleukin 2 (IL-2) production, proliferation, and differentiation to effector cells that migrate to diverse sites to promote pathogen clearance. Activated effector cells are short-lived, although a proportion survive as memory T cells which persist as heterogeneous subsets based on migration, tissue localization, and self-renewal capacities. Each memory subset can participate in maintaining long-term immunity and recall protective responses, although their origin and lineage relationship remains unresolved.
Because humans experience a relatively long lifespan, the critical role of T cells in immunity needs to be studied in the context of different life stages (Fig. 1). In early life (infancy and early childhood), the majority of T cells are naïve T cells newly emerged from the thymus, with Treg cells also significantly represented. During this formative stage when the greatest number of new antigens are encountered, naïve T cells play key protective roles in fending off pathogens, Treg cells are critical for developing tolerance to innocuous and ubiquitous antigens, and long-term reserves of memory T cells are established. Memory T cells accumulate with antigen experience during childhood, with the level of memory T cell accumulation plateauing in adulthood and maintained over decades (Saule et al., 2006). The change in T cell predominance from naïve to memory after childhood and the relative stability of immunity over decades of adulthood suggests changing roles for T cells in adults compared to children (Fig. 1). In adulthood, fewer new antigens are encountered and tolerance establishment may be less prevalent, with the role of T cells shifting to maintain homeostasis and immunoregulation in the context of repeat and chronically encountered antigens, with surveillance for tumors also important during this period. At the later stages of life, there are well-documented immunosenescent changes (for a review, see (Goronzy and Weyand, 2017)), including increased inflammation and a decline in T cell functionality, contributing to immune dysregulation and associated pathology.
Figure 1. Overview of changing role of T cells in at distinct life stages.
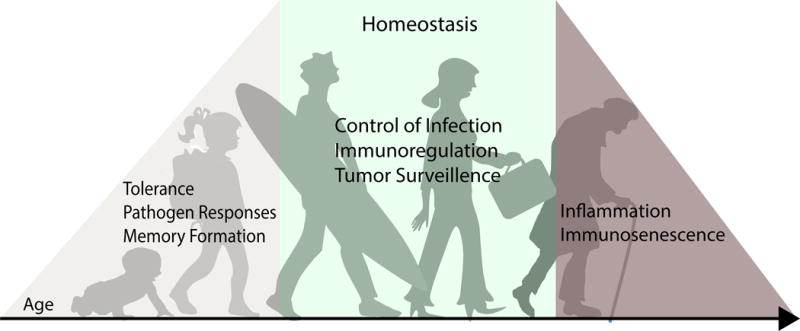
In early years, when humans encounter many antigens for the first time, T cells mediate pathogen clearance for multiple acute infections, develop memory responses, and establish tolerance to innocuous foreign antigens. After childhood, the T cell compartment is more stable with fewer acute infections and reduced generation of memory. During many decades of adult life, T cells maintain homeostasis in tissues by controlling chronic infections, surveilling for cancer cells, and maintaining proper immunoregulation. Finally, in advanced age there is a well-documented decline in T cell function and a corresponding increased susceptibility to infection, cancer, and autoimmunity.
The role of T cells in immune responses and at different life stages is not uniform across the body. T cells populate virtually every organ and tissue in the body including primary and secondary lymphoid tissue, mucosal and barrier sites, exocrine organs, fat, and even the brain and central nervous system (CNS). In terms of numbers, the majority of T cells in the human body are likely found within lymphoid tissues (bone marrow, spleen, tonsils, and an estimated 500-700 lymph nodes) with large numbers also present in mucosal sites (lungs, small and large intestines) and skin, with estimates of 2–3% of the total T cell complement found in human peripheral blood (Clark, 2010; Ganusov and De Boer, 2007). In early life, newly generated naïve T cells and Treg cells populate major lymphoid and mucosal sites in the body and memory T cells begin to develop largely in mucosal sites such as small intestine and lung (Thome et al., 2016a). After childhood, memory T cells are the predominant subset throughout the body; however, the accumulation of memory in lymphoid tissues occurs at a slower rate during childhood and reaches a lower maximum frequency compared with mucosal and barrier sites (Thome et al., 2014). These findings suggest distinct roles for T cells in immunity not only at different life stages, but in specific anatomic compartments.
In this review, we will consider the development, function, and differentiation of human T cells within the complexities of the human condition, involving long-term survival with multiple environmental, pathogenic, and benign antigens through many routes and tissue sites of exposure. We will discuss these multiple aspects of human T cell responses starting from T cell development, the role of naïve and regulatory T cells, differentiation to effector and memory subsets and how tissue localization impacts T cell function and maintenance. We will also discuss how all of these aspects of human T cells function at distinct stages of life from infancy through adulthood, and how specific sites experience different rates of immunosenescence relating to T cell function and persistence.
Thymopoiesis
The thymus is the primary site of T cell development, where progenitors from the bone marrow lacking CD4+ and CD8+ coreceptor expression undergo T cell receptor (TCR) rearrangement to generate CD4+CD8+ double positive (DP) thymocytes. DP cells undergo selection giving rise to CD4+ or CD8+ single positive (SP) thymocytes that ultimately emerge into the periphery as naïve T cells exhibiting CD45RA+CCR7+ phenotypes. Newly developed Treg cells, defined as CD4+CD25+ cells expressing the Foxp3 transcription factor (Hori et al., 2003), represent 9–10% of human CD4+ SP thymocytes (Watanabe et al., 2005), and express naïve CD45RA+CCR7+ phenotypes (Seddiki et al., 2006). Most of our knowledge of thymopoiesis, the genesis of new T cells and mechanisms for their selection and development derive from studies in mouse models using thymectomy, bone marrow reconstitution, and genetic manipulation (for reviews, see (Germain, 2002; Klein et al., 2014)). In humans, similar manipulations are not possible; however, “natural” experiments involving thymectomy and thymic transplantation and studies of recent thymic emigrants (RTE) provide key insights into human thymopoiesis and how it differs from mice.
Unlike mice who are born lymphopenic, with T cells populating secondary lymphoid organs only at the end of gestation, humans are born with a full complement of T cells (Burt, 2013). In utero, human T cell progenitors are detected in fetal thymii as early as 9 weeks of gestation, with mature T cells appearing in the thymus by 12–13 weeks and the spleen and lymph nodes (LNs) by 24 weeks of gestation (Haynes et al., 1988). Human Treg cells also develop early in fetal life and are detected in thymii at 12 weeks and in LN at 14 weeks (Cupedo et al., 2005; Michaelsson et al., 2006). It was established some time ago that neonatal thymectomy in mice results in profound immunodeficiency and multi-organ lymphocytic infiltration (Dalmasso et al., 1963; Sakaguchi et al., 1982), due to defects in development of both naïve T cells and Treg cells, respectively (Sakaguchi et al., 1995). By contrast, neonatal thymectomy in humans, which is performed during cardiac surgery to repair congenital abnormalities, does not impair survival or result in deleterious consequences. Adults who underwent neonatal thymectomy (many now in their third-to-fourth decade of life) do not experience increased incidence of infections (Mancebo et al., 2008; Wells et al., 1998), and remain healthy despite having more extensive declines in naïve T cell frequencies with age compared to control individuals (Prelog et al., 2009; van den Broek et al., 2016). Moreover, neonatally thymectomized adults do not have increased incidences of autoimmunity or allergy compared to aged-matched controls (Silva et al., 2017), consistent with their maintenance of normal blood Treg cell frequencies and numbers (Silva et al., 2016). However, infants born with a congenital defect in Treg cell development (based on deletion or mutation of the Foxp3 transcription factor) present with Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) syndrome manifested by systemic autoimmunity and multi-organ infiltrates (Bennett et al., 2001). Therefore, the critical events in T cell development commence prior to birth in humans, and humans are born with a T cell complement sufficient for anti-pathogen immunity and immunoregulation–at least in the developed world.
T cell development in the thymus involves rigorous selection events as determined in mouse models with newly generated DP thymocytes undergoing positive selection for self human leukocyte antigen (HLA) recognition based on interactions with thymic epithelial cells (non-hematopoietic) and negative selection to remove strongly self-reactive clones through interactions with thymic DC. Treg cells undergo a different type of selection, and tend to be more self-reactive (Stritesky et al., 2012). It is unclear whether thymic selection events occur similarly in humans, but novel insights have emerged from studies of thymus transplantation–a rare surgery used to reconstitute the T cell compartment in infants with complete DiGeorge syndrome who lack a functional thymus (Hudson et al., 2007). Transplantation of fully allogeneic thymus tissues from unrelated infants resulted in thymopoiesis, generation of polyclonal functional naïve T cells and anti-pathogen immunity, enabling these individuals who otherwise would have died of infections to survive (Markert et al., 2010; Markert et al., 2003). Interestingly, thymic recipients are tolerant of self and the thymic transplant (they require no immunosuppression) and also generate Treg cells with diverse repertoires (Chinn et al., 2013; Markert et al., 2010). Based on mouse studies, the generation of functional immunity requires positive selection on thymic epithelial cells (TEC); however, in human thymic transplants TEC are of donor origin, yet functional T cells emerge that can respond to antigens presented by the host antigen presenting cells (APCs) (Li et al., 2011). Whether human thymocyte selection is more permissive than mouse, occurring through interactions with donor epithelium and/or thymic APC is not known but suggests that rules for selection in mice do not apply wholly to humans.
How long does thymopoiesis persist in humans? The thymus is largest at birth, and age-related changes in the thymus including a reduction in thymic volume, loss of epithelial cells, increase in perivascular space, and replacement of thymic tissue by fat begin during childhood (Haynes et al., 2000). Thymic function can be assessed in peripheral T cells based on markers of new thymic emigrants including surface CD31 expression (Junge et al., 2007; Tanaskovic et al., 2010) and TCR excision circles (TREC) as products of gene rearrangement (Hazenberg et al., 2002; Hazenberg et al., 2001). There are well-documented decreases in CD31 expression and TREC content in peripheral blood T cells with age (Douek et al., 1998; Jamieson et al., 1999; Junge et al., 2007). The greatest decline in thymopoiesis was generally believed to occur during puberty; however, more recent studies suggest that this steep reduction may occur later in life. In human thymic tissue obtained from organ donors aged several months to >70 years, DP thymocytes (indicating ongoing selection) were detected at the expected frequency (60-80%) in active thymus tissue from donors <40 years of age, while few DP cells were detected in thymus tissues of adults >40 years of age (Thome et al., 2016b). Similarly, TREC levels of naïve T cells from human blood, spleen, and LN exhibited a steep reduction in individuals over 40 years of age (Douek et al., 1998; Thome et al., 2016b). Residual thymic activity can still persist beyond the fifth decade of life, as RTE-like cells can be detected in peripheral blood (Jamieson et al., 1999) and in renal transplant recipients following T cell depletion therapy (Gurkan et al., 2010). Together, these findings suggest that while thymic function effectively ceases after the fourth decade of life, persistence of residual thymus tissue in some individuals may be amenable to regeneration therapies to rejuvenate the human immune system.
Naïve and Treg cell maintenance
In light of declining thymic output, how do adults maintain a functional and diverse naïve T cell repertoire for responding to new antigens? Studies examining T cell turnover in human volunteers and mice administered deuterated water demonstrated important differences between the two species regarding thymopoiesis and naïve T cell maintenance. In both young and old mice, the majority of naïve T cells derive from thymic output, with minimal peripheral division of naïve T cells, while in humans, the majority of naïve T cells derived from peripheral turnover, even in younger adults with active thymic output (den Braber et al., 2012; Vrisekoop et al., 2008). These studies also showed that the while the average life span of mouse naïve T cells is only 6–10 weeks, in humans, individual naïve T cells can persist 5–10 years (den Braber et al., 2012; Vrisekoop et al., 2008). The intrinsic long lifespan and turnover of human naïve T cells can account for their persistence well into old age.
A mechanistic view of human naïve T cell maintenance has emerged from studies using high throughput DNA sequencing for assessing TCR repertoires and through examination of naïve T cells in tissue sites. TCR sequencing can measure clonal diversity and quantitate expanded clones. Human naïve T cells exhibit a highly diverse TCR encompassing up to 100 million different specificities (Qi et al., 2014). Naïve phenotype cells in the blood of elderly individuals (70–85 years old) still maintain a diverse repertoire of unique TCRβ amino acid sequences that while reduced compared to that of young adults (Qi et al., 2014), suggests that humans are capable of responding to novel antigens even at advanced ages. Study of naïve T cells in circulation and multiple tissue sites obtained from organ donors has enabled a novel assessment of how naïve T cells are maintained over the lifespan (Thome et al., 2016a; Thome et al., 2016b; Thome et al., 2014). Naïve T cells comprise a significant proportion (20–50%) of total T cells within multiple lymph nodes for decades after cessation of thymic output. Naïve T cells also maintain functionality with age, with no overt functional differences between naïve T cells from younger individuals with active thymic output and older individuals with no thymic output (Thome et al., 2016b). TCR repertoire analysis of lymphoid naïve and memory T cells via CDR3 sequencing revealed high diversity (and corresponding low clonality) of CD4+ and CD8+ naïve compared to the corresponding memory subset in spleen and multiple LN (Thome et al., 2016b), indicating that naïve T cells in tissues maintain their capacity to respond to diverse antigens.
Treg cells also emerge from the thymus and exist in high frequencies in tissues during early life; however their persistence and frequency differs from naïve T cells. The early establishment of Treg cells in utero is followed by elevated levels in early life with to 10–30% of all CD4+T cells in blood, lymphoid tissue, and mucosal sites being Treg cells compared to less than 5% in adults (Mold et al., 2008; Thome et al., 2016a). Despite differences in overall frequency, pediatric and adult Treg cells share similar features. Treg cells maintain a TCR repertoire entirely distinct from corresponding non-Treg cell populations, with minimal overlap of TCRβ sequences between these subsets in umbilical cord blood and in adult peripheral blood (Golding et al., 2017). At all ages, Treg cells in blood and lymphoid tissues are CD45RA+CCR7+, indicating a naïve phenotype, while Treg cells in mucosal sites are CD45RA− and express CD45RO, like conventional memory T cells (Thome et al., 2016a). Functionally, pediatric Treg cells in LN can express increased levels of FoxP3 compared to adult LN Treg cells (Thome et al., 2016a); however, more studies are needed to assess how early life Treg cells may differ from those which persist in later life.
The decline in Treg cell frequency in humans begins earlier in childhood than the decline in conventional naïve populations (Thome et al., 2016a), suggesting both thymic and peripheral mechanisms at play. Studies in mice along with complementary analyses in humans indicates that age-associated reductions in Treg cell generation is due to interactions between thymic output, peripheral induction and maintenance. In mice, mature Treg cells migrate back to the thymus and suppress thymic production of Treg cells but not conventional naïve T cells (Thiault et al., 2015). Mature Treg cells were also detected in human pediatric thymii (Thiault et al., 2015), suggesting a similar mechanism controlling Treg cell production in humans. Compared with naïve T cells, human Treg cells exhibit higher turnover, as measured by Ki67 expression (Silva et al., 2016; Thome et al., 2016a), which could also contribute to their declining frequency with age. Treg cell frequency may also be affected by changes in dendritic cell (DC) populations. In a recent study, human fetal DC were shown to promote Treg cell induction more readily than adult DCs (McGovern et al., 2017). Together, these results suggest multiple interacting mechanisms for control of Treg cell frequencies after childhood, consistent with the optimal window for tolerance induction occurring early in life.
T cell effector and memory differentiation
Peripheral T cell differentiation has been extensively characterized in mouse infection models and occurs in 3 phases: clonal expansion, in which activated pathogen-specific T cells expand and differentiate into effector T cells that mediate infection clearance; contraction, in which the majority of effector T cells die by apoptosis following infection; and a memory phase, in which a fraction of these primed T cells persist as long term memory T cells that protect against subsequent infection (Gourley et al., 2004; Kaech and Wherry, 2007). In humans, studying T cell differentiation to effector and memory T cell fates requires specific cohorts that are challenged and followed over time. Elegant studies using the live-attenuated Yellow fever virus vaccination (YFV-7D) show activated CD4+ and CD8+T cells co-expressing CD38 and HLA-DR detectable in the blood within 2–3 days; YFV-specific CD8+ T cells are subsequently detectable in blood and exhibit peak responses after 14–21 days (Akondy et al., 2015; Blom et al., 2013; Miller et al., 2008; Wieten et al., 2016a). As in mouse infection models, the magnitude of the effector CD8+ T cell response correlates to the initial viral load (Akondy et al., 2015), and contracts rapidly, returning to near baseline levels by 30 days (Kohler 2012, Miller 2008, Akondy 2015). Virus-specific memory T cells subsequently persist in frequencies reduced from the effector response (5–6%) (DeWitt et al., 2015; Miller et al., 2008; Wieten et al., 2016b), and are detectable 25 years after vaccination (Fuertes Marraco et al., 2015; Wieten et al., 2016b). Similar kinetics of effector expansion and memory development were observed following smallpox (vaccinia) and influenza vaccination (Miller et al., 2008), with vaccinia-specific memory T cells detected after many decades (Hammarlund et al., 2003). Together, these studies indicate that human effector T cell expansion, contraction, and memory formation following viral infection is similar in kinetics and magnitude to many acute viruses investigated in mice.
While the majority of effector T cells contract rapidly and are not present in significant proportions at steady state, a population of terminal effector cells (Temra) exhibiting CD45RA+CCR7− phenotypes can persist in circulation. Temra cells are mostly present within the CD8+ T cell lineage, exhibit high capacity for IFNγ production and low proliferative capacity, and their frequency in blood is correlated with persistent CMV infection (Larbi and Fulop, 2014). The proportion of CD8+ Temra cells in blood and bone marrow increases with age, specifically in CMV seropositive donors (Di Benedetto et al., 2015; Gordon et al., 2017), suggesting a role for Temra cells in persistent infections. CD4+ Temra cells are rarely detected, but expansion of CD4+Temra cells with cytotoxic function occurs in individuals infected with Dengue virus and is associated with protection (Weiskopf et al., 2015). These findings suggest that certain viruses trigger terminal effector differentiation and Temra cell formation, which may relate to antigen load or persistence. Interestingly, Temra cells appear to be specific to humans, with no clear correlate in mouse infection models.
Memory T cell heterogeneity and tissue distribution
Memory T cells represent a major circulating population in human blood and are subdivided based on phenotype into central memory (Tcm, CD45RA−CCR7+), effector-memory (Tem, CD45RA−CCR7−), and stem-cell memory (Tscm, CD45RA+CCR7+CD95+CD122+) T cells (Gattinoni et al., 2011; Sallusto et al., 1999). Functionally, both Tcm and Tem cells are both capable of producing IL-2 and effector cytokines upon stimulation; however, Tcm cells exhibit lymphoid homing profiles, and high proliferative capacity, while Tem cells produce more effector cytokines (Gattinoni et al., 2011; Sallusto et al., 1999; Willinger et al., 2005). Tscm cells are a relatively rare subset with high proliferative and self-renewal capabilities, but no effector function (Gattinoni et al., 2011). Models for the linear and bifurcated generation of human memory T cell subsets have been previously inferred from functional analysis, differentiation markers, and analysis of cell division (Ahmed et al., 2009). However, recent studies using epigenetic and transcriptional analysis have provided new insights into their differentiation hierarchy. For CD4+T cells, epigenomic profiling based on assessment of methylation status, DNA accessibility, and histone modifications revealed a progressive loss of DNA methylation, of key genes that control memory development, in the order naïve – Tcm – Tem cells (Durek et al., 2016). A linear progression of differentiation for human CD8+ T cells in the order naïve—Tscm—Tcm—Tem cells was suggested by methylation analysis and chromatin accessibility (Abdelsamed et al., 2017; Moskowitz et al., 2017). However, these lineage analyses are based on the fraction and subsets of T cells that emerge into circulation and not the majority populations contained within lymphoid and peripheral tissues sites.
The identification in mouse infection models of heterogeneous tissue distribution of memory T cells and distinct features of T cells in specific sites (Bingaman et al., 2005; Masopust et al., 2001; Masopust et al., 2004) suggested that circulating T cells in humans may not be representative of their counterparts in tissues. However, human tissue samples for research purposes are often limited to surgical explants from disease-affected patients or isolated biopsies, and are derived from low numbers of individuals. More recently, the use of organ donor tissues has enabled assessment of T cell subset distribution, compartmentalization, and function across blood and multiple lymphoid and mucosal sites in healthy humans of all ages (Carpenter et al., 2017; Sathaliyawala et al., 2013; Thome et al., 2014). Analysis of T cell subsets in tissues over six decades of adult life revealed that Tem cells were the predominant population in lungs, small and large intestines, spleen, and blood, and 30–50% of the LN T cell complement, while naïve T cells were primarily found in blood, spleen, and LN (Sathaliyawala et al., 2013; Thome et al., 2014). For CD4+ T cells, Tcm cells are present in similar frequencies (average 20%) in lymphoid sites with lower frequencies in mucosal tissues and few Temra cells, while for CD8+ T cells, significant fractions (~30%) of Temra cells were found within blood and blood-rich tissues (spleen, bone marrow, and lung) but not in other sites, with only low frequencies of CD8+ Tcm-phenotype cells in blood and lymphoid sites (Gordon et al., 2017; Thome et al., 2014). Notably, these subset frequencies were quite stable across highly diverse donors, suggesting tissue-specific compartmentalization of human T cell subsets.
Tissue resident memory T (Trm) cells
It has recently been established in mouse models that a large fraction of memory T cells in tissues comprise a distinct subset, designated tissue resident memory T (Trm) cells (for reviews about mouse Trm cells, see (Mueller and Mackay, 2016; Schenkel and Masopust, 2014)). Mouse Trm cells have been described in diverse types of tissues including barrier sites, mucosal tissues, liver, salivary glands, and brain, where they are retained and do not recirculate as shown by parabiosis and in vivo labeling studies (Jiang et al., 2012; Mueller and Mackay, 2016; Steinert et al., 2015; Teijaro et al., 2011). Functionally, mouse Trm cells mediate rapid in situ protection against diverse viral, bacterial, and parasite infections, and are more effective than circulating memory T cells in pathogen clearance (Fernandez-Ruiz et al., 2016; Gebhardt et al., 2009; Glennie et al., 2015; Jiang et al., 2012; Sakai et al., 2014; Schenkel et al., 2014; Teijaro et al., 2011; Thom et al., 2015). The clinical importance of Trm cells is also underscored by the fact that mouse Trm cells can be generated in response to vaccines, non-infectious allergens, autoantigens, and tumors (Hondowicz et al., 2016; Morawski et al., 2017; Shin and Iwasaki, 2012; Zens et al., 2016).
Phenotypically, mouse Trm cells express the early T cell activation marker CD69 (Anderson et al., 2014; Mueller and Mackay, 2016; Turner et al., 2014), which can promote retention through sequestration of the sphingosine-1-S1PR1 (Mackay et al., 2015; Shiow et al., 2006). CD8+Trm cells in skin and mucosal sites also co-express CD103 in addition to CD69 (Bergsbaken and Bevan, 2015; Mueller and Mackay, 2016). While CD69 expression is a common feature of both CD4+ and CD8+Trm, CD69−Trm cells defined based on tissue retention properties have been identified in mouse tissues (Steinert et al., 2015). Whether CD69 is required for Trm formation also remains a matter of debate; CD69-deficient CD8+T cells exhibit reduced Trm formation in skin (Mackay et al., 2015), while CD69−/− CD4+T cells can generate Trm in lymphoid sites (Ugur et al., 2014).
Recent studies have identified cells with a Trm phenotype in multiple human tissues. Analysis of memory T cells from healthy organ donors and tissue explants revealed that CD69 is expressed by the majority of CD4+ and CD8+ memory T cells in multiple sites including mucosal tissues (lungs, small and large intestines, genital mucosa), skin, lymphoid sites (spleen, LN, BM), and exocrine tissues (pancreas, salivary glands), while blood memory T cells are largely CD69− (Booth et al., 2014; Clark et al., 2006; Gordon et al., 2017; Kumar et al., 2017; Okhrimenko et al., 2014; Pallett et al., 2017; Radenkovic et al., 2017; Swaims-Kohlmeier et al., 2016; Thome et al., 2014; Trimble et al., 2010; Watanabe et al., 2015; Wong et al., 2016; Woon et al., 2016). CD103 is expressed by a subset of CD69+ memory CD8+ T cells in barrier and oral-gastro mucosal sites, but not significantly by lymphoid memory CD8+ T cells or by CD4+ memory T cells in any tissue (Kumar et al., 2017; Thome et al., 2014; Watanabe et al., 2015), suggesting that CD69 is the primary marker distinguishing tissue from circulating memory T cells i. Although CD69 can be upregulated following T cell activation (Cibrian and Sanchez-Madrid, 2017), CD69+ memory T cells in tissues do not exhibit features of activation such as HLA-DR, CD38, or CD25 expression (Kumar et al., 2017; Okhrimenko et al., 2014; Thome et al., 2014; Woon et al., 2016). Together, these findings indicate that human tissue memory T cells exhibit features of resting memory T cells along with CD69, and in some cases CD103 expression.
Defining human Trm as a distinct subset is supported by transcriptional profiling studies of phenotypic subsets. Two recent studies showed that human tissue memory T cells expressing the Trm cell markers CD69 and/or CD103 are a transcriptionally distinct memory subset in humans, with key homologies to mouse Trm cells (Hombrink et al., 2016; Kumar et al., 2017). Notably, a core signature defining human Trm cells has been established through whole transcriptome profiling combined with phenotypic and functional validations in multiple tissues sites for both CD4+ and CD8+ T cells (Kumar et al., 2017) (Fig. 2). Human CD69+ (or CD103+) tissue memory T cells relative to circulating (CD69− CD103−) Tem cells cells in tissue and blood, share key transcriptional features of mouse Trm cells including downregulation of the homing receptor S1PR1 and its associated transcription factor KLF2 (Hombrink et al., 2016; Kumar et al., 2017; Woon et al., 2016), shown to be required for Trm cells formation in mice (Skon et al., 2013). Like mouse Trm cells, human Trm cells also exhibit upregulation of CXCR6 and ITGA1 (CD49a), a collagen binding integrin, and downregulation of SELL (CD62L) and CX3CR1 to avoid egress cues (Hombrink et al., 2016; Kumar et al., 2017; Wong et al., 2016). However, the transcriptional regulators of human and mouse Trm cells may be different. In mice, the transcription factor Hobit is exclusively expressed by and required for the development of Trm cells following infection (Mackay et al., 2016), while in humans, Trm phenotype cells express low-to-negligible levels of Hobit transcripts (Kumar et al., 2017; Vieira Braga et al., 2015).
Figure 2. Defining features of human Trm cells.
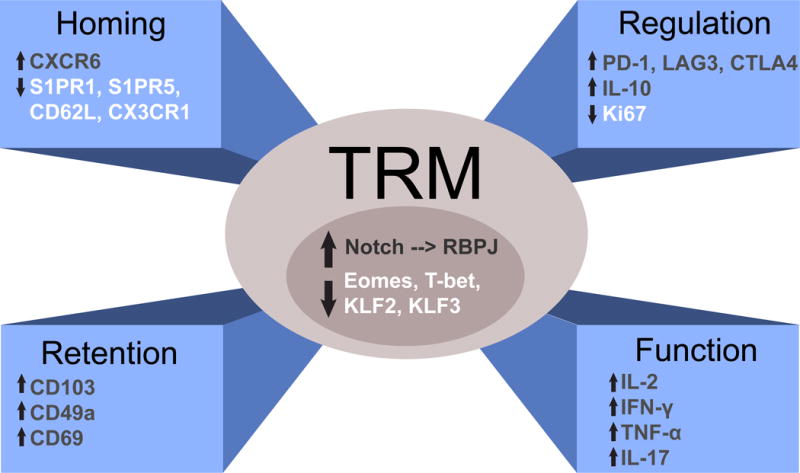
Trm cells have a unique profile of transcription factor expression, surface expression of homing receptors and adhesion markers to maintain tissue residency, and a distinct functional profile with increased expression and ability to secrete both pro and anti-inflammatory cytokines. Trm cells also upregulate a number of inhibitory genes and exhibit reduced proliferative turnover compared to circulating memory T cell counterparts.
Functional aspects of the core signature reveal a dual role for human Trm cells encompassing both protection and regulation (Fig 2B). Human Trm cells produce increased levels of cytokines associated with protective immunity such as IFN-γ, IL-17, TNF-α, and IL-2 compared to circulating Tem cells, but also produce higher levels of the immunoregulatory cytokine IL-10 (Hombrink et al., 2016; Kumar et al., 2017; Pallett et al., 2017; Watanabe et al., 2015). Moreover, human Trm cells express surface receptors known to potently inhibit T cell function including PD-1, LAG3, and CTLA-4 (Hombrink et al., 2016; Kumar et al., 2017; Woon et al., 2016), and also CD101 (Kumar et al., 2017), a molecule associated with inhibition of proliferation (Soares et al., 1998). Interestingly, human Trm cells also exhibit reduced expression of the proliferation marker Ki67 compared with circulating Tem cells (Kumar et al., 2017). We propose that these dual protective and regulatory capabilities are critical for long term maintenance; Trm cells exist in a quiescent state to promote longevity, have anti-inflammatory/regulatory function to prevent unnecessary activation and tissue damage, but also retain the ability to respond quickly upon pathogen invasion.
In addition to the core signature, there is evidence for heterogeneity within human Trm cells. CD103 expression can vary on human CD8+ Trm cells, and in mice, CD103+ and CD103− Trm cells are developmentally distinct subsets (Bergsbaken and Bevan, 2015; Kumar et al., 2017). A recent study examining Trm cells from human skin found that the CD49a+CD103+ subset of CD8+ Trm cells represented a subset with superior cytotoxic abilities compared with CD49a− counterparts (Cheuk et al., 2017). Furthermore, Trm cells may adopt tissue-specific functional or migration capacities, which will be an important area for future studies.
Human Trm cells likely have key roles in mediating protective responses and maintaining long term immunity. Indirect evidence that Trm cells are the critical protective subset derives from studies of patients given alemtuzumab, an agent which profoundly depletes circulating T cells while preserving Trm cells as assessed in skin (Clark et al., 2012). Remarkably, these patients do not experience high rates of infection (Clark et al., 2012) and transplant recipients, who are also treated with depletional agents and other types of systemic immunosuppression, do not succumb to infections, suggesting memory T cells residing within tissues can adequately control ongoing and new infections. Further, the presence of CD8+ resident T cells in human skin is associated with immune responses to herpes simplex virus (HSV) (Zhu et al., 2007; Zhu et al., 2013), analogous to findings in mice which showed skin Trm-mediated protection to HSV (Gebhardt et al., 2009; Gebhardt et al., 2011). These early findings suggest that targeting Trm cells may be an effective strategy to promote vaccine-mediated protection, based on promising results in mouse models (Shin and Iwasaki, 2012; Zens et al., 2016).
One potential mechanism for how human Trm cells may be optimized for protection is in their maintenance of T cell clones specific to pathogens encountered at their site of residence. For example, studies have found biased maintenance of influenza-specific CD8+T cells within the human lung Trm subset (Purwar et al., 2011; Turner et al., 2014), hepatitis B virus (HBV)-specific CD8 T cells within liver CD69+ memory T cells (Pallett et al., 2017), and EBV specific CD8+ Trm cells in the spleen and tonsils (Woon et al., 2016). The tissue distribution of T cells specific for systemic viruses that infect and/or persist in multiple sites is more complex. CMV-specific T cells exhibit different distribution patterns with predominance in either blood, bone marrow (majority of donors), or lung and lung LNs, with higher frequencies of total and activated virus specific T cells being associating with lower viral loads. (Gordon et al., 2017). Bone marrow was also found to be enriched compared to blood for specificities to multiple systemic pathogens (Okhrimenko et al., 2014), suggesting compartmentalization of long-lived memory populations in the bone marrow. Taken together, these findings suggest specific generation and/or retention of memory T cells at the infection site.
In humans, a lifetime of exposure to pathogens and commensal microbiota likely has a profound impact on the development and maintenance of human Trm cells in ways that do not occur in conventional mouse models. For example, the fraction of Trm phenotype cells in tissues is consistently higher in humans than in mice, and memory T cells with a canonical Trm cell phenotype (CD69+CD49a+CD103+ S1PR1loCD62Llo) can be found in human lymphoid tissue (Kumar et al., 2017) but are not typically present in mouse lymphoid sites. The cumulative effects of multiple pathogen exposure on establishment of Trm cells in tissues is also suggested by studies of “dirty” pet store mice which contain increased frequencies of CD69+ T cells within tissues compared to conventional mice in spf (specific pathogen free) facilities (Beura et al., 2016). In addition, the accumulation of Trm cells with age occurs more rapidly in intestines, followed by lungs (Thome et al., 2014), where multiple commensal organisms are also present. Taken together, these results suggest that the rate and extent of Trm cell establishment in tissues may be directly related to antigen load at specific sites.
Tissue residency and other subsets
Tissue residency may not be unique to memory T cells. There is evidence that naïve T cells can persist in tissues, and particularly within LN. Clonal analysis of naïve T cells in spleen and LN of individuals revealed no overlap between the TCR repertoires of naïve T cells from different lymphoid sites, regardless of age (Thome et al., 2016b). Even naïve T cell clones that were modestly expanded were largely limited to a single lymphoid site, in contrast with expanded memory populations which showed high overlap between sites (Thome et al., 2016b). These data suggest that naïve T cells take up long term residence in lymph nodes where they can expand in situ, with lymph nodes serving as reservoirs for their maintenance. Given that naïve T cells lack expression of canonical Trm cell markers, we propose that retention mechanisms for naïve T cells in LN may be more dependent on cytokine signaling rather than specific cell-cell interactions.
The concept of tissue residency has also been described for Treg cells. In mice, tissue Treg cells in fat, lung, and muscle serve key roles in metabolic homeostasis and tissue repair and exhibit distinct phenotypes and transcriptional profiles compared to lymphoid Treg cells (for a review, see (Panduro et al., 2016)). In human tissues, a proportion of Treg cells (40–50%) express the Trm cell marker CD69 (Thome et al., 2016a). Treg cells residing in human skin also express skin-homing receptors CLA, CCR4, and CCR6 (Clark and Kupper, 2007). Increased proportions of Treg cells which differentially produce IL-17 have been identified in psoriatic skin lesions compared to unaffected skin (Sanchez Rodriguez et al., 2014), suggesting roles for Treg cells in controlling local homeostasis. Treg cells have also been identified in human fat with decreased Treg cells correlating with obesity (Feuerer et al., 2009). Human tissue Treg cells in LN may preserve homeostasis as their depletion results in increased T cell proliferation and cytokine production ex vivo (Peters et al., 2013; Thome et al., 2016a). More studies are required to dissect the importance and functional role of human tissue Treg cells in maintaining homeostasis.
T cell subsets over the human lifespan
As described in the sections above, human T cell development, differentiation and maintenance are all dynamic processes with changes occurring over the course of human life. Combining T cell subset frequency with age along with their anatomic localization as diagrammed schematically in Fig. 3, reveals that tissue sites exhibit different qualitative and quantitative changes in the T cell compartment with age. The thymus is the immune organ exhibiting the earliest functional role in production of new T cells during fetal life, yet undergoes the fastest rate of decline compared to other lymphoid sites. Notably, the contribution of thymopoiesis to new naïve T cells (and Treg cells) is highest during infancy, declining in early childhood with conversion of active thymus tissue to fat. During adulthood, thymic output is very low and continues until the fourth or fifth decade of life, when it ceases altogether.
Figure 3. Distinct changes in human T cell subsets with age in diverse tissue sites.
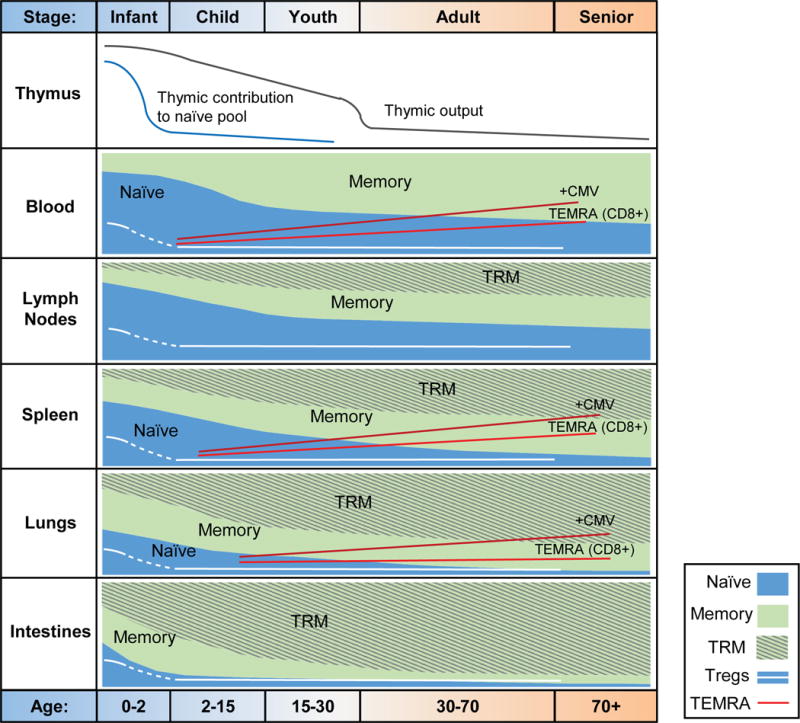
The T cell compartment in humans undergoes major changes from infancy to old age, with key differences by tissue site. Thymic output declines from a very early age and by middle adulthood there is negligible thymic output as measured by peripheral TREC content or DP thymocytes. After early childhood, the relative contribution of the thymus to the peripheral pool of naïve T cells is low due to peripheral mechanisms for naïve T cell maintenance. In all tissues, there is a shift from naïve to memory predominance that occurs with this transition observed during childhood in barrier tissues such as lung and intestines with only very low levels of naïve T cells persisting in adulthood, and much later in adult years in lymphoid sites with lymph nodes exhibiting the slowest transition to memory T cell predominance. Resident memory T cells comprise a varying fraction of the memory compartment in tissues (highest in barrier tissues, lower in lymph nodes); however, the development of Trm cells lags slightly behind the development of memory. Terminally differentiated T cells that re-express CD45RA (Temra) comprise an important fraction of CD8+ T cells in blood, spleen, and lungs, with increased frequencies in old age and with CMV infection.
In the periphery, new naïve T cells emerging from the thymus populate blood and multiple mucosal and lymphoid tissue sites in early life (Thome et al., 2016a). These sites exhibit global and tissue-specific changes in T cell subset composition with age (Fig. 3). In all sites, there is a progressive reduction in naïve T cells along with a compensatory increase in memory subsets from childhood throughout adulthood; however the rate of this change is fastest in mucosal sites, intermediate in spleen and slowest in lymph nodes and blood. Notably, early memory T cells appear primarily in intestines and lungs, with memory T cells predominating in these sites by late childhood (Fig. 3). These mucosal memory T cells exhibit Trm cell phenotypes, which develop gradually in the first two years of life, but rapidly accumulate to become the predominant subset throughout all ages of adulthood. In spleen and lymph nodes, the loss of naïve and accumulation of memory with age is more gradual, with an average of 40–50% memory T cells adopting Trm cell properties in these sites. However, LN exhibit the slowest decline in naïve T cell frequency and accumulation of memory T cells with age, compared to spleen and blood (Thome et al., 2016b)(Saule et al., 2006). CD4+ Treg cells in blood and tissue sites are highest in frequency during early life, declining during childhood, with relatively low, but stable proportions (1–8%) maintained in blood and tissues in adults (Thome et al., 2016a). CD8+ Temra cell populations tend to increase with age in blood and blood-rich sites such as BM, spleen and lungs with a steeper increase in individuals with persistent CMV infection (Gordon et al., 2017) (Fig. 3). Thus, age-associated changes in the T cell compartment exhibit site-specific and subset-specific dynamics.
What are the potential implications of this differential immunological aging of T cells in various sites? The rapid accumulation of memory T cells in mucosal sites likely reflects the high antigen load encountered, particularly for new antigens during infancy and early childhood. Spleen is also a site for diverse antigen encounter via bloodborne antigens, and accumulates both circulating and resident memory T cells. By contrast, lymph nodes, which receive antigenic signals via DC migrating from tissues, are likely to experience lower antigen loads compared to other sites. The early accumulation of Trm cells in mucosal and barrier sites can act to control antigen load and inflammation, and in turn limit DC maturation and migration to draining LNs. In this way, in situ responses in the sites of pathogen entry prevent LN involvement and provide a protective tissue niche for maintenance of naïve and resting memory populations. Evidence from mouse infection models further suggest that Trm cells can promote a generalized anti-pathogen environment in the tissue by local cytokine production and immune cell activation (Ariotti et al., 2014; Schenkel et al., 2014). We propose that the high Trm cell content in mucosal and barrier tissues during adulthood promotes a similar protective environment and limits pathogen spread, without the need to input from circulating and lymphoid reservoirs. It is for this reason that adults can tolerate immunosuppression for transplantation and autoimmune diseases and chemotherapy for cancer without succumbing to multiple infections.
There are also site-specific and temporal dynamics to Treg cells which may impact immunoregulation at different life stages. The higher prevalence of Treg cells early in life compared to older children and young adults suggests a distinct early role for immunoregulation during a period of new antigen exposure. Following this initial immunoregulatory phase, memory T cells and specifically Trm cells in tissues, may also provide regulatory roles, particular in tissues as Trm cells exhibit regulatory capacities like IL-10 production and low proliferation (Kumar et al., 2017). The role of Treg cells in adult immune responses is challenging to assess, as frequency estimates in disease rely only on circulating Treg cells; however, altered frequencies and functionality of circulating Treg cells have been associated with human autoimmune diseases including type 1 diabetes, multiple sclerosis, rheumatoid arthritis, and inflammatory bowel disease (for reviews see (Bacchetta et al., 2007; Dejaco et al., 2006)). Analysis of tissue-specific regulation by Trm cells and tissue Treg cells in future studies will be important for understanding mechanisms for controlling T cell homeostasis at different life stages.
Concluding Remarks
While traditional studies on human T cells have focused on the blood as the most highly accessible site, more recent studies have revealed profound anatomic compartmentalization of T cell subsets. Notably, newly defined subsets of tissue-resident memory T cells and tissue localization of other subsets indicate anatomic complexity of the T cell response. T cell subsets have specialized functions which are associated with the life stage and anatomic compartment, with different sites exhibiting distinct kinetics of changes with age. Molecular, phenotypic and functional profiling on the single cell level will be an important focus for future studies to dissect how alterations in tissue-based immunity may promote or be associated with disease processes, including autoimmune and inflammatory diseases and cancer. Understanding mechanisms for tissue specialization of T cell responses, including potential epigenetic changes that are driven by the tissue environment will also reveal how long term health and immune homeostasis is maintained in the face of diverse antigens, persistent and newly encountered pathogens that are an integral part of the human condition.
Acknowledgments
This work was supported by NIH grants AI06697 and AI128949 awarded to D.L.F. We wish to thank Michelle Miron for critical reading of this manuscript and Dr. Tomer Granot, Ms. Elana Bell, and Joseph Bell for help with the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, Triplett BM, Sekaly RP, Youngblood B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017;214:1593–1606. doi: 10.1084/jem.20161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol. 2009;9:662–668. doi: 10.1038/nri2619. [DOI] [PubMed] [Google Scholar]
- Akondy RS, Johnson PL, Nakaya HI, Edupuganti S, Mulligan MJ, Lawson B, Miller JD, Pulendran B, Antia R, Ahmed R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc Natl Acad Sci U S A. 2015;112:3050–3055. doi: 10.1073/pnas.1500475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346:101–105. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- Bacchetta R, Gambineri E, Roncarolo MG. Role of regulatory T cells and FOXP3 in human diseases. The Journal of allergy and clinical immunology. 2007;120:227–235. doi: 10.1016/j.jaci.2007.06.023. quiz 236-227. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. 2015;16:406–414. doi: 10.1038/ni.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingaman AW, Patke DS, Mane VR, Ahmadzadeh M, Ndejembi M, Bartlett ST, Farber DL. Novel phenotypes and migratory properties distinguish memory CD4 T cell subsets in lymphoid and lung tissue. Eur J Immunol. 2005;35:3173–3186. doi: 10.1002/eji.200526004. [DOI] [PubMed] [Google Scholar]
- Blom K, Braun M, Ivarsson MA, Gonzalez VD, Falconer K, Moll M, Ljunggren HG, Michaelsson J, Sandberg JK. Temporal dynamics of the primary human T cell response to yellow fever virus 17D as it matures from an effector- to a memory-type response. J Immunol. 2013;190:2150–2158. doi: 10.4049/jimmunol.1202234. [DOI] [PubMed] [Google Scholar]
- Booth JS, Toapanta FR, Salerno-Goncalves R, Patil S, Kader HA, Safta AM, Czinn SJ, Greenwald BD, Sztein MB. Characterization and functional properties of gastric tissue-resident memory T cells from children, adults, and the elderly. Front Immunol. 2014;5:294. doi: 10.3389/fimmu.2014.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt TD. Fetal regulatory T cells and peripheral immune tolerance in utero: implications for development and disease. Am J Reprod Immunol. 2013;69:346–358. doi: 10.1111/aji.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DJ, Granot T, Matsuoka N, Senda T, Kumar BV, Thome JJC, Gordon CL, Miron M, Weiner J, Connors T, et al. Human immunology studies using organ donors: impact of clinical variations on immune parameters in tissues and circulation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2017 doi: 10.1111/ajt.14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity. 2017;46:287–300. doi: 10.1016/j.immuni.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn IK, Milner JD, Scheinberg P, Douek DC, Markert ML. Thymus transplantation restores the repertoires of forkhead box protein 3 (FoxP3)+ and FoxP3- T cells in complete DiGeorge anomaly. Clin Exp Immunol. 2013;173:140–149. doi: 10.1111/cei.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibrian D, Sanchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol. 2010;130:362–370. doi: 10.1038/jid.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, Kupper TS. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176:4431–4439. doi: 10.4049/jimmunol.176.7.4431. [DOI] [PubMed] [Google Scholar]
- Clark RA, Kupper TS. IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood. 2007;109:194–202. doi: 10.1182/blood-2006-02-002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4:117ra117. doi: 10.1126/scitranslmed.3003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen IR. Activation of benign autoimmunity as both tumor and autoimmune disease immunotherapy: a comprehensive review. J Autoimmun. 2014;54:112–117. doi: 10.1016/j.jaut.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol. 2005;35:383–390. doi: 10.1002/eji.200425763. [DOI] [PubMed] [Google Scholar]
- Dalmasso AP, Martinez C, Sjodin K, Good RA. Studies on the Role of the Thymus in Immunobiology; Reconstitution of Immunologic Capacity in Mice Thymectomized at Birth. J Exp Med. 1963;118:1089–1109. doi: 10.1084/jem.118.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology. 2006;117:289–300. doi: 10.1111/j.1365-2567.2005.02317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mogling R, de Boer AB, Willems N, Schrijver EH, Spierenburg G, Gaiser K, et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36:288–297. doi: 10.1016/j.immuni.2012.02.006. [DOI] [PubMed] [Google Scholar]
- DeWitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, Sanders C, Utsugi H, Warren EH, McElrath J, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol. 2015;89:4517–4526. doi: 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Benedetto S, Derhovanessian E, Steinhagen-Thiessen E, Goldeck D, Muller L, Pawelec G. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: results from the Berlin BASE-II Study. Biogerontology. 2015;16:631–643. doi: 10.1007/s10522-015-9563-2. [DOI] [PubMed] [Google Scholar]
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- Durek P, Nordstrom K, Gasparoni G, Salhab A, Kressler C, de Almeida M, Bassler K, Ulas T, Schmidt F, Xiong J, et al. Epigenomic Profiling of Human CD4+ T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity. 2016;45:1148–1161. doi: 10.1016/j.immuni.2016.10.022. [DOI] [PubMed] [Google Scholar]
- Evans CJ, Ho Y, Daveson BA, Hall S, Higginson IJ, Gao W, project G.U.C Place and cause of death in centenarians: a population-based observational study in England, 2001 to 2010. PLoS Med. 2014;11:e1001653. doi: 10.1371/journal.pmed.1001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, Lau LS, Mollard V, Cozijnsen A, Collins N, et al. Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity. 2016;45:889–902. doi: 10.1016/j.immuni.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Abed Maillard S, Montandon N, Rufer N, Waldvogel S, Delorenzi M, Speiser DE. Long-lasting stem cell-like memory CD8+ T cells with a naive-like profile upon yellow fever vaccination. Sci Transl Med. 2015;7:282ra248. doi: 10.1126/scitranslmed.aaa3700. [DOI] [PubMed] [Google Scholar]
- Ganusov VV, De Boer RJ. Do most lymphocytes in humans really reside in the gut? Trends Immunol. 2007;28:514–518. doi: 10.1016/j.it.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]
- Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2:309–322. doi: 10.1038/nri798. [DOI] [PubMed] [Google Scholar]
- Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212:1405–1414. doi: 10.1084/jem.20142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding A, Darko S, Wylie WH, Douek DC, Shevach EM. Deep sequencing of the TCR-beta repertoire of human forkhead box protein 3 (FoxP3)+ and FoxP3- T cells suggests that they are completely distinct and non-overlapping. Clin Exp Immunol. 2017;188:12–21. doi: 10.1111/cei.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CL, Miron M, Thome JJ, Matsuoka N, Weiner J, Rak MA, Igarashi S, Granot T, Lerner H, Goodrum F, Farber DL. Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J Exp Med. 2017;214:651–667. doi: 10.1084/jem.20160758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Successful and Maladaptive T Cell Aging. Immunity. 2017;46:364–378. doi: 10.1016/j.immuni.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley TS, Wherry EJ, Masopust D, Ahmed R. Generation and maintenance of immunological memory. Semin Immunol. 2004;16:323–333. doi: 10.1016/j.smim.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, Ames S, Lerner S, Ebcioglu Z, Nair V, et al. Immune reconstitution following rabbit antithymocyte globulin. Am J Transplant. 2010;10:2132–2141. doi: 10.1111/j.1600-6143.2010.03210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu Rev Immunol. 2000;18:529–560. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Martin ME, Kay HH, Kurtzberg J. Early events in human T cell ontogeny. Phenotypic characterization and immunohistologic localization of T cell precursors in early human fetal tissues. J Exp Med. 1988;168:1061–1080. doi: 10.1084/jem.168.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenberg MD, Otto SA, de Pauw ES, Roelofs H, Fibbe WE, Hamann D, Miedema F. T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. Blood. 2002;99:3449–3453. doi: 10.1182/blood.v99.9.3449. [DOI] [PubMed] [Google Scholar]
- Hazenberg MD, Verschuren MC, Hamann D, Miedema F, van Dongen JJ. T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. J Mol Med (Berl) 2001;79:631–640. doi: 10.1007/s001090100271. [DOI] [PubMed] [Google Scholar]
- Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, Brasser G, Jongejan A, Jonkers RE, Nota B, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol. 2016;17:1467–1478. doi: 10.1038/ni.3589. [DOI] [PubMed] [Google Scholar]
- Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, Keitany GJ, Garza EN, Fraser KA, Moon JJ, et al. Interleukin-2-Dependent Allergen-Specific Tissue-Resident Memory Cells Drive Asthma. Immunity. 2016;44:155–166. doi: 10.1016/j.immuni.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Hudson LL, Louise Markert M, Devlin BH, Haynes BF, Sempowski GD. Human T cell reconstitution in DiGeorge syndrome and HIV-1 infection. Semin Immunol. 2007;19:297–309. doi: 10.1016/j.smim.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson BD, Douek DC, Killian S, Hultin LE, Scripture-Adams DD, Giorgi JV, Marelli D, Koup RA, Zack JA. Generation of functional thymocytes in the human adult. Immunity. 1999;10:569–575. doi: 10.1016/s1074-7613(00)80056-4. [DOI] [PubMed] [Google Scholar]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge S, Kloeckener-Gruissem B, Zufferey R, Keisker A, Salgo B, Fauchere JC, Scherer F, Shalaby T, Grotzer M, Siler U, et al. Correlation between recent thymic emigrants and CD31+ (PECAM-1) CD4+ T cells in normal individuals during aging and in lymphopenic children. Eur J Immunol. 2007;37:3270–3280. doi: 10.1002/eji.200636976. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see) Nat Rev Immunol. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017;20:2921–2934. doi: 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbi A, Fulop T. From “truly naive” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. 2014;85:25–35. doi: 10.1002/cyto.a.22351. [DOI] [PubMed] [Google Scholar]
- Li B, Li J, Devlin BH, Markert ML. Thymic microenvironment reconstitution after postnatal human thymus transplantation. Clin Immunol. 2011;140:244–259. doi: 10.1016/j.clim.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, Gebhardt T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. 2015;194:2059–2063. doi: 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- Mancebo E, Clemente J, Sanchez J, Ruiz-Contreras J, De Pablos P, Cortezon S, Romo E, Paz-Artal E, Allende LM. Longitudinal analysis of immune function in the first 3 years of life in thymectomized neonates during cardiac surgery. Clin Exp Immunol. 2008;154:375–383. doi: 10.1111/j.1365-2249.2008.03771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol. 2010;135:236–246. doi: 10.1016/j.clim.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert ML, Sarzotti M, Ozaki DA, Sempowski GD, Rhein ME, Hale LP, Le Deist F, Alexieff MJ, Li J, Hauser ER, et al. Thymus transplantation in complete DiGeorge syndrome: immunologic and safety evaluations in 12 patients. Blood. 2003;102:1121–1130. doi: 10.1182/blood-2002-08-2545. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Usherwood EJ, Cauley LS, Olson S, Marzo AL, Ward RL, Woodland DL, Lefrancois L. Activated primary and memory CD8 T cells migrate to nonlymphoid tissues regardless of site of activation or tissue of origin. J Immunol. 2004;172:4875–4882. doi: 10.4049/jimmunol.172.8.4875. [DOI] [PubMed] [Google Scholar]
- McGovern N, Shin A, Low G, Low D, Duan K, Yao LJ, Msallam R, Low I, Shadan NB, Sumatoh HR, et al. Human fetal dendritic cells promote prenatal T-cell immune suppression through arginase-2. Nature. 2017;546:662–666. doi: 10.1038/nature22795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol. 2006;176:5741–5748. doi: 10.4049/jimmunol.176.10.5741. [DOI] [PubMed] [Google Scholar]
- Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, Lee TH, Nixon DF, McCune JM. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science. 2008;322:1562–1565. doi: 10.1126/science.1164511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski PA, Qi CF, Bolland S. Non-pathogenic tissue-resident CD8+ T cells uniquely accumulate in the brains of lupus-prone mice. Sci Rep. 2017;7:40838. doi: 10.1038/srep40838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, Buenrostro JD, Weyand CM, Greenleaf WJ, Goronzy JJ. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol. 2017;2 doi: 10.1126/sciimmunol.aag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- Okhrimenko A, Grun JR, Westendorf K, Fang Z, Reinke S, von Roth P, Wassilew G, Kuhl AA, Kudernatsch R, Demski S, et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci U S A. 2014;111:9229–9234. doi: 10.1073/pnas.1318731111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, Burton AR, Stegmann KA, Schurich A, Swadling L, et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med. 2017;214:1567–1580. doi: 10.1084/jem.20162115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016;34:609–633. doi: 10.1146/annurev-immunol-032712-095948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, Koenen HJ, Fasse E, Tijssen HJ, Ijzermans JN, Groenen PJ, Schaap NP, Kwekkeboom J, Joosten I. Human secondary lymphoid organs typically contain polyclonally-activated proliferating regulatory T cells. Blood. 2013;122:2213–2223. doi: 10.1182/blood-2013-03-489443. [DOI] [PubMed] [Google Scholar]
- Prelog M, Keller M, Geiger R, Brandstatter A, Wurzner R, Schweigmann U, Zlamy M, Zimmerhackl LB, Grubeck-Loebenstein B. Thymectomy in early childhood: significant alterations of the CD4(+)CD45RA(+)CD62L(+) T cell compartment in later life. Clin Immunol. 2009;130:123–132. doi: 10.1016/j.clim.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6:e16245. doi: 10.1371/journal.pone.0016245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, Goronzy JJ. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A. 2014;111:13139–13144. doi: 10.1073/pnas.1409155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenkovic M, Uvebrant K, Skog O, Sarmiento L, Avartsson J, Storm P, Vickman P, Bertilsson PA, Fex M, Korgsgren O, Cilio CM. Characterization of resident lymphocytes in human pancreatic islets. Clin Exp Immunol. 2017;187:418–427. doi: 10.1111/cei.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA. Decade in review-cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol. 2014;11:630–632. doi: 10.1038/nrclinonc.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in postthymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. J Exp Med. 1982;156:1565–1576. doi: 10.1084/jem.156.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer-Barber KD, Masopust D, Barber DL. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol. 2014;192:2965–2969. doi: 10.4049/jimmunol.1400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions [see comments] Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, Yang SH, Anthony BA, Sverdrup FM, Krow-Lucal E, et al. Memory regulatory T cells reside in human skin. J Clin Invest. 2014;124:1027–1036. doi: 10.1172/JCI72932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saule P, Trauet J, Dutriez V, Lekeux V, Dessaint JP, Labalette M. Accumulation of memory T cells from childhood to old age: central and effector memory cells in CD4(+) versus effector memory and terminally differentiated memory cells in CD8(+) compartment. Mech Ageing Dev. 2006;127:274–281. doi: 10.1016/j.mad.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, Nanan R, Fazekas de Saint Groth B. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood. 2006;107:2830–2838. doi: 10.1182/blood-2005-06-2403. [DOI] [PubMed] [Google Scholar]
- Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature. 2012;491:463–467. doi: 10.1038/nature11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Silva SL, Albuquerque A, Amaral AJ, Li QZ, Mota C, Cheynier R, Victorino RMM, Pereira-Santos MC, Sousa AE. Autoimmunity and allergy control in adults submitted to complete thymectomy early in infancy. PLoS One. 2017;12:e0180385. doi: 10.1371/journal.pone.0180385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva SL, Albuquerque AS, Serra-Caetano A, Foxall RB, Pires AR, Matoso P, Fernandes SM, Ferreira J, Cheynier R, Victorino RM, et al. Human naive regulatory T-cells feature high steady-state turnover and are maintained by IL-7. Oncotarget. 2016;7:12163–12175. doi: 10.18632/oncotarget.7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LR, Tsavaler L, Rivas A, Engleman EG. V7 (CD101) ligation inhibits TCR/CD3-induced IL-2 production by blocking Ca2+ flux and nuclear factor of activated T cell nuclear translocation. J Immunol. 1998;161:209–217. [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, Masopust D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 2015;161:737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaims-Kohlmeier A, Haaland RE, Haddad LB, Sheth AN, Evans-Strickfaden T, Lupo LD, Cordes S, Aguirre AJ, Lupoli KA, Chen CY, et al. Progesterone Levels Associate with a Novel Population of CCR5+CD38+ CD4 T Cells Resident in the Genital Mucosa with Lymphoid Trafficking Potential. J Immunol. 2016;197:368–376. doi: 10.4049/jimmunol.1502628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaskovic S, Fernandez S, Price P, Lee S, French MA. CD31 (PECAM-1) is a marker of recent thymic emigrants among CD4+ T-cells, but not CD8+ T-cells or gammadelta T-cells, in HIV patients responding to ART. Immunol Cell Biol. 2010;88:321–327. doi: 10.1038/icb.2009.108. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiault N, Darrigues J, Adoue V, Gros M, Binet B, Perals C, Leobon B, Fazilleau N, Joffre OP, Robey EA, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol. 2015;16:628–634. doi: 10.1038/ni.3150. [DOI] [PubMed] [Google Scholar]
- Thom JT, Weber TC, Walton SM, Torti N, Oxenius A. The Salivary Gland Acts as a Sink for Tissue-Resident Memory CD8(+) T Cells, Facilitating Protection from Local Cytomegalovirus Infection. Cell Rep. 2015;13:1125–1136. doi: 10.1016/j.celrep.2015.09.082. [DOI] [PubMed] [Google Scholar]
- Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, Granot T, Griesemer A, Lerner H, Kato T, Farber DL. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med. 2016a;22:72–77. doi: 10.1038/nm.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, Sempowski GD, Shen Y, Farber DL. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol. 2016b;1:aah6506. doi: 10.1126/sciimmunol.aah6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJC, Yudanin NA, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, Farber DL. Spatial Map of Human T Cell Compartmentalization and Maintenance over Decades of Life. Cell. 2014;159:814–828. doi: 10.1016/j.cell.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimble CL, Clark RA, Thoburn C, Hanson NC, Tassello J, Frosina D, Kos F, Teague J, Jiang Y, Barat NC, Jungbluth AA. Human papillomavirus 16-associated cervical intraepithelial neoplasia in humans excludes CD8 T cells from dysplastic epithelium. J Immunol. 2010;185:7107–7114. doi: 10.4049/jimmunol.1002756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, Farber DL. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014;7:501–510. doi: 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugur M, Schulz O, Menon MB, Krueger A, Pabst O. Resident CD4+ T cells accumulate in lymphoid organs after prolonged antigen exposure. Nat Commun. 2014;5:4821. doi: 10.1038/ncomms5821. [DOI] [PubMed] [Google Scholar]
- van den Broek T, Delemarre EM, Janssen WJ, Nievelstein RA, Broen JC, Tesselaar K, Borghans JA, Nieuwenhuis EE, Prakken BJ, Mokry M, et al. Neonatal thymectomy reveals differentiation and plasticity within human naive T cells. J Clin Invest. 2016;126:1126–1136. doi: 10.1172/JCI84997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira Braga FA, Hertoghs KM, Kragten NA, Doody GM, Barnes NA, Remmerswaal EB, Hsiao CC, Moerland PD, Wouters D, Derks IA, et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur J Immunol. 2015;45:2945–2958. doi: 10.1002/eji.201545650. [DOI] [PubMed] [Google Scholar]
- Vrisekoop N, den Braber I, de Boer AB, Ruiter AF, Ackermans MT, van der Crabben SN, Schrijver EH, Spierenburg G, Sauerwein HP, Hazenberg MD, et al. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Proc Natl Acad Sci U S A. 2008;105:6115–6120. doi: 10.1073/pnas.0709713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. 2015;7:279ra239. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D, Bangs DJ, Sidney J, Kolla RV, De Silva AD, de Silva AM, Crotty S, Peters B, Sette A. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc Natl Acad Sci U S A. 2015;112:E4256–4263. doi: 10.1073/pnas.1505956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells WJ, Parkman R, Smogorzewska E, Barr M. Neonatal thymectomy: does it affect immune function? J Thorac Cardiovasc Surg. 1998;115:1041–1046. doi: 10.1016/S0022-5223(98)70403-9. [DOI] [PubMed] [Google Scholar]
- Wieten RW, Goorhuis A, Jonker EF, de Bree GJ, de Visser AW, van Genderen PJ, Remmerswaal EB, Ten Berge IJ, Visser LG, Grobusch MP, van Leeuwen EM. 17D yellow fever vaccine elicits comparable long-term immune responses in healthy individuals and immune-compromised patients. J Infect. 2016a;72:713–722. doi: 10.1016/j.jinf.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Wieten RW, Jonker EF, van Leeuwen EM, Remmerswaal EB, Ten Berge IJ, de Visser AW, van Genderen PJ, Goorhuis A, Visser LG, Grobusch MP, de Bree GJ. A Single 17D Yellow Fever Vaccination Provides Lifelong Immunity; Characterization of Yellow-Fever-Specific Neutralizing Antibody and T-Cell Responses after Vaccination. PLoS One. 2016b;11:e0149871. doi: 10.1371/journal.pone.0149871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175:5895–5903. doi: 10.4049/jimmunol.175.9.5895. [DOI] [PubMed] [Google Scholar]
- Wong MT, Ong DE, Lim FS, Teng KW, McGovern N, Narayanan S, Ho WQ, Cerny D, Tan HK, Anicete R, et al. A High-Dimensional Atlas of Human T Cell Diversity Reveals Tissue-Specific Trafficking and Cytokine Signatures. Immunity. 2016;45:442–456. doi: 10.1016/j.immuni.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Woon HG, Braun A, Li J, Smith C, Edwards J, Sierro F, Feng CG, Khanna R, Elliot M, Bell A, et al. Compartmentalization of Total and Virus-Specific Tissue-Resident Memory CD8+ T Cells in Human Lymphoid Organs. PLoS Pathog. 2016;12:e1005799. doi: 10.1371/journal.ppat.1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zens KD, Chen JK, Farber DL. Vaccine-Generated Lung Tissue-Resident Memory T cells Provide Heterosubtypic Protection to Influenza Infection. J Clin Invest Insight. 2016;1:e85832. doi: 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, Wald A, Corey L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med. 2007;204:595–603. doi: 10.1084/jem.20061792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, Jin L, Diem K, Koelle DM, Wald A, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature. 2013;497:494–497. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]