

Abstract
There is a critical need for new mechanism-of-action drugs that reduce the burden of obesity and associated chronic metabolic comorbidities. A potentially novel target to treat obesity and type 2 diabetes is nicotinamide-N-methyltransferase (NNMT), a cytosolic enzyme with newly identified roles in cellular metabolism and energy homeostasis. To validate NNMT as an anti-obesity drug target, we investigated the permeability, selectivity, mechanistic, and physiological properties of a series of small molecule NNMT inhibitors. Membrane permeability of NNMT inhibitors was characterized using parallel artificial membrane permeability and Caco-2 cell assays. Selectivity was tested against structurally-related methyltransferases and nicotinamide adenine dinucleotide (NAD+) salvage pathway enzymes. Effects of NNMT inhibitors on lipogenesis and intracellular levels of metabolites, including NNMT reaction product 1-methylnicotianamide (1-MNA) were evaluated in cultured adipocytes. Effects of a potent NNMT inhibitor on obesity measures and plasma lipid were assessed in diet-induced obese mice fed a high-fat diet. Methylquinolinium scaffolds with primary amine substitutions displayed high permeability from passive and active transport across membranes. Importantly, methylquinolinium analogues displayed high selectivity, not inhibiting related SAM-dependent methyltransferases or enzymes in the NAD+ salvage pathway. NNMT inhibitors reduced intracellular 1-MNA, increased intracellular NAD+ and S-(5′-adenosyl)-L-methionine (SAM), and suppressed lipogenesis in adipocytes. Treatment of diet-induced obese mice systemically with a potent NNMT inhibitor significantly reduced body weight and white adipose mass, decreased adipocyte size, and lowered plasma total cholesterol levels. Notably, administration of NNMT inhibitors did not impact total food intake nor produce any observable adverse effects. These results support development of small molecule NNMT inhibitors as therapeutics to reverse diet-induced obesity and validate NNMT as a viable target to treat obesity and related metabolic conditions. Increased flux of key cellular energy regulators, including NAD+ and SAM, may potentially define the therapeutic mechanism-of-action of NNMT inhibitors.
Keywords: obesity, therapeutics, nicotinamide N-methyltransferase, inhibitors, weight loss
Graphical abstract
1. INTRODUCTION
Obesity is a major public health problem around the world that is linked to severe comorbid disease conditions, physical impairment, high mortality rates, and compromised quality of life.[1–4] Obesity is characterized by the buildup of excessive body fat and extreme dysregulation in whole-body energy expenditure, glucose, hormone, and lipid homeostasis that typically present as adverse metabolic disorders.[4, 5] Additionally, the physiological, metabolic, and psychological changes that accompany obesity are major factors in the development of type 2 diabetes (T2D), cardiovascular disease (CVD) (e.g., coronary heart disease, dyslipidemia, hypertension),[6, 7] stroke, inflammation, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), osteoarthritis, sleep apnea, and several obesity-linked cancers (e.g., colorectal, breast, kidney, prostate).[6, 8] Lifestyle modifications including diet and exercise may help reverse obesity and improve chronic disease biomarkers (e.g., T2D),[9] but are largely ineffective in achieving sustained weight loss and glycemic control.[10] Pharmacological treatments for obesity exist, but unfortunately most approved anti-obesity drugs have only modest efficacy and/or produce severe adverse effects (e.g., cardiovascular risks, central nervous system effects). Thus, there is a critical need for more effective pharmacological interventions that improve long-term management of obesity and its comorbidities.[11]
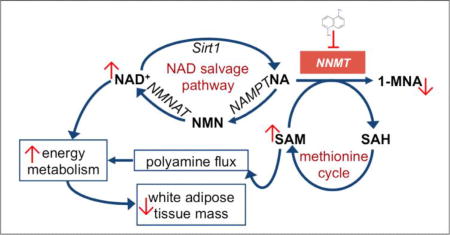
Recently, nicotinamide-N-methyltransferase (NNMT) has emerged as a novel mechanism-of-action target in the adipose tissue to treat obesity and associated T2D.[12–15] NNMT is a cytosolic enzyme with a newly identified role in modulating cellular energy homeostasis by jointly regulating nicotinamide (NA) and S-(5′-adenosyl)-L-methionine (SAM) flux within the critical intracellular nicotinamide adenine dinucleotide (NAD+) salvage pathway and methionine cycle, respectively.[15] NNMT expression is upregulated in the white adipose tissue (WAT) of obese and diabetic mice[12] and has significantly higher activity in the WAT compared to its activity in the brown adipose tissue, liver, and lungs of diet-induced obese mice.[16] Furthermore, plasma levels of the NNMT reaction product 1-methylnicotinamide (1-MNA) correlate with adipose NNMT expression, individuals’ body mass index (BMI), and waist circumference, suggesting the target to be clinically relevant.[13, 14] Importantly, mice fed a high-fat diet and treated with antisense oligonucleotides (ASOs) that reduced adipose NNMT expression were protected from diet-induced obesity (DIO) and showed reduced adiposity compared to control animals.[12]
Using structure-guided design and binding calculations, we recently generated potent small molecule NNMT inhibitors around a methylquinolinium (MQ)-scaffold.[17] In the present study, we extend these findings to show that the small molecule NNMT inhibitors are highly membrane-permeable, selective inhibitors, which reduce intracellular 1-MNA levels and prevent lipogenesis in vitro. These amenable properties demonstrated for the small molecules led us to conduct a proof-of-concept in vivo study in diet-induced obese mice to test the hypothesis that the most potent inhibitor when administered systemically, would reverse obesity by causing substantial loss of body weight and adiposity without causing any observable adverse effects.
2. MATERIALS and METHODS
2.1. Chemicals
NNMT inhibitors and standards for LC/MS/MS studies were purchased from established commercial suppliers or synthesized in-house by established synthetic schemes as described previously.[17] SAM, NA, 1-MQ, 1,8-diMQ, NAD+, and 6-chloro nicotinamide (6-CN) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 1-MNA and S-(5′-adenosyl)-L-methionine (SAH) were obtained from Cayman Chemical Company (Ann Arbor, MI, USA).
2.2 Parallel artificial membrane permeability assay (PAMPA)
Passive membrane transport properties were measured using a 96-well pre-coated PAMPA plate system with membrane pore size 0.4 µm (Gentest™, Corning; Bedford, MA, USA). Briefly, 1 mM stock solution of each compound was prepared in deionized water, diluted to a final concentration of 400 µM in PBS (Sigma Aldrich; St. Louis, MO), and placed in the plate bottom well (donor well). After 4 h incubation at room temperature, the sample concentration in the donor and acceptor wells were measured using a UV-Vis spectrophotometer (Beckman, DU640) set at the wavelength corresponding to the maximum absorption of each compound. Compound concentration in the donor and acceptor wells were calculated from calibration curves spanning 400 – 3.125 µM. Samples were tested in triplicates in three separate experiments.
2.3. Bi-directional permeability assay with Caco-2 cells
Compounds were tested in a Caco-2 cell bi-directional permeability assay using an established contract research organization (Cyprotex; Watertown, MA, USA). Briefly, Caco-2 cells were seeded in 96-well plates and allowed to grow in culture media for three weeks, feeding at 2-day intervals. To ensure a well-defined Caco-2 cell monolayer prior to initiation of experiments, aliquots of the cell buffers were analyzed by fluorescence to determine the transport of the impermeable dye Lucifer yellow. For apical to basolateral (A→B) and basolateral to apical (B→A) permeability, compounds were added at 10 µM concentration to the apical (A) side and basolateral (B) side, respectively, and the corresponding amount of permeation was determined by measuring compound concentration on the B or A side. The A-side buffer contained 100 µM Lucifer yellow dye, in transport buffer (1.98 g/L glucose in 10 mM HEPES, 1× Hank’s balanced salt solution, pH 7.4), and the B-side buffer was transport buffer at pH 7.4. Caco-2 cells were incubated with these buffers for 2 h, and the receiver side buffer was removed for analysis by LC/MS/MS (using bucetin as an analytical internal standard). Data were expressed as permeability (Papp) calculated using the following formula:
Efflux Ratio (Re) was calculated using the formula:
2.4. MTT cell viability assay
3T3-L1 pre-adipocytes cells (catalog CL-173, American Type Culture Collection; Manassas, VA, USA) were seeded at a density of 2 × 103 cells per well in 96-well plates, cultured with standard culture media [DMEM, 4.5 g/L glucose, L-glutamine, sodium pyruvate (Mediatech Inc.; Tewksbury, MA, USA), 10% FBS (Sigma Aldrich; St. Louis, MO, USA), 1% antibiotic-antimycotic solution (Mediatech Inc.; Tewksbury, MA, USA)], and grown for 48 h until > ~90% confluent. Cells were treated for 24 h with 0.1–600 µM NNMT inhibitors in cell culture media. A 24 h time point was chosen based on a previous report of using this time period for transfecting or treating 3T3-L1 cells with NNMT anti-sense oligonucleotides or a small molecule NNMT product inhibitor (1-MNA), respectively, for phenotypic measures.[12] MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (ATCC; Manassas, VA, USA) was added to each well and assayed according to the manufacturer’s instructions. Absorbance corresponding to the amount of formazan dye produced by treated cells was normalized to that produced by control (untreated) cells to calculate % viable cells in the treated samples.
2.5. Differentiation of 3T3-L1 pre-adipocytes
3T3-L1 pre-adipocytes cells were cultured with standard culture media (DMEM, 4.5 g/L glucose, L-glutamine, sodium pyruvate, 10% FBS, 1% antibiotic-antimycotic solution) and grown for 48 h before initiating differentiation using the manufacturer's suggested protocol and modified from previous published work.[18] Briefly, standard culture media was supplemented with scheduled addition of adipogenic agents [3-isobutyl-1methyl xanthine (IBMX), Sigma Aldrich; MO, USA), dexamethasone (Sigma Aldrich; MO, USA), insulin (Gibco Life Technologies Inc.; Grand Island, NY, USA)] over 10 days to promote differentiation of 3T3-L1 fibroblasts into adipocytes; a combination of 1mM IBMX, 1 µM dexamethasone, and 10 µg/ml of insulin in media were added to fully confluent 3T3-L1 fibroblasts for three days (days 0–3) to initiate differentiation. At day 3, the media was replaced with culture media supplemented with insulin (10 µg/ml). After day 6, cells were maintained in culture media until described experiments were begun (days 8–10).
2.6. Quantitative measurement of NNMT reaction product 1-MNA in cultured cells
Cellular 1-MNA concentrations were determined using an ultra-sensitive high-resolution AB Sciex 6500 Q-trap mass spectrometer coupled to an Agilent 1260 ultra-high pressure liquid chromatography (LC/MS/MS) system. Using multiple reaction monitoring (MRM) positive ion mode, the 1-MNA NNMT reaction product was quantified from peak area ratios using AB Sciex Analyst and MultiQuant 2.1 software and the parent precursor and Q3 masses set to m/z 137.1 and 94.1, respectively. Fragment ions at m/z of 92.1 and 77.9 were additionally used for the detection and confirmation of 1-MNA, respectively. Processing of undifferentiated 3T3-L1 pre-adipocytes (day 0) and differentiated adipocytes (day 10) were optimized for recovery and reproducibility of 1-MNA levels across cultured batches of 3T3-L1 cells (~passages 7–8) and the 1-MNA levels were compared between the pre-adipocytes and adipocytes. To determine the effect of NNMT inhibitor on NNMT activity in the pre-adipocytes and differentiated adipocytes (8×104 cells/well seeded prior to beginning differentiation), cells were treated with 30 µM inhibitor for 24 h. Similarly, to compare the relative effects of multiple NNMT inhibitors on NNMT activity in cultured adipocytes, differentiated adipocytes in 6-well plates were treated with 10 µM test compound for 24 h. Following treatment, media was replaced with 80% (v/v) methanol (cooled to −80°C) containing 500 nmol 6-chloronicotinamide (6-CN) as an internal standard (IS) to extract cellular metabolites. Adherent cells were scrapped, then centrifuged at 4°C and 13000 g for 15 min, and the resulting supernatants processed using established protocols.[19] Intracellular levels of 1-MNA and as well as the IS were determined from LC/MS/MS peak areas. Data were subsequently normalized to the IS peak area and transformed as % control values for cross-sample comparisons. The above procedure was repeated with inhibitor concentrations spanning 0.3 – 60 µM to determine the effective concentration (EC50) required to inhibit 50% NNMT activity in cultured adipocytes. Choice of inhibitor concentrations and time period was chosen based on the results from the MTT studies.
2.7. Quantitative measurement of selected metabolites in cultured cells
The relative levels of selected metabolites (NA, SAM, SAH, NAD+) regulated by cellular energy expenditure pathways associated with NNMT were simultaneously detected using LC/MS/MS and MRM ratios. Sample processing was performed as described above. Parent precursor masses of 124.0, 399.3, 385.1, and 665.1 Da and Q3 masses set to m/z 80.0, 250.1, 136.0, and 136.0 were used for the quantitation of NA, SAM, SAH, and NAD+, respectively.
2.8. Selectivity of NNMT inhibitors
Test compounds were screened in biochemical assays for activity against three structurally similar methyltransferases, including catechol-O-methyltransferase (COMT), DNA (cytosine-5)-methyltransferase 1 (DNMT1), and protein arginine methyltransferase 3 (PRMT3). Additional biochemical assays were used to test the ability of compounds to inhibit nicotinamide phosphoribosyl transferase (NAMPT) and NAD+-dependent protein deacetylase sirtuin 1 (SIRT1), two enzymes in the NAD+ biosynthesis/salvage pathway. All assays were performed by Reaction Biology Corporation (RBC; Malvern, PA, USA) and complete assay details are noted below. For each test compound, IC50 values were calculated from dose-response curves established with 10 concentrations of a half-log dilution series. For each assay, established enzyme specific inhibitors were included as positive controls for enzyme function and assay reproducibility. IC50 values were determined by non-linear least-squares fitting of a 4-parameter dose-response curve to collected data points (Graphpad Prism 7.0; La Jolla, CA, USA).
2.8.1. DNMT1 activity assay
A radiometric assay was performed by RBC using 100 µM – 5 nM SAH as an inhibitor positive control. The analogues, 1,8-diMQ and 5-amino-1MQ were tested at concentrations from 200 µM – 10 nM and 600 µM – 10 nM, respectively. Reactions were performed with 0.001 mg/ml DNA substrate Poly(dI-dC), 1 µM radiolabelled S-adenosyl-L-[methyl-3H] methionine (SAM) co-substrate, and recombinant human DNMT1 enzyme. Activity was monitored via quantification of radiolabeled reaction product DNA 5-[methyl-3H]-cytosine.
2.8.2. PRMT3 activity assay
A radiometric assay was performed by RBC using 100 µM – 5 nM SAH as an inhibitor positive control. The analogues, 1,8-diMQ and 5-amino-1MQ were tested at concentrations from 200 µM – 10 nM and 600 µM – 10 nM, respectively. Reactions were performed with 5 µM histone H3 (histone L-arginine) substrate, 1 µM radiolabeled S-adenosyl-L-[methyl-3H] methionine (SAM) co-substrate, and recombinant human PRMT3 enzyme. Activity was monitored via quantification of radiolabeled reaction product histone [methyl-3H]-L-arginine.
2.8.3, COMT activity assay
A radiometric assay was performed by RBC using 1 µM – 50 pM tolcapone as an inhibitor positive control. The analogues, 1,8-diMQ and 5-amino-1MQ were tested at concentrations from 200 µM – 10 nM and 600 µM – 10 nM, respectively. Reactions were performed with 0.5 µM catechol substrate COMT-S01, 1 µM radiolabelled S-adenosyl-L-[methyl-3H] methionine (SAM) co-substrate, and recombinant human COMT enzyme. Activity was monitored via quantification of methylated catechol reaction product (guaiacol [methyl-3H]).
2.8.4. NAMPT activity assay
A fluorometric assay was performed by RBC using 1 µM – 50 pM FK866 as an inhibitor positive control. The analogue 5-amino-1MQ was tested at concentrations from 600 µM – 30 nM. Reactions were performed with 2 µM nicotinamide and 30 µM phosphoribosyl pyrophosphate (PRPP) in the presence of 1mM ATP and recombinant human NAMPT enzyme. Activity was monitored using fluorescence detection and quantification of the nicotinamide mononucleotide (NMN) reaction product.
2.8.5. SIRT-1 activity assay
A fluorometric assay was performed by RBC using 100 µM – 5 nM suramin sodium as an inhibitor positive control. The analogue 5-amino-1MQ was tested at concentrations from 600 µM – 30 nM. Reactions were performed with 50 µM RHKKAc, a fluorogenic peptide substrate from p53 residues 379–382, 500 µM NAD+ co-substrate, and recombinant human SIRT-1 (NAD+-dependent) enzyme. Activity was monitored by the formation of a fluorescent product (coumarin) generated by a two-step coupled reaction that involved deacetylation of substrate by SIRT-1 followed by secondary release of the fluorophore.
2.9. Efficacy of NNMT inhibitor 5-amino-1MQ in diet-induced obese (DIO) mice
17-week old, male DIO C57Bl/6 mice that have been fed high-fat diet (HFD) for 11 weeks (starting at week 6) were purchased from Jackson Labs (Jackson Laboratory; Bar Harbour, ME, USA). Mice were initially group housed (three/cage) and allowed to acclimate to the colony environment maintained at a constant temperature (21–23°C) and humidity (40–50%) on a 12-hour light-dark cycle (lights on 0600–1800 h). Upon arrival, mice were continued to be fedHFD (Open Source Diets formula D12451 from Research Diets Inc.; New Brunswick, NJ, USA), containing 45% energy from fat. Water was available ad libitum. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals[20] and with approval from the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. Following acclimation for seven days, mice were single-housed and maintained on HFD for 4 additional weeks. Mice were intermittently handled, with body weights and food intake (hopper weights) measured 2–3 times per week. After being fed HFD for a total of 16-weeks (an appropriate rodent model of DIO and comparable to human obesity [21]) and reaching pre-arrival body weights (~38g), mice were randomized into balanced control and treatment cohorts (n=9/cohort), with similar group average body weight and standard deviation. Mice in the vehicle cohort received three subcutaneous (SC) saline (1 ml/kg) injections/day (~0930, 1330, 1730 h) and mice in the treatment cohort received three SC injections of the NNMT inhibitor 5-amino-1MQ at a dose of 20 mg/kg/injection for a total dose of ~34 mg/kg/day of the parent compound (calculated according to free weight) for 11 days. The dose chosen was based on an initial dose escalation study (ranging from 10 mg/kg/day to a total dose of 150 mg/kg/day) in DIO mice (n=2); a total dose of 60 mg/kg/day was well tolerated with no observable adverse effects. Body weight and food intake were measured every other day. On day 12, mice were subjected to a 4 h fast period, then deeply anesthetized using isoflurane and trunk blood was collected by decapitation. Plasma was separated from every sample and the samples were submitted to Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL; College Station, TX, USA) for plasma lipid-panel measurements (total cholesterol and triglycerides). Triglycerides values were not included for analysis since the measurements were confounded by sample hemolysis that interfered with the triglyceride reagent in the assay. Epididymal fat pads (epididymal white adipose tissue; EWAT) were excised from every mouse, weighed, and fixed in 10% buffered formalin for further processing.
2.10. Histological analysis
Formalin-fixed EWAT samples were paraffin embedded, sectioned (4 µM), and stained with hematoxylin and eosin (H&E). Images were obtained at 20× magnification using a light microscope (Leica DM LB) and digitally photographed for automated image analysis. Images were analyzed using the “Adiposoft” plug-in software in ImageJ (NIH).[22] Briefly, images were converted to 8-bit images and scald to 0.366 microns per pixel (corresponding to 20× magnification on the Leica microscope). Minimum and maximum diameter parameters were assigned to identify appropriate cells for the automated adipocyte area calculations, and cells along the boundary of the images were excluded from analyses. Three to five images/sample were analyzed, with automated analysis confirmed by visual inspection. Images corresponding to each sample were averaged to obtain the mean adipocyte area (µm2) per sample and combined to calculate group mean values for control (vehicle-treated EWAT samples) and treatment (NNMT inhibitor-treated EWAT samples) cohorts.
2.11. Effect of NNMT inhibitor on adipocyte differentiation quantitated with oil red O staining
3T3-L1 cells were cultured in 60 mm diameter dishes (8.4×104 cells/dish) and treated with NNMT inhibitor dissolved in culture media with/without adipogenic factors (1 mM IBMX, 1 µM dexamethasone, 10 µg/ml of insulin) during each of the scheduled media changes during the differentiation process (described above). On day 9 post-differentiation, cells were subjected to quantitative oil red O (Thermo Fisher Scientific; Waltham, MA, USA) staining as adapted and modified from published protocols.[12] Briefly, cells were washed twice with PBS, fixed with 10% formalin for 30 min at room temperature, and stained with oil red O working solution (~0.2% oil red O in 99% isopropanol) for 30 min. Cells were then washed five times or with sterile water until unincorporated oil red O stain was completely removed. Images of oil red O staining in control and inhibitor-treated cells were digitally photographed using a light microscope (Olympus BX41; Tokyo, Japan). After image capture, 2-propanol (3.5 mL) was added to each dish for 10 min to dissolve the oil red O stain and absorbance was quantified in a plate reader set at 492 nm wavelength. To ensure the absorbance from oil red O staining was within the linear detection range of the plate reader, a calibration curve was established for oil red O staining in adipocytes using a previously described protocol.[23]
2.12. Statistical analysis
Statistical analysis for two-group comparisons was conducted using unpaired Student’s t-test. A one-way analysis of variance (ANOVA) with Dunnett’s posthoc test was used to compare multiple groups (different inhibitor treatments or concentration effects in cellular assessments) to controls. Daily NNMT inhibitor effects on body weight measures in DIO mice was analyzed using a repeated measures two-way ANOVA with Sidak’s multiple comparison posthoc test. All statistical analyses were performed using Graphpad Prism 7.0 with an experiment-wise error rate of α = 0.05.
3. RESULTS
3.1. NNMT inhibitors display high membrane permeability
Compounds spanning ~100-fold IC50 values for NNMT inhibition were selected on the basis of positional substitutions around the N-methylated quinolinium scaffold[17] to obtain an estimate of drug-like oral absorption/bioavailability properties and guide the choice of inhibitors for in vitro and in vivo phenotypic studies. Tables 1 and 2 summarize passive membrane diffusion and active transport membrane permeability, respectively, for select small molecule NNMT inhibitors for which structure activity relationships had been previously developed.[17] 1-MNA, a product inhibitor of NNMT[12, 24, 25] exhibited no passive permeability (Table 1). Similarly, the quinolinium containing parent analogue 1-MQ also lacked passive diffusion properties (Table 1), suggesting that the lipophilicity and drug-like permeability properties of analogues within the methylquinolinium series had to be improved via chemical modification. To this end, we synthesized a number of permethylated quinolinium analogues[17] guided by in silico calculation of partition coefficient (clogP).[26] Addition of hydrophobic methyl group substitutions around the quinolinium scaffold (previously shown to negatively impact NNMT inhibitory activity)[17] only slightly improved membrane permeability via passive transport as indicated by the low, but non-zero, permeability values for 1,8-diMQ and 1,2,4,8-tetraMQ (Table 1). In contrast, positional polar amine substitutions around the quinolinium core not only improved NNMT inhibition as noted previously,[17] but also enabled favorable passive and active transport across membranes (Tables 1 and 2). Specifically, 5-amino-1MQ and 7-amino-1MQ exhibited high passive and active transport across membrane, with no detectable efflux observed in the Caco-2 cell assay. In contrast, the 2,3-diamino substitution in the 1MQ scaffold (2,3-diamino-1MQ) displayed high passive permeability (Table 1), but moderate bi-directional active transport with moderate efflux ratio (Table 2). Consistent with the PAMPA measurements, the 1,8-diMQ analogue exhibited very low bi-directional transport in the Caco-2 cell assay (Table 2).
Table 1.
NNMT inhibitor permeability from passive transport across membranes as measured using PAMPA
| Name | IC50 (µM)a | Flux (cm/s) | Permeability Classificationb |
|---|---|---|---|
| Quinoline (highly permeable)c | ND | 33.9E-06 | High permeability |
| 1-methylnicotinamide (1-MNA) | 9.0 | 0 | No permeability |
| 1-methylquinolinium (1-MQ) | 12.1 | 0 | No permeability |
| 1,8-diMQ | 1.8 | 8.63702E-07 | Low permeability |
| 1,2,4,8-tetraMQ | 109.2 | 6.98184E-07 | Low permeability |
| 5-amino-1MQ | 1.2 | 3.01472E-06 | High permeability |
| 3-amino-6-fluoro-1MQ | 1.2 | 1.07832E-06 | Moderate permeability |
| 7-amino-1MQ | 2.6 | 2.05476E-06 | High permeability |
| 2,3-diamine-1MQ | 2.8 | 3.89795E-06 | High permeability |
Table 2.
Active transport across cell membranes and drug efflux ratios for NNMT inhibitors determined using Caco-2 assay
| Name | Mean A➔B Papp 10−6 cm/s |
Mean B➔A Papp 10−6 cm/s |
Efflux Ratio (Re) |
Classification |
|---|---|---|---|---|
| Ranitidinea | 0.192 | 1.44 | 11.9 | Low permeability (control) |
| Talinolola | 23.5 | 15.7 | 0.673 | High permeability (control) |
| Warfarina | 0.0701 | 5.01 | 73.2 | High efflux (control) |
| 1,8-diMQ | BLQ | 1.78 | NC | Low permeability |
| 5-amino-1MQ | 34.2 | 45.2 | 1.33 | High permeability (no efflux) |
| 7-amino-1MQ | 26.0 | 39.6 | 1.52 | High permeability (no efflux) |
| 2,3-diamine-1MQ | 5.27 | 21.2 | 4.03 | Moderate permeability (moderate efflux) |
Standard controls used in the assay based on permeability classifications
BLQ: No peak detected in receiver side sample for A ➔ B transport
NC: not calculable
3.2. Effects of NNMT inhibitors on 3T3-L1 cell viability
The cytotoxic effects of three membrane-permeable NNMT inhibitors, 5-amino-1MQ, 7-amino-1MQ, and 2,3-diamino-1MQ were evaluated in 3T3-L1 pre-adipocytes. Treatment of cells with 10 µM 5-amino-1MQ or 7-amino-1MQ and 300 µM 2,3-diamino-1MQ for a 24 h period did not impact cell viability (Figure 1). 5-amino-1MQ and 7-amino-1MQ produced modest cytotoxicity relative to untreated cells (P < 0.01, treated vs. control untreated cells) at concentrations ranging from 100–300 µM. All three compounds displayed ~40% cytotoxicity at the highest concentration tested (P < 0.001, 600 µM-treated cells vs. control untreated cells).
Figure 1.

Effect of NNMT inhibitors on 3T3-L1 cell viability. Data represent average viability of 3T3-L1 cells normalized to untreated (control) cells at different concentrations of 5-amino-1MQ (open bar), 7-amino-1MQ (closed bar), and 2,3-diamino-1MQ (hatched bar) (n = 3–6); error bars represent standard error of the mean (SEM). Treatment of 3T3-L1 pre-adipocytes with NNMT inhibitors for 24 h did not impact cell viability up to a concentration of 100 µM. P > 0.05, not significant vs. untreated 3T3-L1 cells (0 µM). **, P < 0.01; ***, P < 0.001 vs. untreated 3T3-L1 cells (0 µM) analyzed by one-way ANOVA with Dunnett’s posthoc.
3.3. Differentiated 3T3-L1 adipocytes provide a relevant cell-based system to validate NNMT inhibitor mechanism-of-action
To determine if differentiated 3T3-L1 adipocytes could be utilized as a cell-based system for mechanism-of-action and phenotypic characterization of NNMT inhibitors, we measured the expression levels of NNMT and used LC/MS/MS to assess the levels of NNMT reaction product 1-MNA in fully differentiated adipocytes (day 9–10 post-differentiation) and undifferentiated pre-adipocytes (day 0). NNMT protein expression was found to be ~37-fold higher in the adipocytes (day 9) vs pre-adipocyte (P < 0.0001, Figure 2A). Similarly, 1-MNA levels normalized to total cellular protein were ~7.5-fold higher in adipocytes compared to preadipocytes (P < 0.05, pre-adipocytes vs. adipocytes, Figure 2B), suggesting relatively higher activity of the NNMT enzyme in the fully differentiated adipocytes. NNMT inhibition using 5-amino-1MQ (30µM concentration) in both the pre-adipocytes (P < 0.01, treated pre-adipocytes vs. untreated controls) and the adipocytes (P < 0.05, treated adipocytes vs. untreated controls) resulted in significant reduction in the intracellular levels of 1-MNA (Figure 2C).
Figure 2.

Relative NNMT protein expression and reaction product 1-MNA levels in the pre-adipocytes vs adipocytes, and the mechanistic validation of NNMT inhibitors in adipocytes. Expression levels of NNMT (normalized to β-actin) in pre-adipocytes (Day 0) and fully differentiated adipocytes (Day 9) (a). Relative levels of 1-MNA in the pre-adipocytes (closed bar) and differentiated adipocytes (checkered bar) per µg of total cellular protein (b). Effects of 5-amino-1MQ (30 µM) (open bar) treatment for 24 h on the intracellular levels of the NNMT reaction product 1-MNA in pre-adipocytes (closed bar) and differentiated adipocytes (checkered bar) (c). Relative effect of NNMT inhibitors on the intracellular levels of 1-MNA in the adipocytes (inhibition of 1-MNA compared at 10 µM concentration of each inhibitor treated for 24 h) (d). Chemical structure and activity of NNMT inhibitor 5-amino-1-methylquinolinium. IC50 for the lead value was determined using a biochemical assay[17] and EC50 was determined from data in Figure 3f (e). Dose-response curve showing intracellular 1-MNA levels in adipocytes following treatment with varied 5-amino-1MQ concentrations (f). Data points represent average 1-MNA levels normalized to an internal standard and transformed to %control values ± SD (n = 2 replicates per data point). The goodness-of-fit R2 between fitted curves and data was 0.94. #, P < 0.05 vs. control pre-adipocytes; *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. control adipocytes; ^, P < 0.01, vs. 5-amino-1MQ (10 µM)-treated adipocytes analyzed by Student’s t-test or one-way ANOVA with Dunnett’s posthoc where appropriate.
3.4. NNMT inhibitors decrease production of 1-MNA in differentiated adipocytes
The relative effectiveness of NNMT inhibitors to lower 1-MNA levels in the differentiated adipocytes were compared at a single concentration of 10 µM (concentration well below the cytotoxic concentration range for NNMT inhibitors). Treatment of adipocytes with membrane-permeable NNMT inhibitors for 24 h resulted in a significant reduction in cellular 1-MNA levels, relative to the levels of 1-MNA in untreated control adipocytes (F(5,6) = 42.64, P < 0.0001) (Figure 2D). Dunnett’s posthoc tests revealed that all membrane-permeable NNMT inhibitors tested significantly decreased 1-MNA levels in the adipocytes relative to control (5-amino-1MQ, P < 0.001; 3-amino-6-fluoro-1MQ, P < 0.01; and 2,3-diamino-1MQ, P < 0.05 vs. control untreated adipocytes, respectively). In contrast, the poorly membrane-permeable NNMT inhibitor 1,2,4,8-tetraMQ did not significantly decrease intracellular 1-MNA levels compared to untreated controls (P > 0.05, n.s.). 5-amino-1MQ, an analogue from our initial series of NNMT inhibitors with low IC50 value (IC50 = ~1 µM;[17]), and high cell membrane permeability (Table 2), produced the greatest reduction of intracellular 1-MNA levels at a concentration of 10 µM among tested inhibitors (Figure 2D). Based on these results, we monitored changes in intracellular 1-MNA in response to 24 h treatment with varied 5-amino-1MQ concentrations. 5-amino-1MQ showed concentration-dependent inhibition of NNMT in fully differentiated adipocytes that could be fit to a 3-parameter sigmoidal dose-response curve with a calculated EC50 = 2.3 +/− 1.1 µM (Figures 2E, 2F; goodness-of-fit R2 = 0.94). At inhibitor concentrations ranging from 10 – 60 µM, the relative intracellular 1-MNA levels stabilized at ~40% the level observed for untreated adipocytes; concentrations greater than 60 µM were not tested due to known cytotoxic effects in 3T3-L1 cells (see Figure 1).
3.5. NNMT inhibition increases intracellular concentrations of NAD+ and SAM in differentiated adipocytes
Figure 3A outlines the major elements of the mammalian NAD+ salvage pathway using NA as the starting substrate.[27] Since the NNMT inhibitor 5-amino-1MQ significantly reduced intracellular 1-MNA concentrations (Figures 2B and 2C), we hypothesized that NNMT inhibition in adipocytes would increase intracellular concentrations of the co-substrates NA and SAM and shunt more NA into the NAD+ salvage cycle. A one-way ANOVA revealed an almost significant main effect of NNMT inhibitor treatment on intracellular NAD+ levels (F(5,6) = 4.131, P = 0.0568) (Figure 3B); treatment of the adipocytes with the NNMT inhibitor 5-amino-1MQ resulted in a concentration-dependent increase in the NAD+ levels with concentrations in the range of 1–60 µM resulting in ~1.2–1.6-fold increase in NAD+ levels relative to control adipocytes. Dunnett’s posttests revealed a significant increase in NAD+ levels at the 10 µM inhibitor concentration (P < 0.05 vs. control; Figure 3B). Similarly, a one-way ANOVA revealed a significant main effect of NNMT inhibition on intracellular SAM levels (F(5.5) = 7.35, P = 0.0236) in the adipocytes (Figure 3B). Dunnett’s posttests revealed a significant increase in the intracellular SAM levels at the higher inhibitor concentration relative to control adipocytes (30 µM, P < 0.05; 60 µM, P = 0.06). However, no statistically significant main effect of NNMT inhibitor treatment were observed for the intracellular levels of NA (F(5,6) = 1.031, P > 0.05) and (F(5,6) = 0.334, P > 0.05) SAH (Figure 3B).
Figure 3.

Effects of NNMT inhibitor on intracellular levels of NAD+ salvage pathways methionine cycle metabolites. Schematic illustration of pathways regulated by NNMT, including the NAD+ biosynthesis salvage pathway starting from NA as a precursor that feeds into energy metabolism, methionine cycle that regulates intracellular SAM concentrations and thus cellular epigenetic modifications and polyamine flux, and clearance of NA by conversion to 1-MNA and excretory products. Pathway enzyme abbreviations include NMNAT (nicotinamide mononucleotide adenylyltransferase), NAMPT (nicotinamide phosphoribosyltransferase), MTase (SAM-dependent methyltransferases), PARPs (poly-ADP-ribose polymerases), and CD38 (cluster of differentiation 38/cyclic ADP ribose hydrolase) (a). Effects of the NNMT inhibitor 5-amino-1MQ on intracellular levels of NAD+, NA, SAM, and SAH metabolites in differentiated adipocytes treated with concentrations 0.3–60 µM for 24 h (b). Data represent mean metabolite levels measured by LC/MS/MS in 5-amino-1MQ-treated adipocytes (open bar) normalized to control untreated adipocyte (closed bar) levels in biological duplicates (± SD). *, P < 0.05 vs. control untreated adipocytes determined by one-way ANOVA analyses followed by Dunnett’s postests comparisons.
3.6. NNMT inhibitors are selective and do not impact related methyltransferases or enzymes in the NAD+ salvage pathway
The selectivity of NNMT inhibitors was confirmed by testing against a panel of structurally similar methyltransferases and two enzymes in the NAD+ salvage pathway (NAMPT and SIRT1; Figure 3). Concentrations of 1,8-diMQ and 5-amino-1MQ ranging from 10 nM to 200 or 600 µM, respectively, did not inhibit DNMT1 or PRMT3 (Figures 4A and 4B). Sigmoidal dose-response curves and reliable estimates of IC50 values based on non-linear least-squares fitting to the available data could not be obtained since no significant inhibition of DNMT1 and PRMT3 was observed at the tested NNMT inhibitor concentrations (Table 3). Additionally, 1,8-diMQ and 5-amino-1MQ showed little inhibition of COMT at maximal tested concentrations of 200 µM (20% inhibition) and 600 µM (10% inhibition), respectively, although no clear trend of concentration-dependent inhibition was observed. As was noted for DNMT1 and PRMT3, sigmoidal dose-response curves and reliable estimates of IC50 values could not be obtained since no significant inhibition was observed at the tested NNMT inhibitor concentrations.
Figure 4.

Normalized response curves for NNMT inhibitors (5-amino-1MQ, open triangle; 1,8-diMQ, open square) against similar methyltransferase enzymes DNMT1 (a), PRMT3 (b), and COMT (c). Normalized response curves for NNMT inhibitor 5-amino-1MQ tested against NAD+ salvage pathway enzymes NAMPT (e) and SIRT1 (f). Data points represent normalized enzyme activity in the respective experiments using 10 concentrations of established enzyme-specific (positive control) and NNMT inhibitors in a 3-fold dilution series. The goodness-of-fit for the positive control inhibitors across all experiments and for 5-amino-1MQ against SIRT1 activity was > 0.95. All other data points could not be used to generate reliable fitted curves and estimates of IC50 values.
Table 3.
Activity for NNMT inhibitors against related methyltransferases and enzymes in the NAD+ salvage pathway
| IC50(µM) | |||
|---|---|---|---|
|
|
|||
| Enzyme | Positive control | 1,8- diMQ |
5-amino- 1MQ |
| DNA (cytosine-5)-methyltransferase 1 | 0.28 ± 0.03 (SAH) | NI | NI |
| Protein arginine methyltransferase 3 | 6.6 ± 1.2 (SAH) | NC | NC |
| Catechol-O-methyltransferase | 0.0009 ± 0.0001 (Talcapone) | NC | NC |
| Nicotinamide phosphoribosyl transferase | 0.0038 ± 0.0001 (FK866) | ND | > 100a |
| NAD+-dependent protein deacetylase sirtuin 1 | 4.3 ± 0.6 (Suramin) | ND | NC |
NI: no inhibition
NC: not calculable
ND: not determined
concentrations above 100 µM could not be tested due to inference in the assay readout signal
5-amino-1MQ did not inhibit NAMPT up to a tested concentration of 100 µM; reliable data could not be obtained at 5-amino-1MQ concentrations above 100 µM due to inference with the NAMPT assay readout signal (Figure 4D, Table 3). However, when the assay was repeated with 5-amino-6-fluoro-1MQ, an analogue of 5-amino-1MQ that did not interfere with the NAMPT assay, no inhibition of NAMPT was observed with analogue concentrations between 30 and 600 µM (data not shown).
5-amino-1MQ did not inhibit SIRT1 concentrations ranging from 10 nM – 300 µM, and minor reduction in SIRT1 activity was observed with 600 µM 5-amino-1MQ (Figure 4E). However, sigmoidal dose-response curves and reliable estimates (i.e., R2 > 0.8) of IC50 values could not be obtained since no significant inhibition was observed with the tested concentrations of 5-amino-1MQ. Taken together, these results suggest high selectivity of the small molecule 5-amino-1MQ analogue at pharmacologically relevant concentrations to NNMT-inhibition.
3.7. NNMT inhibitor caused weight loss and reduced adipose tissue mass in DIO mice
Since in vitro studies showed 5-amino-1MQ to have high cell permeability, enzyme selectivity, and cell culture efficacy, a sub-chronic (11-day) proof-of-concept in vivo study was conducted to test the effect of NNMT inhibition on obesity in HFD fed mice. Three times daily systemic (SC) treatment of DIO mice with 20 mg/kg of 5-amino-1MQ produced a progressive loss of body weight over the treatment period compared to controls (Figure 5A). A repeated-measures two-way ANOVA revealed a significant main effect of the factors treatment (F(1,16) = 12.47, P = 0.0028), time (days) (F(5,80) = 4.437, P = 0.0012), and a significant treatment × time interaction (F(5,80) = 10.89, P < 0.0001). Sidak’s multiple comparison posttests revealed significant differences in body weight between control and treated DIO mice on days 6 (P < 0.01), 9 (P < 0.0001), and 10 (P < 0.0001) (Figure 5A). At the end of the 11-day treatment period, control DIO mice showed a cumulative weight gain of 0.6 ± 0.4 g (~1.4% weight gain from baseline measures), while DIO mice treated with the NNMT inhibitor showed a weight loss of 2.0 ± 0.6 g (~5.1% weight loss from baseline measures) (Figure 5A). Food intake remained the same between the groups suggesting the weight loss effect is primarily related to altered metabolism (F(1,16) = 1.101, P > 0.05; Figure 5B); total cumulative food intake in control and treated DIO mice was 28.1 ± 1.2 g and 26.2 ± 1.4 g, respectively (Figure 5B, inset). Additionally, treatment of DIO mice with the NNMT inhibitor resulted in a substantial ~35% decrease (P < 0.001) in the mass (Figure 5C) and size (Figure 5D) of the EWAT compared with the control DIO mice. Consistent with these results, histological analysis of the EWAT from treated DIO mice had > 30% decrease in adipocyte size (P < 0.05; Figures 5E and 5F) and > 40% decrease in adipocyte volume (data not shown) compared to control DIO mice. Plasma lipid-profile measurements showed that the total cholesterol levels were ~30% lower in treated DIO mice relative to control DIO mice (P < 0.05; Figure 5G). Total cholesterol levels at the end of our study in the control DIO mice were comparable to cholesterol levels reported by the vendor for age-matched DIO mice. In contrast, cholesterol levels in the NNMT inhibitor-treated DIO mice were similar to cholesterol levels reported by the vendor for age-matched normal chow-fed C57Bl/6 mice (www.jax.org/jax-mice-and-services/find-and-order-jaxmice/most-popular-jax-mice-strains/dio-b6).
Figure 5.

Effects of saline or NNMT inhibitor (5-amino-1MQ, 20 mg/kg, t.i.d.) administered SC over a 11-day period in DIO mice on body weight changes from baseline (a), average food intake (g/day) and cumulative food intake across 11-days [inset] (b), epididymal fat-pad weight (c), size of the EWAT (representative images) (d), adipocyte size (µm2) determined in mean number of 20.7 ± 1.8 (DIO, saline) and 28.6 ± 2.3 adipocytes (DIO, 5-amino-1MQ) (e), representative H&E stained images of saline- and 5-amino-1MQ-treated DIO EWAT tissue (scale bar = 200 µm) (f), and total plasma cholesterol levels following a 4-h fasting period (mg/gL) (g). All data points represent the mean values in n = 9 mice/group ± SEM. *, P < 0.05; **, P < 0.01, ***, P < 0.0001 vs. saline-treated DIO analyzed by unpaired Student’s t-test or repeated measures two-way ANOVA with multiple comparisons posthoc tests where applicable.
3.8. NNMT inhibition suppresses lipogenesis in 3T3-L1 cells
In order to determine the effect of NNMT inhibition on adipocyte differentiation and lipogenesis, lipid accumulation was determined in adipocytes following treatment of 3T3-L1 cells with the NNMT inhibitor in media containing adipogenic factors. Treatment with 5-amino-1MQ produced concentration-dependent inhibition of lipid accumulation in differentiating pre-adipocytes (F(3,19) = 39.26, P < 0.0001; Figures 6A and 6B). Concentrations of 30 µM and 60 µM 5-amino-1MQ reduced lipogenesis by 50% and 70%, respectively, compared to control untreated adipocytes (P = 0.0001; Figure 6B). 3T3-L1 cell viability was only slightly reduced at the highest tested concentration of 5-amino-1MQ compared to untreated cell viability (P < 0.05; Figure 6C).
Figure 6.

Effects of 5-amino-1MQ on lipogenesis in differentiating 3T3-L1 cells. Representative images of culture plates (top panels) and microscopic images (20× magnification; scale bar = 50 µm; bottom panels) following oil red O staining of lipid droplets in the control untreated and 5-amino-1MQ (15, 30, and 60 µM)-treated adipocytes (treatment continued throughout the period of differentiation) (a). Lipid accumulation determined by quantification of oil red O staining in 5-amino-1MQ (15, 30, and 60 µM)-treated adipocytes; data points represent average normalized (% untreated control) values (± SEM) in treated adipocyte samples (n = 2 replicates per experiment; experiment performed 3 times) (b). Viability of 3T3-L1 cells treated with 5-amino-1MQ (0.1 – 60 µM); data points represent average normalized (% untreated control) values (± SEM) in treated 3T3-L1 samples (n = 3 replicates per experiment; experiment performed 3 times). ***, P = 0.0001 vs. untreated adipocytes (0 µM); *, P < 0.01 vs. untreated 3T3-L1 cells analyzed by one-way ANOVA with Dunnett’s posthoc tests (c).
4. DISCUSSION
Pharmacological treatment of obesity using target-based approaches to either decrease food intake or increase energy expenditure has been a major area of research in recent years.[11] Here, we demonstrated a novel pharmacological approach using small molecule inhibitors of NNMT, a target enzyme with a newly identified role in energy metabolism,[15] as a potential intervention to prevent adipogenesis and reverse diet-induced obesity. We rationally developed the first series of highly membrane-permeable and selective small molecule inhibitors of NNMT with pronounced efficacy in reducing the levels of the NNMT reaction product 1-MNA in adipocytes. NNMT structure-activity relationships and physical chemical property-based analysis (e.g., logP) guided the synthesis of these membrane-permeable amino-substituted quinolinium analogues, which were validated with the PAMPA and Caco-2 cell-based empirical results, suggesting high oral absorption and bioavailability for these compounds.[28] Mechanisms of active-transport that mediate the intracellular uptake of these small molecules remain to be studied. However, organic cation transporters (OCTs)[29] may likely play role in the active transport of our charged quinolinium-containing quaternary amine NNMT inhibitors across cell membranes since OCTs have been shown to facilitate cellular uptake of several organic cationic drugs (e.g., metformin).[30, 31]
Importantly, quinolinium-based NNMT inhibitors showed exceptional selectivity for NNMT with no apparent activity against either closely-related (i.e., tertiary structure rmsd <3 Å sequence identity >15% relative to NNMT) human methyltransferases (e.g., COMT) or functionally distinct SAM-dependent DNA (e.g., DNMT1) and protein (e.g., PRMT3) methyltransferases with primary and tertiary structures highly similar to NNMT. Additionally, analogues containing the quinolinium scaffold lacked inhibitory activity against enzymes in the NAD+ salvage pathway that bind nicotinamide-containing substrate, including NAMPT[27, 32] and the NAD+-dependent SIRT1 enzyme, which deacetylates NAD+ to produce NA, a product inhibitor of SIRT1.[33] These results suggest that quinolinium-based NNMT inhibitors achieve selectivity by specifically interacting with the NA-binding pocket of NNMT,[17] unlike several known non-selective methyltransferase inhibitors that interact with the SAM-binding pocket, which is highly conserved among SAM-dependent methyltransferases.[34, 35]
Membrane-permeable NNMT inhibitors reduced intracellular 1-MNA levels in a concentration-dependent manner and at pharmacologically relevant concentrations that did not impact cell viability, suggesting these small molecules interact directly with NNMT in cells. Given that NNMT has been reported to be a primary metabolic enzyme[36] and a major clearance pathway for intracellular NA,[15] it is likely that these small molecules are directly and selectively inhibiting the conversion of NA to 1-MNA. We found that intracellular 1-MNA and NNMT protein expression levels were substantially higher in the fully differentiated adipocytes compared to the undifferentiated pre-adipocytes, which is consistent with the differentiation-dependent enhancement in NNMT mRNA levels and enzymatic activity reported previously in the 3T3-L1 cell lines.[37] The higher levels of 1-MNA and NNMT in adipocytes compared to pre-adipocytes may partially account for our observations that increasing concentrations (10 – 60 µM) of NNMT inhibitor did not completely eliminate 1-MNA in adipocytes, but did fully suppress 1-MNA accumulation in undifferentiated pre-adipocytes. In addition, the observed differential 1-MNA steady-state levels following NNMT inhibition may reflect differential transcriptional regulation of cellular metabolic pathways across the various stages of 3T3-L1 differentiation;[38] for example, aldehyde oxidase (AOX)-mediated degradation of 1-MNA may be more pronounced in pre-adipocytes compared to adipocytes, thereby synergistically reducing 1-MNA levels to a greater extent in pre-adipocytes compared to adipocytes. Further studies will examine differentiation-specific differences in the metabolic pathways that directly impact the clearance and/or shunting of NA into the NAD+ salvage pathway.
Our observation that intracellular NA concentrations did not increase following NNMT inhibition extends previous studies that showed NA concentrations were largely unaffected following NNMT knockdown with either adenovirus[39] or anti-sense oligonucleotides (ASOs).[12] It is possible that significant accumulation of NA in adipocytes (and other tissues) is prevented by rapid conversion of NA to NMN and subsequent NAD+ synthesis or transport to the extracellular environment.[27] Importantly, NNMT inhibition potentiated the absolute intracellular concentrations of NAD+ and SAM, without altering the levels of NA and SAH in the adipocytes. These cofactors are critical regulators of cellular energy metabolism, polyamine flux, epigenetic remodeling, and SIRT1 activity in the NAD+ salvage pathway.[12, 27, 40] The increased levels of SAM and NAD+ we observed in cultured adipocytes following NNMT inhibition are consistent with the upregulation of SAM and NAD+ observed in the WAT of DIO mice treated with NNMT ASO, which correlated with increased cellular energy expenditure, reduced adiposity, and protection against diet-induced obesity.[12] In the present study, systemic treatment of DIO mice with a small molecule NNMT inhibitor caused significant loss of body weight and WAT mass, reduction in adipocyte size, and corresponding improvements in the plasma lipid profile (i.e., decreased circulating cholesterol levels). Notably, NNMT inhibition did not impact food intake in the DIO mice nor produce any overt signs of toxicity or adverse behavioral effects at the pharmacologically effective dose. Together, our observations that a small molecule NNMT inhibitor can increase NAD+ and SAM concentrations in differentiated adipocytes and reduce adiposity in DIO mice, suggest that treatment with an NNMT inhibitor effectively reverses obesity via modulation of the NAD+ salvage and SAM-mediated (e.g., polyamine flux) pathways to regulate energy expenditure in the adipose tissue.
A number of studies support NAD+ regeneration as a therapeutic strategy to benefit patients with obesity.[27] For examples, natural NAD+ precursor activators such as nicotinamide riboside (NR) administered via dietary supplements to high-fat fed mice protected against diet-induced obesity, increased energy metabolism, and improved insulin sensitivity;[41] some of these effects were shown to be mediated by NAD+-dependent SIRT1 activation.[42] Similarly, systemic administration of NMN improved glucose tolerance and diet- and age-related insulin resistant conditions.[43, 44] However, dietary supplemental NAD+ precursors (e.g., NR, NMN) require chronic administration and/or very high pharmacological doses to achieve physiologically beneficial enhancements in the NAD+ levels, which potentially limit use in humans.[27] It could be predicted that combined administration of sub-maximal doses of dietary supplements and NNMT inhibitors that function as activators of NAD+ might produce synergistic improvements in diet-induced obesity, and reduce adverse effects associated with chronic high-dose administration of dietary supplemental NAD+ precursors. Studies testing the efficacy of NNMT inhibition on diet-induced obesity, both alone and combination with dietary agents, are currently in progress.
The present study demonstrated that NNMT inhibitor administered during adipocyte differentiation blocked the accumulation of lipid droplets, suggesting suppressed lipogenesis and programed development of mature adipocytes from pre-adipocyte 3T3-L1 precursor cells (i.e., “adipogenesis”). Since 24 h treatment of adipocytes with an NNMT inhibitor increased intracellular levels of SAM and NAD+, it is likely that prolonged exposures to NNMT inhibitor during differentiation may have resulted in significantly enhanced intracellular flux of SAM and NAD+. Increased absolute concentrations of intracellular NAD+ links with activation of NAD+-dependent histone deacetylases (e.g., sirtuins) that also mediate epigenetic regulation of gene expression[40] and adipocyte physiology.[45] Similarly, increased intracellular concentrations of SAM, a universal substrate for SAM-dependent histone methyltransferases, could have profound influence on cellular epigenetic modifications, including transcriptional regulation and the expression of genes that regulate adipogenesis and/or thermogenesis promoting browning of adipocytes (e.g., PPARγ, PRDM16, UCP1, Wnt).[46–48] NNMT protein expression is relatively lower in the murine brown adipose tissue (BAT) compared to the WAT [37] and nnmt (a WAT-selective gene [49, 50]) gene expression has been reported to be lower in the BAT of HFD fed mice compared to normal chow-fed mice.[49] Furthermore, NNMT activity is significantly higher in the white fat compared to brown adipose tissue and the other organs (e.g., liver, lungs) in DIO mice.[16] Hence, treatment with an NNMT inhibitor in DIO mice may less likely impact BAT or other tissue NNMT activity, but could be speculated to modulate thermogenic/adipogenic genes in the WAT. Future studies will validate in vivo modulation of WAT NAD+/SAM and delineate NAD+- and SAM-mediated epigenetic mechanisms that may underlie gene regulation in the presence of NNMT inhibitors.
The current study provides conclusive evidence for the use of small molecule, “drug-like” NNMT inhibitors as a viable therapeutic approach to manage diet-induced obesity and related metabolic comorbidities. Our novel NNMT inhibitors display excellent physical chemical properties (e.g., membrane permeability), selectivity, and cellular inhibitory activity against NNMT, concomitantly enhancing the levels of the cofactors SAM and NAD+ that independently regulate cellular energy metabolism and epigenetic modifications. Importantly, treatment of DIO mice with our NNMT inhibitors did not impact food intake, but produced marked reductions in body weight, WAT mass, adipocyte size, and cholesterol levels with negligible toxicity or observable adverse effects. Future studies will be focused on optimizing the MQ-scaffold lead series to generate more potent NNMT inhibitors with significantly improved “drug-like” ADME profile. Orally bioavailable lead candidates will be tested in a comprehensive doseranging study to confirm efficacy on obesity and co-morbid endpoints.
Acknowledgments
We thank Dr. William Russell (Director, Mass Spectrometry Facility, Department of Biochemistry and Molecular Biology, UTMB) for his assistance in optimizing and running all LC/MS/MS assessments. We thank Dr. Robert Cox and Sam Jacob of the Histopathology Research Laboratory at Shriners Hospital for Children, Galveston for their support in histological processing and light microscopic analysis of the EWAT tissue samples (grant #84060, PI: Dr. Hal Hawkins). We thank Catherine Sampson for her assistance with the mouse blood collection and epididymis fat-pad isolation procedures. This work was supported by the Department of Defense Peer Reviewed Medical Research Program grant PR141776 (S.J.W.), University of Texas Medical Branch Technology Commercialization Award (S.J.W), and NIH grant R01GM112936 (C.C.F).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Kolotkin RL, Meter K, Williams GR. Quality of life and obesity. Obes Rev. 2001;2(4):219–29. doi: 10.1046/j.1467-789x.2001.00040.x. [DOI] [PubMed] [Google Scholar]
- 2.Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378(9793):815–25. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- 3.Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309(1):71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Muniesa P, Martinez-Gonzalez MA, Hu FB, Despres JP, Matsuzawa Y, Loos RJF, Moreno LA, Bray GA, Martinez JA. Obesity. Nat Rev Dis Primers. 2017;3:17034. doi: 10.1038/nrdp.2017.34. [DOI] [PubMed] [Google Scholar]
- 5.Caputo T, Gilardi F, Desvergne B. From chronic overnutrition to metaflammation and insulin resistance: adipose tissue and liver contributions. FEBS Lett. 2017 doi: 10.1002/1873-3468.12742. [DOI] [PubMed] [Google Scholar]
- 6.Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA, Hu FB, Hubbard VS, Jakicic JM, Kushner RF, Loria CM, Millen BE, Nonas CA, Pi-Sunyer FX, Stevens J, Stevens VJ, Wadden TA, Wolfe BM, Yanovski SZ G. American College of Cardiology/American Heart Association Task Force on Practice, S. Obesity. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985–3023. doi: 10.1016/j.jacc.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414(6865):782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 8.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 9.Colberg SR, Sigal RJ. Prescribing exercise for individuals with type 2 diabetes: recommendations and precautions. Phys Sportsmed. 2011;39(2):13–26. doi: 10.3810/psm.2011.05.1909. [DOI] [PubMed] [Google Scholar]
- 10.Fildes A, Charlton J, Rudisill C, Littlejohns P, Prevost AT, Gulliford MC. Probability of an Obese Person Attaining Normal Body Weight: Cohort Study Using Electronic Health Records. Am J Public Health. 2015;105(9):e54–9. doi: 10.2105/AJPH.2015.302773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinussen C, Bojsen-Moller KN, Svane MS, Dejgaard TF, Madsbad S. Emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs. 2017;22(1):87–99. doi: 10.1080/14728214.2017.1269744. [DOI] [PubMed] [Google Scholar]
- 12.Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508(7495):258–62. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannt A, Pfenninger A, Teichert L, Tonjes A, Dietrich A, Schon MR, Kloting N, Bluher M. Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia. 2015;58(4):799–808. doi: 10.1007/s00125-014-3490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M, Li L, Chu J, Zhu B, Zhang Q, Yin X, Jiang W, Dai G, Ju W, Wang Z, Yang Q, Fang Z. Serum N(1)-Methylnicotinamide Is Associated With Obesity and Diabetes in Chinese. J Clin Endocrinol Metab. 2015;100(8):3112–7. doi: 10.1210/jc.2015-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pissios P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol Metab. 2017;28(5):340–353. doi: 10.1016/j.tem.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudolphi B, Zapp B, Kraus NA, Ehebauer F, Kraus B, Kraus D. Body weight predicts Nicotinamide N-Methyltransferase activity in mouse fat. Endocrine Research. 2017:1–9. doi: 10.1080/07435800.2017.1381972. [DOI] [PubMed] [Google Scholar]
- 17.Neelakantan H, Wang HY, Vance V, Hommel JD, McHardy SF, Watowich SJ. Structure-Activity Relationship for Small Molecule Inhibitors of Nicotinamide N-Methyltransferase. J Med Chem. 2017;60(12):5015–5028. doi: 10.1021/acs.jmedchem.7b00389. [DOI] [PubMed] [Google Scholar]
- 18.Zhou QG, Peng X, Hu LL, Xie D, Zhou M, Hou FF. Advanced oxidation protein products inhibit differentiation and activate inflammation in 3T3-L1 preadipocytes. J Cell Physiol. 2010;225(1):42–51. doi: 10.1002/jcp.22189. [DOI] [PubMed] [Google Scholar]
- 19.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7(5):872–81. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(U.S.) IoLARGuide for the Care and Use of Laboratory Animals. 8. National Academies Press; Washington, DC, USA: 2011. [Google Scholar]
- 21.Wang CY, Liao JK. A mouse model of diet-induced obesity and insulin resistance. Methods Mol Biol. 2012;821:421–33. doi: 10.1007/978-1-61779-430-8_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galarraga M, Campion J, Munoz-Barrutia A, Boque N, Moreno H, Martinez JA, Milagro F, Ortiz-de-Solorzano C. Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res. 2012;53(12):2791–6. doi: 10.1194/jlr.D023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraus NA, Ehebauer F, Zapp B, Rudolphi B, Kraus BJ, Kraus D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte. 2016;5(4):351–358. doi: 10.1080/21623945.2016.1240137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aksoy S, Szumlanski CL, Weinshilboum RM. Human Liver Nicotinamide N-Methyltransferase Cdna Cloning, Expression, and Biochemical-Characterization. J Biol Chem. 1994;269(20):14835–14840. [PubMed] [Google Scholar]
- 25.Neelakantan V HV, Wang HL, McHardy SF, Watowich SJ. Noncoupled Fluorescent Assay for Direct Real-Time Monitoring of Nicotinamide N-Methyltransferase Activity. Biochemistry. 2017;56(6):824–832. doi: 10.1021/acs.biochem.6b01215. [DOI] [PubMed] [Google Scholar]
- 26.Hann MM, Keseru GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov. 2012;11(5):355–65. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- 27.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22(1):31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruell J. Membrane-based drug assays. Modern Drug Discovery. 2003:28–30. [Google Scholar]
- 29.Han TK, Everett RS, Proctor WR, Ng CM, Costales CL, Brouwer KL, Thakker DR. Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco-2 cell monolayers and enterocytes. Mol Pharmacol. 2013;84(2):182–9. doi: 10.1124/mol.112.084517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno-Navarrete JM, Ortega FJ, Rodriguez-Hermosa JI, Sabater M, Pardo G, Ricart W, Fernandez-Real JM. OCT1 Expression in adipocytes could contribute to increased metformin action in obese subjects. Diabetes. 2011;60(1):168–76. doi: 10.2337/db10-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Motohashi H, Inui K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013;15(2):581–8. doi: 10.1208/s12248-013-9465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burgos ES, Ho MC, Almo SC, Schramm VL. A phosphoenzyme mimic, overlapping catalytic sites and reaction coordinate motion for human NAMPT. Proc Natl Acad Sci U S A. 2009;106(33):13748–53. doi: 10.1073/pnas.0903898106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai S. The NAD World: a new systemic regulatory network for metabolism and aging--Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys. 2009;53(2):65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8(9):724–32. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 35.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12(6):783–93. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 36.Alston TA, Abeles RH. Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch Biochem Biophys. 1988;260(2):601–8. doi: 10.1016/0003-9861(88)90487-0. [DOI] [PubMed] [Google Scholar]
- 37.Riederer M, Erwa W, Zimmermann R, Frank S, Zechner R. Adipose tissue as a source of nicotinamide N-methyltransferase and homocysteine. Atherosclerosis. 2009;204(2):412–7. doi: 10.1016/j.atherosclerosis.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 38.Hackl H, Burkard TR, Sturn A, Rubio R, Schleiffer A, Tian S, Quackenbush J, Eisenhaber F, Trajanoski Z. Molecular processes during fat cell development revealed by gene expression profiling and functional annotation. Genome Biol. 2005;6(13):R108. doi: 10.1186/gb-2005-6-13-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong S, Moreno-Navarrete JM, Wei X, Kikukawa Y, Tzameli I, Prasad D, Lee Y, Asara JM, Fernandez-Real JM, Maratos-Flier E, Pissios P. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat Med. 2015;21(8):887–94. doi: 10.1038/nm.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janke R, Dodson AE, Rine J. Metabolism and epigenetics. Annu Rev Cell Dev Biol. 2015;31:473–496. doi: 10.1146/annurev-cellbio-100814-125544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15(6):838–47. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imai S, Kiess W. Therapeutic potential of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes. Front Biosci (Landmark Ed) 2009;14:2983–95. doi: 10.2741/3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7(1):78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–36. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, Franczyk MP, Kelly SC, Qi N, Imai S, Yoshino J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016;16(7):1851–60. doi: 10.1016/j.celrep.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JE, Ge K. Transcriptional and epigenetic regulation of PPARgamma expression during adipogenesis. Cell Biosci. 2014;4:29. doi: 10.1186/2045-3701-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okamura M, Inagaki T, Tanaka T, Sakai J. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis. 2010;6(1):24–32. doi: 10.4161/org.6.1.11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo KA, Ng PY, Kabiri Z, Virshup D, Sun L. Wnt inhibition enhances browning of mouse primary white adipocytes. Adipocyte. 2016;5(2):224–31. doi: 10.1080/21623945.2016.1148834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung CM, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, Wagers AJ, Guertin DA. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. 2014;8(1):256–71. doi: 10.1016/j.celrep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song NJ, Choi S, Rajbhandari P, Chang SH, Kim S, Vergnes L, Kwon SM, Yoon JH, Lee S, Ku JM, Lee JS, Reue K, Koo SH, Tontonoz P, Park KW. Prdm4 induction by the small molecule butein promotes white adipose tissue browning. Nat Chem Biol. 2016;12(7):479–81. doi: 10.1038/nchembio.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gopinath VS, Rao M, Shivahare R, Vishwakarma P, Ghose S, Pradhan A, Hindupur R, Sarma KD, Gupta S, Puri SK, Launay D, Martin D. Design, synthesis, ADME characterization and antileishmanial evaluation of novel substituted quinoline analogs. Bioorg Med Chem Lett. 2014;24(9):2046–52. doi: 10.1016/j.bmcl.2014.03.065. [DOI] [PubMed] [Google Scholar]