


Abstract
In humans, peptides derived from dietary proteins and peptide-like drugs are transported via the proton-dependent oligopeptide transporter hPepT1 (SLC15A1). hPepT1 is located across the apical membranes of the small intestine and kidney, where it serves as a high-capacity low-affinity transporter of a broad range of di- and tripeptides. hPepT1 is also overexpressed in the colon of inflammatory bowel disease (IBD) patients, where it mediates the transport of harmful peptides of bacterial origin. Therefore, hPepT1 is a drug target for prodrug substrates interacting with intracellular proteins or inhibitors blocking the transport of toxic bacterial products. In this study, we construct multiple structural models of hPepT1 representing different conformational states that occur during transport and inhibition. We then identify and characterize five ligands of hPepT1 using computational methods, such as virtual screening and QM-polarized ligand docking (QPLD), and experimental testing with uptake kinetic measurements and electrophysiological assays. Our results improve our understanding of the substrate and inhibitor specificity of hPepT1. Furthermore, the newly discovered ligands exhibit unique chemotypes, providing a framework for developing tool compounds with optimal intestinal absorption as well as future IBD therapeutics against this emerging drug target.
Keywords: solute carrier transporter, membrane protein, homology modeling, drug absorption, SLC15, IBD
Graphical abstract
INTRODUCTION
The human peptide transporter hPepT1 (SLC15A1) is a member of the proton-coupled oligopeptide transporter (POT) family and is located in the small intestine, kidney, and pancreas.1–3 hPepT1 is a high capacity, low affinity transporter of di- and tripeptides that are primarily derived from dietary proteins.4 hPepT1 also regulates the intestinal absorption of peptide-like drugs, such as β-lactam antibiotics (cefadroxil) and antiviral drugs (valacyclovir) across the cell membrane.5,6 Rare polymorphisms in hPepT1 can affect therapeutic efficacy of drugs absorbed by this transporter. For example, the hPepT1 mutation F28Y found exclusively in African-Americans leads to reduced substrate uptake.7
Furthermore, hPepT1 was recently shown to be highly expressed in the colon of patients with inflammatory bowel disease (IBD), where it is thought to mediate the intracellular accumulation of bacterial peptides that initiates the intestinal inflammatory response in epithelial cells.8 Potential drugs interacting with hPepT1 can be substrates interacting with an intracellular target or getting absorbed efficiently in the intestines, or inhibitors blocking the transport of harmful bacterial peptides in IBD patients.3 Thus, hPepT1 is an emerging as a protein of the utmost pharmacological importance.9
Understanding cell permeability is essential in drug discovery. Ligand-based approaches have been used to describe the physicochemical properties of a ligand influencing its interactions with membrane transporters.10,11 In contrast, structure-based approaches focus on describing the transporters substrate specificities at a molecular level. The structural elements that determine specificity of hPepT1 transport and inhibition are poorly described. hPepT1 contains two domains, including the transmembrane transport domain as well as a long extracellular region (~200 residues) located between transmembrane helices 9 and 10 of the transport domain. The structures of the extracellular region of hPepT1 and hPepT2 were recently solved providing a starting point to characterize their function.12 Currently, there is no experimentally determined atomic-resolution structure of the transmembrane domain of hPepT1 or other mammalian members of the POT family. However, structures of prokaryotic POT family members have been recently determined in various conformations of the transport cycle.13–18 These structures show that the POT family members adopt the major facilitator superfamily (MFS) fold, consisting of two bundles of six transmembrane helices organized in two inverted pseudorepeats.19 MFS transporters, including POT members, mediate transport of substrates via the rocker-switch mechanism, in which the transporter oscillates back and forth from an outward-facing to an inward-facing V-shaped conformation, passing through intermediate states, such as an occluded conformation.20 Moreover, POT structures in complex with various ligands revealed distinct mode of binding. For example, structures of the Streptococcus thermophiles homologue PepTst in complex with di- and tripeptides show that substrates can adopt distinct orientations within the binding site and that the internal side of the binding site rearranges itself depending on the substrate.16 The relevance of these discoveries to the human homologue hPepT1 is not fully described. The majority of previous models of hPepT1 transport domain relied on distantly related template structures (e.g., of LacY)21 or focused on describing its mode of interaction with the extracellular region.12 The goal of this study is to improve our understanding of the structural basis for the hPepT1 interaction with substrates and inhibitors, and to test our proposed specificity determinants by identifying novel small molecule modulators for this transporter. We first build homology models of hPepT1 transport domain to visualize conformational changes that occur during the transport cycle, and develop hypotheses regarding its ligand-binding and transport determinants. We then perform virtual screening of small molecule libraries against our models, where top-scoring compounds are tested experimentally using cell-based assays. Our experimentally confirmed hits, coupled with calculations with QM-polarized ligand docking (QPLD) and generalized born surface area solvation (MM-GBSA) propose a model for small molecule modulation of hPepT1. Finally, we discuss how the results of this study provide new insights into the inhibition and transport mechanisms of hPepT1, which can potentially guide the development of future drugs targeting this transporter.
MATERIALS AND METHODS
Homology Modeling and Molecular Docking
hPepT1 was modeled based on different template structures (Table 1, Supporting Information).13,15,16,18,22,23 Our initial alignment was generated with Promals3D24 and manually refined based on structural considerations (Supporting Information). Overall, 100–500 models were built, based on the different templates, using various versions of MODELLER25 (Supporting Information). These models were assessed and ranked by the statistical potential Z-DOPE.26 AutoDock Vina27 and OpenEye FRED28 were used to screen the FDA approved drug library and the lead-like subset of the ZINC database,29 respectively (Supporting Information). Although docking cannot accurately rank molecules by binding affinity, it can predict whether a compound is likely to bind the target.30 Therefore, to prioritize molecules for experimental testing, we visually inspected the docking poses of the 200 top-scoring compounds in the lead-like computational screens, removing molecules with a questionable pose and strained conformations. For the FDA approved drugs screen we took a consensus based approach and only analyzed compounds predicted to bind both Models 1 and 3 (Supporting Information). Overall, we selected 22 compounds that interacted with key residues of the binding site or those presenting new chemical scaffolds or exploring putative subpockets of the binding.31–33 Finally, the binding poses of experimentally confirmed hits and four known ligands were optimized with QM-polarized ligand docking implemented in the Schrödinger suite34 (Supporting Information).
Table 1.
hPepT1 Homology Models
| Modela | Binding siteb | Volume (Å3)c |
Z- DOPEd |
PDB ID (Resolution)e |
Identityf | Organismg | Ligandh | Statei |
|---|---|---|---|---|---|---|---|---|
| 1 |

|
141 | +0.12 | 2XUT (3.62 Å) | 34% | So | - | Occluded |
| 2 |

|
183 | −0.13 | 4APS (3.3 Å) | 22% | Sf | - | Unbound Inward-open |
| 3 |

|
291 | −0.18 | 4IKZ (2.4 Å) | 25% | Gk | Alafosfalin | Inward-open |
| 4 |
|
154 | −0.099 | 4D2C (2.47 Å) | 22% | St | Ala-Phe | Inward-open |
| 5 |
|
239 | −0.049 | 4D2D (2.52 Å) | 22% | St | Ala-Ala-Ala | Inward-open |
Model is the nomenclature used in this article.
Binding site shows surface representation of the binding site of the hPepT1 model calculated with POVME 2.0.51
Volume corresponds to the volume of the binding site calculated with the POVME 2.0.
Z-DOPE Score is the normalized atomic-distance dependent statistical potential based on known protein structures calculated by MODELLER.
PDB ID refers to the Protein Data Bank identifier of the template structure with the resolution of the structure in parentheses.
Identity is the percentage sequence identity between hPepT1 and the template structure calculated by HHPred.52
Organism refers to the organism of the template structure transporter: Shewanella oneidensis (So), Streptococcus thermophilus (St), Geobacillus kaustophilus (Gk).
Ligand corresponds to the small molecule bound to the binding site of the template structure.
Conformation refers to the conformation of the model.
Cis Inhibition
The preparation of yeast Pichia pastoris transformants expressing hPepT1 was described previously by our group.35 Uptake measurements were performed by rapidly mixing 20 µL of yeast cell suspension and 30 µL of 100 mM potassium phosphate buffer (pH 6.5, room temperature), which contained 10 µM [3H]glycylsarcosine in the absence and presence of excess (mM) concentrations of potential inhibitors. The reaction was terminated at 2 min, representative of linear uptake, by adding 1 mL of ice-cold buffer. The cell suspension was then passed through HATF filters using a rapid filtration technique, and the filters washed several times with 1.5 mL of ice-cold buffer. The filters were transferred to glass vials containing 6 mL Cytoscint cocktail (MP Biomedicals, Solon, OH) and then left to stand for 24 h at room temperature. Radioactivity was measured on a dual-channel liquid scintillation counter (Beckman LS 6000SC; Beckman Coulter, Fullerton, CA).
Data Analysis
The dose-dependent inhibition of [3H]-glycylsarcosine uptake was best fit to the equation:
where IC50 represents the inhibitor concentration that results in 50% inhibition, I the inhibitor concentration, and s the slope factor. The unknown parameters (IC50 and s) were estimated by nonlinear regression (weighting of unity) using Prism version 7.0 (GraphPad Software, Inc., La Jolla, CA). The quality of fit was evaluated by the SE of parameters, by the coefficient of determination (r2), and by inspection of the residuals.
Electrophysiological Measurement in X. laevis Oocytes
Capped cRNA from hPepT1 cDNA (cloned in pGH19 vector, kindly provided by Dr. Peter S. Aronson, Yale University School of Medicine) was synthesized using Ampli-Cap T7 High Yield Message Maker Kits (Epicenter Biotechnologies, Madison, WI) as previously described.36–38 Mature oocytes (stage V–VI) from X. laevis were isolated by treatment with 1.0 mg/mL collagenase A (Roche Diagnostics GmbH, Mannheim, Germany), manually defolliculated and incubated at 18 °C in modified Barth’s medium supplemented with 50 µg/mL gentamicin as described previously.36 On the following day, oocytes were injected with 50 ng cRNA of hPepT1 (50 nl). H2O injected oocytes served as controls. The experimental protocol for the use of the animals was approved by Toho University Animal Care and Use Committee (approval number: 16–53–258). Electrophysiological studies were performed with the two-electrode voltage clamp (TEVC) technique as described previously. The membrane potential was clamped at −50 mV. The oocyte was always superfused with the perfusion buffer (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes/Tris adjusted to pH 7.5). After the current stabilized, the oocyte was superfused with NaCl buffer (pH 6.0) (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes/Mes adjusted to pH 6.0), followed by the same buffer containing test compound. After application of the test compound, the current reached the maximum and steady state, and the test compound was washed out with the perfusion buffer. The substrate-induced current at an applied voltage was calculated as the difference between the steady-state currents recorded in the absence and presence of substrate.
RESULTS
hPepT1 Homology Modeling
We built homology models of hPepT1 based on prokaryotic POT structures (sequence identities of 22–35%) (Table 1, Materials and Methods Section). Prokaryotic and human POT members, including hPepT1, have similar substrate specificity and predicted transmembrane topology, as well as several highly conserved functionally important regions. For example, three sequence motifs have been identified as a signature of the POT family, including ExxERFxYY (located in TM1), PTR2_1 (TM2-3), and PTR2_2 (TM5). Functional studies using site-directed mutagenesis indicate that these motifs are critical for substrate and ion binding, confirming the evolutionary relationships between the prokaryotic POTs and hPepT1.39,40 Thus, despite sharing distant sequence similarities, the prokaryotic POT members provide, at present, the most suitable templates to model hPepT1.
Currently, there are 16 POT structures in the protein data bank (PDB) that can serve as potential templates for hPepT1 modeling.13,16–18 Here we selected the template structures based on their sequence similarity to hPepT1, quality of the crystal structure, and uniqueness of the conformation, including whether it is ligand-bound or -unbound (Materials and Methods Section). We generated five models in three distinct conformations (Table 1).
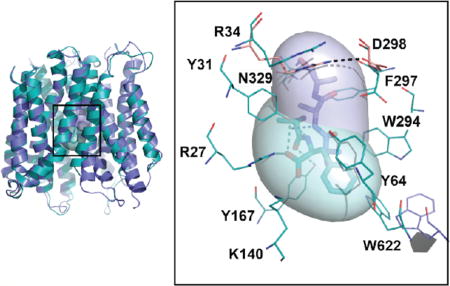
The models include: (i) an occluded conformation based on a homologue from Shewanella oneidensis (PepTSo), which does not have a ligand resolved in the binding site but might represent a ligand-bound conformation17 (Model 1); (ii) an inward-open conformation based on a homologue from Geobacillus kaustrophilus (GkPOT) bound to the dipeptide analog alafosfalin13 (Model 3); and (iii) three models in inward-open conformations based on structures of a homologue from Streptococcus thermophiles (PepTSt), bound to the dipeptide Ala-Phe and the tripeptide Ala-Ala-Ala16 (Models 4,5) (Figure 1A and C), as well as unbound PepTSt structure in this conformation (Model 2).18
Figure 1.

hPepT1 models in the inward-open conformations. (A) hPepT1 inward-open models bound to the dipeptide Ala-Phe (Model 4; cyan) the tripeptide Ala-Ala-Ala (Model 5; purple) are shown in cartoon. (B) Two conformations of the internal gate, constituted by the hairpin loops TM4-5 and TM10-11 are shown in cartoon for Models 4 (cyan) and 5 (purple). (C) The binding site residues of the inward-open conformation of hPepT1 based on the Ala-Phe bound template are shown in cyan lines. An alternative orientation of W622 from the Ala-Ala-Ala bound structure is shown in purple lines. The closed gate, constituted by a salt bridge between R34 and D298 is shown in pink lines, where the coordinates are derived from our model based on the unbound inward-open conformation (Table 1; Model 2). The coordinates of the peptide substrates Ala-Phe and Ala-Ala-Ala, shown in sticks and surface representations, are derived from the corresponding template structures. The hydrogen bonds established between the ligands and hPepT1 binding site are represented in dashed lines.
All models show an MFS fold consisting of two bundles of 6 transmembrane (TM) helices (TM1-TM6 and TM7-TM12) that are related by a pseudo 2-fold symmetry axis, as expected from their template structures (Figure 1A). The location of predicted key residues in the hPepT1 model is consistent with multiple previous studies showing their relevance to hPepT1 function. For example, mutations in conserved tyrosine residues (i.e., Y64, Y91, and Y167) induced alteration of the transport rate or affinity of substrates in hPepT1 (Figure S1).41
We compared the models in the different conformations to identify structural features that might be associated with peptide transport or inhibition. On the cytosolic side of hPepT1, the tips of two hairpin loops (i.e., TM4-TM5 and TM10-TM11) make up the intracellular gate. These loops can bend or straighten, thereby modulating interactions with the ligands (Figure 1A, B). Specifically, in the dipeptide substrate Ala-Phe-bound conformation, the cytoplasmic ends of TM10 and TM11 bend to favor π−π interactions between W622 and the ligand (Model 4; Figure 1C, in cyan); alternatively, in the tripeptide Ala-Ala-Ala-bound conformation the W622 side chain is flipped, thereby facing the opposite side far away from the ligand (Model 5; Figure 1C, in purple). As a result, the hPepT1 binding site can vary in shape and size (Table 1, Figure S1). The occluded conformation has the smallest binding site (141 Å3 for Model 1), whereas the inward-open conformations have larger binding sites (e.g., 291 Å3 for Model 3).
Interestingly, despite the variability in the binding site shape and size, key hPepT1-ligand interactions are conserved among the different conformations. R27, Y31, K140, and E595 form interactions with the ligands that are maintained across all ligand-bound hPepT1 models (Models 3–5; Table 1; Figure S1). The negative charge of E595 of hPepT1 has been previously identified as essential for transport, in agreement with our model.42 Moreover, the conformations of E595 and K140 are almost identical to those of the corresponding residues in their templates structures (Figure 1). For example, K140, and E595 likely form a dipole, enabling the interactions with the carboxylic acid and amine moieties of the N-terminus of the transported substrates, similarly to the corresponding residues in PepTSo (E419 and K127).17
Another structural feature shown in our models is the external gate, constituted by the conserved residues R34 and D298 (Figure 1C). The external gate is also highly conserved, with an acidic residue in the position of D298 observed across the POT family. The corresponding residue in GkPOT (E310) was shown to be involved in proton binding.13 In the unbound hPepT1 inward-open model, the external gate is closed through a salt bridge between R34 and D298 (Model 2, in pink; Figure 1C), whereas in the ligand-bound conformations this salt bridge is disrupted by the ligands (e.g., Model 4, in cyan; Figure 1C).
Structure-Based Ligand Discovery
We investigated which interactions involving the binding site might be needed for a molecule to serve as an hPepT1 inhibitor that binds the transporter, or as a substrate that binds hPepT1 and also gets transported. We conducted virtual screenings of the FDA-approved drugs and lead-like libraries from the ZINC database29 using three distinct models of hPepT1 (i.e., Models 1, 3, and 5; Table 1) (Materials and Methods Section). These models consist of binding sites with unique features and, thus, may cover different fractions of the chemical space in the docking screen. For the FDA-approved drug screen we selected compounds that bind multiple models (Materials and Methods Section). For example, aspartame was tested because (i) it was predicted to bind both Models 1 and 3, together with other known drugs (i.e., Cephadroxil); (ii) it has a peptide-like structure; (iii) its binding pose was similar to those of other peptide and peptide-like ligands. These observations suggested that aspartame might be a hPepT1 substrate. Furthermore, analysis of the literature revealed that a previous study identified aspartame as a hPepT1 inhibitor.43 In addition, for the lead-like screen, we visually inspected the top-ranked 200 molecules (Materials and Methods Section) and we prioritized compounds that were predicted to interact with the conserved binding site residues (i.e., R27, Y31, K140, or E595), as well as those compounds with unique chemotypes or mode of interactions with hPepT1.
Yeast Uptake Studies
Twenty-three compounds were tested to determine whether or not they would inhibit the uptake of the model dipeptide hPepT1 substrate [3H]-glycylsarcosine. Of these compounds, five compounds exhibited substantial inhibitory effects (>25%) and, as a result, underwent dose–response analyses (Figure 2A). Notably, aspartame, a peptide-like compound was the most potent ligand inhibiting the [3H]glycylsarcosine uptake as judged by having the lowest IC50 value (3.9 mM).
Figure 2.

Inhibition studies of five potential hPepT1 substrates on the uptake of GlySar. (A) 10 µM of 3H-GlySar was incubated with a series concentration of inhibitors at room temperature for 2 min, the percent uptake calculated (relative to control), and the results then fit to a dose-dependent inhibition model using Prism version 7.0 (mean ± SE, n = 3). (B) Experimentally confirmed ligands with (a) compound referring to the name of the compound; (b) 2D Sketch representing the two-dimensional structure of the compound; (c) IC50 referring to the experimental IC50 of each compound. The ranking of these compounds in their respective screens is provided in Table S1.
The other four inhibitors were less potent, having IC50 values that were four-times larger (12.0 mM, on average) (Figure 2B). hPepT1 is a low-affinity transporter, and thus, hPepT1 ligands with IC50 values in the mM may still interact with hPepT1 at clinically relevant concentrations.9 Consequently, the ligands discovered in this study have relevant IC50 values. Notably, these four inhibitors are chemically dissimilar from all other known hPepT1 ligands, exhibiting Tanimoto coefficient (Tc) of 0.38–0.43 compared to known hPepT1 ligands, using the Daylight fingerprints (Supporting Information). For example, compounds 3, 7, and 10 consist of carboxylic acid moieties, but lack an α-amino group, thus differing from known peptide-like hPepT1 ligands. Furthermore, compound 39 includes a unique triazole group that can potentially have improved bioavailability compared to known peptide-like drugs (Figure 2B).
Xenopus Oocyte Transport Studies
The transport of compounds 3, 7, 10, and 39 via hPepT1 was monitored directly in hPepT1-expressing oocytes by studying inward currents induced by these compounds under voltage-clamp conditions. This monitoring enables us to distinguish whether test compounds are transportable substrates for hPepT1 or are inhibitors that block the uptake of dipeptides by competing for the substrate-binding site in hPepT1 without being transported across the membrane. Unlabeled glycylsarcosine was used as a positive control and 1 mM concentrations robustly induced inward currents of 300–400 nA (buffer pH 6.0, clamped at −50 mV). In contrast, compounds 3, 7, 10, and 39 did not induce any current (Figure 3B and C). The I–V relationship induced by these compounds exhibited a profile of a moderate negative slope, suggesting that they might inhibit the leak proton through hPepT1. This is because the proton inward leak current flows constitutively through hPepT1, which is driven by the intracellular negative membrane potential even in the absence of a substrate. The feature implies that these compounds are inhibitors (Figure 3B and C). Conversely, aspartame and amino phenyl derivative, 4-amino phenyl acetic acid (4-amino PA, used as positive control) induced significant inward currents (Figure 3A, E, and F), indicating that aspartame and 4-amino PA are transportable substrates via hPepT1. This result confirmed our hypothesis based on the structural models.
Figure 3.

Determination of hPepT1-mediated substrate transport in Xenopus oocyte expression system. (A–C) Currents induced by compounds 3, 7, 10, and 39, glycylsarcosine and aspartame were monitored in hPepT1 cRNA-injected oocytes at pH6.0 using TEVC technique with varying membrane potential. (D and E) Inward currents induced by compounds 3, 7, 10, and 39, aspartame and 4-amino phenyl acetic acid (4-amino PA) were monitored at −50 mV and were expressed as 100% of 1 mM or 250 µM glycylsarcosine induced currents. (F) Tracers of representative 250 µM glycylsarcosine, 30 mM aspartame, and 10 mM 4-amino PA induced currents in hPepT1 cRNA-injected oocytes at pH6.0. Induced currents were monitored at membrane potential clamped at −50 mV.
Mode of Interaction of Substrates and Inhibitors
To rationalize the activities of the newly discovered hPepT1 ligands (i.e., four inhibitors and one substrate), we used QM-polarized ligand docking (QPLD), which increases the accuracy of docking by incorporating the partial charges of the ligands with quantum mechanics calculations.44 We then estimated the binding energies of each compound with the molecular mechanics generalized born surface area (MM-GBSA) solvation method from the Schrödinger package45 (Materials and Methods Section), known to be more accurate than docking scores.8 We docked the five newly discovered ligands as well as four known ligands (i.e., Ala-Phe, Ala-Ala-Ala, valacyclovir, and cephalexin) to all our models and their corresponding templates (representative ligands shown in Figure 4). As expected, the absolute values of the experimental IC50 estimates are not reproduced by the MM-GBSA energy calculations for any of the models. It is noted that computational methods often fail in predicting absolute binding energy due to a variety of approximations made in these calculations,46,47 and also, even if accurate, binding energy does not necessarily translate to being a substrate that get transported through hPepT1.48
Figure 4.

Docking poses of confirmed hPepT1 ligands. The known substrates (A) Ala-Phe and (B) the antiviral valacyclovir are represented in cyan sticks, and (C) the newly characterized hPepT1 substrate (aspartame) and (D) inhibitor (compound 3) are represented in purple sticks. The binding site residues of the dipeptide bound inward-open conformation model (Model 5) are shown in cyan lines. The hydrogen bonds between the ligands and binding site residues are represented in dashed lines.
However, the docking poses of Ala-Phe and Ala-Ala-Ala in the hPepT1 ligand-bound inward-open models (Models 4 and 5) as well as the docking poses of these ligands in their corresponding template PepTSt structures (with the ligands removed) recapitulated the binding poses in the bound X-ray structures. These ligand poses also resulted in the lowest predicted binding energies (i.e., Figure S2). Taken together, these results suggest that using the QPLD and MM-GBSA is appropriate to analyze the binding poses of the new ligands in the inward-open models, Models 4 and 5. Thus, we compared the predicted binding energies of the newly discovered ligands in these models. For the newly discovered ligands, the most similar trends between the IC50 of the compound and the predicted binding energies were observed for the inward-open, Model 5 (Figure S2), suggesting that this conformation best captures hPepT1-ligand interactions identified in this screen.
A comparison of this model in complex with the newly identified and previously known hPepT1 ligands show commonalities and differences among substrates and inhibitors. All ligands including both substrates and inhibitors form hydrogen bonds with the side chains of R27, Y31, K140, and Y167, and make π−π interactions with the indole group of W622 (Figure 4). This suggests that interactions with these residues are critical for small molecules to modulate the activity of hPepT1.
Moreover, two key differences are observed between substrates and inhibitors. First, substrates such as the peptide-like compound aspartame and the larger prodrug valacyclovir interact with E595 (Figure 4C and B), similarly to natural substrates (e.g., Ala-Phe; Figure 4A), while the newly discovered inhibitors do not interact with this residue. This suggests that the formation of a dipole moment is essential for transport by hPepT1. Second, the four inhibitors also consisted of aromatic rings predicted to interact with Y31 via π-stacking (Figure 4D). This interaction likely leads to improved inhibition of these hPepT1 inhibitors.
DISCUSSION
Despite the role and relevance of SLC transporters in disease and drug absorption, these proteins have only been recently acknowledged as the most understudied protein targets.49 hPepT1 is the primary di- and tripeptide transporter in the intestines, regulating the absorption of natural peptides and peptide-like drugs.9 The accumulation of structure–function data on various POT members has enabled us to better describe the transport mechanism of this important transporter. Here we modeled the hPepT1 structure and dynamics, and its mode of interaction with ligands, by combining computational modeling with functional assays. The following three major findings are presented in this study:
First, our models in distinct hPepT1 conformations visualize the flexibility of its binding site providing a framework for understanding hPepT1 ligand promiscuity. hPepT1 is a broad specificity transporter, carrying a range of small dipeptides and larger peptide-like drugs, ranging from 100 to 600 Da. Our homology models show two structural features that are associated with hPepT1 binding site’s flexibility: (i) the binding site is enclosed by two dynamic gates and the extracellular gate is constituted by a salt bridge between R34 and D298, with D298 as the putative proton binding site; and the intracellular gate is formed by two hairpin loops (i.e., TM4-TM5 and TM7-TM11) that are enriched with hydrophobic residues (Figure 1); and (ii) hPepT1’s binding site shape and size can vary substantially during transport and inhibition, depending on the ligand bound and the conformation of the transporter (Table 1). For instance, the orientation of the indole group of W622 can alternate, to accommodate the bound substrate, as observed in the inward-open Models 4 and 5, resulting in binding sites with distinct size (i.e., 154 Å3 and 239 Å3, respectively).
This visualization of the binding site flexibility of hPepT1 permitted us to develop hypotheses that better explain its diverse ligand-binding modes and broad specificity. Previously, using the prokaryotic POT structures and binding free-energy calculations, Samsudin et al. suggested that the biophysical features (e.g., charge) of the N-terminus of the transported peptide-like ligand are conserved, whereas the C-terminus of the ligand is more variable.47 Complementarily, our results show that the extracellular region of the binding site binds the N-terminus of the hPepT1 ligands through conserved hydrogen bonds with key residues (e.g., R27, Y31, K140, and Y167). Further, the conformation of the intracellular region of the binding site can vary to accommodate the variable C-terminus of the peptide-like ligands, thereby facilitating the ligand promiscuity of this transporter (Figure 5). These results expand on the previous analyses based on the prokaryotic homologues structures.
Figure 5.

hPepT1 binding site plasticity and ligand promiscuity. hPepT1 binding site is represented in blue and the key residues interacting with the ligands in black lines. (A) When no ligand is bound to hPepT1, the salt bridge between R34 and D298 (i.e., the external gate) is formed. (B) When a peptide binds, the N-terminus of the peptide, represented in red, disrupts the hydrogen bond to interact with R34 and other key residues such as E595 or N329. Additionally, D298 binds the cotransported proton. (C) Conversely, when an inhibitor binds to hPepT1, key interactions essential for transport are lacking, such as the hydrogen bond between the ligand with E595. The flexibility of the internal side of the binding site permits to accommodate the C-terminal side of the peptide or peptide-like substrates and drugs and inhibitors (represented in blue) of various sizes and shapes.
Second, analysis of the hPepT1 models with newly discovered and previously known ligands enable us to propose preliminary principles describing the chemical basis for distinguishing between hPepT1 substrates and inhibitors. We took a chemical biology approach that uses these proposed guidelines to identify four inhibitors and one substrate. In brief, all newly discovered ligands formed interactions with several conserved residues (e.g., R27). However, unlike peptide-like substrates, the newly discovered inhibitors lack an amine group, thus eliminating the interaction with E595. This suggests that the interaction of ligands with both K140 and E595 is essential for a molecule to be transported, in agreement with previous studies.17 Moreover, the newly discovered inhibitors make additional van der Waals interactions with Y31, thereby increasing their binding affinity. Interestingly, the new hPepT1 ligands were discovered by screening against inward-facing conformations, whereas in principle, outward-facing conformations, which are still unknown, would be preferable for designing inhibitors. Notably, it is possible that an inhibitor binds an inward conformation, as for example, was shown in a crystal structure of hGLUT1, a different MFS transporter.50 Based on the current data, we speculate on three potential scenarios of the newly discovered inhibitors’ mechanisms of inhibition: (i) the inhibitors bind to the outward-facing conformation of the transporter, which then changes conformation to the inward-conformation but then the molecules do not unbind, and therefore remain “stuck” in the transporter as it cycles between different conformation states; (ii) the compounds first cross the membrane through diffusion or a different transporter and then bind hPepT1 from the cytosol; (iii) the binding site of an outward conformation is similar to that of the inward conformation and the compounds bind the outward conformation without triggering conformational change. Overall, the new ligands and predicted mode of binding provide a starting point to build structure activity relationship models, to optimize potential substrate-like drugs with optimal oral performance characteristics, or inhibitors preventing the uptake of toxic peptides.
Third, we identified one substrate and four inhibitors at a high hit-rate (22%) for a highly challenging target for structure-based ligand discovery: (i) hPepT1 shares distant sequence similarity to its template structures (sequence identities of 22–34%); (ii) the template structures have been determined in low resolution (2.4–3.62 Å); and (iii) hPepT1 ligands include peptides and peptide-like compounds which are polar molecules with increased degrees of freedom and are, thus, harder for docking programs to handle; (iv) the binding site of hPepT1 is highly flexible and is thought to adopt different conformations based on the ligands it binds (Table 1; Figure 1). Therefore, approaches optimizing the model binding site to enrich for known hPepT1 ligands with docking failed for this target.
Overall, the newly characterized inhibitors interact with hPepT1 at physiologically relevant concentrations, exhibiting unique chemotypes that deviate from the peptidic structure of the known hPepT1 ligands (e.g., compound 39 contains a triazole ring). This identification of new nonpeptide ligands provides useful chemical tools to further characterize this transporter and highlights the increased applicability of structure-based drug design for human SLC transporters.
Supplementary Material
Acknowledgments
The authors declare no competing financial interest. This work has been supported in part by the National Institutes of Health grants R01 GM108911 (to A.S. and C.C.) and R01 GM115481 (to D.E.S.) and by the Department of Defense grant W81XWH-15-1-0539 (to A.S., C.C., and D.E.S.). We thank P.M.U. Ung for technical assistance and maintenance of the computational resources required for this study. We appreciate OpenEye Scientific Software, Inc. for granting us access to its high-performance molecular modeling applications through its academic license program. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
ABBREVIATIONS
- SLC
solute carrier
- IBD
inflammatory bowel disease
- POT
proton-dependent oligopeptide transporter
- PepT1
peptide transporter 1
Footnotes
ASSOCIATED CONTENT
- Template selection, PepT1-template alignment, homology modeling, selecting relevant docking programs, docking with AutoDock Vina, docking with FRED, chemical similarity calculations, docking with QM-polarized ligand docking, free energy estimation with molecular mechanics generalized born surface area solvation, binding sites of the hPepT1 models, MMGBSA calculations and experimental IC50s values, and compounds experimentally tested (PDF)
The authors declare no competing financial interest.
References
- 1.Daniel H, Kottra G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pfluegers Arch. 2004;447(5):610–618. doi: 10.1007/s00424-003-1101-4. [DOI] [PubMed] [Google Scholar]
- 2.Newstead S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta, Gen. Subj. 2015;1850(3):488–99. doi: 10.1016/j.bbagen.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith DE, Clemencon B, Hediger MA. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Aspects Med. 2013;34(2–3):323–36. doi: 10.1016/j.mam.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knutter I, Hartrodt B, Theis S, Foltz M, Rastetter M, Daniel H, Neubert K, Brandsch M. Analysis of the transport properties of side chain modified dipeptides at the mammalian peptide transporter PEPT1. Eur. J. Pharm. Sci. 2004;21(1):61–7. doi: 10.1016/s0928-0987(03)00141-6. [DOI] [PubMed] [Google Scholar]
- 5.Tamai I, Nakanishi T, Hayashi K, Terao T, Sai Y, Shiraga T, Miyamoto K, Takeda E, Higashida H, Tsuji A. The predominant contribution of oligopeptide transporter PepT1 to intestinal absorption of beta-lactam antibiotics in the rat small intestine. J. Pharm. Pharmacol. 1997;49(8):796–801. doi: 10.1111/j.2042-7158.1997.tb06115.x. [DOI] [PubMed] [Google Scholar]
- 6.Wenzel U, Thwaites DT, Daniel H. Stereoselective uptake of beta-lactam antibiotics by the intestinal peptide transporter. British journal of pharmacology. 1995;116(7):3021–7. doi: 10.1111/j.1476-5381.1995.tb15958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderle P, Nielsen CU, Pinsonneault J, Krog PL, Brodin B, Sadee W. Genetic variants of the human dipeptide transporter PEPT1. J. Pharmacol. Exp. Ther. 2005;316(2):636–646. doi: 10.1124/jpet.105.094615. [DOI] [PubMed] [Google Scholar]
- 8.Genheden S, R U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery. 2015;10(5):449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat. Rev. Drug Discovery. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsson P, Kihlberg J. How Big Is Too Big for Cell Permeability? J. Med. Chem. 2017;60(5):1662–1664. doi: 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]
- 11.Matsson P, Bergstrom CA. Computational modeling to predict the functions and impact of drug transporters. In silico pharmacology. 2015;3(1):8. doi: 10.1186/s40203-015-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beale JH, Parker JL, Samsudin F, Barrett AL, Senan A, Bird LE, Scott D, Owens RJ, Sansom MS, Tucker SJ, Meredith D, Fowler PW, Newstead S. Crystal Structures of the Extracellular Domain from PepT1 and PepT2 Provide Novel Insights into Mammalian Peptide Transport. Structure. 2015;23(10):1889–99. doi: 10.1016/j.str.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doki S, Kato HE, Solcan N, Iwaki M, Koyama M, Hattori M, Iwase N, Tsukazaki T, Sugita Y, Kandori H, Newstead S, Ishitani R, Nureki O. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl. Acad. Sci. U. S. A. 2013;110(28):11343–8. doi: 10.1073/pnas.1301079110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guettou F, Quistgaard EM, Raba M, Moberg P, Low C, Nordlund P. Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat. Struct. Mol. Biol. 2014;21(8):728–31. doi: 10.1038/nsmb.2860. [DOI] [PubMed] [Google Scholar]
- 15.Guettou F, Quistgaard EM, Tresaugues L, Moberg P, Jegerschold C, Zhu L, Jong AJ, Nordlund P, Low C. Structural insights into substrate recognition in proton-dependent oligopeptide transporters. EMBO Rep. 2013;14(9):804–10. doi: 10.1038/embor.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyons JA, Parker JL, Solcan N, Brinth A, Li D, Shah ST, Caffrey M, Newstead S. Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep. 2014;15(8):886–93. doi: 10.15252/embr.201338403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, Ingram JC, Carpenter EP, Sansom MS, McPherson MJ, Baldwin SA, Iwata S. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011;30(2):417–26. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solcan N, Kwok J, Fowler PW, Cameron AD, Drew D, Iwata S, Newstead S. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012;31(16):3411–21. doi: 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013;38(3):151–9. doi: 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. The alternating access transport mechanism in LacY. J. Membr. Biol. 2011;239(1–2):85–93. doi: 10.1007/s00232-010-9327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meredith D, Price RA. Molecular modeling of PepT1–towards a structure. J. Membr. Biol. 2006;213(2):79–88. doi: 10.1007/s00232-006-0876-6. [DOI] [PubMed] [Google Scholar]
- 22.Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, Ingram JC, Carpenter EP, Sansom MS, McPherson MJ, Baldwin SA, Iwata S. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011;30(2):417–26. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solcan N, Kwok J, Fowler PW, Cameron AD, Drew D, Iwata S, Newstead S. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012;31(16):3411–21. doi: 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36(7):2295–300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 26.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–24. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009;31(2):455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGann M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011;51(3):578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 29.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45(1):177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–5. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colas C, Grewer C, Otte NJ, Gameiro A, Albers T, Singh K, Shere H, Bonomi M, Holst J, Schlessinger A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015;11(10):e1004477. doi: 10.1371/journal.pcbi.1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colas C, Pajor AM, Schlessinger A. Structure-Based Identification of Inhibitors for the SLC13 Family of Na(+)/Dicarboxylate Cotransporters. Biochemistry. 2015;54(31):4900–8. doi: 10.1021/acs.biochem.5b00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colas C, Ung PM, Schlessinger A. SLC Transporters: Structure, Function, and Drug Discovery. MedChemComm. 2016;7(6):1069–1081. doi: 10.1039/C6MD00005C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colas C, Schlessinger A, Pajor AM. Mapping functionally important residues in the Na+/dicarboxylate cotransporter, NaDC1. Biochemistry. 2017;56:4432. doi: 10.1021/acs.biochem.7b00503. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Chen X, Smith DE. Species-dependent uptake of glycylsarcosine but not oseltamivir in Pichia pastoris expressing the rat, mouse, and human intestinal peptide transporter PEPT1. Drug Metab. Dispos. 2012;40(7):1328–35. doi: 10.1124/dmd.111.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyauchi S, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na(+)-coupled transporter for short-chain fatty acids. J. Biol. Chem. 2004;279(14):13293–6. doi: 10.1074/jbc.C400059200. [DOI] [PubMed] [Google Scholar]
- 37.Fujisawa Y, Kitagawa T, Miyake M, Nara T, Kamo N, Miyauchi S. Measurement of electric current evoked by substrate transport via bi-directional H+/oligopeptide transporter over-expressed in HeLa cells: electrogenic efflux and existence of a newly observed channel-like state. Arch. Biochem. Biophys. 2006;445(1):166–73. doi: 10.1016/j.abb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 38.Miyauchi S, Gopal E, Babu E, Srinivas SR, Kubo Y, Umapathy NS, Thakkar SV, Ganapathy V, Prasad PD. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim. Biophys. Acta, Biomembr. 2010;1798(6):1164–71. doi: 10.1016/j.bbamem.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Steiner HY, Naider F, Becker JM. The PTR family: a new group of peptide transporters. Mol. Microbiol. 1995;16(5):825–34. doi: 10.1111/j.1365-2958.1995.tb02310.x. [DOI] [PubMed] [Google Scholar]
- 40.Paulsen IT, Skurray RA. The POT family of transport proteins. Trends Biochem. Sci. 1994;19(10):404. doi: 10.1016/0968-0004(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 41.Pieri M, Gan C, Bailey P, Meredith D. The transmembrane tyrosines Y56, Y91 and Y167 play important roles in determining the affinity and transport rate of the rabbit proton-coupled peptide transporter PepT1. Int. J. Biochem. Cell Biol. 2009;41(11):2204–13. doi: 10.1016/j.biocel.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Haworth IS, Kulkarni AA, Bolger MB, Davies DL. Mutagenesis and cysteine scanning of transmembrane domain 10 of the human dipeptide transporter. Pharm. Res. 2009;26(10):2358–66. doi: 10.1007/s11095-009-9952-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ekins S, Johnston JS, Bahadduri P, D’Souza VM, Ray A, Chang C, Swaan PW. In vitro and pharmacophore-based discovery of novel hPEPT1 inhibitors. Pharm. Res. 2005;22(4):512–7. doi: 10.1007/s11095-005-2505-y. [DOI] [PubMed] [Google Scholar]
- 44.Cho AE, Guallar V, Berne BJ, Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM)approach. J. Comput. Chem. 2005;26:915–931. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct., Funct., Genet. 2004;55(2):351–67. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 46.Oehme DP, Brownlee RT, Wilson DJ. Effect of atomic charge, solvation, entropy, and ligand protonation state on MMPB(GB)SA binding energies of HIV protease. J. Comput. Chem. 2012;33(32):2566–80. doi: 10.1002/jcc.23095. [DOI] [PubMed] [Google Scholar]
- 47.Samsudin F, Parker JL, Sansom MS, Newstead S, Fowler PW. Accurate Prediction of Ligand Affinities for a Proton-Dependent Oligopeptide Transporter. Cell chemical biology. 2016;23(2):299–309. doi: 10.1016/j.chembiol.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colas C, Smith DE, Schlessinger A. Computing Substrate Selectivity in a Peptide Transporter. Cell Chem. Biol. 2016;23(2):211–3. doi: 10.1016/j.chembiol.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cesar-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, Reithmeier RA, Hepworth D, Hediger MA, Edwards AM, Superti-Furga G. A Call for Systematic Research on Solute Carriers. Cell. 2015;162(3):478–87. doi: 10.1016/j.cell.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 50.Kapoor K, Finer-Moore JS, Pedersen BP, Caboni L, Waight A, Hillig RC, Bringmann P, Heisler I, Muller T, Siebeneicher H, Stroud RM. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. U. S. A. 2016;113(17):4711–6. doi: 10.1073/pnas.1603735113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durrant JD, Votapka L, Sorensen J, Amaro RE. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J. Chem. Theory Comput. 2014;10(11):5047–5056. doi: 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.