

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is characterized by progressive enlargement of kidney cysts leading to chronic kidney disease (CKD) and end-stage renal disease (ESRD). Identification of an early biomarker that can predict progression of CKD is urgently needed. In an earlier Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) study (a prospective, multicenter, observational analysis of 241 patients with ADPKD initiated in 2000), baseline height-adjusted total kidney volume (htTKV) was shown to be associated with development of CKD stage 3 after eight years of follow-up. Here we conducted an extended study and found that in a multivariable logistic regression model, baseline htTKV was shown to be a strong, independent predictor for the development of CKD after a median follow-up of 13 years. The odds ratio of reaching each CKD stage per 100 mL/m increment in htTKV was 1.38 (95% confidence interval 1.19–1.60) for stage 3, 1.42 (1.23–1.64) for stage 4, and 1.35 (1.18–1.55) for stage 5 or ESRD. Baseline htTKV was also associated with relative decreases in the glomerular filtration rate of 30%, and 57% or more. Moreover, the rate of change in htTKV was negatively correlated with the slope of the glomerular filtration rate. While ADPKD genotype was also associated with CKD outcomes, it was not an independent prognostic factor after adjusting for htTKV. Thus, baseline total kidney volume and the rate of kidney growth are strongly associated with the development of advanced stages of CKD. These findings support the use of total kidney volume as a prognostic and potentially monitoring biomarker in ADPKD.
Keywords: ADPKD, chronic kidney disease
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a life-threatening genetic disease primarily affecting adults.1,2 It is caused predominantly by mutations in two genes, PKD1, which accounts for about 80% of cases, and PKD2, which accounts for 15% of cases. In ADPKD, kidney cysts likely begin forming before birth3 and grow exponentially throughout life.4 During this time, cysts progressively compress and injure neighboring structures, including tubules and vasculature, and incite inflammation and eventually interstitial fibrosis. However, the glomerular filtration rate (GFR) is preserved for several decades, likely due to compensatory hyperfiltration of the remaining functional nephrons. Patients eventually reach end-stage renal disease (ESRD) at a median age of 54 years for PKD1 and 74 years for PKD2 mutations5. There is also considerable allelic heterogeneity in the PKD1 mutations, with a poorer prognosis for patients with truncating mutations and with nontruncating mutations that are predicted to be highly pathogenic.6,7
There is currently no therapy approved for the treatment of ADPKD in the United States. Drugs that slow cyst growth are likely to show greatest benefit in childhood or early adulthood when cyst growth has not yet caused irreparable damage.8 Because of the long natural history of the disease, however, therapeutic trials conducted in early disease are unlikely to show improvement in endpoints considered to be clinically meaningful by regulatory agencies, such as doubling of the serum creatinine or end-stage renal disease (ESRD). Conversely, trials of therapy at later stages of the disease when GFR has begun to decline, and irreversible kidney injury may already have supervened, are less likely to show a benefit. For this reason, there is an urgent need for robust biomarkers of early ADPKD that are predictive of later decline in GFR and progression to ESRD.9 Biomarkers that are directly in the causal pathway for the disease have particular value as they can potentially serve as predictors of the efficacy of drug therapy.10
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) is a prospective, longitudinal, observational cohort study of ADPKD that was conceived in 2000.4 A cohort of young adults with well-preserved GFR was selected with the goal of discovering biomarkers in early disease that could predict long-term renal outcomes. CRISP I showed that MRI could be used to accurately quantify total kidney volume (TKV),11 that TKV increased exponentially with time, and that the increase in TKV was accounted for by the increase in total cyst volume, demonstrating that TKV is an informative marker of disease progression.4,12 CRISP I provided the first indication that baseline TKV was weakly associated with a decline in GFR, after just 3 years of follow-up.4 In CRISP II, with 8 years of follow-up available, baseline TKV adjusted for height (htTKV) was shown to be associated with development of CKD stage 3.13
However, it is unknown whether htTKV has prognostic value over longer durations of follow-up and whether it is prognostic of advanced, clinically significant CKD endpoints. Moreover, the relative value of ADPKD genotype as compared to htTKV, as prognostic biomarkers, is unknown. The CRISP study is uniquely positioned to answer these important questions. We report here the outcomes of CRISP III, now with up to 14.5 years of follow-up, and test the association of baseline htTKV and ADPKD genotype with the development of advanced stages of CKD.
RESULTS
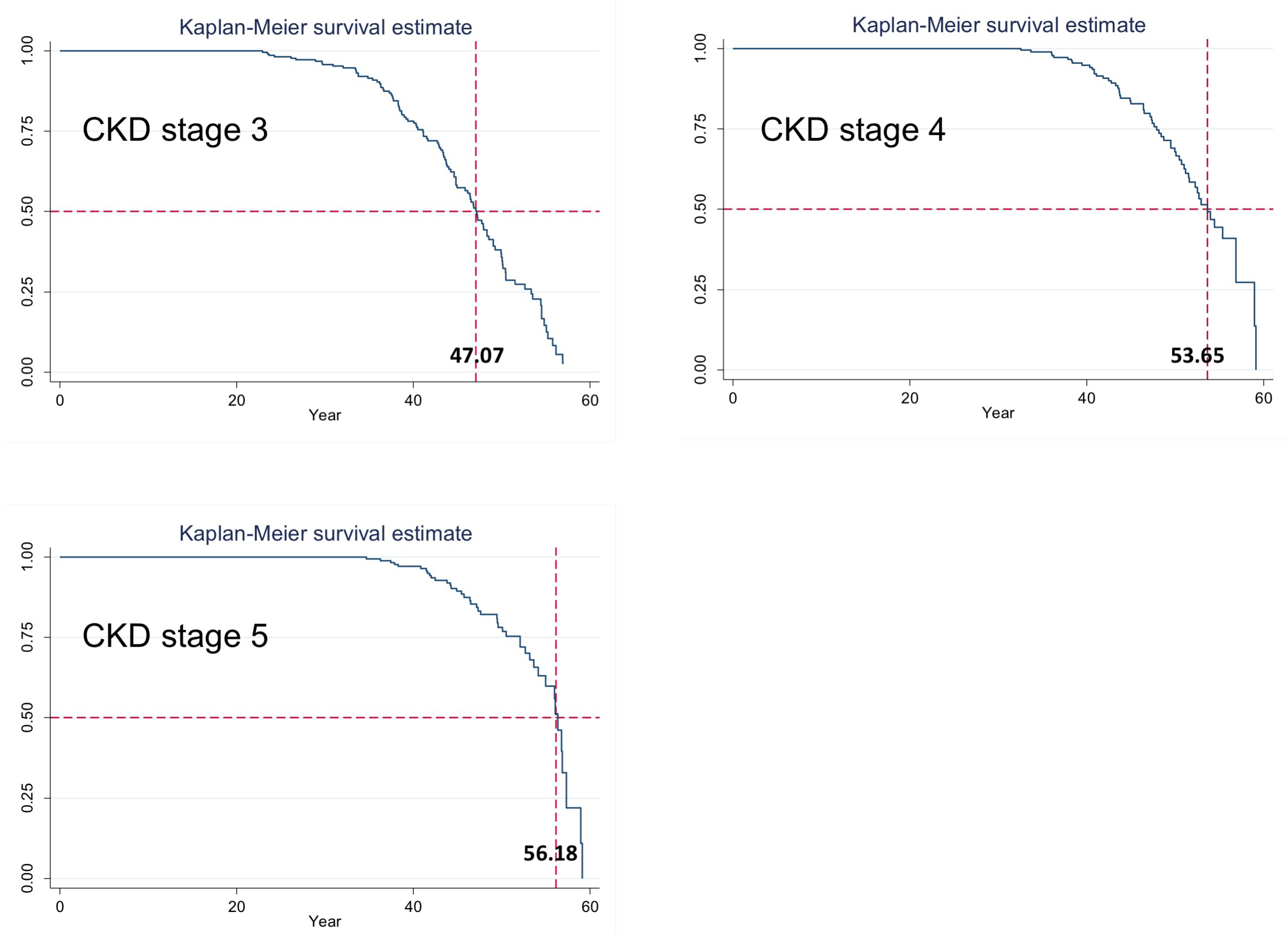
241 patients were originally enrolled in CRISP. Fig. 1 shows the flow of participants and the number included in the current analysis. The baseline characteristics of the primary study cohort of 184 patients is shown in Table 1. The median follow-up duration was 13.0 years (mean 11.3, maximum 14.5). By the end of the follow-up period, 50.0% of subjects had reached CKD stage 3 or higher as defined by measured GFR, with 25.7% reaching CKD stage 4 or higher, and 18.6% reaching CKD stage 5 or ESRD (Table 2). Outcomes were similar when the CKD stages were classified according to eGFR calculated by either MDRD or CKD-EPI equations. The projected median age to reach each CKD stage, determined by Kaplan-Meier survival analysis, was 47.1 years for CKD stage 3, 53.7 years for CKD stage 4, and 56.2 years for CKD stage 5 or ESRD (Supplemental Fig. S1).
Fig. 1.
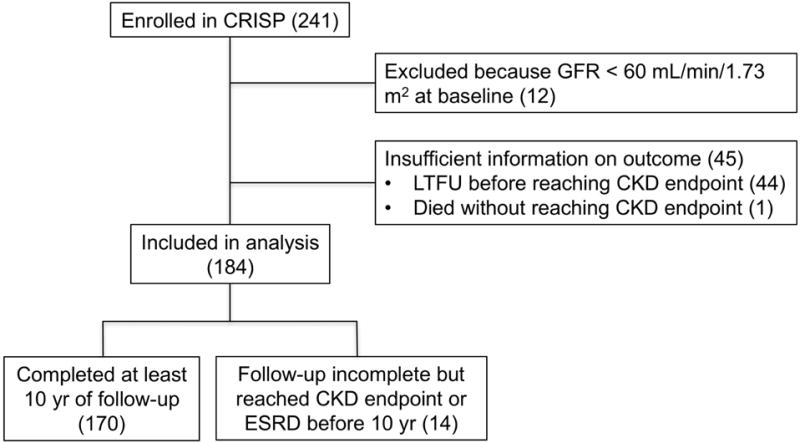
Flow diagram showing the enrolled study participants that were included in the final analysis. Numbers shown are for analysis of the outcome of CKD stage 3, as determined from measured GFR. LTFU, lost to follow-up.
Table 1.
Patient characteristics at baseline
| Characteristics | Study cohorta |
|---|---|
|
| |
| Total patients – no. | 184 |
| Male sex – no. (%) | 76 (41.3) |
| Age – yr | 32.1 (8.9) |
| Race – no. (%) | |
| White | 162 (88.0) |
| African-American | 19 (10.3) |
| Other | 3 (1.7) |
| Hypertension – no. (%) | 75 (41) |
| Height – cm | 172.5 (11.0) |
| BMI – kg/m2 | 25.8 (5.3) |
| Serum creatinine – mg/dL | 0.9 (0.2) |
| Measured GFRb – mL/min/1.73 m2 | 97.6 (23.5) |
| eGFRc – mL/min/1.73 m2 | 93.6 (22.6) |
| htTKV – mL/m | 501.4 (442.9) |
| PKD genotyped – no. (%) | |
| PKD1, truncating (MSG1) and non-truncating MSG2 | 129 (75) |
| PKD1, non-truncating MSG3 | 16 (9.3) |
| PKD2 and NMD | 27(15.7) |
Continuous variables are summarized as mean (S.D.) except for htTKV, which is shown as median (interquartile range).
Measured by iothalamate clearance and corrected for body surface area
Estimated by CKD-EPI formula
Total 182 patients (2 missing data). MSG2, mutation strength group 2; MSG3, mutation strength group 3; NMD, no mutation detected
Table 2.
Distribution of CKD stages at baseline and after 12 years of follow-up
| Renal insufficiency endpoint | Baseline no. (%) | Year 12 no. (%) |
|---|---|---|
| Measured GFR (iothalamate) | ||
| >90 (CKD Stage 1) | 135 (56.0) | 32 (14.2) |
| 60–89 (CKD Stage 2) | 94 (39.0) | 81 (35.8) |
| 30–59 (CKD Stage 3) | 12 (5.0) | 55 (24.3) |
| 15–29 (CKD Stage 4) | 0 (0.0) | 16 (7.1) |
| <15 (CKD Stage 5) or ESRD | 0 (0.0) | 42 (18.6) |
| GFR decreased ≥ 30% | 143 (71.1) | |
| GFR decreased ≥ 57% | 76 (40.4) | |
|
| ||
| eGFR (MDRD formula) | ||
| >90 (CKD Stage 1) | 96 (39.8) | 12(5.2) |
| 60–89 (CKD Stage 2) | 121 (50.2) | 92 (39.7) |
| 30–59 (CKD Stage 3) | 24 (10.0) | 64 (27.6) |
| 15–29 (CKD Stage 4) | 0 (0.0) | 22 (9.5) |
| <15 (CKD Stage 5) or ESRD | 0 (0.0) | 42 (18.1) |
| GFR decreased ≥ 30% | 139 (67.5) | |
| GFR decreased ≥ 57% | 77 (38.7) | |
|
| ||
| eGFR (CKD-EPI formula) | ||
| >90 (CKD Stage 1) | 121 (50.2) | 20 (8.9) |
| 60–89 (CKD Stage 2) | 102 (42.3) | 80 (35.6) |
| 30–59 (CKD Stage 3) | 18 (7.5) | 57 (25.3) |
| 15–29 (CKD Stage 4) | 0 (0.0) | 26 (11.5) |
| <15 (CKD Stage 5) or ESRD | 0 (0.0) | 42 (18.7) |
| GFR decreased ≥ 30% | 140 (70.0) | |
| GFR decreased ≥ 57% | 79 (39.7) | |
GFR values in mL/min/1.73 m2
To examine the relationship between htTKV at baseline and renal function at follow-up, baseline htTKV was plotted against the GFR values at each of the 8 visits over the course of the study (Fig. 2). At baseline, there was a weak negative correlation between htTKV and GFR (r=−0.40). With increasing follow-up time, the correlation grew stronger and the slope became steeper up to Year 8, after which increasing numbers of patients began to reach ESRD. This indicates that GFR declined faster in patients with higher baseline htTKV.
Fig. 2.
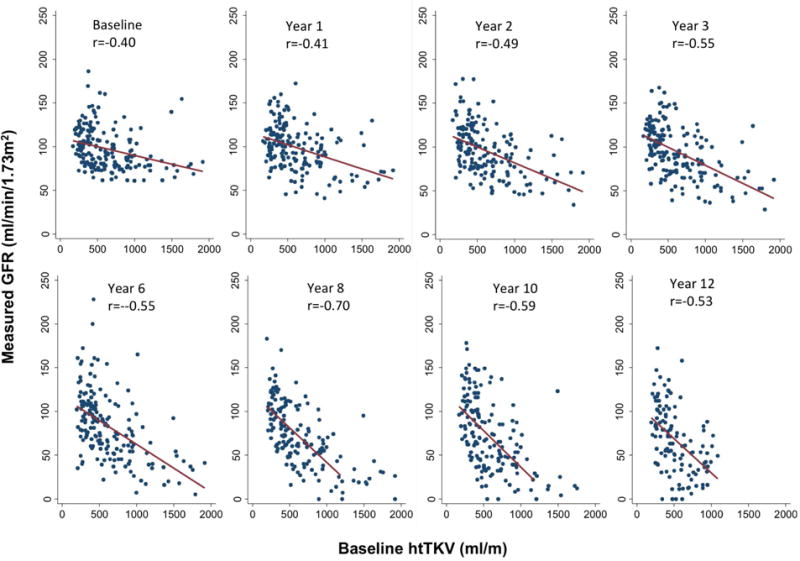
Linear correlations between baseline htTKV and iothalamate GFR during follow-up. Best-fit lines and Spearman’s correlation coefficients (r) were determined at baseline and 7 subsequent visits. htTKV, height-adjusted total kidney volume. P values for all time points < 0.0001.
To test the strength of the association of baseline htTKV with different categories of CKD, we used logistic regression models. Baseline htTKV was a strong independent predictor of the development of advanced stages of CKD both in an unadjusted model, and after multivariable adjustment for baseline age, sex, race, body mass index, and GFR (Table 3). In the adjusted model, the odds ratio of reaching each CKD stage per 100 mL/m increment in htTKV was 1.38 (95% CI 1.19–1.60) for stage 3, 1.42 (95% CI 1.23–1.64) for stage 4, and 1.35 (95% CI 1.18–1.55) for stage 5 or ESRD. The full set of regression coefficients for the multivariable model are listed in Supplemental Table 1. The models were well-calibrated and fit the observed data well, with the Hosmer-Lemeshow goodness-of-fit test yielding non-significant P values of 0.25, 0.79 and 0.45 for the outcomes of CKD stage 3, stage 4 and stage 5 or ESRD, respectively. Similar results were obtained when the GFR was estimated by the MDRD or CKD-EPI equations. A 57% decline in GFR (corresponding to a doubling of serum creatinine) is an established surrogate endpoint for clinical trials of kidney disease, and it has been recently been proposed that a 30% decline in GFR could serve a similar purpose.14 As shown in Table 3, baseline htTKV was strongly associated with the endpoints of a 30% and a 57% decline in GFR from baseline.
Table 3.
Multivariable analyses to evaluate association of baseline htTKV with CKD progression
| Renal insufficiency endpoint |
Unadjusted model |
Adjusted model*
|
|||||
|---|---|---|---|---|---|---|---|
| Measured GFR (iothalamate) | N | OR | 95% CI | P value | OR | 95% CI | P value |
| 30–59 (CKD Stage 3) | 184 | 1.53 | (1.31, 1.77) | <0.001 | 1.38 | (1.19, 1.60) | <0.001 |
| 15–29 (CKD Stage 4) | 179 | 1.52 | (1.33, 1.75) | <0.001 | 1.42 | (1.23, 1.64) | <0.001 |
| <15 (CKD Stage 5) or ESRD | 177 | 1.44 | (1.26, 1.64) | <0.001 | 1.35 | (1.18, 1.55) | <0.001 |
| GFR decreased ≥ 30% | 182 | 1.66 | (1.36, 2.02) | <0.001 | 1.61 | (1.31, 1.98) | <0.001 |
| GFR decreased ≥ 57% | 180 | 1.49 | (1.30, 1.70) | <0.001 | 1.43 | (1.24 1.65) | <0.001 |
| eGFR (MDRD formula) | |||||||
| 30–59 (CKD Stage 3) | 183 | 1.91 | (1.54, 2.37) | <0.001 | 1.69 | (1.35, 2.11) | <0.001 |
| 15–29 (CKD Stage 4) | 181 | 1.51 | (1.32, 1.73) | <0.001 | 1.43 | (1.25, 1.65) | <0.001 |
| <15 (CKD Stage 5) or ESRD | 177 | 1.44 | (1.26, 1.64) | <0.001 | 1.35 | (1.18, 1.55) | <0.001 |
| GFR decreased ≥ 30% | 181 | 1.55 | (1.30, 1.86) | <0.001 | 1.56 | (1.28, 1.91) | <0.001 |
| GFR decreased ≥ 57% | 180 | 1.48 | (1.29, 1.68) | <0.001 | 1.40 | (1.22, 1.60) | <0.001 |
| eGFR (CKD-EPI formula) | |||||||
| 30–59 (CKD Stage 3) | 183 | 1.93 | (1.56, 2.40) | <0.001 | 1.71 | (1.38, 2.14) | <0.001 |
| 15–29 (CKD Stage 4) | 180 | 1.55 | (1.35, 1.79) | <0.001 | 1.47 | (1.27, 1.70) | <0.001 |
| <15 (CKD Stage 5) or ESRD | 177 | 1.44 | (1.26, 1.64) | <0.001 | 1.35 | (1.18, 1.55) | <0.001 |
| GFR decreased ≥ 30% | 181 | 1.54 | (1.29, 1.84) | <0.001 | 1.45 | (1.21, 1.74) | <0.001 |
| GFR decreased ≥ 57% | 180 | 1.46 | (1.28, 1.66) | <0.001 | 1.39 | (1.22, 1.59) | <0.001 |
GFR values in mL/min/1.73 m2
OR, odds ratio of reaching CKD stage per 100 mL/m increment in baseline htTKV
Adjusted for age, sex, race, BMI and baseline GFR
Because we had excluded 12 patients that had baseline CKD stage 3, albeit very mild (median iothalamate GFR 58.1 mL/min/1.73 m2), we also conducted a sensitivity analysis in which we included them in the multivariable logistic regression model. The association between baseline htTKV and CKD endpoints was very similar (“Full cohort” in Supplemental Table 2). In addition, it has been suggested that in ADPKD patients with atypical kidney morphology such as very few, large cysts, marked asymmetry, or renal atrophy, kidney volume might not be as good in predicting disease progression.15 In CRISP, there were only 4 patients that had atypical imaging findings (Class 2 in Irazabal et al.15) and when they were excluded from the logistic regression analysis, the results were essentially unchanged (“Class I patients only” in Supplemental Table 2). Finally, a subset of patients in the CRISP cohort participated in either the HALT-PKD study (59 patients), a trial of angiotensin-converting enzyme inhibitor (ACEI) versus ACEI combined with an angiotensin receptor blocker, and of standard versus intensive blood pressure control,16,17 or in the TEMPO 3:4 study (24 patients), a placebo-controlled trial of tolvaptan.18 To exclude the possibility that modification of the rate of progression of the disease by one of these interventions could have skewed the overall results, we tested the effect of excluding all patients enrolled in these two trials. As shown in Supplemental Table 3, after exclusion of either the HALT-PKD subjects or the TEMPO subjects, the association of htTKV with CKD outcomes remained nearly the same.
Kidney volumes are highly variable in ADPKD. In the CRISP cohort, baseline htTKV ranged from 168 ml/m to 2113 ml/m. To better convey the effect size of baseline htTKV on CKD progression, we divided the cohort into quintiles by baseline htTKV. As compared to the lowest quintile of htTKV, the odds ratio of developing CKD stage 5 or ESRD was 1.4 for quintile 2, 2.1 for quintile 3, 4.1 for quintile 4 and 19.1 for quintile 5 (Table 4). Similarly large effect sizes were evident for other CKD outcomes. In short, htTKV, as measured by MRI at a single time point, is a powerful predictor for the development of advanced stages of CKD over the ensuing 13 years.
Table 4.
Odds ratio by quintile of baseline htTKV
| 1st quintile | 2nd quintile | 3rd quintile | 4th quintile | 5th quintile | |
|---|---|---|---|---|---|
| Mean htTKV within each quintile, mL/m (range) | 260 (168, 312) |
379 (314, 429) |
504 (433, 603) |
738 (605, 908) |
1218 (909, 2113) |
| N | 37 | 37 | 37 | 37 | 36 |
| Odds ratio of reaching each renal insufficiency endpoint (as defined by measured GFR)* | |||||
| 30–59 (CKD Stage 3) | 1.0 | 1.5 | 2.2 | 4.6 | 21.5 |
| 15–29 (CKD Stage 4) | 1.0 | 1.5 | 2.4 | 5.2 | 31.3 |
| <15 (CKD Stage 5) or ESRD | 1.0 | 1.4 | 2.1 | 4.1 | 19.1 |
| GFR decreased ≥ 30% | 1.0 | 1.8 | 3.2 | 9.2 | 101.7 |
| GFR decreased ≥ 57% | 1.0 | 1.5 | 2.4 | 5.4 | 33.5 |
GFR values in mL/min/1.73 m2
Odds ratios are relative to the odds in the 1st quintile, using the adjusted model
If there is a causative relationship between kidney growth and loss of kidney function in ADPKD, we would also expect to observe a correlation between the rate of kidney growth and the rate of decline in GFR. To test this hypothesis, we plotted the kidney growth rate, expressed as the annual percentage change in htTKV, against the annualized rate of change in GFR for each patient over the course of the study. As shown in Fig. 3, there was a negative correlation between the rate of kidney growth and GFR change (r=−0.38, p<0.01).
Fig. 3.
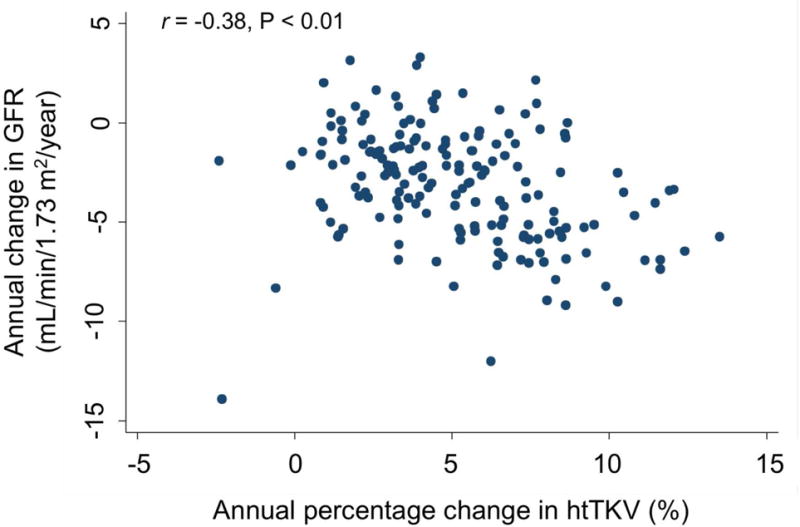
Scatter plots showing the correlation between annual percentage change in htTKV, determined over the course of the entire study (A) or using data from the first 4 years only (B), and the annual change in iothalamate GFR during the study. Spearman’s correlation coefficients (r) are shown.
ADPKD genotype, specifically the causal gene and, in the case of PKD1, the predicted pathogenicity of the allele, is associated with ADPKD disease severity and progression.5–7 To test the association of genotype with CKD progression in CRISP, we categorized the genotypes into 3 groups based on their predicted pathogenicity7 and tested them in our regression model. As shown in Table 5, genotype was a strong predictor of CKD outcomes, after adjustment for baseline age, sex, race, body mass index, and GFR. For example, patients with PKD1 truncating mutations or non-truncating mutations in MSG2 had an odds ratio of reaching CKD stage 5 or ESRD of 7.82 (95% CI 1.62–37.87, P = 0.011), compared to patients with PKD2 mutations or no mutation detected (NMD). The odds ratio of reaching CKD stage 5 or ESRD for patients with PKD1 non-truncating mutations in MSG3 was 6.42 (95% CI 0.82–50.11), but due to the small number of patients in this category, this was not statistically significant (P = 0.076). When ADPKD genotype and htTKV were combined into a single model, the estimated effect size due to genotype was greatly diminished and no longer statistically significant (Table 5).
Table 5.
Models testing the association of genotype with CKD outcomes
| Characteristics | Models with genotypea | Models with both genotype and htTKVa | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| OR | 95% CI | P value | OR | 95% CI | P value | |
|
CKD Stage 3 | ||||||
| htTKVb | 1.36 | 1.16, 1.58 | <0.001 | |||
| ADPKD genotypec | ||||||
| PKD1, MSG1 & MSG2 | 3.75 | 1.52, 9.28 | 0.004 | 1.90 | 0.72, 5.02 | 0.195 |
| PKD1, MSG3 | 3.61 | 0.85, 15.44 | 0.083 | 2.10 | 0.48, 9.23 | 0.326 |
|
CKD Stage 4 | ||||||
| htTKVb | 1.38 | 1.19, 1.60 | <0.001 | |||
| ADPKD genotypec | ||||||
| PKD1, MSG1 & MSG2 | 6.00 | 1.79, 20.16 | 0.004 | 2.49 | 0.67, 9.26 | 0.175 |
| PKD1, MSG3 | 2.57 | 0.43, 15.34 | 0.301 | 1.38 | 0.20, 9.28 | 0.744 |
|
CKD Stage 5 or ESRD | ||||||
| htTKVb | 1.31 | 1.13, 1.51 | <0.001 | |||
| ADPKD genotypec | ||||||
| PKD1, MSG1 & MSG2 | 7.82 | 1.62, 37.87 | 0.011 | 3.33 | 0.64, 17.27 | 0.151 |
| PKD1, MSG3 | 6.42 | 0.82, 50.11 | 0.076 | 3.74 | 0.45, 31.07 | 0.222 |
All models adjusted for sex, race, BMI, baseline age and GFR
Odds ratio of reaching CKD stage per 100 mL/m increment in baseline htTKV
Referenced to baseline genotype group of PKD2 and NMD
To compare the goodness-of-fit of models that included ADPKD genotype and/or htTKV, likelihood ratio tests (LRTs) were conducted (Suppl. Table S4). The LRTs showed that models that included ADPKD genotype and htTKV, either individually or jointly, were significant compared to the base model. HtTKV was also significant versus the model with all other variables, but ADPKD genotype did not add significantly to the model with htTKV already added to the base model (Model 4 vs. Model 3 in Suppl. Table 4). To quantify the added prognostic ability for models that included ADPKD genotype and/or htTKV, the net reclassification index (NRI) was calculated (Fig. 4 and Suppl. Table S5). htTKV had the greatest impact on prognosis with an NRI in the range of 0.20–0.31 when compared to the base model and 0.15–0.27 when compared to the model already including genotype. ADPKD genotype also improved prognosis versus the base model with an NRI of up to 0.18, but the NRI was less than 0.10 when ADPKD genotype was added to a model that already included htTKV.
Fig. 4.
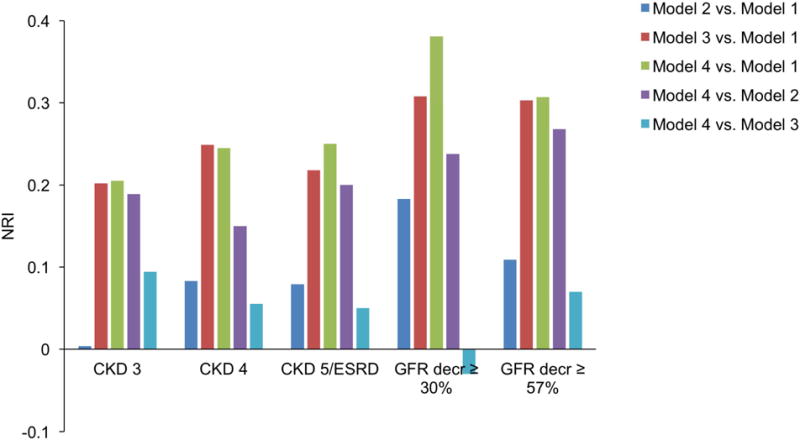
Improvement in the ability of the models to predict various CKD outcomes after addition of htTKV and/or ADPKD genotype, as assessed by the net reclassification index (NRI). Model 1 (Base model): Sex, race, BMI, baseline age and GFR; Model 2: Base model + genotype; Model 3: Base model + htTKV; Model 4: Base model + genotype + htTKV.
Finally, Irazabal et al. recently devised a prognostic marker in ADPKD patients with typical bilateral diffuse cystic kidney disease (Class 1), in which htTKV at a single time point is adjusted for age, thereby estimating the intrinsic rate of kidney growth, and then categorized into one of five subclasses, 1A to 1E.15 Irazabal class was found to be a strong prognostic marker of subsequent eGFR slope. Indeed in our model, Irazabal class improved the prediction of advanced stages of CKD when added to models that included sex, race, BMI, baseline age, GFR and ADPKD genotype (Suppl. Table 6). However, the NRI were no different when compared to the values determined for the equivalent models using htTKV instead (Suppl. Table 5), as indicated by overlapping 95% confidence intervals. This shows that htTKV and Irazabal class have equivalent discriminatory ability.
DISCUSSION
The association between TKV and decline in GFR has been demonstrated now in multiple longitudinal studies of ADPKD patients.4,13,19–27 In one of the earliest, the University of Colorado Natural History Study of ADPKD followed 229 patients for a mean of 7.8 years and showed an association between kidney volume, estimated from ultrasound examination, and the rate of decline in eGFR.21 Subsequent studies, based largely on the findings of CRISP I,4,11 have mostly used MRI to obtain the most accurate estimates of TKV. In a cross-sectional study of 100 patients in the SUISSE ADPKD cohort, htTKV determined by MRI was negatively correlated with eGFR.27 In a large cohort from China, baseline TKV was significantly associated both with baseline eGFR and with the rate of decline in eGFR over 14 months.25 The Mayo Clinic retrospectively studied a longitudinal cohort with a median follow-up of 6 years and showed a strong independent association between baseline htTKV and subsequent decline in eGFR.15
CRISP is unique in being the longest prospectively studied cohort with TKV data determined rigorously by MRI, and GFR measured by iothalamate clearance. By design, it enrolled adult patients that were relatively young and had preserved GFR at baseline, with the goal of identifying early biomarkers that could predict renal outcomes occurring far into the future.4,11 Baseline kidney volume was shown in CRISP I to be associated with decline in GFR at 3 years,4 and in CRISP II with development of CKD stage 3 after 8 years,13 which constitutes the longest duration of follow-up in any study to date. No individual study so far has had sufficient patients and length of follow-up to show an association of TKV with advanced CKD outcomes, including ESRD.
The PKD Outcomes Consortium, which was formed in 2010 to facilitate clinical trial development for ADPKD therapies, aggregated data from CRISP I and II, together with the Colorado, Mayo and Emory registry cohorts into a pooled dataset of 1140 patients that had baseline TKV and eGFR data, representing the largest ADPKD dataset to date. In a Cox model analysis, log-transformed baseline TKV was shown to be associated with increased hazards of 30% and 57% reduction in eGFR, and ESRD.28 On the basis of this data, the FDA qualified TKV as a prognostic biomarker for clinical trial enrichment in patients with ADPKD.29 However, because this was a pooled dataset and included registry data that was not systematically acquired in the setting of a formal clinical study, there was heterogeneity in the modalities used for kidney imaging, varying definitions for covariates and endpoints, and substantial missing data, and the median length of follow-up was only ~5 years.
CRISP III represents the first report of a large, prospectively studied ADPKD cohort with sufficient duration of follow-up to rigorously establish an association between kidney volume and advanced CKD endpoints. These endpoints, which include 57% reduction in GFR, CKD stage 5, and ESRD, are considered important both from a clinical perspective and for the purpose of evaluation of drug efficacy by regulatory agencies.30 Because ESRD, and even a 57% reduction in GFR (which is equivalent to a doubling of serum creatinine concentration), are relatively late events in CKD progression, a 30% reduction in GFR has also been proposed as an alternative end-point.14 We show here that htTKV is also associated with a 30% reduction in GFR.
In addition to demonstrating the prognostic value of htTKV at a single time point, we also show that the rate of change in htTKV is significantly correlated with the rate of change in GFR over the course of the study. A very similar correlation was observed in the HALT Study A control (standard blood pressure) group (r = −0.26, unpublished data), providing strong validation of kidney volume as a biomarker that tracks with change in kidney function over time.
We now know that the increase in TKV in ADPKD is entirely accounted for by the growth in cyst volume.4 Furthermore, there is good experimental evidence to suggest that cyst growth directly causes renal insufficiency. As cysts grow, they progressively obstruct tubules both locally and in upstream nephron units, distort the vasculature and compromise renal blood flow, ultimately leading to inflammation and interstitial fibrosis.31 In animal models, interventions that block cyst growth sufficiently early in the disease can effectively preserve normal renal function.32 Our findings that greater kidney volume temporally precedes and is strongly associated with higher rates of CKD progression, and that the rate of kidney growth is correlated with the rate of decline in GFR, support the hypothesis that cyst growth and hence kidney enlargement is in the causal pathway leading to GFR loss and kidney failure.
ADPKD genotype has also been employed as a prognostic biomarker in combination with clinical factors.33 Because ADPKD genotype affects cyst and hence kidney volume,7,34 it is unclear whether it bears any prognostic information independent of htTKV. We show here that while ADPKD genotype is a prognostic biomarker of advanced CKD outcomes in the CRISP cohort, it is not independently associated with outcomes after adjusting for baseline htTKV. We speculate that this is because htTKV acts as a mediator of the effect of ADPKD genotype on kidney function, but it also reflects other environmental, lifestyle and genetic factors. Addition of ADPKD genotype to a model that already includes htTKV appeared to yield some limited improvement in prognosis, with NRI values of 0.05 to 0.10 for most outcomes. However, the model improvement was not statistically significant, with non-significant likelihood ratio tests for all outcomes. Therefore, ADPKD genotype seems to yield limited benefit when added to htTKV and the other baseline variables. A number of other clinical and biochemical variables have also been suggested as prognostic biomarkers, including proteinuria, hypertension, HDL cholesterol, hematuria, and urinary MCP.35 However each one of these, individually, has limited discriminatory ability and none are superior to htTKV.
In conclusion, our findings show evidence of a strong and consistent association between total kidney volume and the clinical endpoint of ESRD, and support its use as a prognostic biomarker and potentially also as a monitoring biomarker in ADPKD.
CONCISE METHODS
Detailed descriptions of the CRISP study protocol and the baseline characteristics of the cohort have been published previously.4,11 Between January 4, 2001 and October 18, 2002, 241 patients were enrolled at four centers: University of Alabama at Birmingham, Emory University in Atlanta, the University of Kansas Medical Center in Kansas City, and the Mayo Clinic College of Medicine in Rochester, Minnesota. The study was approved by the relevant institutional review board at each site, and all participants gave written informed consent. Patients were eligible if they were 15 to 46 years of age, had received a diagnosis of ADPKD, had an actual or estimated (by the Cockcroft–Gault equation) creatinine clearance of at least 70 ml/min, and had a serum creatinine level of 1.6 mg/dl or less in the case of males and 1.4 mg/dl or less in the case of females. A stated goal of recruitment was to include at least two thirds high-risk individuals to increase the likelihood that disease progression would occur during the study.11 Individuals were considered high risk if hypertension was diagnosed before the age of 35 years, ADPKD was diagnosed in utero or in the first year of life, 24-hour urinary protein excretion was greater than 300 mg/day on any occasion, or a single episode of gross hematuria in men prior to age 30 was present.
Enrolled participants were followed between 2001 and 2005 (CRISP I) with yearly visits. At each visit, TKV was determined from coronal T1- and T2-weighted MRI using a stereologic method,11,36,37 and corrected for height (htTKV, ml/m). Patients were classified into categories according to their predicted rate of kidney growth using the method of Irazabal et al.15 GFR was measured by iothalamate clearance and indexed to body surface area (ml/min/1.73 m2). Blood samples were obtained for determination of serum creatinine at a local laboratory, validated in a duplicate sample at the Cleveland Clinic, and the values used to estimate GFR (eGFR) by the 4-variable Modification of Diet in Renal Disease (MDRD) equation and the CKD-EPI equation.38,39 Screening for PKD1 and PKD2 mutations was performed by denaturing HPLC, followed by direct sequencing, together with screening for larger deletions and/or reverse transcription–PCR to test for abnormal splicing, as previously described.40 PKD1 mutations were classified into 3 mutation strength groups (MSGs): MSG1, truncating mutations; MSG2, strongly predicted nontruncating mutations; and MSG3, weakly predicted nontruncating mutations.7 Two years after completion of the initial study, 201 participants that had not yet reached ESRD were re-enrolled into a 5-year follow-up study (CRISP II).13 Finally, beginning in 2012, 165 participants were re-enrolled in CRISP III. In CRISP II and III, TKV and GFR were measured every 2 years and serum creatinine determined annually.
Both GFR measured by iothalamate clearance and eGFR estimated from serum creatinine were used to classify the outcomes. The primary outcomes for this analysis were CKD stage 3 (GFR less than 60 ml/min per 1.73 m2) with the comparator group being patients with GFR ≥ 60 ml/min per 1.73 m2, CKD stage 4 (GFR less than 30 ml/min per 1.73 m2) vs. GFR ≥30 ml/min per 1.73 m2, and the composite outcome of CKD stage 5 (GFR less than 15 ml/min per 1.73 m2) or ESRD (defined as the need for dialysis or a kidney transplant) vs. GFR ≥15 ml/min per 1.73 m2. Secondary outcomes were relative decreases of 30% and 57% from the individual patient’s baseline GFR. For patients that died, the last available GFR was used for this analysis. To minimize the misclassification of outcomes in patients with incomplete follow-up, we coded patients that did not reach a particular CKD stage, but had no data available from 10 years onwards, as missing data.
Spearman’s correlation coefficients and their corresponding P values were calculated to assess the relationship between baseline htTKV and GFR measured at each visit. Multivariable logistic regression was used to determine the association between baseline htTKV or ADPKD genotype and the development of CKD outcomes at follow-up. ADPKD genotype was grouped into 3 categories: (1) PKD1 MSG1 and MSG2; (2) PKD1 MSG3; (3) PKD2 and no mutation detected (NMD). The third category was treated as the baseline in the regression model. Twelve patients were found to have stage 3 CKD at baseline utilizing iothalamate GFR and were excluded from the main analysis. The multivariable model was adjusted for the following baseline variables: age, sex, race, body mass index and baseline measured GFR. Adjusted odds ratios (ORs) and their 95% confidence intervals (95% CIs) are presented. Statistical significance was tested using the Wald test. Model calibration was evaluated by the Hosmer-Lemeshow goodness-of-fit test.
Likelihood ratio tests (LRTs) were conducted to assess the significance of ADPKD genotype and/or htTKV when added to a base model adjusted for sex, race, BMI and baseline age and measured GFR. The prognostic ability of ADPKD genotype was also compared to htTKV using the net reclassification index (NRI), which assesses the degree to which additional variables improve the prognostic ability of the model. The patients were divided into 3 risk groups by tertiles, based on their predicted probability of CKD. The NRI from the addition of genotype, htTKV or both, to the base model, and from addition of either genotype or htTKV to a model with all other terms, was determined. NRI was determined from the number of subject moving up or down in risk group after reclassification, and calculated as (proportion of cases moving up – proportion of cases moving down) – (proportion of non-cases moving up – proportion of non-cases moving down).41 The 95% confidence intervals for each NRI were determined by bootstrap resampling.
The correlation between the rate of kidney growth and the rate of decline in kidney function was determined as follows. The annual rate of change in GFR was determined individually for each subject from the slope of the linear regression of all the GFR values in the study against age. For patients that reached ESRD, the GFR was assigned a value of 9.5 mL/min/1.73 m2 at time of ESRD, which is the average eGFR at the onset of ESRD for patients with cystic kidney disease in the United States.42 The rate of kidney growth was determined by linear regression of natural log-transformed htTKV against age using either the entire CRISP data set. From the slope, the annual percentage change in htTKV was derived (= exp(slope) − 1). These were then plotted for each subject and Spearman’s correlation coefficient was determined.
Supplementary Material
Supplemental Fig. S1. Projected age at onset of CKD by survival analysis. The figures show Kaplan-Meier plots of age in years against the proportion event-free for each CKD stage, as defined by GFR measured by corrected iothalamate clearance (ml/min/1.73 m2).
{kind=link}
Supplemental Table S1
Acknowledgments
This paper is dedicated to the memory of Dr. Jared J. Grantham, who passed away while this manuscript was being prepared. Dr. Grantham was a founding investigator of CRISP and its scientific and moral compass, and was a key intellectual driving force in developing the studies described in this paper. The CRISP study is supported by cooperative agreements from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (DK056943, DK056956, DK056957, DK056961), and by R01 DK113111. This study was also supported in part by the NIDDK through P30 grants to the Kansas PKD Research and Translation Core Center (DK106912) and the Mayo Translational PKD Center (DK090728), by the National Center for Research Resources General Clinical Research Centers at each institution (RR000039, Emory University; RR00585, Mayo College of Medicine; RR23940, Kansas University Medical Center; RR000032, University of Alabama at Birmingham), and the National Center for Advancing Translational Sciences Clinical and Translational Science Awards at each institution (RR025008 and TR000454, Emory; RR024150 and TR000135, Mayo College of Medicine; RR033179 and TR000001, Kansas University Medical Center; RR025777, TR000165 and TR001417, University of Alabama at Birmingham; RR024153 and TR000005, University of Pittsburgh School of Medicine). The investigators are indebted to the study coordinators in CRISP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
VET, PCH and MM have received research funding from Otsuka Pharmaceuticals. FFR is a consultant for Keryx and Kadmon and has received research funding from Otsuka and Genzyme. ABC is a consultant for Otsuka, Pfizer and Sanofi, and has received research funding from Boston Scientific, Kadmon and Otsuka. All other authors declare no competing interests.
References
- 1.Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. The New England journal of medicine. 2008;359(14):1477–1485. doi: 10.1056/NEJMcp0804458. [DOI] [PubMed] [Google Scholar]
- 2.Ong AC, Devuyst O, Knebelmann B, Walz G, Diseases E-EWGfIK Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385(9981):1993–2002. doi: 10.1016/S0140-6736(15)60907-2. [DOI] [PubMed] [Google Scholar]
- 3.Grantham JJ, Cook LT, Wetzel LH, Cadnapaphornchai MA, Bae KT. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clinical journal of the American Society of Nephrology: CJASN. 2010;5(5):889–896. doi: 10.2215/CJN.00550110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 5.Dicks E, Ravani P, Langman D, Davidson WS, Pei Y, Parfrey PS. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population and family-based cohort followed for 22 years. Clinical journal of the American Society of Nephrology: CJASN. 2006;1(4):710–717. doi: 10.2215/CJN.01581105. [DOI] [PubMed] [Google Scholar]
- 6.Cornec-Le Gall E, Audrezet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. Journal of the American Society of Nephrology: JASN. 2013;24(6):1006–1013. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heyer CM, Sundsbak JL, Abebe KZ, et al. Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27(9):2872–2884. doi: 10.1681/ASN.2015050583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grantham JJ. Rationale for early treatment of polycystic kidney disease. Pediatric nephrology. 2014 doi: 10.1007/s00467-014-2882-8. [DOI] [PubMed] [Google Scholar]
- 9.Alam A, Dahl NK, Lipschutz JH, et al. Total Kidney Volume in Autosomal Dominant Polycystic Kidney Disease: A Biomarker of Disease Progression and Therapeutic Efficacy. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2015;66(4):564–576. doi: 10.1053/j.ajkd.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 10.Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. 2012;31(25):2973–2984. doi: 10.1002/sim.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney international. 2003;64(3):1035–1045. doi: 10.1046/j.1523-1755.2003.00185.x. [DOI] [PubMed] [Google Scholar]
- 12.Perrone R. Imaging progression in polycystic kidney disease. The New England journal of medicine. 2006;354(20):2181–2183. doi: 10.1056/NEJMe068078. [DOI] [PubMed] [Google Scholar]
- 13.Chapman AB, Bost JE, Torres VE, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clinical journal of the American Society of Nephrology: CJASN. 2012;7(3):479–486. doi: 10.2215/CJN.09500911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coresh J, Turin TC, Matsushita K, et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA. 2014;311(24):2518–2531. doi: 10.1001/jama.2014.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging Classification of Autosomal Dominant Polycystic Kidney Disease: A Simple Model for Selecting Patients for Clinical Trials. Journal of the American Society of Nephrology: JASN. 2014 doi: 10.1681/ASN.2013101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. The New England journal of medicine. 2014;371(24):2255–2266. doi: 10.1056/NEJMoa1402685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. The New England journal of medicine. 2014;371(24):2267–2276. doi: 10.1056/NEJMoa1402686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. The New England journal of medicine. 2012;367(25):2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sise C, Kusaka M, Wetzel LH, et al. Volumetric determination of progression in autosomal dominant polycystic kidney disease by computed tomography. Kidney Int. 2000;58(6):2492–2501. doi: 10.1046/j.1523-1755.2000.00433.x. [DOI] [PubMed] [Google Scholar]
- 20.King BF, Reed JE, Bergstralh EJ, Sheedy PF, 2nd, Torres VE. Quantification and longitudinal trends of kidney, renal cyst, and renal parenchyma volumes in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000;11(8):1505–1511. doi: 10.1681/ASN.V1181505. [DOI] [PubMed] [Google Scholar]
- 21.Fick-Brosnahan GM, Belz MM, McFann KK, Johnson AM, Schrier RW. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39(6):1127–1134. doi: 10.1053/ajkd.2002.33379. [DOI] [PubMed] [Google Scholar]
- 22.Tokiwa S, Muto S, China T, Horie S. The relationship between renal volume and renal function in autosomal dominant polycystic kidney disease. Clin Exp Nephrol. 2011;15(4):539–545. doi: 10.1007/s10157-011-0428-y. [DOI] [PubMed] [Google Scholar]
- 23.Thong KM, Ong AC. The natural history of autosomal dominant polycystic kidney disease: 30-year experience from a single centre. Qjm. 2013;106(7):639–646. doi: 10.1093/qjmed/hct082. [DOI] [PubMed] [Google Scholar]
- 24.Higashihara E, Nutahara K, Okegawa T, et al. Kidney volume and function in autosomal dominant polycystic kidney disease. Clin Exp Nephrol. 2014;18(1):157–165. doi: 10.1007/s10157-013-0834-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen D, Ma Y, Wang X, et al. Clinical characteristics and disease predictors of a large Chinese cohort of patients with autosomal dominant polycystic kidney disease. PLoS One. 2014;9(3):e92232. doi: 10.1371/journal.pone.0092232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. Journal of the American Society of Nephrology: JASN. 2015;26(1):160–172. doi: 10.1681/ASN.2013101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petzold K, Poster D, Krauer F, et al. Urinary biomarkers at early ADPKD disease stage. PLoS One. 2015;10(4):e0123555. doi: 10.1371/journal.pone.0123555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.PKD Outcomes Consortium (PKDOC) Qualification of Total Kidney Volume (TKV) as a Prognostic Biomarker for use in Clinical Trials Evaluating Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD) Submission to FDA, March 20th 2014. [Google Scholar]
- 29.Food and Drug Administration. Qualification of Biomarker-Total Kidney Volume in Studies for Treatment of Autosomal Dominant Polycystic Kidney Disease; Draft Guidance for Industry. Federal Register. 80(158):49244–49245. [Google Scholar]
- 30.Thompson A, Lawrence J, Stockbridge N. GFR decline as an end point in trials of CKD: a viewpoint from the FDA. Am J Kidney Dis. 2014;64(6):836–837. doi: 10.1053/j.ajkd.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7(10):556–566. doi: 10.1038/nrneph.2011.109. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. Journal of the American Society of Nephrology: JASN. 2008;19(1):102–108. doi: 10.1681/ASN.2007060688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornec-Le Gall E, Audrezet MP, Rousseau A, et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27(3):942–951. doi: 10.1681/ASN.2015010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17(11):3013–3019. doi: 10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 35.Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clinical journal of the American Society of Nephrology: CJASN. 2011;6(3):640–647. doi: 10.2215/CJN.03250410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bae KT, Commean PK, Lee J. Volumetric measurement of renal cysts and parenchyma using MRI: phantoms and patients with polycystic kidney disease. J Comput Assist Tomogr. 2000;24(4):614–619. doi: 10.1097/00004728-200007000-00019. [DOI] [PubMed] [Google Scholar]
- 37.Bae KT, Tao C, Zhu F, et al. MRI-based kidney volume measurements in ADPKD: reliability and effect of gadolinium enhancement. Clinical journal of the American Society of Nephrology: CJASN. 2009;4(4):719–725. doi: 10.2215/CJN.03750708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 39.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Annals of internal medicine. 2009;150(9):604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18(7):2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 41.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–172. doi: 10.1002/sim.2929. discussion 207–112. [DOI] [PubMed] [Google Scholar]
- 42.US Renal Data Systems. USRDS 2015 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1. Projected age at onset of CKD by survival analysis. The figures show Kaplan-Meier plots of age in years against the proportion event-free for each CKD stage, as defined by GFR measured by corrected iothalamate clearance (ml/min/1.73 m2).
Supplemental Table S1