

Abstract
Methylene blue (MB) is a well-established drug with a long history of use, owing to its diverse range of use and its minimal side effect profile. MB has been used classically for the treatment of malaria, methemoglobinemia, and carbon monoxide poisoning, as well as a histological dye. Its role in the mitochondria, however has elicited much of its renewed interest in recent years. MB can reroute electrons in the mitochondrial electron transfer chain directly from NADH to cytochrome c, increasing the activity of complex IV and effectively promoting mitochondrial activity while mitigating oxidative stress. In addition to its beneficial effect on mitochondrial protection, MB is also known to have robust effects in mitigating neuroinflammation. Mitochondrial dysfunction has been identified as a seemingly unifying pathological phenomenon across a wide range of neurodegenerative disorders, which thus positions methylene blue as a promising therapeutic. In both in vitro and in vivo studies, MB has shown impressive efficacy in mitigating neurodegeneration and the accompanying behavioral phenotypes in animal models for such conditions as stroke, global cerebral ischemia, Alzheimer’s disease, Parkinson’s disease, and traumatic brain injury. This review summarizes recent work establishing MB as a promising candidate for neuroprotection, with particular emphasis on the contribution of mitochondrial function to neural health. Furthermore, this review will briefly examine the link between MB, neurogenesis, and improved cognition in respect to age-related cognitive decline.
Keywords: Methylene Blue, Neuroprotection, Neurogenesis, Neurodegenerative disorders, Cognitive enhancement
1. Introduction
In modern pharmacology, there has been significant emphasis on drug repurposing, the use of previously approved drugs in novel contexts. Repurposing established drugs saves time and money and can lead to clinical translation much more rapidly than a novel therapeutic, due to an established record of extensive safety testing. This strategy is imperative in high-risk research fields, such as neurodegenerative disorders, where pharmaceutical companies may be reluctant to invest large sums of money due to a track record of disappointing high level clinical trials. Conditions like stroke, Alzheimer’s disease (AD), and Parkinson’s disease (PD) are devastating and accompanied with life-long ramifications, leaving patients and families with decreased duration and quality of life. This impact is compounded by the lack of medical options that can slow or reverse the progression of the disorder. These conditions are in dire need of new therapies, and could potentially benefit greatly from the financial and clinical allure of a new application for a time-tested drug.
Methylene blue (MB) is one such example of a promising drug with potential for repurposing. First synthesized in 1876 as a textile dye, MB was investigated for its medicinal applications as early as 1891 [1]. MB has classically been used in the clinic as a potent antimalarial agent, methemoglobinemia treatment, and as a medical staining agent [2]. Evidence has also shown MB to be effective as a prophylactic therapy against vasoplegic syndrome occurring after coronary artery bypass surgery and in septic shock as a result of the vasomodulatory action of MB, which relies on its inhibition of guanylate cyclase (GC) and nitric oxide synthase [3].
While the etiology and progression of the major neurodegenerative disorders varies widely, common to all is mitochondrial dysfunction. In neurodegenerative conditions such as stroke, AD, PD, or traumatic brain injury (TBI), mitochondrial dysfunction and oxidative stress is key to the progressive debilitating nature of the condition [4]. Impairment of the mitochondrial electron transfer chain, through direct damage or insufficient turnover, leads to energy deficits and the release of reactive oxygen species that can cause direct and downstream damage and deleterious effects [5]. As such, targeting and improving mitochondrial health is a prime target for emerging therapies. The mitochondrial mechanism of MB, as well as its established safety record, situates it as a candidate for the treatment of these devastating conditions.
In this review, the basic mechanisms of mitochondrial dysfunction in each of the major neurodegenerative disorders will be discussed, and recent studies examining the effect of MB on each of these will be evaluated. Studies will be highlighted that focus on the emerging role of MB as both an recycling anti-oxidant and an alternative electron carrier in the mitochondrial electron transfer chain in the context of neurodegeneration [5]. When available, combination therapy will be compared to monotherapy. We will also briefly review evidence suggesting a nootropic role for MB.
2. History, Mechanisms, and Current Indications of Methylene Blue
MB was originally synthesized by Hienrich Caro as a textile dye, but it was quickly found to have significant use in the field of medicine [1]. Early pioneering studies involved its use as a medical stain, followed shortly by its application by Ehrlich and Guttman in the treatment of malaria [6]. This effect made it an important drug in many military campaigns, although the side effect of blue urine was not particularly well received. While passing blue urine was undesirable to soldiers, it was of great use in psychiatry; included in medication, MB provided a visibly apparent indicator of compliance [1,7]. Such an indicator was invaluable in the historically harsh treatment of psychiatric patients on early medications that were fraught with undesirable side effects. Through this application in psychiatry, it was eventually discovered that MB alone had antipsychotic effects, leading to the development of the tricyclics. MB was used as a pilot molecule, ushering in a new era of psychopharmacology [1,8]. Some of these effects are known to be due to its function as a powerful monoamine oxidase inhibitor (MAOI), but recent work has implicated the role of mitochondrial mechanisms and metabolic manipulation as the source of at least some of the drug’s beneficial psychiatric effects [9,10]. In recent years, this work has extended the psychiatric application of MB to the treatment of residual bipolar depression [11].
These results imply that MB has bioavailability to the brain, which is corroborated by pharmacokinetic studies [12]. MB is best delivered intravenously, with i.v. administration delivering higher blood concentrations and AUC than oral administration, and has a half-life of approximately 6.6 h. Regardless of route of administration, MB accumulates in significant concentrations in various tissues, with brain tissue concentration of MB as much as tenfold higher than serum levels as early as 1 hour after i.v. administration [12]. Tissue uptake is rapid, with substantial organ accumulation noted after 3 minutes in the lungs, liver, kidneys, and heart [13]. Bioavailability also is modulated by oxidation status, as a stabilized version of the reduced form of MB displays markedly increased brain uptake, which has been factored into a recent clinical trial to be discussed in a subsequent section [14,15].
Currently, the primary medical uses of MB are methemoglobinemia, vasoplegic syndrome, surgical staining, and ifosfamide neurotoxicity [2]. Methemoglobinemia is caused by a prevalence of methemoglobin (met-Hb), a form of hemoglobin (Hb) with wherein the ferrous center of the heme group is oxidized to a ferric state. This can be inherited or induced via exposure to environmental toxins or certain drugs of abuse, such as amyl nitrite [16]. Methemoglobinemia presents as fatigue, headaches, dizziness and bluish skin and can potentially lead to seizures and death if untreated. Once MB is reduced to leucomethylene blue (leucoMB) in the red blood cells, leucoMB can reduce met-Hb to Hb, reoxidizing back to MB [1,17]. This was most famously illustrated in the case of the Blue Fugates, a family in rural Kentucky noted for blue skin as a result of inherited methemoglobinemia. Treatment quickly alleviated the blue hue to their skin, much to their relief [18].
While methemoglobinemia is one of the most common uses of MB, its other applications are invaluable. As a clinical stain, MB delivers striking results in detection of nerves and fistulas and is commonly used in several procedures as well as in histological staining [19]. MB can also be used as an adjunct therapy for chemotherapy with ifosamide, a common chemotherapy agent with detrimental neurological side effects via mitochondrial electron transfer chain (ETC) impairment. As an alternative electron carrier, MB can promote mitochondrial function, limiting the drug’s neurotoxic effects [20]. Finally, vasoplegic syndrome is a life threatening condition occurring after cardiopulmonary bypass, manifesting as significantly reduced arterial pressure, especially prevalent in the case of patients with a history of angiotensin-converting enzyme inhibitors. MB administration can intercede in this condition via inhibition of guanylate cyclase and nitric oxide synthase, increasing arterial pressure [21]. Owing to these important and necessary medical applications, MB is recognized by the World Health Organization as one of the necessary medications needed in a basic healthcare system [22].
In the mitochondria, MB plays a remarkable role, owing to its capacity as a catalytic redox cycler. MB receives electrons from NADH through complex I, converting it to the colorless reduced counterpart leucomethylene blue (leucoMB) (Fig. 1). LeucoMB directly transfers these electrons to cytochrome c, re-oxidizing to MB in the process, ready to begin the cycle anew. Even during complex I inhibition via rotenone MB can bypass ETC blockage at complex I and III, promoting respiration [23] (Fig. 2). Oxidative damage, a cause and consequence of mitochondrial dysfunction, impairs primarily complex IV as well as complex I [24]. This blockage is also bypassed by MB, as it can significantly increase the activity of complex IV. Expression of complex IV subunits is subsequently upregulated, perhaps through induction of nuclear respiratory factor 1 (Nrf1) which was found to be elevated in aged mice treated with MB [25–27]. This increase may be related to PTEN inactivation by modest increases of H2O2 production or by inhibition of GSK-3, increasing NRF1 expression downstream of the NRF2/ARE pathway [27–29].
Figure 1.
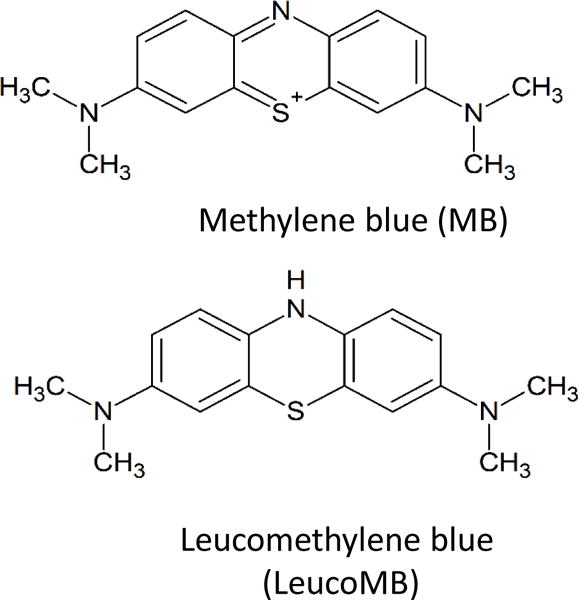
Chemical structure of methylene blue and leucomethylene blue.
Figure 2. Diagram of MB as an alternative mitochondrial electron transporter.
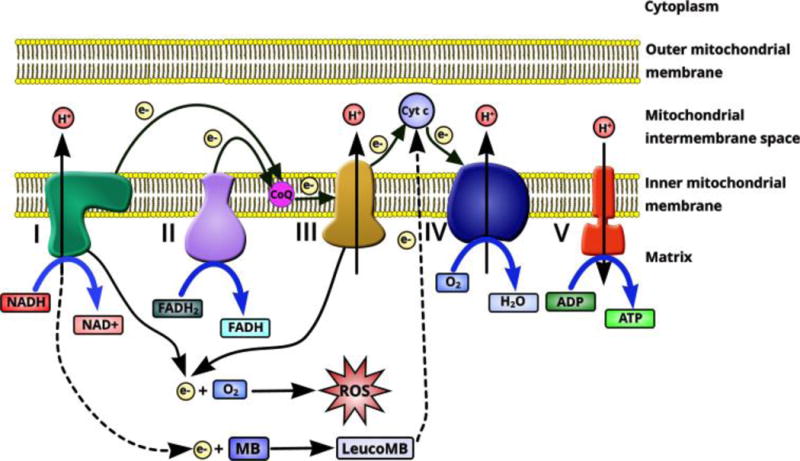
Electrons in the mitochondrial electron transfer chain are transferred from complex I – complex IV, providing the transmembrane potential to drive production of ATP by complex V. Electron leakage from complex I and complex III acts as the main cellular source of ROS production. MB has been demonstrated as an alternative mitochondrial electron transporter to reroute electrons directly from complex I to complex III, avoiding electrons leakage and subsequent ROS production. This significantly facilitates complex IV activity, increasing mitochondrial respiration
This redox cycling can directly protect against oxidative stress in pathological conditions, accompanied with upregulation of nuclear factor (erythroid-derived 2)-like 2/antioxidant response element (Nrf2/ARE) signaling. In physiological conditions, MB likewise upregulates Nrf2/ARE with commensurate increases in antioxidant defense as a result of modest, beneficial increases in H2O2 production [30]. Mechanisms of MB in the context of specific conditions will be discussed at length in following sections.
3. Methylene Blue, Mitochondrial Dysfunction and Neurodegeneration
The brain is remarkably dependent on oxidative metabolism as an energy source, consuming 20% of the body’s glucose and 20% of its oxygen in a resting state [31]. Resting membrane potential maintenance, generation of action potentials, and the postsynaptic actions of glutamate comprise the bulk of this energy demand which is tightly coupled to neuronal activity [32,33]. Considering the striking lack of alternative energy sources in the brain, proper mitochondrial function is imperative to brain health. Dysfunctional mitochondria, conversely, are implicated in several neurodegenerative conditions, playing either a causative or contributing role [4].
The primary function of the mitochondrial ETC is to transfer high energy electrons from food derived energy substrates like NADH to O2 in a stepwise manner. Each step releases energy that is used to transport protons across the inner mitochondrial membrane, establishing the transmembrane potential which drives ATP synthase, the molecular rotor that converts ADP into ATP [23,34]. Proper mitochondrial function is dependent on sequential passage of electrons through each step of the ETC. As the ETC becomes maximally occupied, electron carriers begin to donate electrons to O2 producing detrimental ROS [34].
Superoxide radicals generated by electron leakage are damaging in their own right, but they can react with nitric oxide (NO) generated by nitric oxide synthase to generate highly reactive peroxynitrite (ONOO−). ONOO− can cause extensive damage to cellular lipid components, which particularly affects sensitive oligodendrocytes and their progenitors [35,4]. Superoxide anions can be converted to H2O2 via superoxide dismutase, but this too can be converted into a more dangerous product, hydroxyl radicals [34]. Once generated, these molecules cause largely detrimental modifications to a host of cellular components.
Proteins are frequently the target of free radical attack. Modifications to proteins by ROS/RNS include, but are not limited to: oxidation, nitrosylation, acetylation, and phosphorylation [36]. As the mitochondria are the source of ROS and have several metalloproteins that can catalyze the formation of ROS such as hydroxyl radicals, they are heavily targeted by oxidative damage [34]. Mitochondrial ETC components are targets of oxidative damage, leading to progressive metabolic degradation contributing to neurodegeneration [37] (Fig. 3).
Figure 3. MB in neurodegeneration.
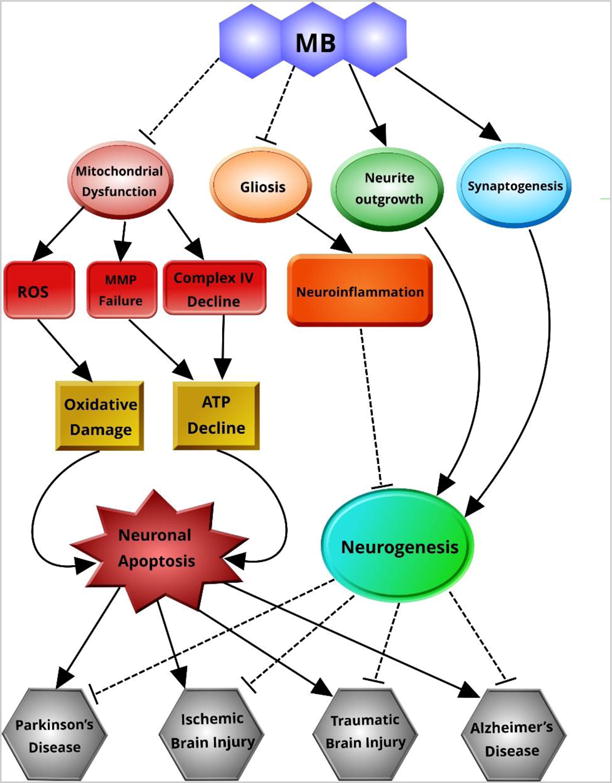
MB can protect against neuronal apoptosis by suppressing mitochondrial dysfunction and subsequent oxidative damage and ATP decline. MB supports neurogenesis by ameliorating neuroinflammation and promoting neurite outgrowth and synaptogenesis. In this manner, MB can prevent neuronal damage and may facilitate neuronal repair.
Mitochondrial function is also dependent on fission/fusion dynamics and mitophagy, the essential components of which can be damaged by nitroxidative damage [38]. Fission/fusion dynamics are regulated by dynamin family GTPases and serve to maintain mitochondrial function by exchange of mitochondrial components and degradation of impaired mitochondria through mitophagy [4]. Oxidative damage to the fusion related protein OPA1 results in translocation to the cytosol followed by mitochondrial fragmentation [39]. Mitochondrial fragmentation is prevalent in several neurodegenerative disorders, as will be discussed in further detail in later sections of this review [40]. Mitochondrial transport is intrinsically linked to fission/fusion dynamics, and is shown to be impaired in neurodegenerative disease [4,41].
All of these detrimental effects are exacerbated by mDNA damage, which leads to damaging mutations that are heterogeneously distributed amongst mitochondria within a single cell. This damage prevents proper mitochondrial repair by preemptively disabling proteins before they are translated. This ensures that damage to the mitochondria is long lasting, and can even spread during fission/fusion events [42]. These mutations are further compounded by impaired fission/fusion dynamics and misregulated mitophagy [34]. Importantly, all of these damages can perpetuate one another in a feed-forward manner, leading to progressive mitochondrial dysfunction that contributes to the inexorable advance of neurodegeneration [38].
3.1 Methylene Blue and Ischemic Brain Injury
As previously noted, the brain is extremely sensitive to oxygen and glucose deprivation as a result of its high energy demands [31]. Ischemia, or cessation of blood flow, to the brain can be either focal or global. Global cerebral ischemia (GCI) is the complete loss of cerebral blood flow (CBF), and occurs during trauma such as cardiac arrest, asphyxiation, cardiac surgery, and hypotensive shock. Cardiac arrest requires rapid intervention; even with resuscitation it is usually fatal, with a mortality rate of over 90%. For those that survive, an array of possible disabilities waits. Survivors must contend with sensorimotor deficits, mood dysregulation, memory loss, and cognitive decline that result from delayed neuronal cell death throughout the brain, specifically in the hippocampus [74,75].
The only currently indicated intervention to prevent GCI induced cell death is therapeutic hypothermia (TH), which slows down oxidative metabolism and potentially mitigates reperfusion induced oxidative stress and mitochondrial derailment [76,77] [78]. Early induction of TH is imperative, as TH delivers diminishing returns with increasing duration after GCI. Moreover, TH induction requires substantial specialized equipment and staff training to prevent significant side effects [79]. This limits deployment, necessitating research into alternative therapies that can be deployed with greater ease and accessibility.
Focal cerebral ischemia, or stroke, occurs nearly every 40 seconds in the United States, and kills or significantly impairs at 60% of patients. Stroke exacts a heavy emotional, medical, and economic toll costing nearly $33 billion every year in the US [80]. There are two main categories of stroke, ischemic or hemorrhagic [80]. Most strokes are ischemic, resulting from the blockage of a cerebral artery, usually via atherothrombosis [80]. The central core of the ischemic region quickly undergoes predominantly necrotic cell death, while the surrounding penumbra region undergoes progressive energy failure, inflammation, and delayed apoptotic cell death [81].
Currently, there is only one indicated intervention in stroke therapy, thrombolysis with tissue plasminogen activator (tPA) [82–84]. This necessitates a need for proper diagnosis via brain imaging techniques, which eats into the 4 hour time window in which infarct core can be salvaged [83]. Since stroke is often not identified with this time window, pharmacological strategies that can minimize or prevent neuronal degeneration are desperately needed, especially those that can be deployed easily and cheaply.
Cerebral ischemia, focal or global, proceeds with two phases of injury: ischemia and reperfusion. During the initial onset of ischemia, energy failure first impairs the maintenance of neuronal membrane potential. Subsequent depolarization triggers excessive glutamatergic release that induces rapid Ca2+ influx. The sharp increase of intracellular Ca2+ levels activates phospholipases, calpains, and cathepsins, increases activity of COX-2 and NOS, and induces the cessation of protein synthesis [85,86]. Meanwhile, as mitochondrial respiration grinds down to a halt, electron leakage generates ROS that damage mitochondrial and cellular components of both neurons and neighboring cells. The vascular endothelium responds to this oxidative damage and local release of inflammatory factors by permeabilizing and compromising the integrity of the blood brain barrier (BBB) [87].
Upon reperfusion, O2 and energy substrates return to the now dysfunctional mitochondria. Respiration resumes within the defective mitochondria, releasing ever increasing levels of ROS, further exacerbating oxidative damage. Permeabilization of the BBB allows for circulating macrophages to infiltrate and perpetuate local inflammation [4]. All the while, microglia and astrocytes respond via glial activation, releasing into the extracellular milieu their own inflammatory factors [88]. Over time, this self-perpetuating cycle eventually manifests as apoptotic cell death in the penumbra in focal ischemia or in particularly sensitive regions, such as the hippocampus, in GCI [89]. Clearly, mitochondrial health is an important contributor to neuronal degeneration. As such, mitochondrial dysfunction has become an attractive target for neuroprotection against ischemic insult.
MB has shown to be neuroprotective in several ischemic injury models, owing in part to its antioxidant capacity and its ability to facilitate alternative mitochondrial transfer. That said, much of the work in the field has focused on MB’s inhibition of NOS activity. Much of this work has been done on piglet models of cardiac arrest in the Wiklund lab. In 2007, they demonstrated that MB administration in the piglet cardiac arrest model could significantly increase survival over 5 hours if delivered 1 minute after resuscitation. Interestingly, they noted that MB crossed the blood brain barrier and later found that it could preserve BBB integrity [90,45]. This group later found that MB treatment increased systemic circulation via NOS inhibition and decreased measures of lipid peroxidation and inflammation. These protective effects were seen both in brain and heart tissue in the porcine model [45]. The neuroprotective effects of MB were amplified significantly when combined with hypothermia in both pigs and rodents [46,47], perhaps due to the maintenance of proper mitochondrial function while slowing down metabolism and inducing hypothermic cellular responses [91]. It is possible that the efficacy of the TH may be enhanced with the addition of a mitochondrial function promoter, especially one that can mitigate oxidative damage. More evidence, however, is required to make a solid conclusion on the prospect of MB+TH combination therapy.
Cell culture studies from other labs have shown that some of the neuroprotective effects of MB are involved with Hif-1α stabilization and phosphorylation of the Akt pathway [44]. Some of the effects may be due to caspase inhibition. MB can also oxidize the functional cysteine residue on both caspase-3 and caspase-6, preventing their proteolytic action [48]. This phenomena could also underlie MB’s neuroprotection, and should be further investigated.
The same mitochondrial neuroprotective mechanisms of MB demonstrated in GCI have likewise displayed efficacy in several animal models of stroke. MB could increase cerebral blood flow in hypoperfused tissue in a permanent MCAO model in rats by blocking the vasomodulatory actions of NO via inhibition of GC and NO synthase, preventing escalating development of the penumbra [52,49]. The underlying mitochondrial mechanism of MB was validated in stroke models, with increased O2 uptake, complex IV activity, and ATP content [54,50,51]. Other work has repeatedly shown that infarct size can be managed or reduced with MB, and that this protection is commensurate with improved behavioral outcomes in rodent models [44,53]. The mechanisms by which this is accomplished are varied.
In an aforementioned study, it was shown that MB managed to rescue mitochondrial structure and mitochondrial membrane potential while preserving mitophagy [51]. Similar studies have related the induction of autophagy by MB with activation of mTOR and AKT phosphorylation [44,92]. Notably, recent work applying MB to subarachnoid hemorrhage, a form of hemorrhagic stroke notably underrepresented in MB research, showed similar increases in AKT phosphorylation and GSK-3β that were accompanied by improved neurological function and reduction of neuroinflammation [55]. Across the board, MB was found to inhibit pro-apoptotic pathways and confer neuroprotection against cell death in the penumbra region, in some cases preserving critical cellular structures like astrocytic endfeet [93,50].
As mentioned earlier, work in our lab found that MB could promote neuroprotection via decreasing caspase activation and protecting mitochondrial membrane potential. The results were also correlated with improved behavioral performance in the Barnes maze, a classic test of hippocampal dependent spatial memory [43]. Our further work applied MB treatment to a rat photothrombotic stroke model. We demonstrated neuroprotection against neuronal cell death as well as promotion of stroke-induced neurogenesis. These results were parallel with decreases in microenvironmental inflammation [54]. We propose that this is a result of the role of mitochondria in neurogenesis, which will be discussed in greater detail in a later section.
Combination therapy is being explored with great interest, despite the inherent complexity of study design. A novel example of this combined normobaric hyperoxia treatment with MB. Combination therapy decreased infarct volume and yielded behavioral improvements in a stroke model beyond that of either treatment alone [56]. Our work found that the addition of MB to therapeutic hypothermia managed to rescue neuronal cell death and behavioral outcomes after extended GCI. Moreover, combination therapy significantly reduced glial activation, inflammation, and caspase 3 apoptotic pathways. This was in conjunction with amelioration of mitochondrial dysfunction. All of these effects were markedly increased in combination therapy in comparison to either monotherapy [46]. It is therefore likely that combination therapy is the key to maximizing the beneficial effects of MB, especially considering the wide range of scope and variety of ischemic brain injury, stroke in particular. If MB does prove successful in the treatment of ischemic brain injury, focal or global, it could open up effective treatment in diverse healthcare settings due to its pervasive distribution worldwide.
3.2 Methylene blue and Alzheimer’s disease
AD is a progressive neurodegenerative disorders affecting 1 in 9 people over 65, leading to memory loss, cognitive decline, severe disability, and death [70]. As it stands, there is no effective treatment for AD that can halt or significantly slow the patient’s inexorable decline. In this regard, this makes AD unique among the US’s major killers, as it is one of the few top causes of death with little preventative or therapeutic measures [70]. Considering the growth of the aged community as a result of better, more accessible healthcare, the importance of therapeutic options to manage AD is only going to become more critical over time. The development of drugs that can target key mechanisms, or multiple mechanisms in the case of MB, is imperative.
AD is classically characterized histologically via the accumulation of senile plaques, amyloid β (Aβ) oligomers, and neurofibrillary tangles (NFT), consisting of hyperphosphorylated tau protein [70]. Aβ is a product of the proteolytic cleavage of membrane-bound amyloid precursor protein (APP) by γ-secretase and β-secretase in succession. APP processing of Aβ can proceed down one of two pathways, the amyloidogenic and non-amyloidogenic pathways [94]. The non-amyloidogenic pathway begins with APP cleavage by α-secretase that yields a soluble N-terminal fragment, sAPP-α, and a membrane-bound C-terminal fragment (CTF-α). CTF-α is further processed by γ-secretase to yield another N-terminal soluble fragment, p3. The soluble fragments generated by this sequence do not form aggregates and are not neurotoxic. The amyloidogenic pathway, however, commences with APP cleavage by β-secretase, generating sAPP-β. γ-secretase cleavage of the remaining CTF-β fragment generates Aβ, which aggregates progressively from oligomers to plaques, which accumulate extracellularly, interfering with cellular function and activating inflammatory pathways [94]. Oligomeric Aβ also accumulates intracellularly, localizing in the mitochondria, contributing to the mitochondrial dysfunction and energy failure characteristic in AD pathology [95].
Classically, the main approach to AD therapeutic research has targeted Aβ. MB has, as recently as 2007, begun to be studied in this capacity. Early work found that MB can promote fibrillization of Aβ, thereby inhibiting the formation of neurotoxic oligomeric Aβ, although later in vitro studies were contradictory [96,67]. Work done in transgenic mice (3xTg-AD) has found that MB supported proteolytic clearance of Aβ by increasing chymotrypsin and trypsin-like proteasome activity in the brain [64]. Decreased deposition of Aβ in the hippocampus and neighboring cortex was observed in another transgenic mouse (APP/PS1) model, and these observations were supported by commensurate protection against cognitive decline in behavioral tasks measuring social interaction, learning and memory, and exploratory activity [61]. Similar results were reported on the transgenic PSAPP mouse wherein the anti-amyloidogenic mechanism was determined to be related to attenuation of β-secretase activity and expression [60].
Mitochondrial dysfunction in early occurrence in AD pathology, present before significant plaque deposition and cognitive decline [97–99]. Respiratory chain activity is hampered, specifically at complexes III and IV, leading to decreased energy metabolism in affected regions [98,100,101]. Soluble Aβ is known to colocalize in the mitochondria, and is imported by the mitochondrial import complex, TIM/TOM [102,103]. Once in the mitochondria, Aβ acts on several molecular targets, including Aβ-binding alcohol dehydrogenase (ABAD) and complex IV, triggering ETC damage and subsequent ROS production leading to a failure of mitochondrial membrane potential [104,63,105,103].
Beyond damaging cellular components, Aβ-induced ROS generation also serves to induce mitochondrial fragmentation as a result of s-nitrosylation of Drp1 [106]. This alteration in mitochondrial dynamics is concomitant with decreases in axonal mitochondrial transport [104] Aβ also leads to increased mitochondrial Ca2+ levels that can induce mPTP opening, releasing cytochrome c and apoptosis inducing factor (AIF) that trigger induction of cell death pathways [97]. These features point to the striking clinical significance of mitochondrial failure in AD as well as a potent target of therapeutics, such as MB.
MB is well known to promote complex IV activity and mitochondrial activity and this effect extends to the AD brain [107,26]. One of MB’s primary mechanisms is promotion of complex IV activity via electron cycling, but a contributing factor is upregulation of heme synthesis [63,107]. In the rat streptozotocin (STZ) model of AD, increases in complex IV and ATP production were reflected in the amelioration of cognitive deficits induced by hippocampal damage [57]. MB has shown to decrease markers of oxidative stress in several AD models through methods of electron cycling and through inhibiting downstream mechanisms and interactions [57,108,109].
One such method is inhibition of Aβ-ABAD binding, preventing the associated ROS output, MMP failure, and subsequent cell death [109–111,103]. Promoting mitochondrial function can prevent cytosolic release of pro-death factors, but MB can take this protective measure one step further by deactivating caspases via oxidation of functional cysteine MB also targets the other hallmarks of the disorder, NFTs and Aβ plaques. Considering the progressive feedback manner of nearly every aspect of AD mechanisms, these broader mechanisms mitigate further mitochondrial impairment.
Another significant feature of AD is the presence of NFTs, aggregates of hyperphosphorylated tau protein (p-tau). Tau is a microtubule associated protein that is highly prevalent in the CNS, stabilizing neuronal microtubules. Tau is a target of phosphorylation for kinases such as glycogen-synthase kinase 3 β (GSK3β), c-Jun kinase (JNK), and cyclin-dependent kinase 5 (cdk5) [112–114]. While phosphorylation of tau is a physiological process, excessive tau phosphorylation is pathological and a contributor to neuronal degradation in AD.
When excessively phosphorylated, tau dissociates from microtubules causing their destabilization and subsequent dissolution. Meanwhile, p-tau itself aggregates into tangles [115]. By disrupting microtubule networks, cellular function and axonal transport is compromised, such as the aforementioned mitochondrial transport deficits [116]. Clearance of via the proteasome is also hampered. Though p-tau is heavily ubiquitinylated, p-tau continues to accumulate in excess of what the proteasome can clear [117,116]. Whether tauopathy precedes Aβ pathology or is a consequence of it is hotly debated, as is its exact contribution to the pathology. Regardless, it is clear that tau aggregation plays a vital role in AD, and targeting its dysfunction is an important topic of study.
Pioneering work in the application of MB to AD was done by Claude Wischik, wherein his laboratory found that MB reverses tau aggregation by blocking tau-tau binding, albeit at doses higher than clinically relevant levels [66]. MB and its derivatives decreased accumulation of tau filaments in an in vitro assay, which was later found to be due to inhibition of filament formation on the first and fourth repeat peptides on the tau microtubule binding domain [68,118]. Unlike other related dyes, MB doesn’t bind to or associate with tau proto-fibrils [119]. It prevents fibrillization by oxidizing cysteine residues in tau, causing them to form a more stable monomer form that is resistant to aggregation [69].
In addition, it was also observed that MB helped promote clearance of tau filaments by inducing autophagy [65]. Multiple studies have found that MB helped reduce tau load in different tauopathy transgenic mouse models over short term and long term treatment and that this clearance was associated with amelioration of cognitive deficits [58,62]. If delivered preemptively, MB can prevent cognitive decline and tau accumulation in transgenic mice expressing pro-aggregant human tau [120].
Studies applying MB to different animal models yielded interesting, albeit somewhat conflicting, results. MB failed to reduce tau burden in a zebrafish model, although it showed promising results in decreasing huntingtin aggregation [121]. In a C. elegans transgenic tauopathy model, MB cleared tau and promoted proper mitochondrial transport that was accompanied by increased motility [122].
MB has been applied to human clinical trials, beginning with a phase 2 clinical trial for MB under the name “Rember.” The trial showed both cognitive and cerebral blood flow improvements for patients with mild to moderate AD [59]. A variant of the molecule, leuco-methylthioninium bis (hydromethanesulfonate), or LMTM, was developed that was more stable in the reduced form [15]. Unfortunately, a subsequent 15 month phase 3 study of LMTM yielded negative results [14]. Another study of LMTM on mild cases of AD is forthcoming. These studies and others may further elucidate protective mechanisms of MB against AD, and may one day lead to clinical translation.
3.3 Methylene Blue and Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder resulting from the death of dopaminergic neurons in the substantia nigra (SN), clinically presenting as tremor, rigidity, impaired movement, flat facial affect, and depression [4]. Occurrence of the disease usually presents after age 50, affecting 100–300 out of every 100,000 people, manifesting motor symptoms in the majority of patients [123]. PD decreases life expectancy of patients, with the age of onset significantly affecting prognosis; those with early onset have decreased life expectancy of 10 years while those with late onset lose around 5 years of life expectancy [124]. Mortality in PD is commonly a result of falls or respiratory infection and edema, often resulting from accidental food aspiration due to difficulty swallowing [125–127].
Parkinson’s treatment usually consists of administration of the dopamine precursor levodopa which decreases rigidity and tremor. Unfortunately, levodopa therapy is concomitant with neurological and behavioral side effects, and ever increasing doses and secondary medications are usually required over the course of treatment [128]. Levadopa, however vital to patients, is a palliative treatment, and doesn’t slow the progression of the disorder. As such, it is critical to develop therapies that can target key mechanisms of neuronal damage to halt or reverse the course of the disease.
The histological hallmark of Parkinson’s disease is the presence of Lewy Bodies and Lewy Neurites, intracellular aggregates of the protein α-synuclein (α-syn) [4]. These aggregates are heavily associated with alterations in the expression of the E3 ubiquitin ligase, Parkin, which causes deficits in mitochondrial quality control by impeding microtubule trafficking [129]. A-syn, when translocated to the mitochondria, can impair the function of both complex I and complex IV of the ETC, leading to progressive mitochondrial dysfunction that is potentiated by deficient mitophagy [130,131]. This ETC dysfunction is the basis upon which two of the most widely used models of PD symptoms are generated: MPTP and rotenone, both of which inhibit complex I [5,132]. These toxins cause characteristic damage to SN dopaminergic neurons and manifest parkinsonian tremor [132]. As with all neurodegenerative diseases with a mitochondrial mechanism, therapeutic strategies targeting such dysfunction offer an alluring line of research.
The seminal work that determined the alternative electron transfer capacity of MB was discovered using a rotenone model of PD. Along with the aforementioned preservation of ETC function, MB treatment was accompanied with behavioral improvements in tremor, locomotion, posture and motor skills [5]. Beyond this study, the evidence of the efficacy on PD is indirect, yet still promising. Both mitophagy and general autophagy are induced by MB administration, which could mitigate the mitochondrial quality control deficits that lead to the progression of the disorder [51,65]. Moreover, the established antioxidant and ETC promoting functions of MB in the mitochondria target the very complexes most targeted by PD [27,5,133].
With its low side effect profile, there lies the possibility that MB delivered prophylactically or at the early stages of PD may prevent the progression of the disorder due to the beneficial effects of the low levels of H2O2 generated by MB in physiological conditions, mediated by the Nrf2/ARE pathway, which is tentatively implicated in PD [134,135,30]. Finally, the MAOI properties of MB may provide added benefits to an ongoing regimen of levodopa, although the safety profile of this combination needs to be carefully investigated [136,128]. The close association of the profile of MB’s mitochondrial effects and the mitopathy present in PD seem promising, highlighting the necessity of applying MB to the various PD chemical and genetic animal models.
3.4 Methylene Blue in Traumatic Brain Injury
In recent years, TBI has gained renewed public interest due to the visibility of sports injuries and soldiers returning from active combat zones worldwide. Traumatic brain injury is a significant cause of death and disability, with nearly 2 million cases annually in the United States [137]. TBI is the result of forceful head trauma, although this simple explanation belies the heterogeneous nature of the condition. TBI injury consists of two phases: primary and secondary. The primary injury is the mechanical insult itself, which causes shearing of axonal tracts, rupture of microvasculature and compression of brain tissue. Secondary injury develops over time, encompassing increasing intracranial pressure, brain edema, excitotoxicity, progressive inflammation, glial activation, metabolic failure, seizures and ischemic damage [138,139]. The disturbances caused by TBI vary based on the intensity of the blow; severe cases may present unconsciousness, long-term memory loss, and seizures, while “mild” TBI can present confusion, attentional or cognitive deficits, and memory retention deficits, although the definition of mild TBI is somewhat blurry [138,140].
Much of the cellular pathology in TBI bears striking similarities to ischemic brain injury, and for good reason. Increased intracranial pressure, misregulated CBF, and compromised microvasculature all serve to limit blood flow and subsequent oxygen delivery to the brain leading to energy failure, excitotoxicity and mitochondrial dysfunction [141,142]. Overproduction of ROS leads to progressive mitochondrial decline and chronic glial activation, serving to generate the familiar pattern of mitochondrial driven neurodegeneration [143,144]. Mechanical disruption of white matter tracts further compounds damage by disrupting axonal mitochondrial transport. Dysfunctional mitochondria post-axotomy release excessive ROS near sensitive, lipid-rich oligodendrocytes causing significant lipid peroxidation and further the progression of gliosis [145]. ROS and regional inflammation cause the permeabilization of the BBB by activating matrix metalloproteinases and decreasing tight junction protein expression. This feeds back and promotes inflammation and infiltration of peripheral macrophages, again upregulating expression and release of inflammatory factors and inflammatory signaling [146].
These processes lead to activation of competing cell death pathways generating death phenotypes on a continuum from apoptosis to necrosis [147]. Targeting these pathways has proven difficult. Therapeutic hypothermia has been applied, much as to GCI. In the case of TBI, it is effective for reducing intracranial pressure, but the same limitations in technique and accessibility apply [148]. Hyperbaric oxygen therapy has likewise been studied with limited efficacy [149]. Significant study has been devoted to anti-oxidant therapy for TBI yielding promising preclinical results. These results, unfortunately, have not translated as well in clinical trials [150]. It is possible that many of the failures of TBI therapies, as well as those for other neurodegenerative conditions, relates to preclinical study design or species difference. In stroke and TBI especially, a major factor may be the heterogeneity of injury. Regardless, the window for effective therapies is wide open and patients are in desperate need.
Few preclinical studies have examined MB therapy after mild TBI, but the present studies are encouraging. Initial studies indicated that MB delivered twice shortly after TBI could decrease infarct volume over multiple time points extending out to 14 days post-TBI. MB mitigated somatosensory deficits in behavioral tests and decreased cell death in the affected cortex [72]. This was corroborated with work showing that MB treatment administered within 15–30 minutes of TBI could reduce cerebral edema, inflammation long term after TBI. Neurological scoring improved with MB, but no differences were seen on specific motor coordination tests [73]. These results are encouraging, but the dosing strategy is unrealistic; it is highly unlikely that a TBI patient will have the opportunity to have near-immediate infusion of MB.
Fortunately, other studies have shown that treatment even 24 hours after TBI can reproduce similar improvements in infarct size and neurological function [71]. These results were mirrored and expanded on in a subsequent study with a multi-treatment paradigm wherein MB treatment reduced edema, long-term gliosis, neuronal cell death, and infarct size while protecting against behavioral deficits. Moreover, these benefits were tied to increased induction of autophagy [70].
These studies and others relating to ischemic injury are heartening, as patients with TBI are in dire need of effective therapies, especially those as inexpensive and accessible as MB. Owing to the diverse nature of head trauma, however, it is unlikely that a single agent will provide the most optimal protection from the constellation of pathologies following TBI. As such, MB will likely prove most effective in a form of combination therapy, as previously described in the context of GCI and stroke. Currently, there have been no such studies, but the growing literature of MB combination therapy in other brain injuries may provide a solid base upon which to design those applicable to TBI. Though these studies will be inherently complex, the prospect of effective management of TBI-induced neuronal damage is too vital to ignore.
4. Methylene Blue in Cognitive Enhancement, Age-Related Cognitive Decline, and Neurogenesis
Cognitive enhancement via pharmaceuticals is an idea that has long captured the public’s attention and imagination. The concept is prevalent in the public consciousness, and entire online communities are centered on the concept and exploration of potential cognitive enhancers, or nootropics. Proposed cognitive enhancers range from amino acid supplements to classically used herbal supplements [151,152]. Clinically prescribed stimulants are often abused, particularly on college campuses, for this purpose, adding to public stigma of legitimate patients and the validity of attention deficit hyperactivity disorder diagnoses at large [153]. Clearly, enhancing cognition is of significant interest by the public, as well as researchers. Improvements to cognition could prove in the favor of public good in terms of productivity and quality of life, especially in respect to aged populations. In light of this, cheaply available compounds that could safely support or improve cognition are ideal candidates. MB, in this regard, is therefore a promising contender.
The concept of cognitive enhancement via mitochondrial modulation has been investigated increasingly in recent years. The general concept is that by improving mitochondrial function and oxidative defenses, neurons can function with improved efficiency and maintain proper health, improving basal function and stymieing cognitive decline associated with age and neurodegeneration [154]. Early work by Gonzalez-Lima has shown that MB improved spatial memory retention alongside long-lasting mitochondrial respiratory function, mediated through complex IV [26]. The long-term upregulation of CCO may be related to increased H2O2 production without superoxide formation, via MB in physiological conditions, leading to upregulation of Nrf2/ARE [135]. In a human study, MB administration increased cerebrovascular reactivity in psychomotor vigilance task and a short-term memory test. This was accompanied with modest improvements in performance on the short-term memory test [155]. These benefits correlated with mitochondrial function are corroborated by experiments showing similarly improved cognition with photobiomodulation, the stimulation of complex IV with transcranial near-infrared laser irradiation [156].
Metabolic derangement is a well-established observation in the brains of the elderly, especially in regions classically associated with higher brain functions [157–159]. Mitochondrial damage accumulates over time and progressively contributes to neuronal decline as one ages, much as in neurodegenerative conditions [158]. These mitochondrial insults include breakdown of the electron transfer chain, deficient mitophagy and a shift towards excessive fission, mtDNA mutations, and excessive ROS production [158]. These features are associated with cognitive deficits, in the elderly just as in those with AD, GCI, major depressive disorder, and other conditions. As such, protecting mitochondrial health may provide some measure of protection against dementia or other forms of age-related cognitive deficits. MB, as detailed previously can stymie or prevent many of these mitochondrial insults and may have potential, in this manner, to protect cognition against the metabolic ravages of advancing age. This possibility, as of yet, has not been investigated formally, and remains an open-ended question and field of scientific inquiry.
Neurogenesis is well accepted to occur through adulthood, both as a physiological process and as a response to injury [160]. After development, neurogenesis is limited to three regions of the brain, the olfactory bulb, the cortical subventricular zone (SVZ), and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG). Impairment of this process is associated with disruption of memory consolidation and mood regulation, and is implicated in neurodegeneration and age-related cognitive decline, as neurogenesis and related processes sharply decline in old age [161]. As such, stimulating and sustaining neurogenesis is an alluring target for age-related dementia, considering the substantial shared features with other pathological forms of neurodegeneration.
Neurogenesis is likewise simultaneously stimulated and inhibited in the conditions proceeding after brain injury [162]. In animal models of stroke, neurogenesis is promoted in both the SVZ and SGZ alongside release of erythropoietin and both vascular and epidermal epithelial growth factors that induce proliferation of neuronal progenitor cells (NPCs) [162–164]. Newly formed neurons migrate to the injured region, and attempt to functionally incorporate into existing neuronal circuitry to facilitate repair [165]. Neural repair by stimulating neurogenesis is appealing, but is limited by the microenvironmental response to injury. Chronic neuroinflammation and glial activation, both features associated with advancing age [166] are shared across most neurodegenerative conditions, and both of these factors stunt neurogenesis [166] [160,167–171]. This deficit in neurogenesis can by mitigated by anti-inflammatory treatments [172]. ROS generated by reactive glia also damage and deplete NPC pools [173]. The addition of an antioxidant, much like the anti-inflammatory blockade, prevented the depletion of newly-formed neurons [174]. In addition, integrity of mitochondria is crucial for neurogenesis, and is thusly upregulated during proliferation and differentiation [175]. Deficits of mitochondria, largely performed in cybrid experiments, display marked impairments in both proliferation and differentiation [176–178].
Few studies have applied MB to neurogenesis, but those that have, as well as the general actions of MB, suggest a potential for beneficial effects. Previously mentioned GCI research applying MB to a piglet model revealed that MB upregulated genes related to neurogenesis, including synaptogenesis, neurite outgrowth, and pro-survival factors [91]. MB downregulated the proliferation of rat NPCs by downregulating cyclin expression and mTOR activity. This seems contradictory, but excessive proliferation depletes the NPC pool, decreasing future capacity for neurogenesis. MB also did not affect neuronal differentiation. These results, however, were observed in post-natal day 1 rats, so it is unclear whether this protective mechanism is relevant in the injured adult brain [179]. Work in our lab found that MB delivered after photothrombotic stroke promoted neurogenesis. Furthermore, it downregulated inflammation, gliosis, and improved mitochondrial function, all features that contribute to neurogenic deficits. This leads us to believe that MB may possibly promote or support neurogenesis after injury by making the local microenvironment more amenable to newly formed neurons while supporting their mitochondrial efficiency [54]. While sustaining the survival of newly formed neurons is tantamount, supporting their migration is likewise imperative to sustain neural repair. Once again, evidence is limited, but recent evidence indicates that MB could modulate the migratory of murine adult neural stem cells in a cell migration assay [180]. Much more evidence is necessary to draw a solid conclusion on the effect and role of MB in promoting adult neurogenesis and mitochondrial health in old age. If validated, MB may serve as a multifaceted tool for preserving cognition and quality of life for the elderly population, a demographic vital in an aging society.
6. Adverse Effects and Contraindications of Methylene Blue
MB clearly shows promise as a therapeutic for many conditions, but, like any drug, it carries with it a set of limitations. The vasomodulatory properties of MB are useful at therapeutic doses, but carry risk at higher concentrations. These risks range from increased blood pressure and vascular resistance to disturbances in cardiac function and rhythmicity [2]. Likewise, MB can influence pulse oximetry measurements, hindering proper diagnostics.
Another toxic effect is the induction of anemia in individuals with glucose-6-phosphate dehydrogenase deficiency. Neonates are also susceptible to anemia and many other adverse effects as a response to MB [2]. In the neonate, MB can induce hyperbilirubinemia, presenting as jaundice [181]. This is rather unfortunate, as the accepted treatment for hyperbilirubinemia is phototherapy, which activates MB’s photodynamic activity. This generates excited oxygen species, which causes significant damage to the epidermis [182]. Thus, in any application, MB should be treated with the care of any other photosensitizer. In the neonate, MB treatment can also lead, paradoxically, to methemoglobinemia [183].
The final, and most clinically relevant in the adult patient, adverse effect is related to the use of common psychiatric medications, selective serotonin reuptake inhibitors (SSRI). MB is a potent MAOI which, in conjunction with an SSRI, can potentiate serotonin syndrome, a life threatening medical emergency [184–186]. Considering the ubiquity of patients on SSRIs, it is prudent to screen patients for these drugs before MB administration. Regardless, that the risks of MB are relatively low and reasonably predictable bodes well for adaptation in the clinic.
7. Conclusions
Neurodegenerative disorders are a daunting challenge in medicine, exacting a heavy toll, both human and financial. The relative scarcity of therapeutic options is intrinsically discouraging, but it is this same vacancy that should spur innovation. The struggle of these patients motivates dedicated investigators, and the untapped nature of this market should encourage investors to support these efforts. Though these ambitions may be dampened by a less than stellar track record, there is hope in lower-risk ventures in the form of repurposed pharmaceuticals, such as MB. The excellent safety record of MB is well established by a century of medical use. Its newly explored role as a mitochondrial enhancer and recycling antioxidant is key to its potential as a therapeutic agent. With encouraging results in preclinical trials of some of the most disabling neurodegenerative conditions, MB should be investigated with renewed enthusiasm in years to come. If these results are validated in future human studies, MB could prove to be a versatile agent that could improve the health of patients suffering from the burden of neurodegenerative disorders and brain injury, giving them and their loved ones relief and a better quality of life.
Table 1.
Summary of effects of MB on neurodegenerative disorders and brain injury and related mechanisms
| Condition of Interest | Observed Actions of MB | Known Mechanisms of Action |
|---|---|---|
| Global Cerebral Ischemia | ||
| Stroke | ||
| Alzheimer’s Disease |
|
|
| Parkinson’s Disease |
|
|
| Traumatic Brain Injury |
Acknowledgments
This work was supported by Research Grant NS086929 from the National Institute of Neurological Disorders and Stroke, National Institutes of Health, USA, and an American Heart Association Grant-in-Aid 15GRNT25240004.
Footnotes
Conflict of interest
The authors confirm that their contributions to this article are free from conflict of interest.
References
- 1.Schirmer RH, Adler H, Pickhardt M, Mandelkow E. Lest we forget you–methylene blue…. Neurobiology of aging. 2011;32(12):2325 e2327–2316. doi: 10.1016/j.neurobiolaging.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Ginimuge PR, Jyothi SD. Methylene blue: revisited. Journal of anaesthesiology, clinical pharmacology. 2010;26(4):517–520. [PMC free article] [PubMed] [Google Scholar]
- 3.Stawicki SP, Sims C, Sarani B, Grossman MD, Gracias VH. Methylene blue and vasoplegia: who, when, and how? Mini reviews in medicinal chemistry. 2008;8(5):472–490. doi: 10.2174/138955708784223477. [DOI] [PubMed] [Google Scholar]
- 4.Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain research. 2016;1637:34–55. doi: 10.1016/j.brainres.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang SH, Li W, Sumien N, Forster M, Simpkins JW, Liu R. Alternative mitochondrial electron transfer for the treatment of neurodegenerative diseases and cancers: Methylene blue connects the dots. Prog Neurobiol. 2015 doi: 10.1016/j.pneurobio.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guttman PE. On the Action of Methylene Blue on Malaria. In: Himmelwelt F, editor. The Collected Papers of Paul Ehrlich: Chemotherapy, vol III. Elsivier; 1891. pp. 15–20. [Google Scholar]
- 7.Schaefer B, E P. Natural Products in the Chemical Industry. Springer; 2015. Illustrated edn. [Google Scholar]
- 8.Hughes DE, L E. The Use of Methylene Blue as a Seditive. In: Curtin Roland G, D EH., editors. Philadelphia Hospital Reports. Vol. 4. Detre & Blackburn: 1901. pp. 272–282. [Google Scholar]
- 9.Gillman PK. CNS toxicity involving methylene blue: the exemplar for understanding and predicting drug interactions that precipitate serotonin toxicity. J Psychopharmacol. 2011;25(3):429–436. doi: 10.1177/0269881109359098. [DOI] [PubMed] [Google Scholar]
- 10.Eroglu L, Caglayan B. Anxiolytic and antidepressant properties of methylene blue in animal models. Pharmacological research. 1997;36(5):381–385. doi: 10.1006/phrs.1997.0245. [DOI] [PubMed] [Google Scholar]
- 11.Alda M, McKinnon M, Blagdon R, Garnham J, MacLellan S, O’Donovan C, Hajek T, Nair C, Dursun S, MacQueen G. Methylene blue treatment for residual symptoms of bipolar disorder: randomised crossover study. The British journal of psychiatry: the journal of mental science. 2017;210(1):54–60. doi: 10.1192/bjp.bp.115.173930. [DOI] [PubMed] [Google Scholar]
- 12.Peter C, Hongwan D, Kupfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56(3):247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- 13.DiSanto AR, Wagner JG. Pharmacokinetics of highly ionized drugs. 3. Methylene blue–blood levels in the dog and tissue levels in the rat following intravenous administration. J Pharm Sci. 1972;61(7):1090–1094. doi: 10.1002/jps.2600610711. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JM, Harrington CR, Wischik CM. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–2884. doi: 10.1016/S0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J Pharmacol Exp Ther. 2015;352(1):110–118. doi: 10.1124/jpet.114.219352. [DOI] [PubMed] [Google Scholar]
- 16.Coleman MD, Coleman NA. Drug-induced methaemoglobinaemia. Treatment issues. Drug safety. 1996;14(6):394–405. doi: 10.2165/00002018-199614060-00005. [DOI] [PubMed] [Google Scholar]
- 17.Bradberry SM. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicological reviews. 2003;22(1):13–27. doi: 10.2165/00139709-200322010-00003. [DOI] [PubMed] [Google Scholar]
- 18.Cawein M, Behlen CH, 2nd, Lappat EJ, Cohn JE. Hereditary Diaphorase Deficiency and Methemoglobinemia. Archives of internal medicine. 1964;113:578–585. doi: 10.1001/archinte.1964.00280100086014. [DOI] [PubMed] [Google Scholar]
- 19.Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of Methylene Blue in the nervous system. Medicinal research reviews. 2011;31(1):93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alici-Evcimen Y, Breitbart WS. Ifosfamide neuropsychiatric toxicity in patients with cancer. Psycho-oncology. 2007;16(10):956–960. doi: 10.1002/pon.1161. [DOI] [PubMed] [Google Scholar]
- 21.Shanmugam G. Vasoplegic syndrome–the role of methylene blue. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2005;28(5):705–710. doi: 10.1016/j.ejcts.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 22.2015 WHO Model List of Essential Medicines. 2015 http://www.who.int/medicines/publications/essentialmedicines/en/
- 23.Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. The Journal of biological chemistry. 2011;286(18):16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. Journal of neurotrauma. 2007;24(5):772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 25.Wong-Riley MT. Bigenomic regulation of cytochrome c oxidase in neurons and the tight coupling between neuronal activity and energy metabolism. Advances in experimental medicine and biology. 2012;748:283–304. doi: 10.1007/978-1-4614-3573-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacology, biochemistry, and behavior. 2004;77(1):175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Gureev AP, Syromyatnikov MY, Gorbacheva TM, Starkov AA, Popov VN. Methylene blue improves sensorimotor phenotype and decreases anxiety in parallel with activating brain mitochondria biogenesis in mid-age mice. Neurosci Res. 2016;113:19–27. doi: 10.1016/j.neures.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Wu HM, Lee CG, Hwang SJ, Kim SG. Mitigation of carbon tetrachloride-induced hepatic injury by methylene blue, a repurposed drug, is mediated by dual inhibition of GSK3beta downstream of PKA. Br J Pharmacol. 2014;171(11):2790–2802. doi: 10.1111/bph.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor KM, Subbaram S, Regan KJ, Nelson KK, Mazurkiewicz JE, Bartholomew PJ, Aplin AE, Tai YT, Aguirre-Ghiso J, Flores SC, Melendez JA. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280(17):16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 30.Stack C, Jainuddin S, Elipenahli C, Gerges M, Starkova N, Starkov AA, Jove M, Portero-Otin M, Launay N, Pujol A, Kaidery NA, Thomas B, Tampellini D, Beal MF, Dumont M. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum Mol Genet. 2014;23(14):3716–3732. doi: 10.1093/hmg/ddu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends in neurosciences. 2013;36(10):587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32(7):1222–1232. doi: 10.1038/jcbfm.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du F, Zhu XH, Zhang Y, Friedman M, Zhang N, Ugurbil K, Chen W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(17):6409–6414. doi: 10.1073/pnas.0710766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth AD, Nunez MT. Oligodendrocytes: Functioning in a Delicate Balance Between High Metabolic Requirements and Oxidative Damage. Advances in experimental medicine and biology. 2016;949:167–181. doi: 10.1007/978-3-319-40764-7_8. [DOI] [PubMed] [Google Scholar]
- 36.Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, Hardwick JP. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox biology. 2014;3:109–123. doi: 10.1016/j.redox.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. Journal of the neurological sciences. 2005;233(1–2):145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. The Journal of pharmacology and experimental therapeutics. 2012;342(3):619–630. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanderson TH, Raghunayakula S, Kumar R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Molecular and cellular neurosciences. 2015;64:116–122. doi: 10.1016/j.mcn.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain research reviews. 2011;67(1–2):103–118. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Giannoccaro MP, La Morgia C, Rizzo G, Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society. 2017;32(3):346–363. doi: 10.1002/mds.26966. [DOI] [PubMed] [Google Scholar]
- 43.Lu Q, Tucker D, Dong Y, Zhao N, Zhang Q. Neuroprotective and Functional Improvement Effects of Methylene Blue in Global Cerebral Ischemia. Mol Neurobiol. 2016;53(8):5344–5355. doi: 10.1007/s12035-015-9455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryou MG, Choudhury GR, Li W, Winters A, Yuan F, Liu R, Yang SH. Methylene blue-induced neuronal protective mechanism against hypoxia-reoxygenation stress. Neuroscience. 2015;301:193–203. doi: 10.1016/j.neuroscience.2015.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiklund L, Basu S, Miclescu A, Wiklund P, Ronquist G, Sharma HS. Neuro- and cardioprotective effects of blockade of nitric oxide action by administration of methylene blue. Ann N Y Acad Sci. 2007;1122:231–244. doi: 10.1196/annals.1403.016. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Yang R, Li P, Lu H, Hao J, Tucker D, Zhang Q. Combination Treatment with Methylene Blue and Hypothermia in Global Cerebral Ischemia. Mol Neurobiol. 2017 doi: 10.1007/s12035-017-0470-1. [DOI] [PubMed] [Google Scholar]
- 47.Wiklund L, Zoerner F, Semenas E, Miclescu A, Basu S, Sharma HS. Improved neuroprotective effect of methylene blue with hypothermia after porcine cardiac arrest. Acta Anaesthesiol Scand. 2013;57(8):1073–1082. doi: 10.1111/aas.12106. [DOI] [PubMed] [Google Scholar]
- 48.Pakavathkumar P, Sharma G, Kaushal V, Foveau B, LeBlanc AC. Methylene Blue Inhibits Caspases by Oxidation of the Catalytic Cysteine. Sci Rep. 2015;5:13730. doi: 10.1038/srep13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weingartner R, Oliveira E, Oliveira ES, Sant’Anna UL, Oliveira RP, Azambuja LA, Friedman G. Blockade of the action of nitric oxide in human septic shock increases systemic vascular resistance and has detrimental effects on pulmonary function after a short infusion of methylene blue. Braz J Med Biol Res. 1999;32(12):1505–1513. doi: 10.1590/s0100-879x1999001200009. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Z, Watts LT, Huang S, Shen Q, Rodriguez P, Chen C, Zhou C, Duong TQ. The Effects of Methylene Blue on Autophagy and Apoptosis in MRI-Defined Normal Tissue, Ischemic Penumbra and Ischemic Core. PLoS One. 2015;10(6):e0131929. doi: 10.1371/journal.pone.0131929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Y, He YL, Zhao T, Huang X, Wu KW, Liu SH, Zhao YQ, Fan M, Wu LY, Zhu LL. Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol Med. 2015;21:420–429. doi: 10.2119/molmed.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez P, Jiang Z, Huang S, Shen Q, Duong TQ. Methylene blue treatment delays progression of perfusion-diffusion mismatch to infarct in permanent ischemic stroke. Brain Res. 2014;1588:144–149. doi: 10.1016/j.brainres.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen Q, Du F, Huang S, Rodriguez P, Watts LT, Duong TQ. Neuroprotective efficacy of methylene blue in ischemic stroke: an MRI study. PLoS One. 2013;8(11):e79833. doi: 10.1371/journal.pone.0079833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed ME, Tucker D, Dong Y, Lu Y, Zhao N, Wang R, Zhang Q. Methylene Blue promotes cortical neurogenesis and ameliorates behavioral deficit after photothrombotic stroke in rats. Neuroscience. 2016;336:39–48. doi: 10.1016/j.neuroscience.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H, Li J, Wang Z, Feng M, Shen Y, Cao S, Li T, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3beta/MEF2D signaling pathway. Brain Behav Immun. 2017 doi: 10.1016/j.bbi.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez P, Zhao J, Milman B, Tiwari YV, Duong TQ. Methylene blue and normobaric hyperoxia combination therapy in experimental ischemic stroke. Brain Behav. 2016;6(7):e00478. doi: 10.1002/brb3.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L, Qin L, Lu HL, Li PJ, Song YJ, Yang RL. Methylene blue improves streptozotocin-induced memory deficit by restoring mitochondrial function in rats. Brain Res. 2017;1657:208–214. doi: 10.1016/j.brainres.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 58.Melis V, Magbagbeolu M, Rickard JE, Horsley D, Davidson K, Harrington KA, Goatman K, Goatman EA, Deiana S, Close SP, Zabke C, Stamer K, Dietze S, Schwab K, Storey JM, Harrington CR, Wischik CM, Theuring F, Riedel G. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015;26(4):353–368. doi: 10.1097/FBP.0000000000000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 60.Mori T, Koyama N, Segawa T, Maeda M, Maruyama N, Kinoshita N, Hou H, Tan J, Town T. Methylene blue modulates beta-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice. J Biol Chem. 2014;289(44):30303–30317. doi: 10.1074/jbc.M114.568212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paban V, Manrique C, Filali M, Maunoir-Regimbal S, Fauvelle F, Alescio-Lautier B. Therapeutic and preventive effects of methylene blue on Alzheimer’s disease pathology in a transgenic mouse model. Neuropharmacology. 2014;76(Pt A):68–79. doi: 10.1016/j.neuropharm.2013.06.033. [DOI] [PubMed] [Google Scholar]
- 62.Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7(12):e52389. doi: 10.1371/journal.pone.0052389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis. 2010;20(Suppl 2):S439–452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]
- 64.Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8(4):609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93(20):11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46(30):8850–8860. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 68.Hattori M, Sugino E, Minoura K, In Y, Sumida M, Taniguchi T, Tomoo K, Ishida T. Different inhibitory response of cyanidin and methylene blue for filament formation of tau microtubule-binding domain. Biochemical and biophysical research communications. 2008;374(1):158–163. doi: 10.1016/j.bbrc.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Crowe A, James MJ, Lee VM, Smith AB, 3rd, Trojanowski JQ, Ballatore C, Brunden KR. Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J Biol Chem. 2013;288(16):11024–11037. doi: 10.1074/jbc.M112.436006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao M, Liang F, Xu H, Yan W, Zhang J. Methylene blue exerts a neuroprotective effect against traumatic brain injury by promoting autophagy and inhibiting microglial activation. Mol Med Rep. 2016;13(1):13–20. doi: 10.3892/mmr.2015.4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Talley Watts L, Long JA, Boggs RC, Manga H, Huang S, Shen Q, Duong TQ. Delayed Methylene Blue Improves Lesion Volume, Multi-Parametric Quantitative Magnetic Resonance Imaging Measurements, and Behavioral Outcome after Traumatic Brain Injury. J Neurotrauma. 2016;33(2):194–202. doi: 10.1089/neu.2015.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, Shen Q, Duong TQ. Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma. 2014;31(11):1063–1071. doi: 10.1089/neu.2013.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma. 2015;32(2):127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mangus DB, Huang L, Applegate PM, Gatling JW, Zhang J, Applegate RL., 2nd A systematic review of neuroprotective strategies after cardiac arrest: from bench to bedside (Part I – Protection via specific pathways) Medical gas research. 2014;4:9. doi: 10.1186/2045-9912-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider A, Bottiger BW, Popp E. Cerebral resuscitation after cardiocirculatory arrest. Anesthesia and analgesia. 2009;108(3):971–979. doi: 10.1213/ane.0b013e318193ca99. [DOI] [PubMed] [Google Scholar]
- 76.Kim YM, Yim HW, Jeong SH, Klem ML, Callaway CW. Does therapeutic hypothermia benefit adult cardiac arrest patients presenting with non-shockable initial rhythms?: A systematic review and meta-analysis of randomized and non-randomized studies. Resuscitation. 2012;83(2):188–196. doi: 10.1016/j.resuscitation.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 77.Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bbttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez-Nunez A, Sellke FW, Spaulding C, Sunde K, Vanden Hoek T. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication: a scientific statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke (Part II) International emergency nursing. 2010;18(1):8–28. doi: 10.1016/j.ienj.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WG, Billi J, Bottiger BW, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Carli P, Atkins D. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108(1):118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- 79.Scirica BM. Therapeutic hypothermia after cardiac arrest. Circulation. 2013;127(2):244–250. doi: 10.1161/CIRCULATIONAHA.111.076851. [DOI] [PubMed] [Google Scholar]
- 80.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 81.Victor Li, X B, Paul Szelemej, Jiming Kong. Delayed Neuronal Death in Ischemic Stroke: Molecular Pathways. In: Balestrino DM, editor. Advances in the Preclinical Study of Ischemic Stroke. InTech. [Google Scholar]
- 82.Kim J, Park JE, Nahrendorf M, Kim DE. Direct Thrombus Imaging in Stroke. Journal of stroke. 2016;18(3):286–296. doi: 10.5853/jos.2016.00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kirmani JF, Alkawi A, Panezai S, Gizzi M. Advances in thrombolytics for treatment of acute ischemic stroke. Neurology. 2012;79(13 Suppl 1):S119–125. doi: 10.1212/WNL.0b013e3182695882. [DOI] [PubMed] [Google Scholar]
- 84.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. The New England journal of medicine. 1995;333(24):1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 85.Ferrer I. Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis. 2006;21(Suppl 2):9–20. doi: 10.1159/000091699. [DOI] [PubMed] [Google Scholar]
- 86.Siesjo BK. Cell damage in the brain: a speculative synthesis. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1981;1(2):155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- 87.Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic conditions. Current neurovascular research. 2008;5(1):71–81. doi: 10.2174/156720208783565645. [DOI] [PubMed] [Google Scholar]
- 88.Giulian D, Li J, Leara B, Keenen C. Phagocytic microglia release cytokines and cytotoxins that regulate the survival of astrocytes and neurons in culture. Neurochemistry international. 1994;25(3):227–233. doi: 10.1016/0197-0186(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 89.Bothe HW, Bosma HJ, Hofer H, Hossmann KA, Angermeier WF. Selective vulnerability of hippocampus and disturbances of memory storage after mild unilateral ischemia of gerbil brain. Stroke. 1986;17(6):1160–1163. doi: 10.1161/01.str.17.6.1160. [DOI] [PubMed] [Google Scholar]
- 90.Sharma HS, Miclescu A, Wiklund L. Cardiac arrest-induced regional blood-brain barrier breakdown, edema formation and brain pathology: a light and electron microscopic study on a new model for neurodegeneration and neuroprotection in porcine brain. J Neural Transm (Vienna) 2011;118(1):87–114. doi: 10.1007/s00702-010-0486-4. [DOI] [PubMed] [Google Scholar]
- 91.Martijn C, Wiklund L. Effect of methylene blue on the genomic response to reperfusion injury induced by cardiac arrest and cardiopulmonary resuscitation in porcine brain. BMC medical genomics. 2010;3:27. doi: 10.1186/1755-8794-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie L, Li W, Winters A, Yuan F, Jin K, Yang S. Methylene blue induces macroautophagy through 5′ adenosine monophosphate-activated protein kinase pathway to protect neurons from serum deprivation. Frontiers in cellular neuroscience. 2013;7:56. doi: 10.3389/fncel.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fang Q, Yan X, Li S, Sun Y, Xu L, Shi Z, Wu M, Lu Y, Dong L, Liu R, Yuan F, Yang SH. Methylene Blue Ameliorates Ischemia/Reperfusion-Induced Cerebral Edema: An MRI and Transmission Electron Microscope Study. Acta neurochirurgica Supplement. 2016;121:227–236. doi: 10.1007/978-3-319-18497-5_41. [DOI] [PubMed] [Google Scholar]
- 94.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. Journal of cell science. 2007;120(Pt 23):4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 95.Arrazola MS, Silva-Alvarez C, Inestrosa NC. How the Wnt signaling pathway protects from neurodegeneration: the mitochondrial scenario. Frontiers in cellular neuroscience. 2015;9:166. doi: 10.3389/fncel.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Irwin JA, Wong HE, Kwon I. Different fates of Alzheimer’s disease amyloid-beta fibrils remodeled by biocompatible small molecules. Biomacromolecules. 2013;14(1):264–274. doi: 10.1021/bm3016994. [DOI] [PubMed] [Google Scholar]
- 97.Supnet C, Bezprozvanny I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2010;20(Suppl 2):S487–498. doi: 10.3233/JAD-2010-100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Human molecular genetics. 2006;15(9):1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 101.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. European journal of nuclear medicine and molecular imaging. 2005;32(4):486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 102.Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochimica et biophysica acta. 2010;1802(1):11–19. doi: 10.1016/j.bbadis.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 104.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Human molecular genetics. 2011;20(23):4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pratico D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends in pharmacological sciences. 2008;29(12):609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 106.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Atamna H. Amino acids variations in amyloid-beta peptides, mitochondrial dysfunction, and new therapies for Alzheimer’s disease. Journal of bioenergetics and biomembranes. 2009;41(5):457–464. doi: 10.1007/s10863-009-9246-2. [DOI] [PubMed] [Google Scholar]
- 108.Violet M, Chauderlier A, Delattre L, Tardivel M, Chouala MS, Sultan A, Marciniak E, Humez S, Binder L, Kayed R, Lefebvre B, Bonnefoy E, Buee L, Galas MC. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiology of disease. 2015;82:540–551. doi: 10.1016/j.nbd.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Zakaria A, Hamdi N, Abdel-Kader RM. Methylene Blue Improves Brain Mitochondrial ABAD Functions and Decreases Abeta in a Neuroinflammatory Alzheimer’s Disease Mouse Model. Molecular neurobiology. 2016;53(2):1220–1228. doi: 10.1007/s12035-014-9088-8. [DOI] [PubMed] [Google Scholar]
- 110.Lim YA, Grimm A, Giese M, Mensah-Nyagan AG, Villafranca JE, Ittner LM, Eckert A, Gotz J. Inhibition of the mitochondrial enzyme ABAD restores the amyloid-beta-mediated deregulation of estradiol. PloS one. 2011;6(12):e28887. doi: 10.1371/journal.pone.0028887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stern DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19(6):597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 112.Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Frontiers in pharmacology. 2015;6:321. doi: 10.3389/fphar.2015.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla F, Gallego-Gomez JC, Kosik KS, Cardona-Gomez GP. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(42):13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. Journal of neurochemistry. 2008;104(6):1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. International journal of molecular sciences. 2014;15(3):4671–4713. doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP. Tau phosphorylation affects its axonal transport and degradation. Neurobiology of aging. 2013;34(9):2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sulistio YA, Heese K. The Ubiquitin-Proteasome System and Molecular Chaperone Deregulation in Alzheimer’s Disease. Molecular neurobiology. 2016;53(2):905–931. doi: 10.1007/s12035-014-9063-4. [DOI] [PubMed] [Google Scholar]
- 118.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. The Journal of biological chemistry. 2005;280(9):7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 119.Lira-De Leon KI, Garcia-Gutierrez P, Serratos IN, Palomera-Cardenas M, Figueroa-Corona Mdel P, Campos-Pena V, Meraz-Rios MA. Molecular mechanism of tau aggregation induced by anionic and cationic dyes. Journal of Alzheimer’s disease: JAD. 2013;35(2):319–334. doi: 10.3233/JAD-121765. [DOI] [PubMed] [Google Scholar]
- 120.Hochgrafe K, Sydow A, Matenia D, Cadinu D, Konen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E, Mandelkow EM. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta neuropathologica communications. 2015;3:25. doi: 10.1186/s40478-015-0204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiology of disease. 2010;39(3):265–271. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 122.Fatouros C, Pir GJ, Biernat J, Koushika SP, Mandelkow E, Mandelkow EM, Schmidt E, Baumeister R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Human molecular genetics. 2012;21(16):3587–3603. doi: 10.1093/hmg/dds190. [DOI] [PubMed] [Google Scholar]
- 123.Driver JA, Logroscino G, Gaziano JM, Kurth T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology. 2009;72(5):432–438. doi: 10.1212/01.wnl.0000341769.50075.bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baumann CR. Epidemiology, diagnosis and differential diagnosis in Parkinson’s disease tremor. Parkinsonism & related disorders. 2012;18(Suppl 1):S90–92. doi: 10.1016/S1353-8020(11)70029-3. [DOI] [PubMed] [Google Scholar]
- 125.Martinez-Ramirez D, Almeida L, Giugni JC, Ahmed B, Higuchi MA, Little CS, Chapman JP, Mignacca C, Wagle Shukla A, Hess CW, Hegland KW, Okun MS. Rate of aspiration pneumonia in hospitalized Parkinson’s disease patients: a cross-sectional study. BMC neurology. 2015;15:104. doi: 10.1186/s12883-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tjaden K. Speech and Swallowing in Parkinson’s Disease. Topics in geriatric rehabilitation. 2008;24(2):115–126. doi: 10.1097/01.TGR.0000318899.87690.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iwasaki S, Narabayashi Y, Hamaguchi K, Iwasaki A, Takakusagi M. Cause of death among patients with Parkinson’s disease: a rare mortality due to cerebral haemorrhage. Journal of neurology. 1990;237(2):77–79. doi: 10.1007/BF00314665. [DOI] [PubMed] [Google Scholar]
- 128.Lewitt PA. Levodopa for the treatment of Parkinson’s disease. The New England journal of medicine. 2008;359(23):2468–2476. doi: 10.1056/NEJMct0800326. [DOI] [PubMed] [Google Scholar]
- 129.Arduino DM, Esteves AR, Cortes L, Silva DF, Patel B, Grazina M, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Human molecular genetics. 2012;21(21):4680–4702. doi: 10.1093/hmg/dds309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. The Journal of biological chemistry. 2008;283(14):9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elkon H, Don J, Melamed E, Ziv I, Shirvan A, Offen D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. Journal of molecular neuroscience: MN. 2002;18(3):229–238. doi: 10.1385/JMN:18:3:229. [DOI] [PubMed] [Google Scholar]
- 132.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochimica et biophysica acta. 2010;1802(1):29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 133.Lee KK, Boelsterli UA. Bypassing the compromised mitochondrial electron transport with methylene blue alleviates efavirenz/isoniazid-induced oxidant stress and mitochondria-mediated cell death in mouse hepatocytes. Redox biology. 2014;2:599–609. doi: 10.1016/j.redox.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Todorovic M, Wood SA, Mellick GD. Nrf2: a modulator of Parkinson’s disease? J Neural Transm (Vienna) 2016;123(6):611–619. doi: 10.1007/s00702-016-1563-0. [DOI] [PubMed] [Google Scholar]
- 135.Tretter L, Horvath G, Holgyesi A, Essek F, Adam-Vizi V. Enhanced hydrogen peroxide generation accompanies the beneficial bioenergetic effects of methylene blue in isolated brain mitochondria. Free radical biology & medicine. 2014;77:317–330. doi: 10.1016/j.freeradbiomed.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 136.Delport A, Harvey BH, Petzer A, Petzer JP. Azure B and a synthetic structural analogue of methylene blue, ethylthioninium chloride, present with antidepressant-like properties. Life sciences. 2014;117(2):56–66. doi: 10.1016/j.lfs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 137.Yonutas HM, Vekaria HJ, Sullivan PG. Mitochondrial specific therapeutic targets following brain injury. Brain research. 2016;1640(Pt A):77–93. doi: 10.1016/j.brainres.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 138.Haddad SH, Arabi YM. Critical care management of severe traumatic brain injury in adults. Scandinavian journal of trauma, resuscitation and emergency medicine. 2012;20:12. doi: 10.1186/1757-7241-20-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chesnut RM. Secondary brain insults after head injury: clinical perspectives. New Horiz. 1995;3(3):366–375. [PubMed] [Google Scholar]
- 140.Kimbler DE, Murphy M, Dhandapani KM. Concussion and the adolescent athlete. The Journal of neuroscience nursing: journal of the American Association of Neuroscience Nurses. 2011;43(6):286–290. doi: 10.1097/JNN.0b013e31823858a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rosenfeld JV, Maas AI, Bragge P, Morganti-Kossmann MC, Manley GT, Gruen RL. Early management of severe traumatic brain injury. Lancet. 2012;380(9847):1088–1098. doi: 10.1016/S0140-6736(12)60864-2. [DOI] [PubMed] [Google Scholar]
- 142.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. British journal of anaesthesia. 2007;99(1):4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 143.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. The American journal of the medical sciences. 2015;350(2):132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 144.Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatric disease and treatment. 2015;11:97–106. doi: 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maxwell WL. Development of Concepts in the Pathology of Traumatic Axonal and Traumatic Brain Injury. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): 2015. (Frontiers in Neuroengineering). [Google Scholar]
- 146.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Molecular neurobiology. 2015;51(3):966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ahmed AI, Bullock MR, Dietrich WD. Hypothermia in Traumatic Brain Injury. Neurosurgery clinics of North America. 2016;27(4):489–497. doi: 10.1016/j.nec.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 149.Bennett MH, Trytko B, Jonker B. Hyperbaric oxygen therapy for the adjunctive treatment of traumatic brain injury. The Cochrane database of systematic reviews. 2012;12:CD004609. doi: 10.1002/14651858.CD004609.pub3. [DOI] [PubMed] [Google Scholar]
- 150.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. BioMed research international. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suliman NA, Mat Taib CN, Mohd Moklas MA, Adenan MI, Hidayat Baharuldin MT, Basir R. Establishing Natural Nootropics: Recent Molecular Enhancement Influenced by Natural Nootropic. Evidence-based complementary and alternative medicine: eCAM. 2016;2016:4391375. doi: 10.1155/2016/4391375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Camfield DA, Stough C, Farrimond J, Scholey AB. Acute effects of tea constituents L-theanine, caffeine, and epigallocatechin gallate on cognitive function and mood: a systematic review and meta-analysis. Nutrition reviews. 2014;72(8):507–522. doi: 10.1111/nure.12120. [DOI] [PubMed] [Google Scholar]
- 153.Varga MD. Adderall abuse on college campuses: a comprehensive literature review. Journal of evidence-based social work. 2012;9(3):293–313. doi: 10.1080/15433714.2010.525402. [DOI] [PubMed] [Google Scholar]
- 154.Gonzalez-Lima F, Barksdale BR, Rojas JC. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochemical pharmacology. 2014;88(4):584–593. doi: 10.1016/j.bcp.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 155.Rodriguez P, Zhou W, Barrett DW, Altmeyer W, Gutierrez JE, Li J, Lancaster JL, Gonzalez-Lima F, Duong TQ. Multimodal Randomized Functional MR Imaging of the Effects of Methylene Blue in the Human Brain. Radiology. 2016;281(2):516–526. doi: 10.1148/radiol.2016152893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barrett DW, Gonzalez-Lima F. Transcranial infrared laser stimulation produces beneficial cognitive and emotional effects in humans. Neuroscience. 2013;230:13–23. doi: 10.1016/j.neuroscience.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 157.Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017 doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grimm A, Friedland K, Eckert A. Mitochondrial dysfunction: the missing link between aging and sporadic Alzheimer’s disease. Biogerontology. 2016;17(2):281–296. doi: 10.1007/s10522-015-9618-4. [DOI] [PubMed] [Google Scholar]
- 159.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8(4):393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Voloboueva LA, Giffard RG. Inflammation, mitochondria, and the inhibition of adult neurogenesis. Journal of neuroscience research. 2011;89(12):1989–1996. doi: 10.1002/jnr.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132(4):645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 162.Kernie SG, Parent JM. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis. 2010;37(2):267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang RL, Zhang ZG, Zhang L, Chopp M. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience. 2001;105(1):33–41. doi: 10.1016/s0306-4522(01)00117-8. [DOI] [PubMed] [Google Scholar]
- 164.Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(19):7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nature medicine. 2002;8(9):963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 166.Bettio LEB, Rajendran L, Gil-Mohapel J. The effects of aging in the hippocampus and cognitive decline. Neurosci Biobehav Rev. 2017;79:66–86. doi: 10.1016/j.neubiorev.2017.04.030. [DOI] [PubMed] [Google Scholar]
- 167.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta neuropathologica. 2003;106(6):518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 168.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer research. 2003;63(14):4021–4027. [PubMed] [Google Scholar]
- 169.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nature medicine. 2002;8(12):1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 170.Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1999;19(1):87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- 171.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 172.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 173.Torroglosa A, Murillo-Carretero M, Romero-Grimaldi C, Matarredona ER, Campos-Caro A, Estrada C. Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of epidermal growth factor receptor and phosphoinositide-3-kinase/Akt pathway. Stem Cells. 2007;25(1):88–97. doi: 10.1634/stemcells.2006-0131. [DOI] [PubMed] [Google Scholar]
- 174.Fike JR, Rola R, Limoli CL. Radiation response of neural precursor cells. Neurosurgery clinics of North America. 2007;18(1):115–127. doi: 10.1016/j.nec.2006.10.010. x. [DOI] [PubMed] [Google Scholar]
- 175.Cordeau-Lossouarn L, Vayssiere JL, Larcher JC, Gros F, Croizat B. Mitochondrial maturation during neuronal differentiation in vivo and in vitro. Biology of the cell. 1991;71(1-2):57–65. doi: 10.1016/0248-4900(91)90051-n. [DOI] [PubMed] [Google Scholar]
- 176.Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez JA, Trevelyan AJ, Turnbull DM, Lightowlers RN. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell proliferation. 2009;42(4):413–424. doi: 10.1111/j.1365-2184.2009.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Papa S, Petruzzella V, Scacco S, Vergari R, Panelli D, Tamborra R, Corsi P, Picciariello M, Lambo R, Bertini E, Santorelli FM. Respiratory complex I in brain development and genetic disease. Neurochemical research. 2004;29(3):547–560. doi: 10.1023/b:nere.0000014825.42365.16. [DOI] [PubMed] [Google Scholar]
- 178.Wong A, Cavelier L, Collins-Schramm HE, Seldin MF, McGrogan M, Savontaus ML, Cortopassi GA. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Human molecular genetics. 2002;11(4):431–438. doi: 10.1093/hmg/11.4.431. [DOI] [PubMed] [Google Scholar]
- 179.Xie L, Choudhury GR, Wang J, Park Y, Liu R, Yuan F, Zhang CL, Yorio T, Jin K, Yang SH. Methylene blue promotes quiescence of rat neural progenitor cells. Frontiers in cellular neuroscience. 2014;8:315. doi: 10.3389/fncel.2014.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.van der Ven AT, Pape JC, Hermann D, Schloesser R, Genius J, Fischer N, Mossner R, Scherbaum N, Wiltfang J, Rujescu D, Benninghoff J. Methylene Blue (Tetramethylthionine Chloride) Influences the Mobility of Adult Neural Stem Cells: A Potentially Novel Therapeutic Mechanism of a Therapeutic Approach in the Treatment of Alzheimer’s Disease. J Alzheimers Dis. 2017;57(2):531–540. doi: 10.3233/JAD-160755. [DOI] [PubMed] [Google Scholar]
- 181.Crooks J. Haemolytic jaundice in a neonate after intra-amniotic injection of methylene blue. Archives of disease in childhood. 1982;57(11):872–873. doi: 10.1136/adc.57.11.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Porat R, Gilbert S, Magilner D. Methylene blue-induced phototoxicity: an unrecognized complication. Pediatrics. 1996;97(5):717–721. [PubMed] [Google Scholar]
- 183.Spahr RC, Salsburey DJ, Krissberg A, Prin W. Intraamniotic injection of methylene blue leading to methemoglobinemia in one of twins. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics. 1980;17(5):477–478. doi: 10.1002/j.1879-3479.1980.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 184.Wolvetang T, Janse R, Ter Horst M. Serotonin Syndrome After Methylene Blue Administration During Cardiac Surgery: A Case Report and Review. Journal of cardiothoracic and vascular anesthesia. 2016;30(4):1042–1045. doi: 10.1053/j.jvca.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 185.Hencken L, To L, Ly N, Morgan JA. Serotonin Syndrome Following Methylene Blue Administration for Vasoplegic Syndrome. Journal of cardiac surgery. 2016;31(4):208–210. doi: 10.1111/jocs.12705. [DOI] [PubMed] [Google Scholar]
- 186.Smith CJ, Wang D, Sgambelluri A, Kramer RS, Gagnon DJ. Serotonin syndrome following methylene blue administration during cardiothoracic surgery. Journal of pharmacy practice. 2015;28(2):207–211. doi: 10.1177/0897190014568389. [DOI] [PubMed] [Google Scholar]