

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal lung disease manifested by overtly scarred peripheral and basilar regions and more normal-appearing central lung areas. Lung tissues from macroscopically normal-appearing (IPFn) and scarred (IPFs) areas of explanted IPF lungs were analyzed by RNASeq and compared with healthy control (HC) lung tissues. There were profound transcriptomic changes in IPFn compared with HC tissues, which included elevated expression of numerous immune-, inflammation-, and extracellular matrix-related mRNAs, and these changes were similar to those observed with IPFs compared to HC. Comparing IPFn directly to IPFs, elevated expression of epithelial mucociliary mRNAs was observed in the IPFs tissues. Thus, despite the known geographic tissue heterogeneity in IPF, the entire lung is actively involved in the disease process, and demonstrates pronounced elevated expression of numerous immune-related genes. Differences between normal-appearing and scarred tissues may thus be driven by deranged epithelial homeostasis or possibly non-transcriptomic factors.
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a distinct disorder within the broad group of diseases termed interstitial lung diseases (ILD), an inclusive group of lung disorders characterized by inflammation and/or fibrosis of the lung parenchyma [1-3]. As the most severe form of ILD, IPF causes substantial patient morbidity and mortality, has a median survival of approximately three years, and has limited proven efficacious therapies. Lung transplantation remains the only viable intervention in end-stage lung disease due to IPF.
As a fibrotic lung disease, IPF histologically demonstrates a pattern of lung injury termed usual interstitial pneumonia (UIP), which is characterized by dense regions of scarring, interspersed regions of relatively normal lung architecture, fibroblastic foci, patchy inflammatory cell infiltration, and honeycomb (cystic) change. The fibroblastic foci contain alpha-smooth muscle actin (α-SMA)-expressing myofibroblasts. These histologic observations support the commonly accepted paradigm that IPF is a disease of excess extracellular matrix (ECM) accumulation and dysregulated mesenchymal cell proliferation [4].
Despite intense research effort over the past several decades, the pathobiological mechanisms of IPF are not fully understood. As part of this effort, numerous transcriptomic profiling studies of lung tissues from patients with IPF have been performed using several approaches, including serial analysis of gene expression (SAGE) [5], microarray analysis [6-12], RNASeq [13-16], and single-cell RNASeq [17]. These studies have revealed a wealth of phenomenological information with important mechanistic implications, stimulating and focusing research on several specific pathophysiological mechanisms of IPF, such as disturbances in expression of genes associated with extracellular matrix, inflammation and immunity, and pulmonary epithelia. Subsequent studies have more specifically focused on contributions from epithelial disturbances [17], including those affecting expression of surfactants, cilium-associated genes, and mucins, including MUC5B [11, 18]; matrix metalloproteinases, including MMP7 [8, 19]; and immune inflammation involving T cells, B cells, macrophages [20-23], and numerous cytokines and chemokines [24]. The majority of molecular studies have focused on the most scarred areas of the lung since these areas are usually more accessible by standard surgical biopsy. However, the heavily scarred areas likely represent late stages of disease in which initial pathobiological mechanisms have dissipated.
IPF is characterized by scarring, but the IPF lung consistently demonstrates substantial geographic heterogeneity. Radiologic and gross pathology observations often show severe scarring of the lung in predominantly peripheral and basilar areas, whereas central and apical areas appear normal and seemingly unaffected. We recently analyzed and quantified histologic findings in macroscopically normal-appearing lung tissue in patients with IPF [25] and found that these areas exhibited patterns of lung injury termed organizing pneumonia ([OP], characterized by basophilic-staining deposits of ECM containing spindle-shaped fibroblasts or myofibroblasts), and nonspecific interstitial pneumonia ([NSIP], characterized by diffuse interstitial inflammation and/or fibrosis that occur in a spatially uniform pattern throughout the lung). Additionally, α-SMA-expressing myofibroblasts were present in the OP foci in the normal-appearing areas, similar to α-SMA-expressing myofibroblasts found in fibroblastic foci in scarred areas of UIP. Based on these findings, we concluded that lung injury is widespread throughout the IPF lung, extending beyond peripheral and basilar regions, and hypothesized a continuum of lung injury responses in IPF, with macroscopically normal-appearing areas representing earlier stages of disease and macroscopically scarred areas representing more advanced stages [25]. Transcriptomic profiles of macroscopically normal-appearing and scarred regions of IPF lungs have not been comparatively analyzed. Studying areas of the IPF lung that appear to represent earlier stages in the course of injury (macroscopically normal-appearing yet with microscopic evidence of lung injury) may allow for the detection of key mechanisms in disease pathogenesis.
In this study, we hypothesized that our recently reported histological abnormalities in macroscopically normal-appearing lung tissue in IPF [25] are accompanied by substantial transcriptomic disturbances. To challenge this hypothesis and thus gain a deeper understanding of the IPF disease process, we assessed transcriptomic profiles of macroscopically normal-appearing IPF lung tissues, and compared these profiles with transcriptomes of macroscopically scarred IPF lung tissues and healthy control lung tissues. It appears intuitive that macroscopically normal-appearing tissue represents earlier stages of disease, as opposed to densely scarred areas which represent later stages of disease. The scope of this study was to gain better insight into disease pathobiology in macroscopically normal-appearing (though microscopically abnormal [25]) areas, and such scope is consistent with the universal movement to understand and treat diseases at their earliest stages.
2. Materials and Methods
2.1. Patients and Controls
This study was reviewed and approved by the University of Maryland Institutional Review Board, and all experiments were performed in accordance with relevant guidelines and regulations. Patients agreed to participate in the study and provided informed consent. From a group of 41 patients who had undergone lung transplantation in 2016 at the University of Maryland, we obtained lung explants from three patients with idiopathic pulmonary fibrosis (IPF). IPF was diagnosed based on standard published criteria [2, 26], which included clinical idiopathic disease (no identifiable evidence of auto-immune or connective tissue disease, pulmonary drug toxicity, or significant environmental, avocational, or occupational exposure), chest computed tomography (CT) findings of a probable or definite usual interstitial pneumonia (UIP) pattern, and definite UIP histology in lung explant analysis. These three patients did not manifest clinical or histologic evidence of an acute exacerbation of IPF and had not undergone extracorporeal membrane oxygenation (ECMO) treatment prior to transplantation. Clinical information, including demographics, smoking history, environmental and occupational history, pharmacologic treatment, pulmonary function tests, computed tomography of the chest, right heart catheterization data, and duration of illness prior to transplantation, was reviewed and documented for each of the three patients.
For controls, lungs from three previously healthy persons that had been harvested as transplant donor lungs but ultimately not utilized for transplantation were used for healthy control (HC) tissues.
2.2. Macroscopic and Microscopic Tissue Evaluation
A detailed macroscopic (gross) and microscopic (histologic) evaluation of each IPF lung explant and each HC donor lung was performed by members of the research team (NWT, ILG) and by a clinical pathologist with extensive experience in interstitial lung disease (APB), as demonstrated in Figure 1 and as previously described [25]. Macroscopically, the external surface of each explant was examined, and subsequently the lung was cut sagittally resulting in two gross sections of the explant that revealed the internal parenchyma of the lung. Following sagittal sectioning, macroscopically normal-appearing (IPFn) and macroscopically scarred (IPFs) areas were identified. Macroscopically normal-appearing tissue was identified grossly as areas similar in color and consistency to normal lung tissue, which was dark red in color due to normal tissue perfusion, had a soft consistency to finger palpation, was often located in central areas of the lung parenchyma, and had no visual evidence of cyst formation. Macroscopically scarred tissue was identified grossly as areas pale in color due to poor or absent perfusion (scar), had a firm consistency to finger palpation, was most often located in peripheral subpleural areas of the lung parenchyma, and contained numerous visible small cysts (honeycombing). The macroscopically normal-appearing and macroscopically scarred areas were usually well-delineated (Figure 1), which allowed for straight-forward sectioning of individual areas. For the purposes of this study, tissue samples were obtained only from those normal-appearing and scarred areas that were ≥ 1 cm2, so that each area could be reliably and accurately identified and sampled. Following macroscopic assessment of the normal-appearing and scarred areas, numerous tissue samples from these areas were obtained for molecular studies and paraffin sectioning.
Figure 1.
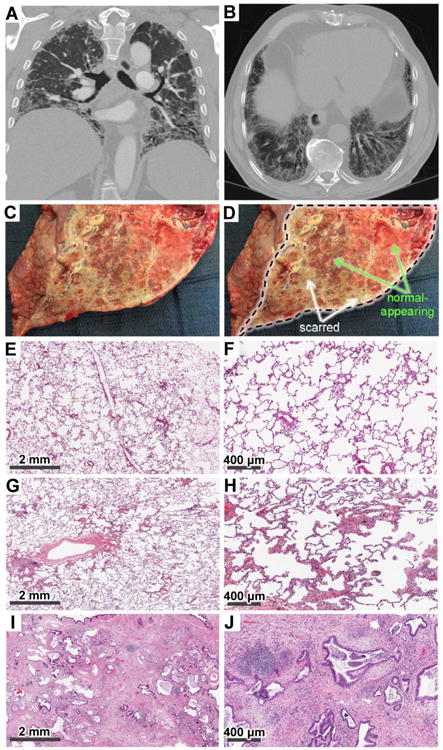
Representative radiology (A, B), gross pathology (C, D), and histology (E–J) findings in patients with IPF (A–D, G–J) and healthy controls (E, F). A, B. Radiologic images in the IPF patients demonstrate reticulation, honeycombing, traction bronchiectasis, and volume loss in a predominantly peripheral and basilar distribution, consistent with advanced pulmonary fibrosis. C, D. Gross appearance of a sagittally cut lung explant from a patient with IPF (C) and the same image with markings superimposed (D). The cut plane revealing the internal parenchyma of the lung is demarcated by the black dotted line in panel D, whereas selected macroscopically normal-appearing and macroscopically scarred areas are indicated with green and white arrows, respectively. Also note the cobblestone appearance of the pleural surface on the left side of panels C and D outside of the dissection area. E, F. Low- (E) and high-magnification (F) histologic images of normal lung parenchyma from HC lung tissue. G, H. Low- (G) and high-magnification (H) histologic images from macroscopically normal-appearing IPF lung areas (IPFn) demonstrate largely preserved pulmonary microarchitecture, but scattered areas of organizing pneumonia and non-specific interstitial pneumonia are also present. I, J. Low- (I) and high-magnification (J) histologic images from macroscopically scarred IPF lung areas (IPFs) demonstrate dense areas of scarring, collapse of secondary lobules, architectural remodeling, honeycombing (cysts lined with ciliated respiratory epithelium and goblet cells), fibroblastic foci, and lymphocyte aggregates.
Following procurement of the samples based on their macroscopic appearance, the microscopic (histologic) findings were assessed in selected samples. Macroscopically normal-appearing tissue demonstrated microscopic signs of lung injury as previously described [25], including microscopic organizing pneumonia and cellular non-specific interstitial pneumonia [25]. Macroscopically scarred areas demonstrated findings of advanced pulmonary fibrosis as previously described [25], characterized by dense regions of scarring, fibroblastic foci, patchy inflammatory cell infiltration, and honeycomb (cystic) change.
A similar detailed macroscopic (gross) and microscopic (histologic) evaluation of each HC donor lung was performed. Evaluation of the HC lungs and tissue revealed normal gross appearance of the lungs on macroscopic examination, and normal lung tissue histologically without signs of organizing pneumonia or non-specific interstitial pneumonia on microscopic examination.
Microscopic paraffin tissue sections from the IPF explants and the HC donor lungs were scanned into a digital pathology system (Aperio Digital Pathology, Leica Biosystems, Buffalo Grove, IL, USA) to assist with histologic analyses of the tissue samples.
2.3 Primary Fibroblast Cell Cultures
Primary adult pulmonary fibroblasts were expanded in cell culture from IPFn and IPFs areas of lung explants from the three patients with IPF. Fibroblasts were cultured in DMEM (Corning Cellgro, Manassas, VA) supplemented with 10% bovine calf serum (BCS; HyClone Laboratories, GE Healthcare Life Sciences, Logan, UT), 2 mM L-glutamine (Gemini BioProducts, West Sacramento, CA), 1 mM sodium pyruvate, MEM nonessential amino acids solution, and antibiotic-antimycotic (10,000 units/ml penicillin, 10,000 μg/ml streptomycin, and 25 μg/ml amphotericin B), all from GIBCO Life Technologies (Thermo Fisher Scientific, Waltham, MA). Fibroblast cultures were maintained in T75 culture flasks (Nest Biotechnology, Rahway, NJ) in a humidified atmosphere with 5% CO2 at 37°C. These cultures were used for RNASeq analyses at passage 3. The mesenchymal nature of these cultures was validated by the characteristic cell morphology and RNASeq results that showed high expression levels of COL1A1, COL1A2, COL3A1, and VIM, but very low expression levels of an epithelial marker, E-cadherin, and an endothelial marker, PECAM1.
2.4. RNA Extraction and Sequencing
Numerous tissue samples for RNA extraction were obtained from macroscopically normal-appearing and macroscopically scarred areas from the IPF explants, and from the healthy control donor lungs. Tissue samples for RNA analysis obtained from the three healthy control lungs were labeled HC1 – HC3, and from the three IPF explants were labeled IPF1 – IPF3. Tissue samples were numbered sequentially. For healthy controls, the first digit in the numbering indicates the donor, and the second digit identifies a separate tissue sample, with a total of eight such tissue samples obtained: HC11, HC12, HC21, HC22, HC23, HC31, HC32, and HC33. From the IPF explants, tissue samples were obtained from both macroscopically normal-appearing (IPFn) areas and from macroscopically scarred (IPFs) areas (Figure 1), which were labeled sequentially, with the first digit in the numbering indicating the explant, and the second digit identifying a separate tissue sample. Overall, ten tissue samples were obtained from macroscopically normal-appearing areas (IPF1n1 – IPF1n4, IPF2n1 – IPF2n3, IPF3n1 – IPF3n3), and eight tissue samples were obtained from scarred areas (IPF1s1 – IPF1s3, IPF1s1 – IPF2s4, IPF3s1). A total of 26 tissue samples were thus obtained from the HC lungs and the IPF explants for RNA analysis.
Separate RNASeq analyses were performed with the cultured primary fibroblasts (described above) derived from IPFn and IPFs tissues from the three IPF patients. IPFn and IPFs fibroblast cultures were numbered IPFn1 – IPFn3 and IPFs1 – IPFs3 to match the numbering of the corresponding tissue samples.
Total cellular RNA was isolated using TRIzol reagent (Invitrogen Thermo Fisher, Waltham, MA). The integrity and purity of the total RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA) and a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE). Total RNA was used to generate 5 μg of cDNA using the SMARTer PCR cDNA Synthesis Kit (Clontech Laboratories, Mountain View, CA).The resulting cDNA was fragmented using a Bioruptor (Diagenode, Denville, NJ), profiled using an Agilent 2100 Bioanalyzer or Agilent 2200 TapeStation, and subjected to Illumina library preparation using SPRIWorks HT (Beckman Coulter, Indianapolis IN). The quality, quantity, and size distribution of the Illumina library was determined using an Agilent Bioanalyzer or Tapestation. Library preparation for RNASeq was performed using the Illumina TruSeq Low Throughput (LT) RNA Sample Preparation Protocol. Short-read sequencing was performed on an Illumina HiSeq 2500, generating 100–125-bp paired-end libraries with an average of 40 million paired reads per sample, and then on an Illumina HiSeq 2500. The raw RNASeq reads (fastq files) for each sample were checked using FastQC software (Babraham Institute, Cambridge, UK) to check the quality of the data and then aligned to the reference human genome (hg19 / GRCh37 assembly) using STAR software [27]. Transcript abundance was then estimated as counts per million (cpm) values using the htseq-count script of the open source Python package HTSeq 0.6.1p2 [28, 29]. The RNASeq data have been deposited in the Gene Expression Omnibus database (accession number GSE99621; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99621).
2.5. RNASeq Analysis
Normalization of counts, principal component analysis (PCA), the likelihood ratio test (LRT), and pair-wise analyses of differential gene expression between tissue groups were performed using the DESeq2 package of Bioconductor [30, 31]. PCA was performed based on the 500 most variable genes, in order to generate an overview of the underlying structure of the dataset and assess separation of sample groups. LRT was used to assess differences in gene expression across all three groups of samples (HC, IPFn, IPFs), and the Wald test was used for pair-wise comparisons between groups (HC vs IPFn, HC vs IPFs, HC vs IPF combined, IPFn vs IPFs). For LRT and each of the pair-wise comparisons, genes with unreliably low expression levels (mapped read counts <50) were filtered out. Then, LRT and pair-wise comparisons were performed using DESeq2, with false discovery rate-adjusted p-value < 0.05 and fold difference ≥ 2.0 used to complete selection of differentially expressed genes.
Hierarchical clustering of genes and samples was performed based on log2-transformed normalized counts using Morpheus [32]. Spearman rank correlation with average linkage was used for clustering. In all cluster heatmaps, rows represented genes and columns represented tissue samples. Heatmaps were color-coded by rows, so that the minimum and maximum expression values of each gene were used to define the color scheme for the corresponding row. This method of color-coding was chosen to emphasize expression differences of each gene across tissue samples. Each cluster heatmap is accompanied with a corresponding supplementary dataset, as indicated in the Results, listing numerical values of log2-transformed normalized counts. Gene ontology enrichment analysis and visualization of differentially expressed gene lists was performed using the gene ontology (GO) analysis tool GOrilla [33-35]. Venn diagrams were constructed using BioVenn [36, 37].
2.6. RT-qPCR
Total RNA was reversed-transcribed into cDNA using the SuperScript First-Strand synthesis kit from Invitrogen Life Technologies (Carlsbad, CA). RT-qPCR was performed with SYBR Green PCR Master Mix (Life Technologies, Thermo Fisher Scientific) with validated primers that were obtained from Qiagen (Valencia, CA). The levels of gene expression relative to 18S rRNA or GAPDH mRNA were calculated using the using the 2−ΔΔCt method. Differences between sample groups were calculated using a two-tailed Student's t-test and a Mann-Whitney U-test. Violin plots were constructed using BoxPlotR [38].
3. Results
3.1. Characteristics of Patients, Controls, and Lung Tissues
From a group of 41 patients who had undergone lung transplantation in 2016 at the University of Maryland, we obtained lung explants from three patients who met established criteria for idiopathic pulmonary fibrosis (IPF) [2, 26]. Each of the three patients had idiopathic disease (no identifiable evidence of autoimmune or connective tissue disease, pulmonary drug toxicity, or significant environmental, occupational or avocational exposures), had chest computed tomography (CT) findings of a probable or definite usual interstitial pneumonia (UIP) pattern, and had definite UIP histology on lung explant histologic evaluation. Representative radiologic and histologic images from the three patients are shown in Figure 1, and the clinical, physiologic, radiologic, and histologic characteristics of each patient, all consistent with advanced lung disease and end-stage pulmonary fibrosis, are shown in Supplementary Table 1. None of the patients had clinical or histologic manifestations of an acute exacerbation of IPF, nor had they undergone extracorporeal membrane oxygenation (ECMO) treatment prior to transplantation.
Lungs from three previously healthy persons that had been harvested as transplant donor lungs, but ultimately not utilized for transplantation, were used as healthy control (HC) lung tissues. Demographic and clinical characteristics of the control lung donors are shown in Supplementary Table 1.
Numerous lung tissue samples were obtained from each IPF explant and each HC donor lung (Supplementary Table 1). From the IPF explants, tissue samples were obtained from macroscopically normal-appearing (IPFn) and macroscopically scarred (IPFs) areas, as explained in detail in the Materials and Methods, as shown in Figure 1, and as previously described [25]. For RNASeq analyses, ten tissue samples were obtained from IPFn areas and eight tissue samples were obtained from IPFs areas from the three IPF explants, with each numbered sequentially (see Materials and Methods for details of tissue sample numbering). For some analyses, the IPFn and IPFs groups of samples were pooled into a single group termed IPF combined. From the three HC donor lungs, eight tissue samples were obtained for RNASeq analyses. Overall, a total of 26 tissue samples were obtained from the IPF explants and the HC donor lungs for transcriptomic analyses.
3.2. Overall Assessment of Transcriptomic Differences Across All Three Tested Lung Tissue Sample Groups
Initial exploratory assessment of the RNASeq dataset was performed using principal component analysis (PCA) based on the 500 most variable genes. Principal components 1 and 2 accounted for 43% and 21% of the variance, respectively, and showed a clear separation of lung samples from healthy controls (HC) and samples obtained from the lungs of patients with IPF (Figure 2A, Supplementary Figure 1). Samples from macroscopically normal-appearing (IPFn) and macroscopically scarred (IPFs) areas of the IPF lungs separated also, but less distinctly and with noticeable overlap between these two groups: samples IPF3s1, IPF2s2, and IPF2s4 localized in the IPFn region of the PCA plot. An alternative depiction of this PCA (Supplementary Figure 1) revealed proximity of tissue transcriptomes from separate donors in each tissue group: some of the samples from separate donors localized closer in PCA space than the samples derived from the same donor. The exploratory power of higher-number principal components was substantially lower because of their small contribution to variance (7.7% and 7.1% for principal components 3 and 4, respectively), and therefore they were not used.
Figure 2.
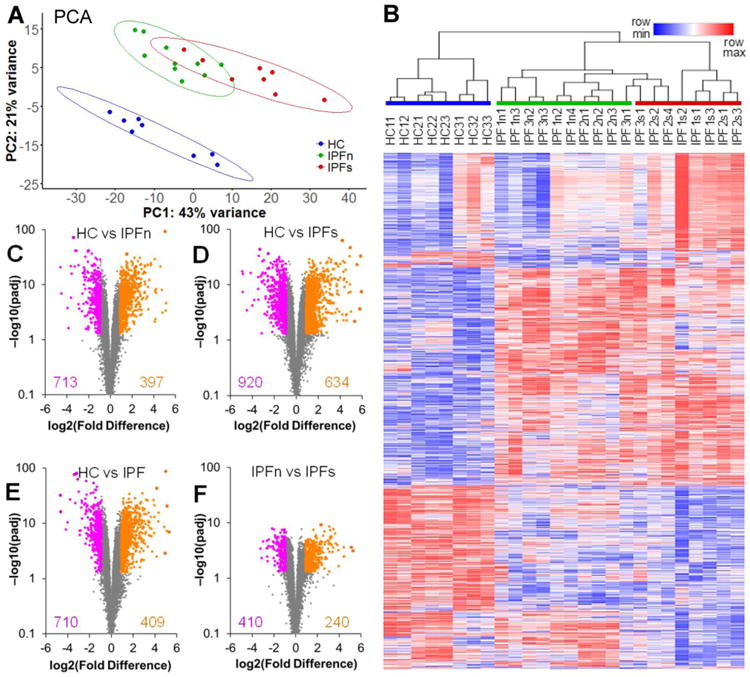
Overview of the differential expression of genes between lung tissue samples from patients with IPF and healthy controls. A. Two-dimensional principal component analysis of the RNASeq transcriptome dataset for lung tissue samples in this study reveals a clear separation of the samples from healthy controls (HC) and samples from patients with IPF. The separation of IPF samples from macroscopically normal-appearing (IPFn) and scarred (IPFs) areas is less pronounced, with some overlap present. Data ellipses for sample groups are plotted assuming a t-distribution at the confidence level 0.90 B. Unsupervised clustering of log2-transformed normalized counts (see Supplementary Dataset 1 for numerical values), using Spearman rank correlation with average linkage, of tissue samples based on 2,099 differentially expressed genes (see text for selection criteria). The genes themselves were also clustered using the same procedure. Note the varying extent of overlap between the sample groups (HC, IPFn, and IPFs highlighted with colored bars above the sample labels) and the clusters: the HC samples clustered separately from the IPF samples, whereas some of the IPFs samples clustered together with the IPFn. C-F. Volcano plots [−log10(padj) vs log2(Fold Difference)] of genes in the HC vs IPFn (C), HC vs IPFs (D), HS vs IPF combined (E), and IPFn vs IPFs (F) comparisons. The total numbers of genes meeting the selection criteria for elevated (purple) or reduced (orange) expression are indicated in each panel. See Supplementary Tables 3-6 for numerical values of RNASeq counts for differentially expressed genes in each of these of these comparisons.
To assess differential gene expression across the three tested groups of samples (HC, IPFn, IPFs), the DESeq2 likelihood ratio test (LRT) was performed, and genes were identified for which p values adjusted for false discovery rate (padj) were <0.05. This set was further narrowed by selecting genes for which the fold-difference of mean counts in pair-wise comparisons of sample groups was ≥2.0 in at least one of the four comparisons (HC vs IPFn, HC vs IPFs, HC vs IPF combined, IPFn vs IPFs). The selected 2,099 genes (Supplementary Dataset 1) were used for unsupervised clustering of tissue samples, revealing distinct subsets (Figure 2B). Of note, samples from HC clustered separately from IPF samples. IPFn and IPFs samples also clustered separately, though less distinctly, with some of the IPFs samples clustering together with IPFn samples (Figure 2B). Such clustering indicated that differences between HC and IPF samples were more pronounced than differences between IPFn and IPFs samples, thus echoing the observations gained from PCA analysis (Figure 2A).
To further explore the validity of this notion, pairwise DESeq2 analyses using the Wald test were performed, comparing HC vs IPFn (Supplementary Dataset 2 and Figure 2C), HC vs IPFs (Supplementary Dataset 3 and Figure 2D), HC vs IPF combined (pooled IPFn and IPFs samples, Supplementary Dataset 4 and Figure 2E), and IPFn vs IPFs (Supplementary Dataset 5 and Figure 2F). These analyses revealed large numbers of differentially expressed genes with remarkably low padj values in the HC vs IPFn, HC vs IPFs, and HC vs IPF combined comparisons, but fewer differentially expressed genes with not as remarkably low, though still significant, padj values in the IPFn vs IPFs comparison (Figure 2C-F). These findings echoed the observations from PCA analysis (Figure 2A) and the LRT with subsequent unsupervised clustering (Figure 2B) that differences between HC and IPF samples were more pronounced than differences between IPFn and IPFs samples. These combined observations indicate that macroscopically normal-appearing areas, which we previously reported to be microscopically abnormal [25], manifested substantial transcriptomic abnormalities.
3.3 Similar Transcriptomic Changes in Macroscopically Normal-Appearing and Scarred IPF Lung Tissues
A previously unreported observation presented in Figure 2 was that IPFn tissues were substantially transcriptomically different from HC tissues. There were 713 genes with significantly elevated and 397 genes with significantly reduced expression in IPFn compared with HC tissues (Supplementary Dataset 2, Figure 2C, and Supplementary Figure 2). These differences were similar to those observed in IPFs compared with HC tissues: there were 920 genes with elevated and 634 genes with decreased expression in the IPFs group (Supplementary Dataset 3, Figure 2D, and Supplementary Figure 3). The somewhat greater number of differentially expressed genes in the HC vs IPFs comparison than in the HC vs IPFn comparison is not surprising, considering that scarred areas likely represent a more advanced stage in the disease process, with more profound changes in cellular composition and in the regulation of gene expression. The comparison of HC with the combined IPF group (IPFn and IPFs samples pooled together) revealed 710 genes with elevated and 409 genes with reduced expression in the combined IPF group (Supplementary Dataset 4, Figure 2E, and Supplementary Figure 4).
To focus on gene expression that was similarly changed in both IPFn and IPFs, an overlap gene set was created (Supplementary Dataset 6, Figure 3, Supplementary Figure 5) that contained genes significantly different (elevated or reduced expression) from HC at both earlier stages of the disease (IPFn) and later stages of the disease (IPFs). This overlap gene set contained 458 genes with elevated expression and 291 genes with reduced expression in both the HC vs IPFn and the HC vs IPFs comparisons. Gene ontology (GO) analyses of genes with similarly elevated expression in this overlap group revealed highly significant enrichment of genes associated with connective tissue, and immune and inflammatory activation (Figure 3A and more detailed analysis below). Genes with reduced expression in this overlap group (Figure 3B) were associated with solute carrier membrane transport and metabolic processes, though the statistical significance of pathway enrichment for these genes with reduced expression was lower than for the genes with elevated expression.
Figure 3.
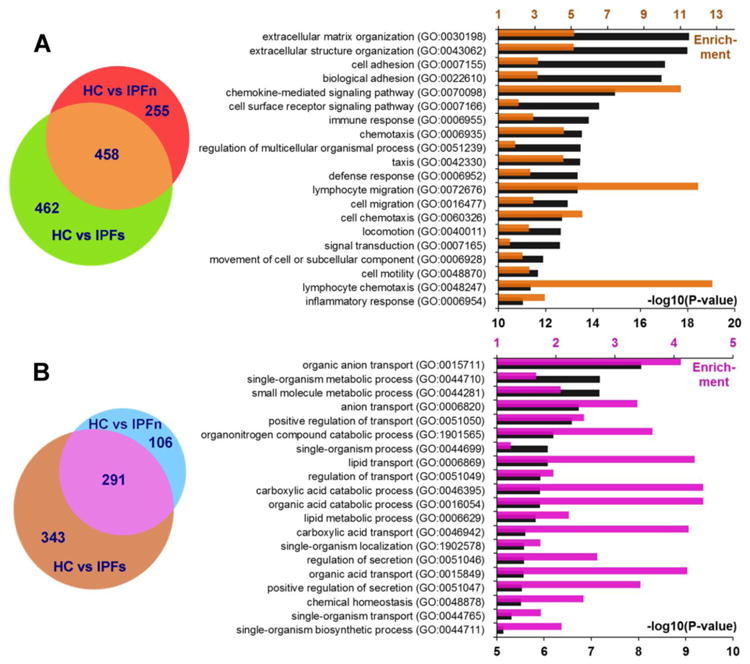
Elevated (A) or reduced (B) differential expression of genes in IPFn and IPFs tissues, each compared with HC samples. Among differentially expressed genes, 458 were similarly elevated and 291 similarly decreased in both the IPFn and IPFs groups, thus defining the overlap group, which was further considered in gene ontology enrichment analyses. The bar graphs represent the top 20 pathways with the lowest P-values for elevated and reduced gene expression patterns. Colored bars (upper horizontal axes) indicate pathway enrichment, and black bars (lower horizontal axes) indicate –log10(P-values) for the pathways indicated on the vertical axes. Note that the statistical significance of pathway enrichment was substantially higher for the overlap group with elevated expression in IPF tissues (A). See Supplementary Dataset 6 for numerical values of RNASeq counts for genes that were similarly elevated and similarly reduced in both IPFn and IPFs tissues compared with HC samples.
3.4 Transcriptomic Differences between Macroscopically Normal-Appearing and Macroscopically Scarred IPF Areas
To assess transcriptomic changes associated with a more advanced stage of disease in the overtly scarred areas of the lungs, direct comparative analyses were performed between IPFn and IPFs tissues (Supplementary Dataset 5, Figures 2F and Figure 4). These analyses revealed 410 genes with elevated and 240 genes with reduced expression in IPFs compared with IPFn tissues. These differences were not quite as pronounced in terms of number of genes, fold difference, or magnitude of statistical significance as was observed with the HC vs IPFn or HC vs IPFs comparisons.
Figure 4.
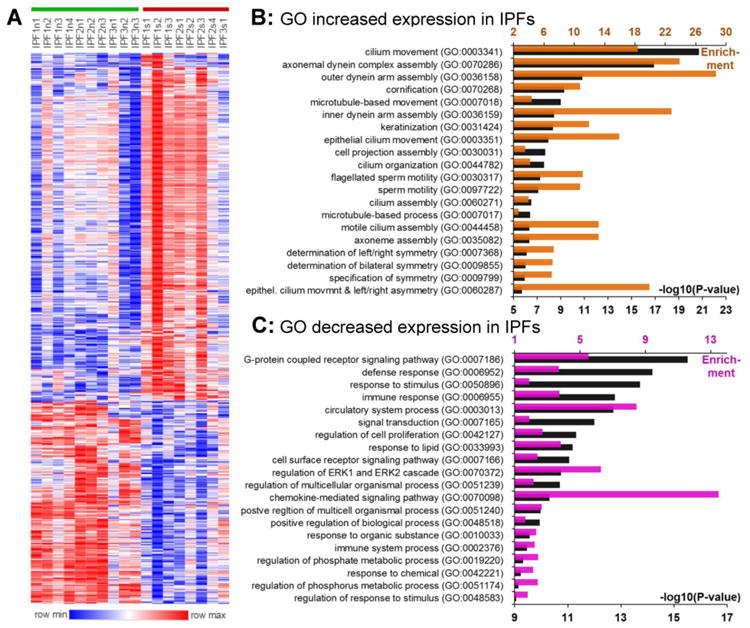
Differential expression of genes between IPFn and IPFs tissues. The volcano plots for this comparison are shown in Figure 2F and the numerical values of RNASeq counts are included in Supplementary Dataset 5. A. Unsupervised clustering of log2-transformed normalized counts using Spearman rank correlation with the average linkage of 650 differentially expressed genes (410 elevated and 240 reduced in IPFs vs IPFn). B. Gene ontology enrichment in the gene set with elevated expression in IPFs; the top 20 pathways with the lowest P-values are included. C. Gene ontology enrichment in the gene set with reduced expression in IPFs; the top 20 pathways with lowest P-values are included. In panels B and C, the colored bars (upper horizontal axes) indicate pathway enrichment and black bars (lower horizontal axes) indicate – log10(P-values) for the pathways indicated on the vertical axes.
Using GO analysis, the top 20 enriched pathways involving genes with elevated expression in IPFs tissues were, rather remarkably, cilia- and flagella-related (Figure 4B). These observations are consistent with the light microscopy findings of widespread ciliated respiratory epithelium lining honeycombed cysts throughout the UIP fibrotic lung, and with recent reports regarding contributions from mucociliary abnormalities to IPF pathobiology [1-3, 11, 39, 40]. Genes with decreased expression in IPFs compared with IPFn tissues (Figure 4C) were related to signal transduction, metabolism, and defense response, arguably suggesting that scarred tissues represent less active disease and a transition towards resolution of injury with overt scar deposits.
Considering that fibroblasts are commonly thought to be a cell type centrally involved in the IPF disease process, separate experiments were performed with cultured primary fibroblasts derived from IPFn and IPFs tissues from the three IPF explants. Comparison of transcriptomes between three IPFn fibroblast cultures and three IPFs fibroblast cultures revealed merely 25 genes for which padj values were <0.05 and the fold difference was ≥2.0 (Supplementary Figure 6). All 25 genes demonstrated elevated expression in the IPFs group, with fold-difference not exceeding 4.5, and the majority of genes encoding ribosomal L and S proteins. Such minimal transcriptomic differences between IPFn and IPFs lung fibroblasts (25 differentially expressed genes, Supplementary Figure 6) were in strong contrast to the pronounced transcriptomic differences between the corresponding lung tissues from which they were derived (650 differentially expressed genes, Figures 2F and Figure 4, Supplementary Dataset 5). These findings suggest that the substantial differences in gene expression profiles between IPFn and IPFs tissues are due to cell types other than fibroblasts.
3.5 Detailed View of Changes in Expression of Genes in Selected Pathways
GO enrichment analyses (Figures 3, 4) suggested that the most prominently enriched pathways across IPFn and IPFs tissues compared to HC were those associated with elevated expression of genes related to extracellular matrix, immunity, and inflammation (Figure 3A), whereas in IPFs compared to IPFn tissues, enriched genes were those associated with elevated expression in ciliated epithelium-related pathways (Figure 4B). Therefore, a detailed view of changes in the expression of genes identified by GO analyses associated with these selected pathways is shown in Figure 5. Specific connective tissue-related genes with elevated expression in IPFn and IPFs included alpha-smooth muscle actin (ACTA2), fibrillin (FBN1), fibronectin (FN), tenascin C (TNC), osteopontin (SPP1), the chains of highly abundant structural collagens (COL1A1, COL1A2, COL3A1, COL4A1) and other collagens, multiple matrix metalloproteinases (MMPs), and Wilms tumor protein (WT1) (Figure 5A). There was a remarkably extensive increase in expression of numerous immunity- and inflammation-related genes, including multiple cytokines, chemokines, and their receptors in IPFn and IPFs (Figure 5B). Differences between IPFn and IPFs tissues were most notable for a broad increase in IPFs mRNA levels for proteins abundantly present in ciliated epithelia, specifically for ciliary and flagellar components (Figure 5C).
Figure 5.

Differential expression of selected genes associated with extracellular matrix (A), inflammation and immunity (B), and cilia, flagella, and pulmonary epithelia (C). These gene lists were suggested by GO analyses and further modified based on the existing literature regarding involvement of related pathways in mechanisms of lung fibrosis. The numerical values of expression levels can be found by searching Supplementary Dataset 1 for the genes of interest.
3.6. Validation of RNASeq Data
We compiled a list of genes that were most prominently elevated or reduced in comparisons between the studied groups of lung tissues (Supplementary Dataset 7). These genes were selected independently of their gene ontology assignment but based on the magnitude and statistical significance of expression differences. Subsequent experiments were focused on validation, using RT-qPCR as an independent technique, of the observed differences in mRNA levels of selected genes that have been less well characterized for their potential involvement in IPF. These included receptor–ligand pair apelin receptor (APLNR) – apelin (APLN), a tight junction protein claudin 2 (CLDN2, which is also known for its expression in pulmonary macrophages [41]), lysosome-associated membrane protein LAMP5, α-smooth muscle actin (ACTA2), and the enzymes acidic chitinase (CHIA) and flavin-containing monooxygenase 5 (FMO5). The RT-qPCR data for each gene were normalized to levels of 18S rRNA [42-44] (Figure 6) and to GAPDH (Supplementary Figure 7). Differences in expression levels of these mRNAs measured by RT-qPCR mirrored those observed by RNASeq (Figure 6, Supplementary Figure 7).
Figure 6.
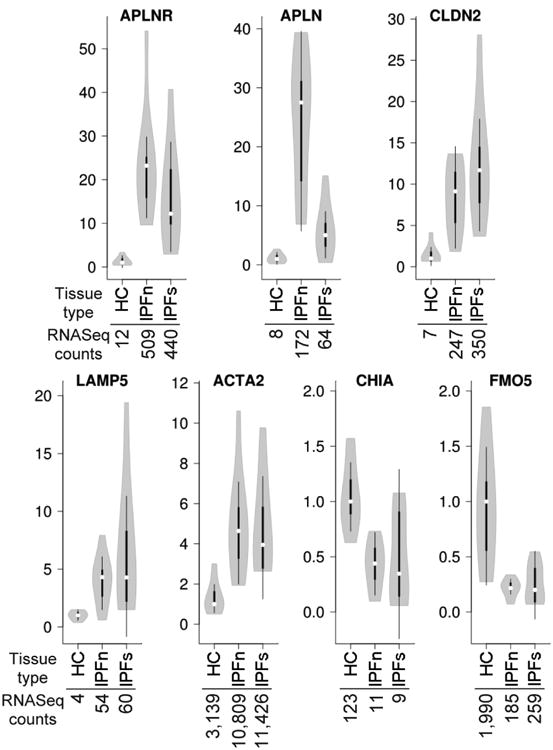
Confirmation of selected RNASeq findings by RT-qPCR. The HC, IPFn, and IPFs mRNA samples used for the RNASeq analyses described above were reverse-transcribed and the cDNAs amplified with primers for the indicated targets. Data for each target were normalized to 18S rRNA and presented in the form of violin plots as fold differences versus the median value in the HC groups. The numbers below the indicated tissue types represent median normalized RNASeq counts from the LRT analysis described in the text. In all cases, IPFn and IPFs were significantly different from their corresponding HC groups according to a two-tailed Student's t-test and a Mann-Whitney U-test.
To additionally validate our transcriptomic observations, we performed immunohistochemical staining on HC, IPFn, and IPFs tissues for DIO2, and observed elevated expression of this protein in IPFn and IPFs tissues, mostly in epithelial and endothelial cells, and in alveolar macrophages (Supplementary Figure 8A). A recent report of others [45] is consistent with our observations. Additional western blotting (Supplementary Figure 8B) and ELISA (Supplementary Figure 8C) tests confirmed elevations of CLDN2 and CCL8, respectively, in IPF tissues compared with HC.
4. Discussion
The primary goal of this study was to gain detailed insight into the pathobiology of IPF through comparative analyses of transcriptomes of lung tissues from macroscopically non-scarred, normal-appearing areas (IPFn) versus clearly diseased and overtly scarred areas (IPFs) of explanted IPF lungs. To our knowledge, this is the first investigation in which such comparisons have been performed. Previous transcriptomic profiling studies compared lung tissues from patients with IPF against tissues from healthy controls [5-17], but without differentiating macroscopically normal-appearing and scarred tissues. We also included healthy control tissues (HC) in our analyses, and performed complex, three-way comparisons (HC vs IPFn, HC vs IPFs, IPFn vs IPFs). This research was prompted by our recent observations of histological abnormalities in normal-appearing lung tissue in patients with IPF [25]. The notion that the macroscopic appearance of a tissue may be misleading in regard to its microscopic and molecular changes is hardly surprising, but the specific details of histologic and transcriptomic changes are likely to provide important pathobiological clues.
Our results confirm many of the previously reported IPF-associated disturbances in gene expression, but the novelty of this work is in revealing the breadth, magnitude, and specific details of transcriptomic changes in macroscopically normal-appearing lung tissues in IPF. Our data indicate that in patients with IPF, the disease process is not limited to the overtly scarred, basolateral areas of the lungs. Macroscopically normal-appearing, more centrally localized, areas are fully engaged in the disease process, manifesting broad transcriptional activation of connective tissue-related genes as well as extensive and exaggerated immune and inflammatory activity. The grossly scarred areas, despite their macroscopic and microarchitectural features reflective of advanced pathological change, exhibit substantial transcriptomic similarities to macroscopically normal-appearing areas.
The observation of increased expression of connective tissue-related mRNAs in pulmonary fibrosis, including mRNAs for collagens, MMP1, MMP7, WT1, and SPP1 (Figure 5A) is not unexpected [8, 19, 46-48]. However, the levels of these mRNAs were significantly increased not only in IPFs, but also in IPFn tissues. By light microscopic criteria, IPFn tissues are not overtly scarred: their alveolar microarchitecture remains preserved and the lung tissue is aerated despite the presence of some histological abnormalities [25]. It therefore follows that production of excess mRNAs for numerous extracellular matrix components is not sufficient to cause a dense scarring phenotype such as that seen in IPFs tissue. These observations support the notion that, in addition to excess deposition of extracellular matrix, other pathophysiological contributors, such as alveolar collapse and subsequent collapse induration, are likely required for formation of the overtly scarred areas of the lungs in pulmonary fibrosis with a UIP pattern [49-51].
Elevations in the expression of numerous immunity- and inflammation-related genes (Figure 5B) have been associated with pulmonary fibrosis in the past [22, 24], including T and B lymphocyte-specific molecules such as the CD3 chains, MS4A1/CD20, CD79A, CD40LG, and FASLG [52-54]. These increases are consistent with the known influx of T and B lymphocytes into the lungs of patients with IPF [52, 53]. Expression levels of mRNAs for cytokines, chemokines, and related genes, including CCL2, CCL8, CCL18, CCL19, CCL22, CCL24, CCR7, CXCL12, CXCL13, CXCL14, CXCR3, IL13RA2, and IL-33 were also elevated in agreement with many previous observations [24, 46, 55-62]. However, we report for the first time that all of these increases in immunity- and inflammation-related genes occurred across IPFn and IPFs tissues, further supporting the notion that macroscopically normal-appearing lung tissue is intensively involved in the disease process.
Despite similar expression of connective tissue-, immunity, and inflammation-related mRNAs in IPFn and IPFs tissues, their microscopic histologic appearance is drastically different, suggesting that the progression to terminal scarring in basolateral areas of IPF lungs is driven by other contributors. Additional factors may play a role, such as alveolar collapse and subsequent collapse induration [49-51], as well as significant epithelial alterations that are suggested by the observed mucociliary signature in IPFs, but not IPFn tissues (Figure 5C). The broad increase in IPFs mRNA levels for proteins abundantly present in ciliated epithelia, more specifically, ciliary and flagellar components, included numerous coiled-coil domain-containing proteins (CCDCs), chains of axonemal dynein and dynein assembly factors, and other proteins involved in maintaining ciliary structure and function. These findings are consistent with and further detail the current view of mucociliary involvement in IPF [11, 39, 40]. The known increases in mRNAs for CLDN2 [41] and MUC5B [18, 39] were also confirmed in IPFs tissues. Expression of several cytokeratins, including KRT5 [63, 64] and KRT14 [65], known for their association with the epithelial progenitor phenotype, were also elevated in IPFs. Simultaneously, expression of several alveolar epithelial-associated genes, including cystic fibrosis transmembrane conductance regulator (CFTR) and surfactant proteins (SFTPs), was decreased. All these epithelial changes are consistent with the notion of alveolar epithelial loss and/or aberrant epithelial repair in IPF, and consistent with the observations of abundant metaplastic mucociliary epithelium lining honeycomb cysts. It is therefore understandable that such changes occur more in IPFs than in IPFn regions of the lungs.
The results of this work, while only associative at the molecular level, provide new insight into the pathobiological mechanisms of IPF at the organ level. The assessment of transcriptomes of lung areas was unbiased by macroscopic appearance (normal-appearing vs overtly scarred) and was a priori agnostic of the intensity of involvement in the disease. This approach resulted in the finding that macroscopically normal-appearing lung tissues in IPF are substantially involved in the disease process. This finding has an important implication for the future development of novel therapeutic modalities: the therapeutic focus needs to be shifted from overtly fibrotic, terminally altered areas of the lungs to attenuating the disease process in regions of the lungs that have not yet irreversibly collapsed and densely scarred. While clearly diseased, based on the transcriptomic changes described above and previously reported histological changes [25], the macroscopically normal-appearing areas of IPF lungs maintain well-preserved alveolar microarchitecture and remain aerated. Slowing the disease process in these areas would hopefully prevent progression towards fundamental loss of pulmonary architecture and irreversible scarring.
There are several potential limitations in this study, one of which is the descriptive nature of the study. Experimental studies in humans pose obvious ethical and regulatory challenges. Short of a clinical trial, studies in humans are usually descriptive and/or associative in nature. However, due to the molecular scope of this study, our results have important mechanistic implications and form the required basis for future mechanistic investigations. Abundant patient-oriented research literature indicates that studies of mechanistically suggestive phenomenology makes subsequent mechanistic research relevant to human health and drug development.
Another potential limitation is the relatively small number of lungs (three patients with end-stage IPF and three healthy controls) used to derive the 26 separate tissue samples. This limitation stems, in part, from the innovative scope of this research: our primary goal was to compare different areas within the same IPF lungs, whereas comparisons against healthy control were secondary. For this work, previously accumulated leftover lung tissues (from previous diagnostic biopsies or lung transplantations) could not be used since such tissues are routinely collected without separating normal-appearing from scarred areas. In our study, each lung explant was processed immediately after explantation for separation of macroscopically normal-appearing and grossly scarred areas, and for subsequent processing of each tissue sample for mRNA, protein, and histological analyses. Understandably, the availability of such lung explants is limited and collection of such samples is more technically challenging than in straightforward studies comparing IPF to control lungs.
This potential limitation regarding the small number of tested lungs is partially assuaged by several factors. This study was designed to analyze multiple tissue samples from IPFn and IPFs areas of each lung to accurately assess the extent of transcriptomic heterogeneity. Despite the well-known clinical heterogeneity of IPF phenotypes, our careful identification and sampling of multiple lung areas resulted in a reasonable degree of homogeneity within the three tested tissue groups (HC, IPFn, IPFs), as well as clear separation between the groups, with both homogeneity within and separation between groups demonstrated by principal component analysis and hierarchical clustering of tissue samples (Figure 2A, B and Supplementary Figure 1). Additionally, false discovery rate-adjusted differences between sample groups were characterized by remarkably low p values for many genes (Figure 2C–F).
It should be noted that two of the IPF patients had received antifibrotic therapy with pirfenidone prior to transplant. Despite antifibrotic therapy, both patients developed clinical end-stage lung disease requiring lung transplantation and demonstrated severe histologic fibrosis on lung explant. IPF patient 2 (IPF2) did receive pirfenidone for seven months, but continued to have progressive disease, requiring lung transplantation merely 15 months subsequent to time of diagnosis. IPF patient 3 (IPF3) received pirfenidone for several months only, which was then discontinued due to disease progression. We cannot completely exclude that RNA expression in these patients could be altered by antifibrotic therapy. Since these therapies are being increasingly utilized, it may be difficult in the future to procure fresh explant lung tissue from patients with IPF in which anti-fibrotic therapies were not administered.
The healthy control donor lungs were obtained from persons significantly younger than the IPF patients, thus making age a possible confounding factor. Using donor lungs harvested but ultimately not used for lung transplantation has its obvious advantages, but donor lungs are not commonly harvested from older individuals. To partially address the possibility of age as a confounding factor on our RNASeq dataset, we considered expression levels of sirtuins, which are known to change in association with aging, but observed no differences between the studied tissue groups. An alternative source of control tissue from age-matching donors would be normal-appearing lung tissues surrounding bronchogenic carcinomas, which are available from surgical lobectomies performed in lung cancer patients. However, this alternative approach would introduce another confounding factor, the so-called “field cancerization,” in which normal tissues surrounding areas of cancer undergo substantial molecular changes [66-68].
The results of this study need to be carefully interpreted, with a clear understanding that an associative link implies, but does not prove, a causal link. More specifically, the magnitude and statistical significance of changes in expression of a specific gene or gene set may be the result, but not the cause, of pathological changes. Additionally hindering the ability to discern causative links is the cross-sectional nature of this study, which did not allow for longitudinal insight into the dynamics of gene expression patterns. Therefore, our findings establish the involvement of immune and inflammatory processes in the pathogenesis of IPF, but do not resolve the longstanding debate about a causative contribution of inflammation and immunity to scarring and to overall disease progression. It is possible that the focus of this debate has been misplaced: instead of considering whether lung inflammation drives fibrosis, it is important to recognize that many of the mediators that activate inflammation and immunity simultaneously stimulate fibrosis [24]. It is therefore important to ask whether targeting pleiotropic regulators with elevated expression in IPF might alleviate both inflammation and fibrosis, and thus be therapeutic.
In summary, macroscopically normal-appearing lung tissues in patients with IPF are actively involved in the disease process and demonstrate substantial transcriptomic similarities with overtly scarred tissues: elevated expression of genes related to extracellular matrix, immunity, and inflammation, and reduced expression of genes related to solute carrier membrane transport and metabolic processes. Pulmonary fibroblasts from macroscopically normal-appearing and scarred areas are transcriptomically identical, suggesting that the differences in gene expression profiles between macroscopically normal-appearing and overtly scarred tissues are due to cell types other than fibroblasts. Macroscopically scarred compared to macroscopically normal-appearing tissues exhibit elevated expression of genes related to mucociliary epithelium. Our overall observations expand and enhance our understanding of the pathobiology of IPF.
Supplementary Material
Macroscopically normal tissue in IPF patients is profoundly involved in the disease
Immune activation is overt in normal-appearing and scarred tissue in IPF lungs
Differences between normal-appearing and scarred tissue involve mostly epithelium
Acknowledgments
This work was supported by the NIH R01HL126897 (SPA) and VA Merit Awards I01CX000101 (IGL) and I01BX002499 (SPA). The authors thank Dr. Paul Todd for his expert editorial support.
Footnotes
Disclosures: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of the presented manuscript or other conflicts of interest to disclose. The views expressed in this article are those of the authors and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or the U.S. Government.
Author Contribution: IGL, NWT, APB, and SPA conceived the idea of the study. JRG, AS, JK, TJF, EJB, SMP, APB, and NWT identified and consented the participants, reviewed clinical, radiological, and pathology information, and contributed to the scientific message of the manuscript. IGL, NWT, and APB collected and processed explanted lungs and isolated lung tissues for subsequent analyses. IGL, AEW, and AC isolated RNA, performed quantitative reverse transcriptase-polymerase chain reactions, and performed primary fibroblast culture experiments. MVS, MLRP, JDH, and SPA performed RNASeq data analyses and statistical comparisons. SPA composed figures and tables. NWT and SPA wrote the manuscript. All authors edited the manuscript and approved its final version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, Hirani N, Hubbard R, Lake F, Millar AB, Wallace WA, Wells AU, Whyte MK, Wilsher ML B.T.S.S.o.C.C. British Thoracic Society Interstitial Lung Disease Guideline Group, A. Thoracic Society of, S. New Zealand Thoracic, S. Irish Thoracic. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(5):v1–58. doi: 10.1136/thx.2008.101691. [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ A.E.J.A.C.o.I.P. Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. American journal of respiratory and critical care medicine. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D A.E.C.o.I.I. Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American journal of respiratory and critical care medicine. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. American journal of respiratory and critical care medicine. 2014;190:867–878. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A. 2000;97:1778–1783. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. American journal of respiratory and critical care medicine. 2006;173:188–198. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, Bisceglia M, Gilbert S, Yousem SA, Song JW, Kim DS, Kaminski N. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2009;180:167–175. doi: 10.1164/rccm.200810-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63:783–794. doi: 10.1002/art.30159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mura M, Anraku M, Yun Z, McRae K, Liu M, Waddell TK, Singer LG, Granton JT, Keshavjee S, de Perrot M. Gene expression profiling in the lungs of patients with pulmonary hypertension associated with pulmonary fibrosis. Chest. 2012;141:661–673. doi: 10.1378/chest.11-0449. [DOI] [PubMed] [Google Scholar]
- 11.Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, Rosen R, Neidermyer AJ, McKean DF, Groshong SD, Cool C, Cosgrove GP, Lynch DA, Brown KK, Schwarz MI, Fingerlin TE, Schwartz DA. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–1121. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DePianto DJ, Chandriani S, Abbas AR, Jia G, N'Diaye EN, Caplazi P, Kauder SE, Biswas S, Karnik SK, Ha C, Modrusan Z, Matthay MA, Kukreja J, Collard HR, Egen JG, Wolters PJ, Arron JR. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. 2015;70:48–56. doi: 10.1136/thoraxjnl-2013-204596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng N, Sanchez CG, Lasky JA, Zhu D. Detecting splicing variants in idiopathic pulmonary fibrosis from non-differentially expressed genes. PLoS One. 2013;8:e68352. doi: 10.1371/journal.pone.0068352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G, Montgomery SB. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One. 2014;9:e92111. doi: 10.1371/journal.pone.0092111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusko RL, Brothers JF, 2nd, Tedrow J, Pandit K, Huleihel L, Perdomo C, Liu G, Juan-Guardela B, Kass D, Zhang S, Lenburg M, Martinez F, Quackenbush J, Sciurba F, Limper A, Geraci M, Yang I, Schwartz DA, Beane J, Spira A, Kaminski N. Integrated Genomics Reveals Convergent Transcriptomic Networks Underlying Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. American journal of respiratory and critical care medicine. 2016;194:948–960. doi: 10.1164/rccm.201510-2026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vukmirovic M, Herazo-Maya JD, Blackmon J, Skodric-Trifunovic V, Jovanovic D, Pavlovic S, Stojsic J, Zeljkovic V, Yan X, Homer R, Stefanovic B, Kaminski N. Identification and validation of differentially expressed transcripts by RNA-sequencing of formalin-fixed, paraffin-embedded (FFPE) lung tissue from patients with Idiopathic Pulmonary Fibrosis. BMC Pulm Med. 2017;17:15. doi: 10.1186/s12890-016-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, Stripp BR, Whitsett JA. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc. 2015;12(2):S193–199. doi: 10.1513/AnnalsATS.201503-110AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, Sciurba F, Dauber J, Selman M, Gochuico BR, Kaminski N. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoyne GF, Elliott H, Mutsaers SE, Prele CM. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol Cell Biol. 2017 doi: 10.1038/icb.2017.22. [DOI] [PubMed] [Google Scholar]
- 21.Homer RJ, Elias JA, Lee CG, Herzog E. Modern concepts on the role of inflammation in pulmonary fibrosis. Archives of pathology & laboratory medicine. 2011;135:780–788. doi: 10.5858/2010-0296-RA.1. [DOI] [PubMed] [Google Scholar]
- 22.Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol. 2008;83:237–244. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 23.Balestro E, Calabrese F, Turato G, Lunardi F, Bazzan E, Marulli G, Biondini D, Rossi E, Sanduzzi A, Rea F, Rigobello C, Gregori D, Baraldo S, Spagnolo P, Cosio MG, Saetta M. Immune Inflammation and Disease Progression in Idiopathic Pulmonary Fibrosis. PLoS One. 2016;11:e0154516. doi: 10.1371/journal.pone.0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luzina IG, Todd NW, Sundararajan S, Atamas SP. The cytokines of pulmonary fibrosis: Much learned, much more to learn. Cytokine. 2015;74:88–100. doi: 10.1016/j.cyto.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 25.Todd NW, Galvin JR, Sachdeva A, Luzina IG, Atamas SP, Burke AP. Microscopic organizing pneumonia and cellular non-specific interstitial pneumonia are widespread in macroscopically normal-appearing lung tissue in idiopathic pulmonary fibrosis. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2016;35:1367–1370. doi: 10.1016/j.healun.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung JH, Chawla A, Peljto AL, Cool CD, Groshong SD, Talbert JL, McKean DF, Brown KK, Fingerlin TE, Schwarz MI, Schwartz DA, Lynch DA. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest. 2015;147:450–459. doi: 10.1378/chest.14-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.HTSeq, http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html.
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DESeq2, https://bioconductor.org/packages/release/bioc/html/DESeq2.html.
- 32.Morpheus, https://software.broadinstitute.org/morpheus/.
- 33.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.GOrilla, http://cbl-gorilla.cs.technion.ac.il/.
- 36.BioVenn, http://www.biovenn.nl/.
- 37.Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.BoxPlotR, http://shiny.chemgrid.org/boxplotr/.
- 39.Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, Yang IV, Schwartz DA. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev. 2016;96:1567–1591. doi: 10.1152/physrev.00004.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, Reynolds SD. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaarteenaho-Wiik R, Soini Y. Claudin-1, -2, -3, -4, -5, and -7 in usual interstitial pneumonia and sarcoidosis. J Histochem Cytochem. 2009;57:187–195. doi: 10.1369/jhc.2008.951566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuchipudi SV, Tellabati M, Nelli RK, White GA, Perez BB, Sebastian S, Slomka MJ, Brookes SM, Brown IH, Dunham SP, Chang KC. 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol J. 2012;9:230. doi: 10.1186/1743-422X-9-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goidin D, Mamessier A, Staquet MJ, Schmitt D, Berthier-Vergnes O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal Biochem. 2001;295:17–21. doi: 10.1006/abio.2001.5171. [DOI] [PubMed] [Google Scholar]
- 44.Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69–81. doi: 10.1016/s0165-022x(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 45.Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, Aurelien N, Arrojo EDR, Gan Y, Graham M, Liu X, Homer RJ, Scanlan TS, Mannam P, Lee PJ, Herzog EL, Bianco AC, Kaminski N. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2017 doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foster MW, Morrison LD, Todd JL, Snyder LD, Thompson JW, Soderblom EJ, Plonk K, Weinhold KJ, Townsend R, Minnich A, Moseley MA. Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res. 2015;14:1238–1249. doi: 10.1021/pr501149m. [DOI] [PubMed] [Google Scholar]
- 47.Sontake V, Shanmukhappa SK, DiPasquale BA, Reddy GB, Medvedovic M, Hardie WD, White ES, Madala SK. Fibrocytes Regulate Wilms Tumor 1-Positive Cell Accumulation in Severe Fibrotic Lung Disease. J Immunol. 2015;195:3978–3991. doi: 10.4049/jimmunol.1500963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2:e251. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galvin JR, Frazier AA, Franks TJ. Collaborative radiologic and histopathologic assessment of fibrotic lung disease. Radiology. 2010;255:692–706. doi: 10.1148/radiol.10090717. [DOI] [PubMed] [Google Scholar]
- 50.Lutz D, Gazdhar A, Lopez-Rodriguez E, Ruppert C, Mahavadi P, Gunther A, Klepetko W, Bates JH, Smith B, Geiser T, Ochs M, Knudsen L. Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am J Respir Cell Mol Biol. 2015;52:232–243. doi: 10.1165/rcmb.2014-0078OC. [DOI] [PubMed] [Google Scholar]
- 51.Coxson HO, Hogg JC, Mayo JR, Behzad H, Whittall KP, Schwartz DA, Hartley PG, Galvin JR, Wilson JS, Hunninghake GW. Quantification of idiopathic pulmonary fibrosis using computed tomography and histology. American journal of respiratory and critical care medicine. 1997;155:1649–1656. doi: 10.1164/ajrccm.155.5.9154871. [DOI] [PubMed] [Google Scholar]
- 52.Todd NW, Scheraga RG, Galvin JR, Iacono AT, Britt EJ, Luzina IG, Burke AP, Atamas SP. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J Inflamm Res. 2013;6:63–70. doi: 10.2147/JIR.S40673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marchal-Somme J, Uzunhan Y, Marchand-Adam S, Valeyre D, Soumelis V, Crestani B, Soler P. Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol. 2006;176:5735–5739. doi: 10.4049/jimmunol.176.10.5735. [DOI] [PubMed] [Google Scholar]
- 54.Kaufman J, Sime PJ, Phipps RP. Expression of CD154 (CD40 ligand) by human lung fibroblasts: differential regulation by IFN-gamma and IL-13, and implications for fibrosis. J Immunol. 2004;172:1862–1871. doi: 10.4049/jimmunol.172.3.1862. [DOI] [PubMed] [Google Scholar]
- 55.Lee JU, Cheong HS, Shim EY, Bae DJ, Chang HS, Uh ST, Kim YH, Park JS, Lee B, Shin HD, Park CS. Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis. Respiratory research. 2017;18:3. doi: 10.1186/s12931-016-0493-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pochetuhen K, Luzina IG, Lockatell V, Choi J, Todd NW, Atamas SP. Complex regulation of pulmonary inflammation and fibrosis by CCL18. Am J Pathol. 2007;171:428–437. doi: 10.2353/ajpath.2007.061167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prasse A, Pechkovsky DV, Toews GB, Schafer M, Eggeling S, Ludwig C, Germann M, Kollert F, Zissel G, Muller-Quernheim J. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. 2007;56:1685–1693. doi: 10.1002/art.22559. [DOI] [PubMed] [Google Scholar]
- 58.Marchal-Somme J, Uzunhan Y, Marchand-Adam S, Kambouchner M, Valeyre D, Crestani B, Soler P. Dendritic cells accumulate in human fibrotic interstitial lung disease. American journal of respiratory and critical care medicine. 2007;176:1007–1014. doi: 10.1164/rccm.200609-1347OC. [DOI] [PubMed] [Google Scholar]
- 59.Lee JU, Chang HS, Lee HJ, Jung CA, Bae DJ, Song HJ, Park JS, Uh ST, Kim YH, Seo KH, Park CS. Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm Med. 2017;17:39. doi: 10.1186/s12890-017-0380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, Hasday JD, Todd NW, Atamas SP. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2013;49:999–1008. doi: 10.1165/rcmb.2013-0093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barnes JC, Lumsden RV, Worrell J, Counihan IP, O'Beirne SL, Belperio JA, Fabre A, Donnelly SC, Boylan D, Kane R, Keane MP. CXCR3 Requirement for the Interleukin-13-Mediated Up-Regulation of Interleukin-13Ralpha2 in Pulmonary Fibroblasts. Am J Respir Cell Mol Biol. 2015;53:217–225. doi: 10.1165/rcmb.2013-0433OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 63.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517:616–620. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Confalonieri M, Buratti E, Grassi G, Bussani R, Chilosi M, Farra R, Abrami M, Stuani C, Salton F, Ficial M, Confalonieri P, Zandona L, Romano M. Keratin14 mRNA expression in human pneumocytes during quiescence, repair and disease. PLoS One. 2017;12:e0172130. doi: 10.1371/journal.pone.0172130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kadara H, Fujimoto J, Yoo SY, Maki Y, Gower AC, Kabbout M, Garcia MM, Chow CW, Chu Z, Mendoza G, Shen L, Kalhor N, Hong WK, Moran C, Wang J, Spira A, Coombes KR, Wistuba Transcriptomic architecture of the adjacent airway field cancerization in non-small cell lung cancer. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju004. dju004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kadara H, Shen L, Fujimoto J, Saintigny P, Chow CW, Lang W, Chu Z, Garcia M, Kabbout M, Fan YH, Behrens C, Liu DA, Mao L, Lee JJ, Gold KA, Wang J, Coombes KR, Kim ES, Hong WK, Wistuba Characterizing the molecular spatial and temporal field of injury in early-stage smoker non-small cell lung cancer patients after definitive surgery by expression profiling. Cancer Prev Res (Phila) 2013;6:8–17. doi: 10.1158/1940-6207.CAPR-12-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kadara H, Wistuba Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc. 2012;9:38–42. doi: 10.1513/pats.201201-004MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.