

Abstract
Hepatocellular Cancer (HCC) is a common and deadly cancer. Most cases of HCC arise in a cirrhotic/fibrotic liver, raising the possibility that the environment plays a paramount role in cancer genesis. Previous studies from our group and others have shown that, in desmoplastic cancers, there is a rich intercellular communication between activated, cancer-associated fibroblasts (CAF) and cancer cells. Moreover, Extracellular Vesicles (EVs), or exosomes, were identified as an important arm of this intercellular communication platform. Last, these studies have shown that EVs can carry miR species in vivo and deliver them to desmoplastic cancers. The precise role played by activated liver fibroblasts/stellate cells in HCC development is insufficiently known. Based on previous studies, it appears plausible that activated fibroblasts produce signals carried by EVs that promote HCC genesis. In the current study, we first hypothesized and then showed that stellate cell-derived EVs 1) can be loaded with a miR species of choice (miR-335-5p); 2) are uptaken by HCC cells in vitro and more importantly in vivo; 3) can supply the miR-335-5p cargo to recipient HCC cells in vitro as well as in vivo; and finally that they 4) inhibit HCC cell proliferation and invasion in vitro as well as induce HCC tumor shrinkage in vivo. Last, we identified mRNA targets for miR-335 that are down-regulated following treatments with EV-miR-335-5p. This study informs novel therapeutic strategies in HCC, whereby stellate cell-derived EVs are loaded with therapeutic nucleic acids and delivered in vivo.
Keywords: Extracellular vesicles, Hepatocellular carcinoma, Cancer-associated fibroblasts, Hepatic stellate cells
Introduction
HCC ranks as the third most common cause of cancer-related death worldwide (1). Importantly, the vast majority of HCCs develop in a background of liver fibrosis or cirrhosis (2). Liver fibrosis is produced when inactive liver fibroblasts (stellate cells) become activated following liver injury, and turn into collagen-producing cells (3). In desmoplastic cancers, cancer associated fibroblasts (CAFs) are loosely defined as fibroblasts found within the tumor mass. These CAF have been implicated as etiologic players both in the cancer genesis and homeostasis (4, 5). These data appear to suggest that liver fibrosis, or activated stellate cells, play a role in the development of HCC, perhaps similar to the role played by CAFs in desmoplastic cancers.
The origin of activated fibroblasts in hepatocellular carcinoma (HCC) remains controversial. Recent studies revealed that multiple origins including activation of stellate cells, or of portal fibroblasts, or trans-differentiation of hepatocytes through Epithelial to Mesenchymal Transition (EMT) (6, 7). In a previous study, we identified microRNA (miR) species downregulated in human liver stellate cells LX2 by co-culturing them with cholangiocarcinoma (CCA) cells (8). Interestingly, there is a significant overlap between the list of miR species found to be downregulated in LX2 cells placed in co-culture with cancer cells (arguably promoting their CAF phenotype) and miR species identified to be down regulated in same cells during the transition from retinol-containing inactive stellate cells to activated, collagen producing cells (9-11). It is therefore formally possible that the transition from inactive stellate cells to activated cells to CAF is a continuum of relevance in HCC development. By extension, this scenario suggests that activated stellate cells/liver fibroblasts are active participants in the development of HCC.
The precise nature of the interactions between liver cancer cells and the rest of the cells in the cancer environment are under intense investigation. For example, in cholangiocarcinoma, we have previously shown that fibroblasts and cancer cells communicate via EVs (8). Moreover, we have shown that stellate cell-derived EVs are uptaken by cancer cells in vitro and in vivo (8). Last, and of high clinical interest, we have shown that we can manipulate in vitro EVs derived from these stellate cells to efficiently carry a miR cargo to CCA in vivo, shrink tumors and increase survival in a rat CCA model (8). We concluded that disrupting the cross-talk between liver fibroblasts/activated stellate cells/CAFs and cancer cells may potentially open a novel avenue to improve clinical outcomes.
The current study is based on the hypothesis that stellate cell-derived EVs can be delivered to HCC cells in vitro and in vivo, and that they can inhibit cancer growth when carrying a therapeutic miR cargo. Here, we demonstrated that EVs loaded with miR-335-5p (EV-miR-335) can decrease cancer growth and invasion in vitro and in vivo. We have also shown that EV-miR-335-5p can be utilized successfully in vivo to induce HCC shrinking. Last, we identified mRNA targets for miR-335 that are down-regulated following treatments with EV-miR-335. These mRNA species are likely downstream effectors of EV-miR-335 treatment.
Materials and Methods
Additional information can be found in the Supplementary Materials and Methods.
Cell lines and co-culture conditions
Four human hepatocellular carcinoma cells: MHCC97H, MHCC97L, HepG2 and Huh7, as well as human hepatic stellate cell LX2 were maintained in Dulbecco's modified Eagle medium (DMEM) (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10 % fetal bovine serum (Invitrogen), 100 U/ml penicillin G, and 100 μg/ml streptomycin (Quality Biological, MD, USA) at 37 °C in a humidified chamber with 95 % air and 5% CO2.
Plasmid transfection and virus infection
pCDH-EF1-MCS-IRES-GFP cDNA Cloning and Expression Vector (HIV) (System Biosciences, CD530A-2-SBI) (7.5 μg/ 10 cm plate), PxPAX2 expressing HIV gag/ pol, Rev and tat (6 μg/10 cm plate, Addgene), and pMD2.G vector expressing VSV.G (2 μg/ 10 cm plate) were transfected into 2, 100 mm culture dishes of 293T cells, using X-tremeGENE HP DNA Transfection reagent (Roche). Seventy-two hours after transfection, supernatant was collected. HCC cells were transduced with the viral supernatant and GFP positive cells were sorted with BD FACSJazz (BD Biosciences).
Establishment of loxp-dsRed-loxp-Stop-eGFP-puro-WPRE constructs for each of the 4 HCC cells lines
PMSC-loxp-dsRed-loxp-Stop-eGFP-puro-WPRE (Addgene plasmid, 7.5 μg/10 cm plate) gag/pol plasmid (6 μg/10 cm plate) (Addgene), and VSV-G vector expressing (2 μg/ 10 cm plate) were transfected into 4, 100 mm culture dishes of 293T cells, using X-tremeGENE HP DNA Transfection reagent (Roche). Seventy-two hours after transfection, supernatant was collected. HCC cells were transduced with virus supernatant. Seventy two hours later, cells were treated with puromycin (9μg/ml for MHCC97H, MHCC97L, HepG2, and 7ug/ml for Huh7) for 2 weeks.
Exosome isolation and characterization
Exosomes were isolated and characterized as described previously (8).
Exosomes transfection with miR-335-5p mimics or miR-NSM
Exosomes from LX2 cells were transfected with miR-335-5p mimics or miR-NSM as described previously (8). Specifically, the EVs were transfected with miR-335-5p or NSM using Lipofectamine RNAiMAX Reagent (ThermoFisher, USA). Specifically, to transfect 30μg exosomes, we used 2 μl Lipofectamine RNAiMAX Reagent diluted in 25μl Opti-MEM Medium (OM), and 1μl miR-335-5p mimic or NSM (10 pmol) diluted in 25μl Opti-MEM Medium. Then we mixed the diluted Lipofectamine RNAiMAX Reagent and diluted miR-335-5p mimic or NSM and incubated for 5 minutes at room temperature. Isolated EVs were diluted in 250ul OM, then the miRNA-lipid complexes were added to the diluted EVs, and incubated for 6 hours at 37°C. Then, EV-miR-335 or EV-NSM were concentrated with Vivaspin 2 purification column (50 kDa, GE Healthcare, UK). Real time PCR with miR-335-5p primers identified in excess of 6,000 fold more miR-335-5p when EVs were present, suggesting that all miR-335-5p is associated with EVs and almost nothing exists outside of EVs (Supplementary Figure S1).
Detection of Cre mRNA expression in EVs
LX2 cells were transfected with Cre as described above. EVs were isolated from cell conditioned media (CM) using high speed centrifugation. RNA was isolated from cells, CM and Supernatant (Sup) using the Trizol reagent (Ambion, Life Technologies) according to the manufacturer's instructions. The amount and purity of isolated RNA was analyzed by the Nanodrop spectrophotometer (Wilmington, DE, USA). Reverse transcription was performed using TaqMan MicroRNA Reverse Transcript kit (Applied Biosystems). The resulting cDNA was then amplified in a PCR reaction using Cre-specific primers (forward primer: 5′ GCCTGCATTACCGGTCGATGC 3′; reverse primer: 5′ GTGGCAGATGGC GCGGCAACA 3′). Thermal cycle conditions used for all reactions were as follows: 5 min at 95°C, followed by 35 cycles consisting of denaturation for 30 sec at 95°C, annealing for 30 sec at 58°C, and extension for 1 min at 72°C. PCR reactions were concluded with incubation for 10 min at 72°C to complete the extension of all synthesized products. PCR products were then visualized on a 1.25% TAE agarose gel (Supplementary Figure S2).
In vitro detection of exosome transfer
MHCC97L cells were stably transfected with PMSC-loxp-dsRed-loxp-Stop-eGFP-puro-WPRE, then, exosomes from LX2-Cre cell or control LX2 cells were added to the medium of 97L-loxp-dsRed-loxp-Stop-eGFP. The medium was not changed for 72h to allow LX2 exosomes carrying Cre to be uptaken by MHCC97L cells. The successful packaging of Cre into EVs by LX2 cells, followed by the successful uptake of these exosomes by MHCC97L and finally utilization of Cre by these cancer cells would result in a deletion event between the 2 loxp sites of MHCC97L. This deletion event would in turn cause the excision of dsRed (red color) and expression of GFP (green color).
In vivo detection of exosome transfer
MHCC97H cells were stably transfected with PMSC-loxp-dsRed-loxp-Stop-eGFP-puro-WPRE as described above. These 97H-loxp cells (2×106) were injected subcutaneously into female NOD scid gamma mice (The Jackson Laboratory) at the age of 6 weeks, to establish tumors. After 1 week, exosomes from LX2-Cre medium were injected intra-tumorally. Seven days later, tumors were harvested. Expression of eGFP and dsRed were tested with western blot assay and immunofluoresence. The described animal procedures have been reviewed and approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University.
Measurement of LX2-exosome-miR-335 effects on MHCC97H spheroids
We generated MHCC97H spheroids by using the hanging drop method, which utilizes spheroids completely embedded in matrigel as described in Supplementary Materials and Methods and in (12). Exosomes were transfected with miR-335-5p mimics or miR-NSM as above. Then, 5μg exosomes-miR-335 or exosome-NSM, respectively, per well were added in the medium on the top of spheroid. The invasion areas were measured with Image J software. And the invasion index was calculated by invasion area divide the area of 24h spheroid.
Measurement of LX2-exosome-miR-335 effects on MHCC97H cell in vivo
MHCC97H cells (2×106) were injected subcutaneously into female NOD scid gamma mice (Jackson Laboratory) at the age of 6 weeks, to establish tumors. When tumors can be seen and get to length of 5mm, exosomes were transfected with miR-335 or miR-NSM as above. Then, 50μg exosomes per mouse (16.7 pmol miR-mimics) were injected intra-tumorally twice a week for 4 weeks. In order to determine tumor, the greatest longitudinal diameter (length) and the greatest transverse diameter (width) were determined. Tumor volume based on caliper measurements were calculated by the modified ellipsoidal formula (13, 14): Tumor volume = 1/2(length × width2).
Quantitative mRNA RT-PCR using Taqman Human Cancer pathway panel
MHCC97H and MHCC97L cells were transfected with miR-335-5p mimic or. Total RNA was extracted from cultured cells using TRIzol reagent (Ambion by Life Technologies, Carlsbad, California, USA). In addition, RNA was extracted from mouse tumors treated with miR-335 or NSM, respectively. cDNA was synthesized with High-Capacity cDNA Reverse Transcription Kits (ThermoFisher, USA), then quantitative RT-PCR analysis were applied with specific Taqman Human Cancer pathway panels, which contains 624 genes related to DNA repair, angiogenesis, cell adhesion, and ECM, as well as genes involved in the cell cycle and apoptosis that have been found to be differentially expressed in early cancer and metastatic disease.
Immunostaining of ki67 and cleaved caspase-3 on frozen sections
Mouse xenograft tumor tissue were harvested, and then prepared for frozen sectioning. The frozen sections were fixed and stained with ki67 and cleaved caspase-3 antibodies (Sigma, USA). The positive cells were counted with image-J software and relative ratio was calculated by dividing the total cells in the same view, and experiments were repeated three times.
Statistical methods
SPSS software version 17 (IBM Corp., Armonk, NY, USA) was used to perform statistical tests. Data are expressed as mean ± SD. Paired Student's t test was used to test the differences in sample means for data with normally distributed means. Statistical significance was assumed at a p value of <0.05.
Results
Co-culturing HCC cells with fibroblasts leads to down-regulation of miR-335-5p within fibroblasts, as well as in HCC cells
MiR-335 has been implicated as a tumor suppressor miR species in HCC (15). In addition, outside the liver, miR-335 over-expression suppresses metastatic colonization in human breast cancer lines. Conversely, the inhibition of miR-335 has the opposite effect, enhancing metastasis (16). We concluded that, in some human cancers, and more importantly, in HCC, miR-335 acts as a tumor suppressor. In addition, further literature review uncovered a report of gradual miR-335 downregulation along the continuum from inactive to activated human stellate cells (11). These 2 lines of reported involvement of miR-335 in liver diseases suggested a scenario where liver fibrosis (activation of stellate cells) and cancer development may be characterized by downregulation of miR-335 both in stellate cells as well as in epithelial cancer cells. We have chosen to study miR-335-5p due to these its reported cancer suppressing and anti-fibrotic effects. In the current study, we focused on the delivery of miR-335-5p to HCC cells by utilizing EVs. First, we assessed baseline levels of miR-335 in LX2, as well as in several HCC cell lines when cultured individually. We found that the level of miR-335 was higher in LX2 cultured by themselves vs. HepG2, MHCC97L, MHCC97H and Huh7 liver cancer cells cultured by themselves, respectively (Figure 1A). Next, we investigated how miR-335-5p levels change when co-culturing fibroblast cells (LX2) with each of the four cancer cell lines. HCC cells were stably transduced with pCDH-EF1-MCS-IRES-GFP lentivirus. These HCC cells expressed GFP for the purpose of FACS sorting after direct co-culture with LX2 cells. Each of these 4 HCC cell lines were then directly co-cultured with LX2 cells for 14 days. After co-culturing, HCC cells and LX2 cells were separated by sorting according to GFP positivity. Of note, previous published literature demonstrating downregulation of miR-335-5p in HCC as well as in activated stellate cells had not investigate if the downregulation of miR-335-5p occurs concurrently when these 2 cell types are in direct contact (such as is the case in vivo or in direct co-culture conditions). We hypothesized that an etiologic role for miR-335-5p in HCC development (in particular in a background of fibrosis) might be accompanied by simultaneous downregulation both in cancer cells and stromal cells. Remarkably, we found that miR-335 levels are down-regulated in each of the 4 HCC cell lines upon co-culturing with LX2 cells (Figure 1B, C, D, and E). Moreover, we noted that miR-335-5p levels were also down-regulated in LX2 cells when co-cultured with any of the HCC cell types (Figure 1F). These findings suggest that the symbiotic cancer cell-CAF environment appears to benefit from the down-regulation of miR-335-5p. Another possible implication is that miR-335-5p can be actively shuttled between the 2 cell types and therefore, the levels are in equilibrium. By extension, if these hypotheses are correct, the consequence would be that up-regulating miR-335-5p in either of the 2 cell types would have similar phenotypic effects.
Figure 1. Co-culturing HCC cells with fibroblasts leads to down-regulation of miR-335-5p within fibroblasts, as well as in HCC cells.
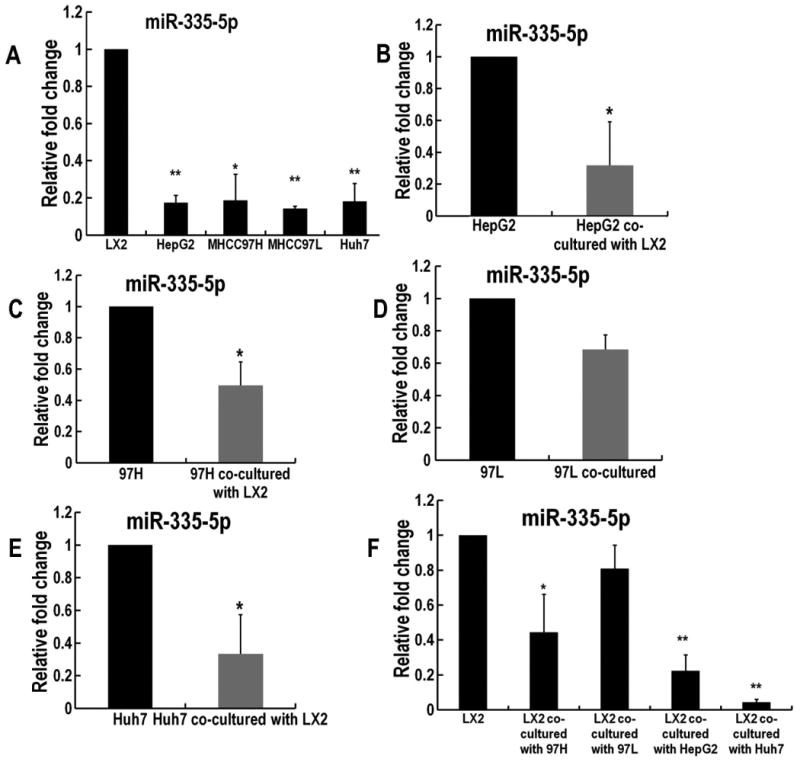
Panel A. Shown are baseline levels of miR-335-5p in LX2, as well as in several HCC cell lines when cultured individually. RNU6B was utilized as internal reference. All the reactions were run in triplicate. * P<0.05 and ** P<0.01 comparing with LX2 group. Panels B-E. These panels demonstrate downregulation of miR-335-5p in each of the 4 HCC cell lines upon their individual co-culture with LX2 cells. * P<0.05 comparing with each cell line group. Panel F. MiR-335-5p levels were also downregulated in LX2 cells when co-cultured with any of the HCC cell types * P<0.05, ** P<0.01 comparing to LX2 cell before co-culturing.
Over-expression of miR-335-5p in HCC cells inhibits their proliferation and invasion
In order to investigate the potential role of miR-335-5p in HCC growth and metastasis, HepG2, MHCC97H, MHCC97L and Huh7 cells were transfected with miR-335-5p mimics and negative control (NSM). Next, we determined cancer cell proliferation by MTT assay and invasion by transwell assay. The proliferation assays showed that miR-335-5p restoration markedly inhibited cell growth in each of the 4 HCC cell lines tested (Figure 2A) when compared with control NSM transfected cells. The transwell assay demonstrated significant cell invasion inhibition in the miR-335-5p transfected cells compared with their control counterparts (Figure 2B, C). We conclude that miR-335-5p exerts its anti-cancer effects through inhibiting the growth as well as the invasion of HCC cells.
Figure 2. Over-expression of miR-335-5p in HCC cells inhibits their proliferation and invasion.
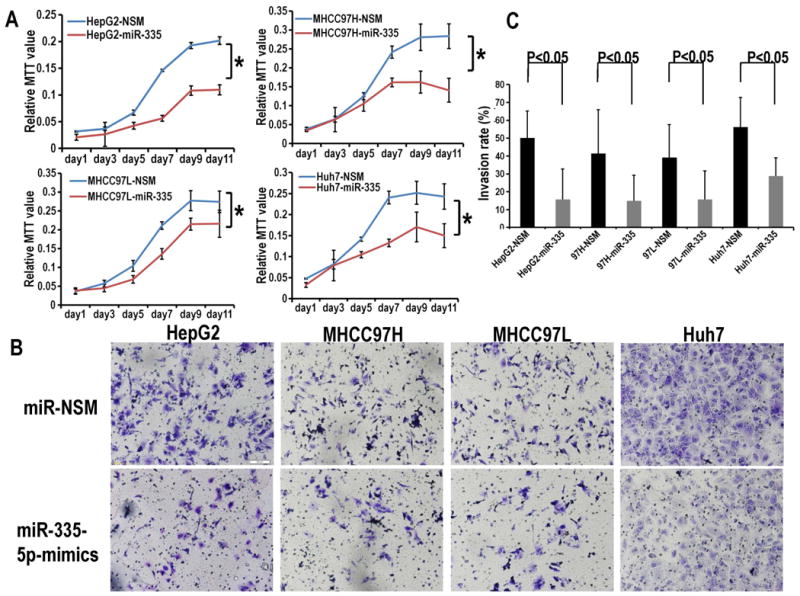
Panel A. MTT proliferation assays showed that miR-335-5p restoration markedly inhibited cell growth vs. NSM transfected cells in each of the 4 HCC cell lines tested. All the reactions were run in triplicate. Absorbance was read at 570nm and subtracts background absorbance measured at 690 nm using an Envision 2104 multipliable Plate Reader. Data are represented as mean ± SD, and analyzed with two-way ANOVAs in SPSS software. Panel B. The transwell assays demonstrated significant cell invasion inhibition in the miR-335-5p mimic-transfected cells compared with their counterparts. Panel C. Cells from five random fields in invasion assay were counted at X10 magnification with Image J software. Error bars represent the SE for n=5 experiments carried out in triplicate. The p-values were obtained using the Student's paired t-test, two-tailed.
Up-regulation of miR-335-5p within fibroblasts is sufficient to inhibit neighboring cancer cell proliferation, invasion and motility
We transfected LX2 cells with miR-335-5p or NSM, respectively, then co-cultured these cells with each of the following 4 HCC cell lines: HepG2, MHCC97H, MHCC97L and Huh7. The number of cancer cells after 3 days was then assessed. We found that each of the 4 cancer cell lines proliferated less when co-cultured with LX2 cells over-expressing miR-335 vs. control (Figure 3A). We further explored the invasion ability of cancer cells when co-cultured with LX2-miR-335-5p or LX2-NSM. We found that co-culturing with LX2 cells transfected with miR-335-5p, cancer cells invaded less vs. control. (Figure 3B, C). Similar results were obtained in a scratch assay, suggesting that the motility of HCC cells, including HepG2 (Figure 4A, B), MHCC97H (Figure 4C, D), MHCC97L (Figure 4C, D), as well as Huh7 (Figure 4E, F) is inhibited when each individual cancer cell line is co-cultured with LX2 cells over-expressing miR-335. We concluded that up-regulation of miR-335-5p in fibroblasts is sufficient to inhibit growth and invasion in neighboring cancer cells. This observation raises an important question – is the observed phenotype due, at least in part, to miR transfer from fibroblasts to cancer cells?
Figure 3. Upregulation of miR-335-5p in fibroblasts inhibits neighboring cancer cell proliferation and invasion.
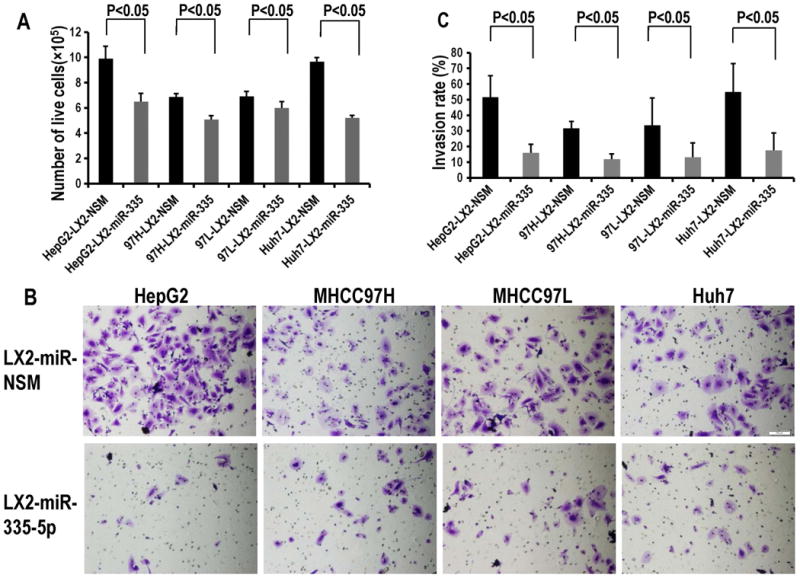
Panel A. Upregulation of miR-335-5p mimic in LX2 cells inhibits neighboring cancer cell growth. All the experiments were done in triplicate. Panel B. Cancer cells co-cultured with LX2 cells transfected with miR-335-5p invaded less vs. control cancer cells. Panel C. The invasion rates from Panel B were quantified as in Figure 2, Panel C.
Figure 4. Over-expression of miR-335-5p in fibroblasts inhibits cancer cell motility.
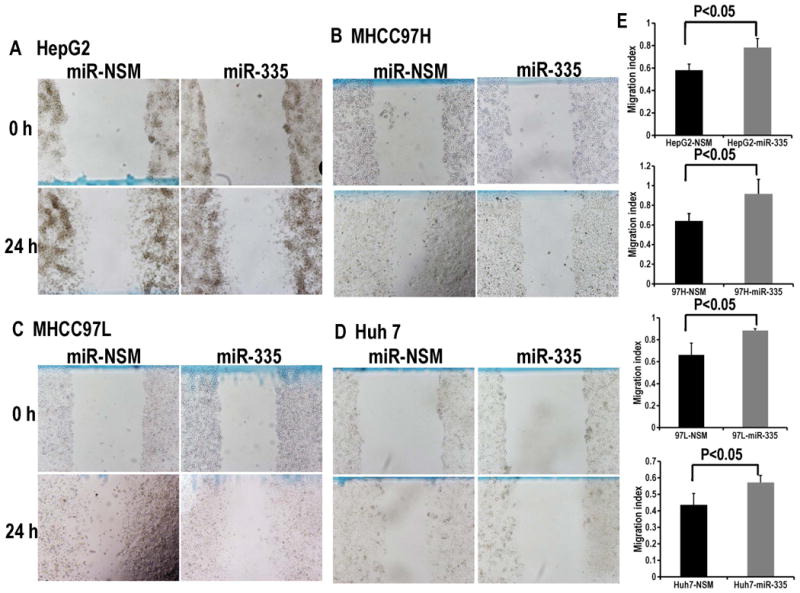
Panels A, B, C and D demonstrate that each of the HCC cell lines display less motility when co-cultured with LX2-miR335-5p prior to their plating for the scratch assay. Panel E shows the quantification of motility from Panels A-D. Note that the higher numbers (height of bars in the figure) for miR-335 treated cancer cells signify a wider wound at 24h, which translates into less motility. Bars represent the migration index for each treatment arm (NSM vs. miR-335), expressed as a value of scratch area at 24h divided the area at time 0h. Error bars represent the SE for n=5 experiments carried out in triplicate. The p values were obtained using a 2-tailed Student's paired t-test.
Exosomal transfer from fibroblasts to HCC cells in vitro
It was previously reported by our group, as well as others, that EVs can transfer RNA between cells (17). Data presented above suggested that miR-335-5p might be actively shuttled between fibroblasts and cancer cells in our co-culture system. To investigate this hypothesis, we employed the Cre-loxp system. First, EVs from LX2-Cre cells were isolated. As shown in Supplementary Figure S2 and in previous studies, EVs from cells transfected with a Cre vector express Cre mRNA (18). Next, we characterized LX2-derived EVs and found typical exosomal markers (Figure 5A). To further characterize these particles, we utilized Nanoparticle Tracking Analysis (NTA). We found that the mean size of isolated EVs was approximately 100 nM, suggestive of typical size of exosomes (Figure 5B). To determine whether miR-335-5p can be directly transferred from fibroblasts to HCC cells via EVs, we transfected HCC cells with PMSC-loxp-dsRed-loxp-Stop-eGFP-puro-WPRE retrovirus. After transfection and selection, HCC cells demonstrated, as expected, fluorescence in the red, but not in the green channels (Supplementary Figure S3.). As an intermediary step, and to verify that LX2-Cre release EV-Cre in co-culture, we directly co-cultured LX2-Cre cells with HCC cells expressing loxp-dsRed-loxp-Stop-eGFP. We surmised that if EVs carry Cre from LX2 cells to co-cultured HCC cells, then we would note a change in fluorescence from red to green. Indeed, after 1 week of co-culture, we noted that select cancer cells in culture switched fluorescence from the red to the green spectrum (Supplementary Figure S4.) Last, we directly added exosomes isolated from LX2-Cre to MHCC97L-loxp-dsRed-loxp-Stop-eGFP for 1 week. We noted that select cancer cells have switched from red to green, demonstrating that 1) EVs carrying Cre were uptaken by these cancer cells and 2) the EV cargo had been utilized within cancer cells (Figure 5C). We therefore concluded that EVs released from fibroblasts have the ability to shuttle their cargo from cells of origin (fibroblasts) to neighboring target cells (cancer cells).
Figure 5. Exosomes transfer nucleic acid cargo from fibroblasts to HCC cells in vitro and in vivo.
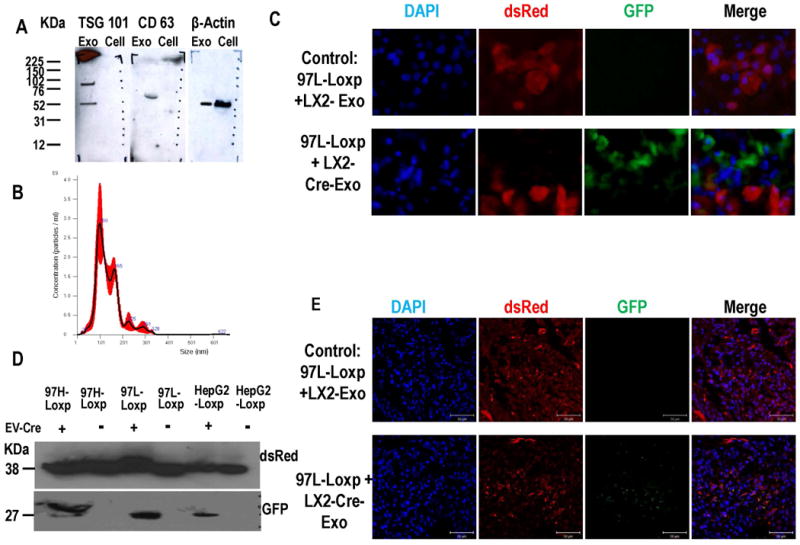
Panel A. LX2-derived EVs (Exo) display typical exosomal markers (CD63 and TSG101). β-actin was utilized as internal control. Panel B. Nanoparticle Tracking Analysis (NTA) displayed the size distribution of EVs isolated from LX2 cell cultures. Black line represents mean of five experiments, SD in red. The mean size of isolated EVs was approximately 100 nM, which is the typical size of exosomes. Panel C. Exosomes isolated from LX2-Cre cells were added to cultures of HepG2-loxp-dsRed-loxp-Stop-eGFP and MHCC97L-loxp-dsRed-loxp-eGFP for 1 week. As shown, there were cancer cells that switched colors from red to green to signify that Cre has been transferred by exosomes to the cytoplasm of these cancer cells. Scale bars represent 100 μm. Panel D. Western blot assay for GFP and dsRed in each tumor tissue. dsRed was detected in lysate from all tumors, while GFP was detected only in lysates from mouse tumors treated with EV-Cre. Panel E. Immunofluorescence of GFP and dsRed in tumor tissue frozen slides. When tumor were infected with exosomes isolated from LX2-Cre, the dsRed positive cells were detected. Scale bars represent 50 μm.
Exosomal delivery to HCC cells in vivo
To study the intercellular EV exchange in vivo, we first established xenograft subcutaneous tumors in NOD Scid Gamma (NSG) mice with HepG2-loxp-dsRed-loxp-Stop-eGFP, MHCC97H-loxp-dsRed-loxp-Stop-eGFP and MHCC97L-loxp-dsRed-loxp-Stop-eGFP cells, respectively. After we successfully established tumor xenografts from each of these 3 HCC cell lines, we sought to inject EV directly into these tumors and detect the utilization of EV cargo in the cytoplasm of cancer cells. Last, in order to gain insight into the effects of a low dose of EVs, we decided to perform a single injection of EVs into tumors. To this end, we administered a single dose of exosomes (150 ng) from LX2-Cre by injection directly into the tumor mass. We surmised that if EVs injected in the cancer mass are uptaken by cancer cells, even after a single dose, then we should see at least a few cancer cells switching from red to green fluorescence. Seven days later, the tumor tissue was harvested and proteins were isolated. GFP and dsRed proteins were detected by western blot. As shown in Figure 5D, dsRed was detected in lysate from all tumors, while GFP was detected only in lysates from mouse tumors treated with EV-Cre. These data demonstrate that EV-Cre were uptaken by cancer cells in vivo and their cargo was utilized within these cancer cells. To further verify our findings, we sought to detect GFP by an independent method. To this end, we performed immunofluorescence on cancer tissue slides, which showed that cancer cells within tumors treated with EV-Cre have uptaken and utilized Cre (Figure 5E). In conclusion, 1) we gave mice a single dose of EVs (low dose, through direct tumor injection); 2) to ensure reproducibility, experiments were performed in xenografts established from 3 different cell lines and 3) we detected a change in GFF/dsRed through 2 independent methods (western blotting and immunofluorescence).
MiR-335-5p delivered by EVs inhibit HCC invasion in vitro
While we have shown that 1) cancer cells grow less and invade less when co-cultured with fibroblasts over-expressing miR-335-5p; 2) EVs produced by fibroblasts can successfully deliver their cargo to cancer cells and 3) that cancer cells utilize the cargo delivered by EVs intra-cytoplasmatically, we had yet to demonstrate that miR-335 delivered by EVs had a phenotypic effect on target cancer cells. To this end, we generated cancer cell spheroids by employing the hanging-drop methodology, as described in Supplementary Materials and Methods and in other published protocols (12). The hanging drop methodology was implemented to obtain a 3D pre-clinical representation of cancer invasion capabilities. Spheroids were then treated with EVs-miR-335 and EVs-NSM, respectively. We noted MHCC97H spheroids treated with EV-miR-335-5p invaded the surrounding Matrigel significantly less vs. spheroids treated with the control EV-NSM (Figure 6A, B).
Figure 6. MiR-335-5p delivered by EVs inhibit HCC invasion in vitro and inhibit HCC growth in vivo.
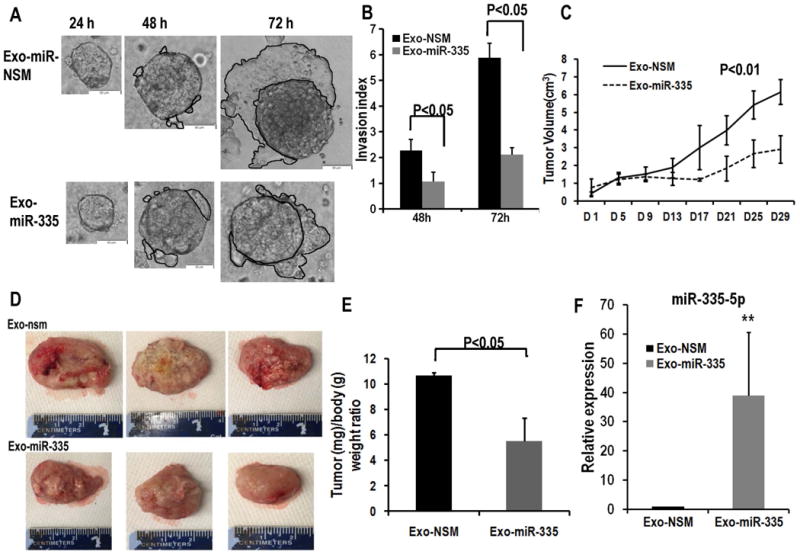
Panel A. Cancer spheroids treated with EV-miR-335-5p invade surrounding matrigel less vs. control cells. Scale bars represent 100 μm. Panel B. The invasion area was measured with Image J software. The invasion index was calculated by the area at 48h and 72h, respectively, divided by the area of the 24h spheroid. Data is presented as mean, and error bars represent the SE for n=5 experiments carried out in triplicate. Panel C. Xenograft tumor volumes were measured twice a week right before EV injection. EV treatment resulted in significantly smaller tumors in the treatment group vs. control. Panel D. Excised xenograft tumors from EV-335-5p and control animals. Tumor size was significantly smaller in animals injected with LX2 cell-derived exosomes loaded with miR-335-5p mimics (Exo-miR-335) when compared with negative control (Exo-NSM). Panel E. Tumor/body weight ratio when animals were euthanized. Data is presented as mean, and error bars represent the SE for n=5 for each group. Panel F. miR-335-5p is expressed at higher values in tumor tissue from mice treated with EV-miR-335-5p vs. control tissue (n=5). Levels of miR-335-5p were approximately 30 fold higher in tumors that were treated with EV-miR-335 vs. control.
MiR-335-5p delivered by EVs inhibits HCC growth in vivo
To further delineate whether EV transfer of fibroblast-derived miR-335-5p could impact HCC growth in vivo, MHCC97H cells (2×106) were injected subcutaneously into 6-week old female NSG mice. When tumors reached 5mm in diameter, EVs that were transiently transfected with miR-335-5p or negative control were quantified. Next, 50 μg EV-miR-335-5p (16.7 pmol of miR-335-5p) per mouse were injected intra-tumorally twice a week for 4 weeks. We found that the EV treatment resulted in significantly smaller tumors in the treatment group vs. control (Figure 6C). Similar findings were found when the animals were euthanized and the tumors measured directly after excision (Figure 6D, E). To verify that, indeed, the levels of miR-335-5p are higher in tumors that grew less vs. control tumors, we quantified its levels by RT-PCR. We found that levels of miR-335-5p were approximately 30 fold higher in tumors that were treated with EV-miR-335 vs. control (Figure 6F).
In vivo treatment of HCC with EV-miR-335 induces less proliferation as well as increased apoptosis
To investigate the mechanism of miR-335-5p meditated tumor inhibition, we assayed for ki67 and caspase-3 in slides prepared from the tumors. As shown in Figure 7, the expression of ki67 was reduced in LX2-Exo-miR group vs. with LX2-Exo-nsm group, and the cleaved caspase-3 expression in LX2-Exo-miR group was increased. These data suggest that EV-miR-335-5p inhibits tumor growth in vivo through decreased proliferation as well as increased apoptosis.
Figure 7. In vivo treatment of HCC with EV-miR-335 induces less proliferation as well as increased apoptosis.
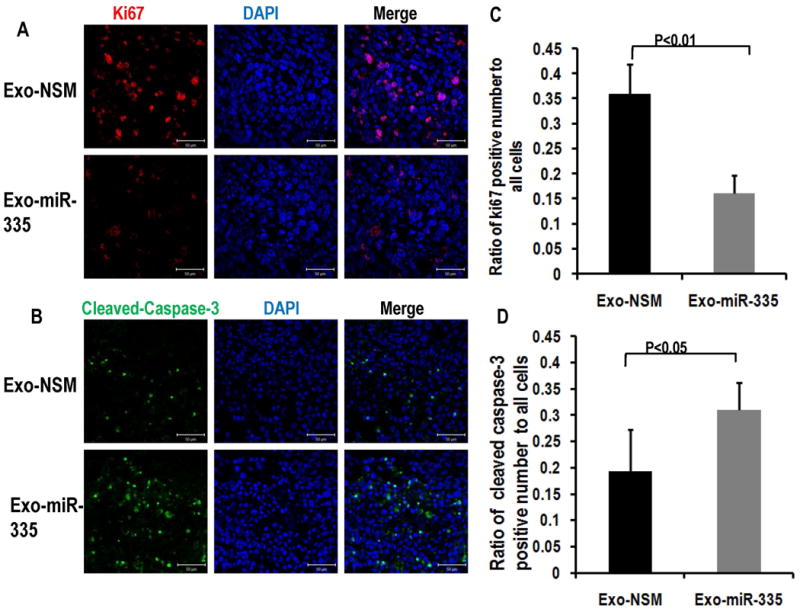
Panel A. demonstrates immunostaining for Ki67 and Panel B. demonstrates Cleaved-Caspase-3 in mice tumors treated by intra-tumoral injection with LX2-derived exosomes carrying miR-335-5p mimics (Exo-miR-335). Scale bars represent 50 μm. Ki67 positive cell or Cleaved-Caspase-3 positive cells and total cells were counted by Image J, and the ratio of Ki67 positive cell or Cleaved-Caspase-3 positive cell as a function of total cells on the same slides were calculated. Data is presented as mean, and error bars represent the SE for 5 slides from 3 different mice for each group. Student t-test was used for statistics. Panels C and D present the quantification of fluorescence from Panels A and B, respectively.
Unbiased identification of miR-335-5p downstream mRNA targets
To determine mRNA targets of miR-335-5p in an unbiased fashion, we transfected MHCC97H and MHCC97L with miR-335-5p mimic or negative control. In addition, mouse xenograft tumors from mice treated with EV-miR-335-5p as well as control, respectively, were collected. A total of 4 mice tumors were used for this experiment. Specifically, two mice were used to establish MHCC97L-derived subcutaneous tumors (one tumor per mouse). One of the tumors was treated with EV-NSM and the other mouse tumor was treated with EV-miR-335-5p. In addition, two mice were used to establish MHCC97H subcutaneous tumors. Similarly, one mouse tumor was treated with EV-NSM and the other with EV-miR-335-5p. Intra tumoral injections with EV-NSM or EV-miR-335-5p were performed twice a week for duration of 4 weeks. RNA from cell lines (a total of 4 samples) and from mouse tumors (a total of 4 samples) were used to identify differentially expressed mRNA species between NSM and miR-335-5p treated samples. The Taqman Human Cancer Panels (Thermofisher Scientific) was used. Bioinformatics analyses identified 13 genes which were consistently and uniformly down-regulated in the 4 RNA samples treated with miR-335-5p. Of note, we utilized this stringent, experimental approach, instead of the bioinformatics-based search engine approach to confer our report a high specificity. Given how strict the determination of these putative targets was (in vitro – Figure 8 A and B, as well as in vivo –Figure 8 C and D, each on 2 separate cell lines), it is likely that these targets are indeed down-regulated by miR-335-5p and involved in its downstream effects. These mRNA species are: CDC42 (Cell division control protein 42 homolog), CDK2 (Cyclin-dependent kinase 2), CSNK1G2 (Casein kinase 1 gamma 2), EIF2C2 (Protein argonaute-2), EIF5 (Eukaryotic translation initiation factor-5), LIMK1 (LIM Domain Kinase 1), NRG1 (Neuregulin 1), PLK2 (Polo Like Kinase 2), RGS19 (G-protein signaling 19), TCF3 (Transcription Factor 3), THBS1 (Thrombospondin 1), YBX1 (Y-Box Binding Protein 1) and ZMYND8 (Human Zinc Finger MYND-type containing 8). The modulation of pathways downstream of miR-335 is likely to be explained, at least in part, by these trancriptomics changes. The exhaustive identification of mechanisms of miR-335 action is beyond the purpose of this manuscript.
Figure 8. Downstream mRNA targets of miR-335-5p were identified by qPCR array.
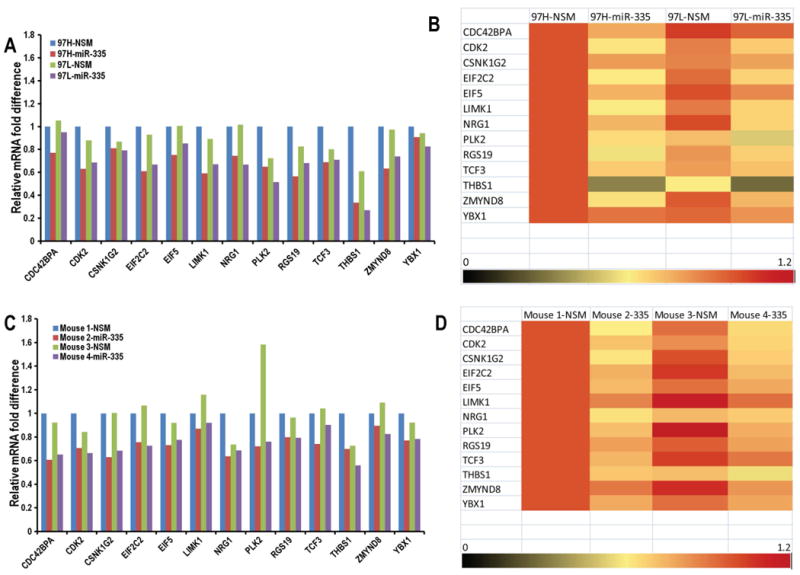
Panels A, B: MHCC97H and MHCC97L cells were transfected with miR-335-5p mimic (97H-miR-335 and 97L-miR-335) or negative control (97H-NSM and 97L-NSM).Thirteen potential mRNA targets of miR-335-5p were identified to be downregulated by miR-335 treatment. Panels C, D: Mouse 1 and mouse 2 are subcutaneous tumors established by injecting MHCC97L, and mouse 3 and mouse 4 are subcutaneous tumors established by injecting MHCC97L. Each of the two pairs was treated with EV-NSM and the other mouse tumor per group was treated with EV-miR-335-5p. We extracted the total RNA from the mice tumor. Real time qPCR array using The Taqman Human Cancer Panels demonstrate the downregulation of these 13 potential mRNA targets of miR-335-5p.
Discussion
The vast majority of HCC cases arise in a background of liver fibrosis/cirrhosis that is rich in activated fibroblasts/stellate cells. Starting with this observation, we hypothesized that stellate cells and cancer cells can communicate. Furthermore, we posited that the communication between stellate cells and cancer cells is done in part via EVs. Here, we found that co-culturing stellate and cancer cells results in downregulation of miR-335-5p in both cell types. Although it was not the purpose of this study to investigate the mechanisms through which this concomitant down-regulation occurs, we did not a similar phenomenon in cholangiocarcinoma (8). It is attractive to hypothesize that the down-regulation of miR-335-5p is conducive to cancer development and/or homeostasis. Conversely, up-regulation of miR-335-5p in cancer cells, or in neighboring fibroblasts, should result in cancer repression. We have noted such cancer repression (invasion as well as growth) when miR-335-5p has been up-regulated either in cancer cells or in fibroblasts.
Previous work in cholangiocarcinoma demonstrates that cancer cells avidly uptake stellate cell/fibroblast-derived EVs (8). While the mechanisms of EV shuttling between cholangiocarcinoma cells and neighboring stromal cells in vivo have not been elucidated to date, this previous report argues that such process occurs and also that it can be utilized/adapted for therapeutics delivery to cancer cells in vivo (8). Previously published work showed that that miR-335-5p is 1) downregulated in liver cancer cells (15); and 2) downregulated in activated, collagen producing stellate cells vs. inactive cells (11). In the current manuscript we noted further downregulated in both cancer cells and stellate cells upon their direct co-culture. These findings further suggest that shuttling of EVs and their cargo between HCC cells and surrounding stromal cells occurs, but the precise mechanisms of such process continue to be elusive to date. Furthermore, it is formally possible that, through a yet to be discovered mechanism, the symbiotic cancer cell-stromal cell environment requires low levels of miR-335-5p for successful cancer genesis and development. In strong support of this hypothesis are our findings that upregulation of miR-335-5p in either cancer cells, or in stromal cells, is sufficient to inhibit HCC growth and/or invasion (Figures 2, 3, 4 and 6).
The observation that the up-regulation of miR-335-5p in fibroblasts results in neighboring cancer cell repression suggests that miR-335-5p might be shuttled between these 2 cell types. Indeed, when miR-335-5p was directly up-regulated in EVs and delivered to cancer cells, a similar effect was noted. Building on these observations, we verified that EVs can be utilized as vehicles to deliver miR-335-5p in vivo to cancer cells, as it has been reported previously by our group in cholangiocarcinoma (8). The fact that we successfully delivered EVs to cancer cells in vivo, that EV content was successfully taken into, and utilized by cancer cells and finally, that the treatment resulted in slower cancer growth suggests that EVs may, indeed, be utilized as cancer therapeutics in HCC.
To obtain an unbiased view on mRNA targets downstream of miR-335-5p, we have performed a quantitative PCR (qPCR) screen. We have used Taqman Human Cancer Panel which has 624 genes related to DNA repair, angiogenesis, cell adhesion, cell cycle and apoptosis. We identified the most likely targets of miR-335-5p as CDC42 (Cell division control protein 42 homolog), CDK2 (Cyclin-dependent kinase 2), CSNK1G2 (Casein kinase 1 gamma 2), EIF2C2 (Protein argonaute-2), EIF5 (Eukaryotic translation initiation factor-5), LIMK1 (LIM Domain Kinase 1), NRG1 (Neuregulin 1), PLK2 (Polo Like Kinase 2), RGS19 (G-protein signaling 19), TCF3 (Transcription Factor 3), THBS1 (Thrombospondin 1), YBX1 (Y-Box Binding Protein 1) and ZMYND8 (human Zinc Finger MYND-type containing 8). Among these genes, CDC 42(19, 20), CDK2(21, 22), EIF2C2(23, 24), EIF5(25, 26), LIMK1(27), NRG1(28), PLK2(29), RGS19(30), TCF3(31), THBS1(32) as well as YBX1(33) have been implicated in hepatocellular carcinoma, and some of them are related to invasion and metastasis. Out of these 13 downstream miR-335-5p targets, CSNK1G2 and ZMYND8 are reported here for the first time as playing a role in HCC.
In the current study we chose to deliver EVs directly to xenografted tumors (via direct intra-tumor injections). Unlike in experiments performed previously in cholangiocarcinoma (8), in this study we chose to circumvent vascular delivery of EVs to tumor site by directly injecting EVs into the tumors. The rationale was 2 fold – first, we focused on the uptake of EVs by cancer cells, once these EVs were delivered to the tumor mass (as opposed to focusing on concentrating EVs from the blood stream to the cancer mass) and second, there is a precedent whereby active chemotherapeutics are given to un-resectable or multifocal HCC patients by intra-arterial delivery to the cancer. This method, trans-arterial chemo embolization (TACE) is an accepted and widely utilized HCC treatment modality (34). Nonetheless, it should be noted that intra-tumoral injections of EVs is merely a model of cancer therapeutics delivery that does not perfectly model intravenous nor intra-arterial delivery. Therefore, further studies must identify if the best delivery method of EVs to HCC patients is via intra-venous delivery, or, as in the case of TACE, via direct delivery to the cancer mass.
There are multiple future directions stemming from this study. For example, we have not aimed at identifying the causal factors resulting in the down-regulation of miR-335-5p in both stellate cells and cancers cells upon their co-culture. In addition, we have not tested EVs produced by other cell types to investigate if the cargo delivery by stellate cell produced EVs is related to the cell of origin. This aspect is important for future developments of EVs as cancer therapeutics. Last, pathways downstream of miR-335-5p have not been exhaustively investigated. While we took first steps towards finding miR-335-5p targets in an unbiased fashion, further work is necessary should miR-335-5p be considered as a potential HCC therapeutics.
Supplementary Material
Figure S1. RT-PCR analysis of EVs transfected with miR-335-5p. A comparison is made between EVs transfected with miR mimics vs. control (PBS with no EVs).
Figure S2. Verification of Cre expression. PCR for Cre mRNA was performed. LX2 cells (Cre negative, control cells) do not show any expression of Cre. Similarly, the EVs obtained from these cells, as well as the supernatant (Sup in the figure) that is left from conditioned media (CM) after EV extraction do not demonstrate any Cre. LX2 cells that express Cre demonstrate the Cre mRNA as shown. In addition, EVs isolated from these cells demonstrate Cre, but not the Sup (CM depleted of EVs). H2O is included as another negative control.
Figure S3. HCC cells transfected with PMSC-Lox-ds Red-Lox-eGFP-puro-WRPE. As expected, these cells demonstrate fluorescence in the red, but not the green channel.
Figure S4. Exosomal transfer from fibroblasts cells to HCC cells in vitro. LX2-Cre cells were added to MHCC97L-Loxp-dsRed-Loxp cells (red) for 1 week. We noted the switch in color from red to green occurring in select cancer cells. These findings argue that Cre was shuttled from LX2 cells to HCC cells. Scale bars represent 50 μm.
Acknowledgments
Financial Support: This work was supported by National Institutes of Health Grants, DK090154, R01CA190040 and R01EB017742 to Florin M. Selaru, and National Institutes of Health Grant R01CA083650 to Alphonse E. Sirica. Fang Wang was supported, in part, by the China Scholarship Council.
List of abbreviations
- HCC
Hepatocellular Cancer
- CAF
cancer-associated fibroblasts
- EVs
Extracellular Vesicles
- EMT
Epithelial to Mesenchymal Transition
- miR
microRNA
- CCA
cholangiocarcinoma
- GFP
green fluorescent protein
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- NSM
nonspecific mimic
- dsRed
Discosoma species red fluorescent protein
- mRNA
messenger RNA
- NSG
NOD Scid Gamma mice
- CDC42
Cell division control protein 42 homolog
- CDK2
Cyclin-dependent kinase 2
- CSNK1G2
Casein kinase 1 gamma 2
- EIF2C2
Protein argonaute-2
- EIF5
Eukaryotic translation initiation factor-5
- LIMK1
LIM Domain Kinase 1
- NRG1
Neuregulin 1
- PLK2
Polo Like Kinase 2
- RGS19
G-protein signaling 19
- TCF3
Transcription Factor 3
- THBS1
Thrombospondin 1
- YBX1
Y-Box Binding Protein 1
- ZMYND8
Human Zinc Finger MYND-type containing 8
References
- 1.Singal AG, El-Serag HB. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015;13:2140–2151. doi: 10.1016/j.cgh.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel) 2015;7:2443–2458. doi: 10.3390/cancers7040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, Chai S, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016;15:1175–1189. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R, Mezey E, et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2016 doi: 10.1002/hep.28735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo CJ, Pan Q, Cheng T, Jiang B, Chen GY, Li DG. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 2009;276:5163–5176. doi: 10.1111/j.1742-4658.2009.07213.x. [DOI] [PubMed] [Google Scholar]
- 10.Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, Wu J, et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol. 2010;298:G101–106. doi: 10.1152/ajpgi.00220.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, Wu CQ, Zhang ZQ, Yao DK, Zhu L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp Cell Res. 2011;317:1714–1725. doi: 10.1016/j.yexcr.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Berens EB, Holy JM, Riegel AT, Wellstein A. A Cancer Cell Spheroid Assay to Assess Invasion in a 3D Setting. J Vis Exp. 2015 doi: 10.3791/53409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31:229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 14.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Li W, Chen C, Pei Y, Long X. MiR-335 acts as a potential tumor suppressor miRNA via downregulating ROCK1 expression in hepatocellular carcinoma. Tumour Biol. 2015;36:6313–6319. doi: 10.1007/s13277-015-3317-2. [DOI] [PubMed] [Google Scholar]
- 16.Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, Schiffelers RM, et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015;161:1046–1057. doi: 10.1016/j.cell.2015.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng L, Tung-Ping Poon R, Yau S, Chow A, Lam C, Li HS, Chung-Cheung Yau T, et al. Suppression of actopaxin impairs hepatocellular carcinoma metastasis through modulation of cell migration and invasion. Hepatology. 2013;58:667–679. doi: 10.1002/hep.26396. [DOI] [PubMed] [Google Scholar]
- 20.Wang R, Zhao N, Li S, Fang JH, Chen MX, Yang J, Jia WH, et al. MicroRNA-195 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting the expression of VEGF, VAV2, and CDC42. Hepatology. 2013;58:642–653. doi: 10.1002/hep.26373. [DOI] [PubMed] [Google Scholar]
- 21.Kim HS, Lee KS, Bae HJ, Eun JW, Shen Q, Park SJ, Shin WC, et al. MicroRNA-31 functions as a tumor suppressor by regulating cell cycle and epithelial-mesenchymal transition regulatory proteins in liver cancer. Oncotarget. 2015;6:8089–8102. doi: 10.18632/oncotarget.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai WL, Hung WY, Wong LL, Zhou Y, Leong VY, Lee JM, Ng IO, et al. The centrosomal protein Tax1 binding protein 2 is a novel tumor suppressor in hepatocellular carcinoma regulated by cyclin-dependent kinase 2. Hepatology. 2012;56:1770–1781. doi: 10.1002/hep.25851. [DOI] [PubMed] [Google Scholar]
- 23.Ye ZL, Huang Y, Li LF, Zhu HL, Gao HX, Liu H, Lv SQ, et al. Argonaute 2 promotes angiogenesis via the PTEN/VEGF signaling pathway in human hepatocellular carcinoma. Acta Pharmacol Sin. 2015;36:1237–1245. doi: 10.1038/aps.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Sheng C, Yin Y, Wen S, Yang G, Cheng Z, Zhu Q. PABPC1 interacts with AGO2 and is responsible for the microRNA mediated gene silencing in high grade hepatocellular carcinoma. Cancer Lett. 2015;367:49–57. doi: 10.1016/j.canlet.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Ge Y, Yan X, Jin Y, Yang X, Yu X, Zhou L, Han S, et al. MiRNA-192 [corrected] and miRNA-204 Directly Suppress lncRNA HOTTIP and Interrupt GLS1-Mediated Glutaminolysis in Hepatocellular Carcinoma. PLoS Genet. 2015;11:e1005726. doi: 10.1371/journal.pgen.1005726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng N, Li Y, Han ZG. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology. 2013;57:1906–1918. doi: 10.1002/hep.26202. [DOI] [PubMed] [Google Scholar]
- 27.Guo H, Lv Y, Tian T, Hu TH, Wang WJ, Sui X, Jiang L, et al. Downregulation of p57 accelerates the growth and invasion of hepatocellular carcinoma. Carcinogenesis. 2011;32:1897–1904. doi: 10.1093/carcin/bgr220. [DOI] [PubMed] [Google Scholar]
- 28.Sakai K, Takeda H, Nishijima N, Orito E, Joko K, Uchida Y, Izumi N, et al. Targeted DNA and RNA sequencing of fine-needle biopsy FFPE specimens in patients with unresectable hepatocellular carcinoma treated with sorafenib. Oncotarget. 2015;6:21636–21644. doi: 10.18632/oncotarget.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhullar KS, Jha A, Rupasinghe HP. Novel carbocyclic curcumin analog CUR3d modulates genes involved in multiple apoptosis pathways in human hepatocellular carcinoma cells. Chem Biol Interact. 2015;242:107–122. doi: 10.1016/j.cbi.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 30.Vedarethinam V, Dhanaraj K, Soundherrajan I, Sivanesan R. Identification of Differential Protein Expression in Hepatocellular Carcinoma Induced Wistar Albino Rats by 2D Electrophoresis and MALDI-TOF-MS Analysis. Indian J Clin Biochem. 2016;31:194–202. doi: 10.1007/s12291-015-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kataoka J, Shiraha H, Horiguchi S, Sawahara H, Uchida D, Nagahara T, Iwamuro M, et al. Loss of Runt-related transcription factor 3 induces resistance to 5-fluorouracil and cisplatin in hepatocellular carcinoma. Oncol Rep. 2016;35:2576–2582. doi: 10.3892/or.2016.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Turpin CP, Wang S. Role of Thrombospondin 1 in Liver Diseases. Hepatol Res. 2016 doi: 10.1111/hepr.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gunasekaran VP, Ganeshan M. Inverse correlation of ribosomal protein S27A and multifunctional protein YB-1 in hepatocellular carcinoma. Clin Biochem. 2014;47:1262–1264. doi: 10.1016/j.clinbiochem.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Brown KT, Do RK, Gonen M, Covey AM, Getrajdman GI, Sofocleous CT, Jarnagin WR, et al. Randomized Trial of Hepatic Artery Embolization for Hepatocellular Carcinoma Using Doxorubicin-Eluting Microspheres Compared With Embolization With Microspheres Alone. J Clin Oncol. 2016;34:2046–2053. doi: 10.1200/JCO.2015.64.0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. RT-PCR analysis of EVs transfected with miR-335-5p. A comparison is made between EVs transfected with miR mimics vs. control (PBS with no EVs).
Figure S2. Verification of Cre expression. PCR for Cre mRNA was performed. LX2 cells (Cre negative, control cells) do not show any expression of Cre. Similarly, the EVs obtained from these cells, as well as the supernatant (Sup in the figure) that is left from conditioned media (CM) after EV extraction do not demonstrate any Cre. LX2 cells that express Cre demonstrate the Cre mRNA as shown. In addition, EVs isolated from these cells demonstrate Cre, but not the Sup (CM depleted of EVs). H2O is included as another negative control.
Figure S3. HCC cells transfected with PMSC-Lox-ds Red-Lox-eGFP-puro-WRPE. As expected, these cells demonstrate fluorescence in the red, but not the green channel.
Figure S4. Exosomal transfer from fibroblasts cells to HCC cells in vitro. LX2-Cre cells were added to MHCC97L-Loxp-dsRed-Loxp cells (red) for 1 week. We noted the switch in color from red to green occurring in select cancer cells. These findings argue that Cre was shuttled from LX2 cells to HCC cells. Scale bars represent 50 μm.