

Abstract
Objective
Aberrant neutrophil extracellular trap (NET) formation has been implicated as a mechanism to induce auto-reactivity in at-risk individuals. The study objective was to assess whether medications implicated in cases of drug-induced autoimmunity (hydralazine and procainamide) and medications less commonly associated with drug-induced autoimmunity (minocycline and clozapine) induce NET formation and/or prevent NET degradation.
Methods
Human neutrophils were incubated with the drugs of interest and resultant NET formation was quantified by fluorescent microscopy. The ability of these drugs to interfere with NET degradation by serum nucleases was assessed. Pathways of drug-induced NET formation were studied with pharmacologic inhibitors of reactive oxygen species (ROS), peptidylarginine deiminases (PADs), and muscarinic receptors, and by assessment of intracellular calcium levels by flow cytometry. To determine if NET protein cargo varies by drug stimuli and/or neutrophil source, proteomic analysis of NET lysates induced by specific medications was compared using neutrophils from healthy donors and from patients with autoimmune diseases.
Results
Hydralazine and procainamide significantly induced NET formation while minocycline and clozapine did not. None of the medications significantly impaired NET degradation. NETosis induced by these drugs required NADPH oxidase and PAD4 activation. Procainamide triggered NETs via muscarinic receptor engagement on neutrophils, while hydralazine modulated calcium release from intracellular stores. Differences in protein cargo, particularly histone content, were observed in NETs induced by hydralazine and procainamide.
Conclusion
Medications commonly implicated in drug-induced autoimmunity trigger NET formation displaying distinct protein cargo, via common and specific pathways. NETosis may play a role in the pathogenesis of drug-induced autoimmunity.
Keywords: neutrophil extracellular traps (NETs), drug-induced vasculitis, drug-induced lupus, systemic lupus erythematosus, ANCA-associated vasculitis, hydralazine, procainamide, minocycline, clozapine
Drug-induced autoimmunity (DIA) is an immune regulated adverse effect triggered by exposure to specific medications. Clinical features of DIA include multisystem organ disease associated with production of various autoantibodies, particularly antinuclear antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA). The pattern of autoantibody production and organ system involvement often mirrors clinical features otherwise specific to systemic lupus erythematosus (SLE) or ANCA-associated vasculitis (AAV). Among the list of medications implicated in DIA, procainamide and hydralazine are considered high-risk for the development of autoimmunity[1]. Procainamide is associated with development of ANA in almost all patients chronically exposed to the drug, and symptoms of a lupus-like syndrome develop in approximately 20% of these patients[2]. Approximately 5% of patients exposed to hydralazine develop autoimmune disease, often characterized by overlap features otherwise seen in idiopathic SLE and AAV[2, 3]. Cases of drug-induced lupus or vasculitis have been reported in association with use of minocycline or clozapine; however, these medications carry substantially lower risk to induce autoimmunity compared to hydralazine or procainamide[3].
Neutrophil extracellular traps (NETs) are web-like structures comprised of nuclear DNA and cytosolic proteins that are extruded from neutrophils in response to specific sterile and non-sterile stimuli[4]. Enhanced NET formation and impaired NET degradation have been implicated in the pathogenesis of SLE and AAV[5]. Specific contents of NETs include double-stranded DNA, histones, myeloperoxidase (MPO), and proteinase-3, which are the major antigenic targets of autoantibodies detected in AAV, SLE, and DIA. Certain medications linked to DIA are known to trigger NET formation. Propylthiouracil, a thyroid medication associated with drug-induced AAV, can induce NET formation and impair NET degradation[6]. Levamisole, an antihelmitic drug, can induce autoimmunity in people inadvertently exposed to the drug via contaminated cocaine. Levamisole can trigger NET formation via stimulation of muscarinic receptors on the surface of neutrophils, and the resultant NETs are toxic to vasculature[7].
The objective of this study was to profile the effect of procainamide, hydralazine, minocycline, and clozapine on NET formation, NET degradation, and NET protein content. The study rationale was to demonstrate that certain medications causally linked to DIA enhance NET formation, providing additional evidence that neutrophils and NETs are involved in the pathogenesis of specific drug-induced and idiopathic autoimmune diseases.
MATERIALS AND METHODS
Human subjects
Healthy volunteers were recruited from the National Institutes of Health (NIH) Blood Bank. Patients with a diagnosis of AAV or SLE were recruited from disease specialty clinics within the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). Patients with AAV or SLE fulfilled respective classification criteria and were studied during a period of clinical remission[8, 9]. All patients provided written informed consent, and samples were collected according to human subject protocols approved by the NIAMS Investigational Review Board.
Neutrophil isolation and stimulation
Neutrophils were isolated from heparinized blood over a density gradient as previously described[10]. Red blood cells were lysed with hypertonic solution, and neutrophils were resuspended in neutrophil medium (RPMI 1640, Life Technologies). Isolated neutrophils (1 × 106 cells/mL) were stimulated with 50nM PMA and with drugs across a range of concentrations including physiologic concentrations when administered to humans: hydralazine (10μM), procainamide (40μM), clozapine (0.8μM), or minocycline (8.5μM) [all reagents from Sigma-Aldrich], and subsequently incubated in poly-L-lysine coated coverslips for four hours at 37°C. Pathways of NET formation were studied by pre-treating neutrophils with 10μM diphenyleneiodonium chloride (DPI; NADPH-oxidase and mitochondrial ROS inhibitor) for 10 min, with 200μM Cl-amidine (peptidylarginine deiminase (PAD) inhibitor) for 15 min, or with 100μM TMB-8 (intracellular calcium antagonist) for 15 min. To assess cholinergic receptor involvement, neutrophils were pre-treated with a generic muscarinic receptor antagonist (100μM atropine) or with antagonists to specific muscarinic subtype receptors: 100nM Telenzepine (muscarinic type 1 (M1) receptor antagonist), 2.5μM Methoctramine (M2 receptor antagonist), or 12.5μM 4-DAMP (M3 receptor angatonist) for 10 min. Telenzepine and 4-DAMP were obtained from Tocris. Atropine and methoctramine were obtained from Sigma-Aldrich.
NET degradation assay
Neutrophils from healthy donors were resuspended in 1 × 106 cells/mL, treated with 2.5μM calcium ionophore (A23187) or 500nM PMA, procainamide, hydralazine, minocycline, or clozapine, and seeded in coverslips for 4 hours at 37°C. Since positively charged residues on hydralazine or procainamide could potentially bind to DNA and prevent NET degradation, an additional dose of the medications was added after 4 hours, to test whether direct interaction between drug-induced NETs and the drugs themselves also contributed to the rate of NET degradation. Drug-induced NETs were exposed to 1% serum from healthy donors and incubated for 16 hours at 37°C. After the incubation, cells were fixed with 4% paraformaldehyde and the degree of NETs was quantified by fluorescence microscopy as below.
Visualization and quantification of NETs by fluorescence microscopy
Neutrophils, seeded in coverslips, were fixed with 4% paraformaldehyde. Cells were stained with antibodies directed against MPO (1:1000, Dako), DNA (1:1000 Hoechst 33342, Life Technologies), as well as secondary antibody Alexa Fluor 555 donkey anti-rabbit (1:400, Life Technologies). After mounting (Prolong, Life Technologies), cells were visualized using a Leica DMI 4000B fluorescent microscope. Images were loaded onto Adobe Photoshop (Adobe Systems) and NET formation was quantified at a 40× magnification by counting the percentage of neutrophils undergoing NET formation averaged over 4 fields of view. NETs were identified as extracellular structures positive for both MPO and DNA.
Calcium release and influx measurements using Indo-1-AM
Neutrophils isolated from healthy volunteers were resuspended in a Ringer’s solution and loaded with Indo-1 (Life Technologies) by incubation with 2.5μM Indo-1-AM for 30min. The cells were washed and resuspended in RPMI and 10% FBS. Immediately prior to the experiment, the cells were resuspended in warm calcium free media containing (mM) 140 NaCl, 5 KCl, 1 MgCl2, 10 Hepes (pH7.4 with NaOH), 10 glucose, 0.2 EGTA. Indo-1 fluorescence was measured using the BD Biosciences LSR Fortessa. Calcium levels were quantified as the ratio of bound violet (BUV 395) versus free blue (DAPI). Baseline calcium levels were measured for 1.5 minutes. Cells were stimulated with: 10μM hydralazine or 40μM procainamide to test for calcium release from intracellular stores. The SERCa (sarco/endoplasmic reticulum Ca2+-ATPase) inhibitor thapsigargin (1 μM) was used as a positive control to induce calcium release. Data were collected for 1–5 minutes after addition of each compound. The resulting data were analyzed using FlowJo version 9.9.3. At least four independent experiments were performed per condition.
TLR ligand screening
Activation of TLRs can trigger NET formation. Because the chemical structures of hydralazine and procainamide resemble nucleotides, TLR ligand screening (InvivoGen) was performed to assess whether hydralazine or procainamide activate TLR pathways, as described (http://www.invivogen.com/custom-tlr-screening). Screening was performed using a panel of HEK293-TLR-Blue clones engineered to express only a single specific TLR and a SEAP-reporter plasmid activated with NF-κB. Cells incubated with TLR specific ligands were used as a positive control. Cell activation was evaluated as an increase in SEAP activity measured as absorbance at OD650 nm, using Quanti-Blue reagent according to the manufacturer’s protocol.
Proteomic analysis of NETs
To determine if specific drugs can affect the contents of NETs, neutrophils isolated from 5 healthy donors were stimulated with procainamide, hydralazine, or 5μg/mL of lipopolysaccharide (LPS), and the protein content of resultant NETs was compared across the stimuli. Since most healthy people do not develop DIA when exposed to hydralazine or procainamide, genetic susceptibility factors may influence the contents of drug-induced NETs. To model potential additional susceptibility factors, neutrophils prone to NETosis were isolated from 4 patients with SLE and 4 patients with AAV during clinical remission and stimulated with procainamide, hydralazine, or allowed to generate spontaneously in absence of additional stimuli. NET lysates were isolated as previously described[11]. For each stimulation experiment, a standardized concentration of NET proteins was sent for proteomic analysis to the Protein Characterization Laboratory (Frederick National Laboratory for Cancer Research, Frederick, MD).
Statistical analyses
All statistical analyses were performed using GraphPad Prism version 6. Differences in distributions were compared using the Kruskal-Wallis test with post-hoc Dunn’s test for group comparisons or the Mann-Whitney U test for pairwise comparisons. For the proteomics data, spectral counts were considered equivalent to protein counts and were compared across the groups using one-way ANOVA and p values were adjusted using Bonferroni’s correction for multiple comparisons. A p value <0.05 defined statistical significance for all experiments except the proteomic analyses where a p value of <0.002 defined statistical significance.
RESULTS
Procainamide and hydralazine trigger NET formation
Neutrophils were treated with procainamide, hydralazine, clozapine, and minocycline. After four hours of stimulation, procainamide and hydralazine induced significant NET formation when compared to unstimulated conditions, whereas clozapine and minocycline did not (Figures 1A and 1B). To address whether drugs that induce NETs also impair NET degradation, drug-induced NETs were exposed in vitro to 1% serum from healthy volunteers for 16 hours. In each experiment, sera significantly degraded NETs with no differences observed when comparing induction with the ionophore A23187 (known to induce NET formation)[12], hydralazine, or procainamide (Figure 1C). Stimulation with minocycline, clozapine, or adding an additional dose of the drugs after the 4-hour stimulation did not result in protection of NETs from subsequent degradation (data not shown). These results indicate that procainamide and hydralazine promote NET formation without interfering with NET degradation.
Figure 1.
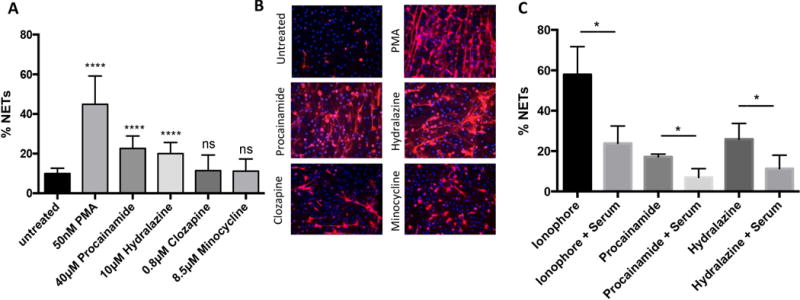
Neutrophils treated with procainamide (Pro) and hydralazine (Hyd) undergo neutrophil extracellular trap (NET) formation. A, Quantification of NET formation after treatment with drugs. B, Fluorescence microscopy of NETs induced by the drugs after 4 hour incubation. C, Quantification of NET degradation after exposure with 1% serum from healthy donors. NETs were identified as extracellular structures positive for both MPO (red) and DNA (blue). Results in A and B are representative of 6 independent experiments. Results in C are representative of 5 independent experiments. **** = P < 0.0001 by Kruskal-Wallis test with Dunn’s post hoc test or Mann-Whitney U test. DPI = diphenyleneiodonium chloride; IO = calcium ionophore (A23187); Cl-am = Cl-amidine.
Procainamide and hydralazine induce NET formation in a ROS and PAD4 dependent manner
To investigate whether NETs induced by procainamide or hydralazine were dependent on formation of ROS and PAD4 activation, neutrophils were pre-treated with DPI and the pan-PAD inhibitor Cl-amidine, respectively. NET formation induced by procainamide or hydralazine was significantly inhibited in the presence of DPI and Cl-amidine (Figure 2A), implicating the production of ROS and activation of PAD4 in drug-induced NETosis.
Figure 2.
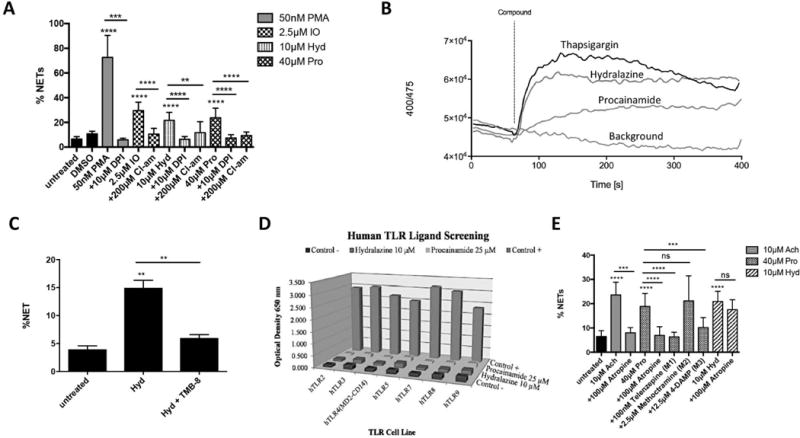
Mechanisms of NET formation. A, Quantification of NET formation after exposing drug-induced NETs to diphenyleneiodonium chloride (DPI) or to Cl-amidine (peptidylarginine deiminase inhibitor). B, Measurement of calcium flux using Indo-1-AM. Neutrophils from donors were treated in calcium-free media with drug compound [thapsigargin 0.5μM, hydralazine 10μM, or procainamide 40μM]. Calcium levels were quantified as the ratio of bound violet (BUV 395) versus free blue (DAPI) and compared to background. C, Quantification of NET formation after exposing hydralazine-induced NETs to TMB-8, an intracellular calcium antagonist. D, Human Toll-like receptor (TLR) ligand screening to assess activation of TLR pathways by procainamide or hydralazine. E, Quantification of NET formation after exposing drug-induced NETs to a muscarinic receptor antagonist (atropine) or to specific muscarinic subtype receptors antagonists (Telenzepine (muscarinic type 1 (M1) receptor antagonist), Methoctramine (M2 receptor antagonist), or 4-DAMP (M3 receptor antagonist)). ACH= acetylcholine. Results in A represent 5 independent experiments. Results in B are the average of 4 independent experiments with 3–7 replicates per experiment. Results in C and D represent 5 independent experiments. ** = P < 0.01; *** = P < 0.001 **** = P < 0.0001 by Kruskal-Wallis test with Dunn’s post hoc test or Mann-Whitney U test.
Activation of PAD4 is a calcium-dependent process, and hydralazine can modulate intracellular calcium levels in cardiac myocytes[13]. We therefore investigated whether stimulation with hydralazine or procainamide causes neutrophils to release calcium from intracellular stores. In calcium-free media, intracellular calcium levels rapidly and transiently increased upon exposure of neutrophils to hydralazine or thapsigargin (p<0.05 for max calcium peak compared to background). Exposure of neutrophils to procainamide resulted in a slower nonspecific rise in intracellular calcium that did not significantly differ from background (Figure 2B). Inhibition of intracellular calcium release with TMB-8 significantly abrogated hydralazine-induced NETosis (Figure 2C). These results demonstrate that hydralazine induces calcium release, providing a mechanism for hydralazine-induced PAD4 activation.
Since NET formation can be initiated by stimulation of different membrane and intracellular neutrophil receptors, we determined whether procainamide or hydralazine could induce TLR activation. Screening analysis showed that procainamide and hydralazine did not stimulate any human TLRs (Figure 2D). Levamisole, a known cause of DIA, can trigger NET formation via engagement of muscarinic receptors on the surface of neutrophils[7]. NETs induced by procainamide, but not hydralazine, were inhibited by atropine (non-specific muscarinic receptor antagonist) and by M1/M3 muscarinic subtype receptor antagonists (Figure 2E). These findings indicate that procainamide engages neutrophil muscarinic receptors to trigger NET formation.
Procainamide and hydralazine induce distinct protein cargo in the NETs
Since hydralazine and procainamide induced NETs via different mechanisms, we assessed whether specific drug stimuli could influence NET contents. The protein content of NETs induced by procainamide or hydralazine induced was studied using neutrophils from healthy donors (Figure 3A). There were no significant differences in levels of expression on NETs among neutrophil proteins originating from the nuclear, cytosolic, and cytoskeletal compartments. Amongst granular proteins, NETs induced by hydralazine contained significantly more azurocidin and less lactoferrin compared to NETs induced by procainamide, but there was considerable variability amongst different samples.
Figure 3.
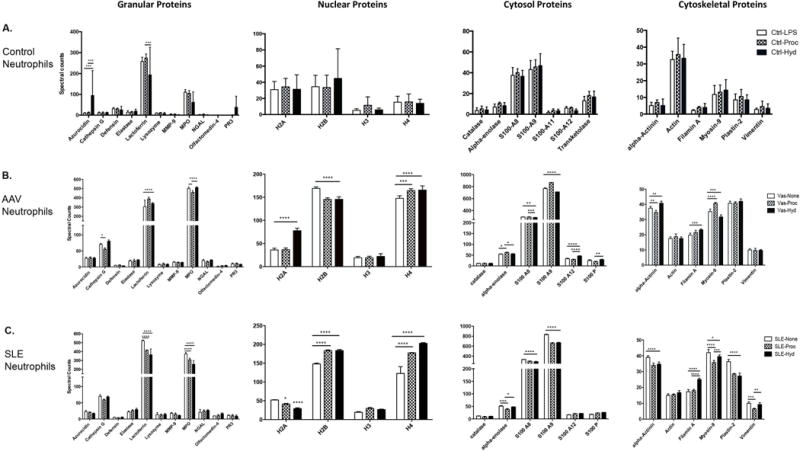
Procainamide and hydralazine induce NETs comprised of distinct proteins. A–C, Differences in NET protein cargo (granular, nuclear, cytosolic and cytoskeletal) in neutrophils from healthy controls (n=5), patients with ANCA-associated vasculitis (AAV) (n=4), and patients with systemic lupus erythematosus (SLE) (n=4) exposed to lipopolysaccharide (LPS), procainamide (Proc), Hydralazine (Hyd), or no additional stimuli (none). * = P < 0.05; ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001 by two-way analysis of variance test. A p<0.002 defines statistical significance per Bonferroni correction for multiple comparisons.
Greater differences in protein content of NET cargo were observed in neutrophils from patients with autoimmune diseases (Figures 3B and 3C). The most significant differences were seen among nuclear proteins externalized in NETs. Patterns of histone abundance depended upon both stimulus (spontaneous, hydralazine, procainamide) and underlying disease (SLE, AAV). Of the cytosolic proteins, S100 calcium-binding protein A9 (S100A9), S100A8, and S100A12 were all differentially expressed in drug-induced NETs compared to spontaneous NETs. With regards to cytoskeletal proteins externalized in NETs, differential expression of alpha-actinin, filamin A, myosin-9, plastin-2, and vimentin depended again on stimulus and underlying disease. In the granular compartment, significant differences were observed for lactoferrin and MPO. Notably, higher amounts of MPO were observed in spontaneous NETs isolated from patients with AAV or SLE.
When comparing NET contents in patients with AAV versus SLE, there was a significantly greater amount of lactoferrin, H2A, S100A8, and S100A9 in NETs from unstimulated neutrophils in SLE and significantly greater MPO, H2b, H4, and S100A12 in NETs from unstimulated neutrophils in AAV. With respect to histones, stimulation with procainamide or hydralazine resulted in significantly greater amounts of H2b, H3, and H4 in patients with SLE versus AAV. There was significantly more H2A in AAV when neutrophils were stimulated with hydralazine and significantly more H2A in SLE when neutrophils were stimulated with procainamide.
DISCUSSION
NETosis is a distinct form of neutrophil cell death that differs morphologically from apoptosis, necrosis, and other types of cell death. The extent of known stimuli that can trigger NET formation and the subcellular events of NETosis remain to be fully elucidated. The present study expands the list of medications known to induce NETs. Procainamide and hydralazine, two medications definitively implicated in cases of DIA, trigger NET formation, while minocycline and clozapine, two medications less commonly implicated in DIA, do not induce NETs. These observations, in combination with the previously reported associations between propythiouracil, levamisole, and NETosis, strongly suggest that NETosis is involved in the pathogenesis of DIA, possibly through the externalization and modification of intracellular autoantigens.
A few cellular events are considered important in NET formation, namely the production of ROS and histone modifications mediated by PAD enzymes[5]. Generation of superoxide by the NADPH oxidase process is a critical step in NET formation induced by procainamide or hydralazine. PAD4 activation, a calcium-dependent process, mediates citrullination of histones, which promotes chromatin decondensation. Inhibition of PADs abrogates NETs induced by procainamide and hydralazine.
Modulation of calcium flux is a known trigger of NETosis, and both calcium ionophore and thapsigargin can induce NETs[12]. Although the precise mechanism of action remains unknown, hydralazine is thought to function as an antihypertensive medication by altering the calcium balance within vascular smooth muscle cells[13]. Short-term exposure of neutrophils to hydralazine resulted in a transient increase in intracellular calcium levels that mirrored the kinetics of calcium flux induced by thapsigargin, and calcium antagonists inhibited hydralazine-induced NETosis. These findings suggest that hydralazine triggers NET formation in part by triggering calcium release from intracellular stores.
The present study provides additional evidence that cholinergic receptors can affect neutrophil function. We previously demonstrated that neutrophils possess muscarinic receptors that can be activated by acetylcholine or levamisole to induce NET formation and that neutrophils from patients with SLE possess a greater abundance of surface cholinergic receptors[7]. In this study, we demonstrate that procainamide also triggers muscarinic receptors on neutrophils to promote NETosis. Procainamide is an antiarrhythmic agent that blocks sodium channels but can interact with muscarinic receptors[14]. These findings suggest that cholinergic properties of specific medications may contribute to the risk of drug induced-autoimmunity.
The present study demonstrates that protein content within NETs varies by source of stimulation and by source of donor neutrophils. In a previous study using similar methodology, NET protein content differed when neutrophils from healthy donors were stimulated by rheumatoid arthritis-associated autoantibodies versus various cytokines[11]. In this study, there were few differences in NET content when stimulating neutrophils from healthy donors and greater differences in NET proteins when neutrophils from patients with AAV or SLE were tested. Since, most patients who are exposed to medications associated with DIA do not develop overt signs of autoimmunity, additional susceptibility factors are likely involved. Although we did not directly study neutrophils from patients with DIA, a limitation of the current study, we did observe differences in NET content when specific medications were used to stimulate neutrophils prone to NETosis from patients with other autoimmune diseases. External factors (e.g. environmental exposures to certain medications) and intrinsic host factors (e.g. donor source of neutrophils) may both contribute to disease susceptibility in cases of DIA.
Whether protein content of NETs influences patterns of disease-associated autoantibodies or clinical features of DIA warrants future investigation. In addition to differences in protein abundance within NETs, post-translational modification of proteins during NETosis and drug-protein adduct effects may contribute to pathogenesis of DIA and should be explored in future studies. Historically, investigations into the mechanisms of DIA have focused upon the effects of specific medications on the adaptive immune system. Inhibition of DNA methylation in T cells has been proposed as a potential mechanism underlying drug-induced autoimmunity[15]. Whether epigenetic modifications of neutrophils contribute to drug-induced autoimmunity warrants investigation.
The present study demonstrates that neutrophils, in addition to mononuclear cells, may contribute to DIA. Medications commonly implicated in DIA can trigger NET formation through common and specific pathways. Elucidating the mechanisms of DIA may provide key insights into causal factors of autoimmunity.
Acknowledgments
The authors are grateful to Dr. Venkataraman Subramanian, PhD and Dr. Paul Thompson for providing Cl-amidine and to Ann Biehl for calculating the relevant physiologic concentrations of the drugs used in the experiments.
This research was supported through the Intramural Research Program at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS, ZIAAR041199). JAIC is supported by a postbaccalaureate intramural research training award.
Footnotes
Financial supports of conflicts disclosure: The authors report no conflicts of interest with regard to the work.
DR. PETER C GRAYSON (Orcid ID: 0000-0002-8269-9438)
References
- 1.Borchers AT, Keen CL, Gershwin ME. Drug-induced lupus. Annals of the New York Academy of Sciences. 2007;1108:166–82. doi: 10.1196/annals.1422.019. [DOI] [PubMed] [Google Scholar]
- 2.Hess E. Drug-related lupus. The New England journal of medicine. 1988;318(22):1460–2. doi: 10.1056/NEJM198806023182209. [DOI] [PubMed] [Google Scholar]
- 3.Pendergraft WF, 3rd, Niles JL. Trojan horses: drug culprits associated with antineutrophil cytoplasmic autoantibody (ANCA) vasculitis. Current opinion in rheumatology. 2014;26(1):42–9. doi: 10.1097/BOR.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 5.Grayson PC, Kaplan MJ. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. Journal of leukocyte biology. 2016;99(2):253–64. doi: 10.1189/jlb.5BT0615-247R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakazawa D, Tomaru U, Suzuki A, Masuda S, Hasegawa R, Kobayashi T, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2012;64(11):3779–87. doi: 10.1002/art.34619. [DOI] [PubMed] [Google Scholar]
- 7.Carmona-Rivera C, Purmalek MM, Moore E, Waldman M, Walter PJ, Garraffo HM, et al. A role for muscarinic receptors in neutrophil extracellular trap formation and levamisole-induced autoimmunity. JCI Insight. 2017;2(3):e89780. doi: 10.1172/jci.insight.89780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis and rheumatism. 1990;33(8):1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 9.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis and rheumatism. 1997;40(9):1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 10.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. Journal of immunology. 2010;184(6):3284–97. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Science translational medicine. 2013;5(178):178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS letters. 2010;584(14):3193–7. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Kao YH, Cheng CC, Chen YC, Chung CC, Lee TI, Chen SA, et al. Hydralazine-induced promoter demethylation enhances sarcoplasmic reticulum Ca2+ -ATPase and calcium homeostasis in cardiac myocytes. Lab Invest. 2011;91(9):1291–7. doi: 10.1038/labinvest.2011.92. [DOI] [PubMed] [Google Scholar]
- 14.Fields JZ, Roeske WR, Morkin E, Yamamura HI. Cardiac muscarinic cholinergic receptors. Biochemical identification and characterization. The Journal of biological chemistry. 1978;253(9):3251–8. [PubMed] [Google Scholar]
- 15.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. Journal of immunology. 1988;140(7):2197–200. [PubMed] [Google Scholar]