

Abstract
Background
Previous ex vivo studies using truncated VWF (Von Willebrand Factor) suggest that domain-level molecular architecture may control platelet-GpIbα binding function.
Objective
We determined if this is the case with multimeric VWF in vivo.
Methods
Full-length human VWF (‘hV’) was modified with a 22-amino acid mucinous stretch at either the N-terminus of VWF-A1 to create ‘hNV’ or C-terminus to yield ‘hCV’. This extends the physical distance between VWF-A1 and the adjacent domains by ~6nm. Similar mucin inserts were also introduced into a human-murine chimera (‘h[mA1]V’) where murine-A1 replaced human-A1 in hV. This yielded ‘h[mA1]NV’ and ‘h[mA1]CV’, with N- and C-terminal inserts. The constructs were tested ex vivo and in vivo.
Results
Mucin insertion at the N-terminus, but not C-terminus, in both types of constructs resulted in >50-fold increase in binding to immobilized GpIbα. N-terminal insertion also resulted in greater shear-induced platelet activation, more thrombus formation on collagen, enhanced platelet accumulation and slower platelet translocation on immobilized VWF in microfluidics assays. Hydrodynamic injection based expression of h[mA1]NV, but not h[mA1]V or h[mA1]CV, in VWF−/− mice caused profound thrombocytopenia, reduced VWF plasma concentrations, lower multimer distribution, and incessant tail bleeding that is reminiscent of VWD Type-2B. Platelet plugs were noted in the portal veins and hepatic arteries. An anti-D’D3 mAb DD3.3 that displays enhanced binding to VWF containing the N-terminal mucin insert also exhibited increased binding to wild-type VWF under shear and upon ristocetin addition.
Conclusion
Conformation changes at the VWF D’D3-A1 interface may be a key regulator of thrombosis in vivo.
Keywords: blood platelets, thrombosis, von Willebrand Factor, hemodynamics, microfluidics
INTRODUCTION
Von Willebrand factor (VWF) is a large, multimeric plasma glycoprotein that is secreted into circulation from endothelial cells, platelets and megakaryocytes [1, 2]. It contains a globular, compact head-structure with the D’D3-A1-A2-A3-D4 domains, followed by an extended arm that contains six C-type domains and the cysteine knot [3, 4].
The A1-domain of VWF regulates thrombosis and hemostasis in part by binding the platelet GpIbα under fluid shear [5, 6]. A number of ex vivo studies using truncated, VWF mutants show that the structural domains adjacent to A1, particularly the D’D3 and A2-domains, regulate the rates of platelet binding [7-10]. Such domain-level interactions may be important since the VWF-A1 crystal structure itself is not very different (root mean square deviation<2Å), between wild-type A1 and VWD mutants that display altered A1-GpIbα binding affinity [11]. The most profound change noted here relates to the position of the VWF-A1 N-(amino acids/aa 1261-1271) and C-terminal (aa 1459-1468) peptides [12, 13]. These peptides are proximal to the β-finger binding region in the unliganded A1-domain but are displaced in the VWF-A1 co-crystal with GpIbα [13].
Consistent with the notion that structural features N-terminus to VWF-A1 regulate GpIbα binding, a dimeric protein containing A1-CK binds GpIbα better than another dimeric protein containing D’D3 [9]. This implies that the D’D3-domain may shield A1-GpIbα binding. Additionally, the binding of the VWF propeptide to VWF-D’D3 hinders functional A1-GpIbα interactions [14], with D’D3-domain deletion enhancing this molecular binding [8]. Deletion of the ‘N-terminal flanking peptide’ of VWF-A1 (amino acid/aa 1238-1260) also enhances platelet binding, particularly in assays where VWF is immobilized on plastic or collagen [8, 15]. Modifying the O-glycans also reduces platelet adhesion [16]. Besides, features at the N-terminal, the A2-domain located at the C-terminus has also been implicated to bind VWF-A1 in surface plasmon resonance studies [10], and molecular dynamics simulations suggest that VWF-A2 may reduce GpIbα-A1 binding [17]. The above studies have some limitations: i. They rely on VWF deletion mutations, rather than the full multimeric protein. ii. They focus individually on structural features at either the N- or C-terminal alone, and thus a systematic side-by-side comparison of the relative impact of the D’D3 and A2-domains in regulating A1-GpIbα binding remains unknown. iii. They rely on in vitro measurements, and thus the role of domain-level interactions in controlling bleeding and thrombosis in the more complex in vivo physiological milieu remains undetermined.
To address these shortcomings, the current study ‘minimally’ perturbed human VWF by introducing a short 22 amino acid peptide from human CD43 as a physical spacer, at either the N- or C-terminus of VWF-A1 in multimeric VWF. This insert has 10 putative O-glycosylation sites and thus it is expected to act as a stiff spacer that extends the physical distance between VWF-A1, and either D’D3- or A2-domains by up to ~6nm (3Å/residue) [18, 19]. In vitro and in vivo functional studies with these constructs suggest that structural features N-terminus to the A1-domain, but not the C-terminus, is the major feature regulating platelet adhesion and thrombosis. Hydrodynamic injection of the VWF construct with the N-terminal spacer (between D’D3 and A1) into mice resulted in profound thrombocytopenia and bleeding diathesis that resembles VWD Type 2B. A screen of several anti-D’D3 mAbs identified a clone DD3.3, that displays higher binding to VWF protein with N-terminal spacer. The binding of mAb DD3.3 to VWF was augmented by hydrodynamic shear and ristocetin, suggesting that structural changes at the D’D3-A1 interface may be physiologically relevant and may contribute to ‘VWF activation’.
MATERIAL AND METHODS
Materials
Function-blocking mouse anti-human IgGs used include anti-VWF-A1 mAb AVW-3 (GTI Diagnostics) and anti-GpIbα clone AK2 (Millipore) which inhibit A1-GpIbα binding; and anti-VWF-D’D3 mAb DD3.1 which blocks both VWF-propeptide binding to mature VWF and A1-GpIbα interactions [14]. Additional anti-D’D3 mAbs were produced by immunizing BALB/c mice with human-D’D3 [14].
To create recombinant VWF, wild-type human VWF (‘hV’) in pCDNA3.1 vector was used as starting material [4]. Complementary oligos encoding for 22 amino acids (ESTSEPLSSKMYTTSITSDPKI/V) of the mucin-rich repeat of CD43 were annealed and inserted either just after aa1263 in VWF-A1 in ‘hV’ (after the ‘N-terminal flanking peptide’) or following aa1476 [4]. The resulting proteins with N- and C-terminal inserts are called ‘hNV’ and ‘hCV’, respectively. Additionally, for some studies, the CD43-insert was also placed following aa1238 (before the ‘N-terminal flanking peptide’) to yield hNV[1238], and aa1458 to obtain hCV[1458]. A human-murine chimeric VWF where the murine-A1 domain replaced human VWF-A1 (‘h[mA1]V’) was also cloned into the pLive vector (Mirus-Bio). Insertion of the CD43 mucin insert at the N- (after aa1263) and C-terminus (after aa1476) of [mA1] in this vector yielded ‘h[mA1]NV’ and ‘h[mA1]CV’. All proteins were expressed by transient transfection of HEK-293T cells over-expressing furin in serum-free media [20], and concentrated 50-100 fold to 200-400μg/mL for functional studies.
Human subjects protocols were approved by the UB Health Science Institutional Review Board, and animal protocols were approved by the UB Institutional Animal Care and Use Committee.
Concentration determination and western blotting
VWF concentration was determined using a flow cytometer-bead sandwich assay, using multimeric VWF purified from humate-P (CSL Behring) as a standard [4]. Human-murine VWF chimera levels in mouse plasma was determined using calibration curves generated by spiking VWF−/− mouse plasma with human plasma pooled from 5 normal, adult volunteers.
VWF multimer distribution was assayed using 0.6-1.2% agarose gel electrophoresis [21]. To assay VWF susceptibility to proteolysis by ADAMTS13, 10μg/mL VWF was incubated with 10U/mL ADAMTS13 (WHO standard) and 1.6M urea at 37°C for 24h. Western blotting of cleaved VWF products was assayed under standard reducing conditions using 4%-8% SDS-PAGE followed by detection using anti-VWF Ab (Dako).
Functional assays
Washed platelets were purified as described earlier [14, 20]. In vitro functional assays measured: i. The apparent dissociation constant (KD) of VWF-GpIbα binding using ELISA [8]. Here, KD measures the avidity of multimeric VWF binding to GpIbα-Fc immobilized at ~100 sites/μm2 ; ii. Shear-induced platelet activation (SIPAct) using a cone-plate viscometer to apply shear and Annexin-V to quantify cell activation [20]; iii. Platelet accumulation (bound cells/mm2) and cell translocation velocity (μm/s) on substrates bearing VWF using a microfluidic flow cell [8, 22]; and iv. Thrombus formation (% surface area coverage and fluorescence intensity) on 20μg/mL equine collagen Type-I using a microfluidics assay [8].
Conformation sensitive anti-D’D3 mAbs
ELISA measured the binding of a panel of anti-D’D3 mAbs to substrates immobilized with 10μg/mL human VWF constructs: hV, hNV or hCV [8]. mAb (DD3.3) binding hNV at higher levels compared to hV and hCV, were further tested under physiological shear using a cone-plate viscometer. Here, 10μg/mL hV (recombinant wild-type VWF) was captured onto 3μm polystyrene beads bearing anti-VWF Ab (Dako) over 30min at room temperature, blocked with PBS containing 1%BSA and washed to remove unbound VWF. The beads were then subjected to fluid shear at 0-9600 s−1 for 5min, in the presence of 10μg/mL anti-VWF FITC (Dako) to measure total bound VWF and 20 μg/mL DD3.3-Alexa 647 to detect protein confirmation change. The fluorescence intensity of anti-D’D3 mAb binding was normalized to anti-VWF FITC signal. In independent runs, the anti-D’D3 mAb DD3.5 replaced DD3.3 Alexa 647. Here, bead-bound DD3.5 was quantified after shear stoppage, using a secondary anti-mouse Alexa 647 Ab.
Mouse studies
50μg of VWF cDNA in pLive vector were hydrodynamically injected into tail vein of 8-12 week old VWF−/− mice of either sex using a 26½ gauge needle. Injection volume was 0.1mL DNA in sterile PBS/g mouse and injection time was ~5s [23, 24]. Blood was collected at various times thereafter using retro-orbital bleeds for measurement of: i. complete blood count, ii. plasma VWF levels using the cytometer-bead assay, iii. bilirubin, based on plasma 450nm absorbance with respect to standard curve generated by diluting bilirubin (Sigma) into VWF−/− plasma, and/or iv. flow cytometry based platelet counts. All mouse tail bleed assays were performed at 48h. Here, mice anesthetized using isoflurane were placed in a restraining device. 4mm of the distal tail was cut using a sharp scalpel, and the amputated tail was quickly immersed in a pre-weighted tube containing physiological saline at 37°C. Bleeding time was measured from the moment of transection until first arrest of bleeding. Observations were stopped at 30min. when bleeding did not cease. The bleeding volume was obtained from the change in weight of the pre-weighted tube.
Standard paraffin-embedded H&E, trichrome staining and immunohistochemistry were performed. In some runs, to stain for VWF expression and platelet thrombi, the rodent was perfusion-fixed at 48h [25]. These sections were stained with 1:100 rhodamine-conjugated wheat germ agglutinin for cell membrane, 1:1000 DyLight-488 conjugated anti-platelet Ab (#X488, Emfret Analytics), and 1:100 anti-VWF Ab (Dako) followed by secondary goat anti-rabbit Alexa 647 Ab to detect chimeric-VWF. Images were acquired using Zeiss LSM510 meta NLO scanning confocal microscope (40X/0.55 N.A objective).
Statistics
All data are presented as mean + SD for >3 independent experiments. Student’s two-tailed t-test was performed for dual comparison. One-way analysis of variance (ANOVA) followed by the Tukey post-test was performed for multiple comparisons. P<0.05 was considered significant.
RESULTS
Mucin insert at the N-, but not C-, terminus of VWF-A1 augments binding to GpIbα
The 22aa CD43 mucin insert was placed at the N-terminus of the A1-domain to create hNV, or C-terminus to create hCV (Fig. 1A). When resolved using SDS-agarose gel, hV and hCV had a comparable multimer distribution with hNV tending to produce smaller units (Fig. 1B). Under reducing conditions, the molecular mass of hNV and hCV monomers was ~12kDa greater than that of hV, consistent with the notion that the mucin insert is extensively glycosylated (Fig. 1C). Here, the 22aa acids themselves contribute 2kDa to the mass, with each additional O-glycan adding ~1kDa, as these are commonly either disialylated T-antigen (Neu5Acα2-3Galβ1-3[Neu5Acα2-6]GalNAcα) or core-2 (Neu5Acα2-3Galβ1-3[Neu5Acα2-3Galβ1-4GlcNAcβ1-6]GalNAcα) structures. Increased glycosylation in mucin-modified VWF (hNV and hCV) was also evident in a peanut agglutinin binding assay (Supplemental Fig. S1). Incubation of the constructs with ADAMTS13 for 24h in the presence of urea revealed that hCV was somewhat less susceptible to proteolysis compared to hNV and hV. Thus, the C-terminal mucin may partially inhibit A2-domain cleavage (Fig. 1D).
Figure 1. Human VWF variant expression and function.
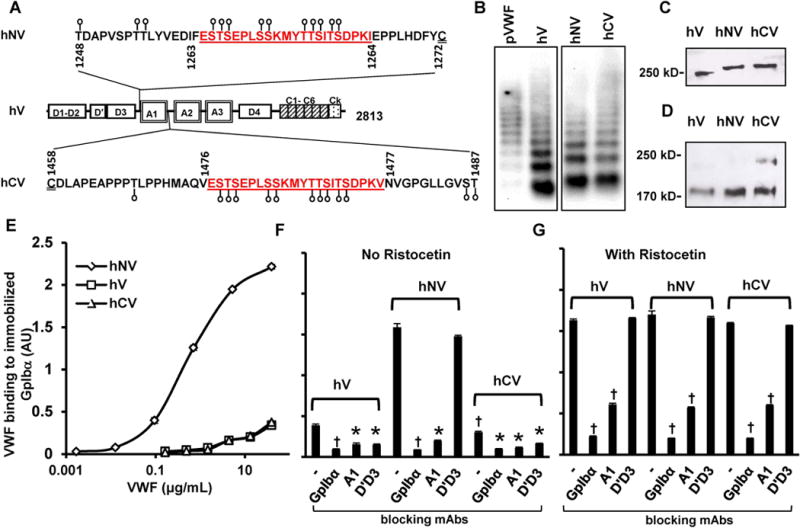
A. The A1-domain of wild-type human VWF (‘hV’) is flanked by a few potential O-linked glycosylation sites at both the N- (aa 1248-1271) and C-terminus (1459-1487). These O-glycans are depicted by lollipop symbols. A 22 amino acid mucin-repeat motif from human CD43 (underlined) replaced Ser1263 to generate the VWF-variant ‘hNV’. Insertion of the same 22 aa sequence at the C-terminus in place of Thr1477 resulted in ‘hCV’. B. Agarose gel electrophoresis-western blotting of non-reduced VWF variants expressed in HEK293T cells. pVWF is human plasma VWF from humate-P. C. 8% SDS-PAGE analysis of VWF variants showing the higher molecular mass of hNV and hCV. D. hCV cleavage by ADAMTS13 is partly reduced compared to other proteins after 24h digestion. E. Binding of different concentrations of VWF variants to 4 μg/mL immobilized GpIbα-Fc in microtiter plates. hNV binding is remarkably higher. F-G. Binding of VWF variants to immobilized GpIbα-Fc in the absence (F) or presence of ristocetin (G). 20 μg/mL of indicated blocking mAbs was added in some cases: anti-GpIbα mAb AK2; anti-VWF A1-domain clone AVW-3; and anti-VWF D’D3 mAb DD3.1. All ELISA data are from 2-4 independent runs, each with 2-3 repeats. Error bars are too small to be visible in some cases. †P<0.05 with respect to all other treatments. *P<0.05 with respect to all other treatments except that treatments indicated by * are not different from each other.
Static microtiter plate studies measured soluble VWF binding to immobilized GpIbα-Fc. Here, hNV displayed higher binding for immobilized GpIbα (KD,app~0.37μg/mL) compared to hV and hCV (Fig. 1E). hV and hCV binding affinity did not reach saturation up to 40μg/mL. Thus, increasing the physical spacing between the D’D3 and A1-domains dramatically decreased the apparent dissociation constant (KD) by >100-fold. The binding of all VWF variants to immobilized GpIbα was specific, and could be blocked by mAbs against either GpIbα (AK2) or VWF A1-domain (AVW-3) (Fig. 1F). The anti-D’D3 mAb DD3.1 blocked hV and hCV binding to GpIbα, but had no effect on hNV-GpIbα binding. Increasing the spacing between D’D3 and A1 may prevent steric mAb blocking observed with wild-type VWF. Mucin insertion between VWF-A1 and -A2 in hCV had no effect on DD3.1 inhibition. Upon ristocetin addition, all constructs bound GpIbα equally well (Fig. 1G). While, blocking mAbs against GpIbα and VWF-A1 were effective, the anti-D’D3 mAb DD3.1 was now ineffective. Overall, mucin insertion between D’D3 and A1 released steric blocking by VWF-D’D3, thus promoting A1-GpIbα binding.
N-terminal mucin insert enhances platelet activation
We determined if N-terminal mucin insertion enhances platelet activation in the SIPAct assay by promoting VWF-GpIbα binding. Here, washed human platelets were shear mixed with the different recombinant VWF-variants at 9600/s in a cone-plate viscometer. Annexin-V binding to platelets was used as a marker of cell activation (Fig. 2). Here, hNV triggered more rapid and higher platelet activation compared to hV or hCV (Fig. 2A), with ~80% platelet activation being observed within 60s. Whereas DD3.1 inhibited SIPAct triggered by hV or hCV by ~70% (Fig. 2B, 2D), this mAb did not reduce platelet activation triggered by hNV (Fig. 2C). Blocking mAbs against GpIbα and VWF-A1 abolished SIPAct in all cases (Fig. 2E). hNV increased SIPAct without altering the critical shear threshold required for platelet activation (Supplemental Fig. S2). This is consistent with the notion that SIPAct is a two-step process, with VWF-GpIbα binding being separable from subsequent mechanotransduction [21]. Overall, the physical spacing between D’D3 and A1 is a critical determinant for solution based platelet activation.
Figure 2. Shear induced platelet activation (SIPAct).
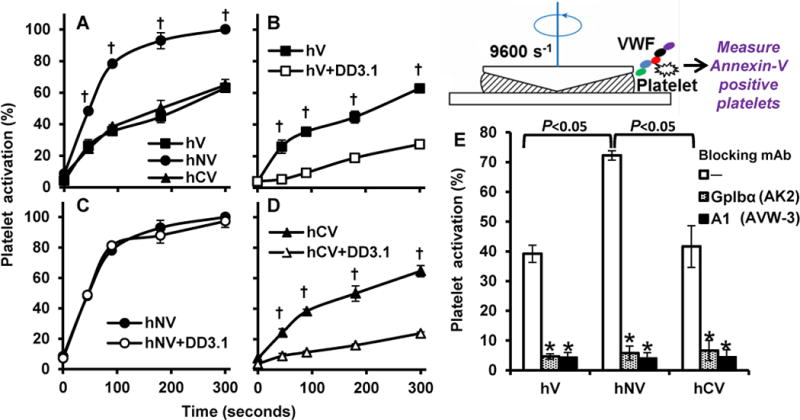
SIPAct studies were performed by shearing 107/mL CD31-labeled washed platelets with 20μg/mL recombinant VWF variants at 9600 s−1 (~100 dyn/cm2 shear stress) in a cone-plate viscometer. Blocking mAbs against GpIbα (AK-2), VWF-A1 (AVW-3) and VWF-D′D3 (DD3.1) was added in some cases for 10 min prior to shear application. A. Platelet activation induced by human VWF variants was measured based on % platelets binding greater than basal levels of Annexin-V. B-D. SIPAct in the presence or absence of DD3.1 for hV (B), hNV (C) and hCV (D). E. SIPAct at 3 min in the presence of various blocking mAbs. Data are mean + SD from 4 independent experiments. Statistical symbols used are same as in Fig. 1. hNV induced greater platelet activation compared to other proteins.
VWF-D’D3 regulates thrombus formation and platelet translocation
Thrombus formation on fibrillar collagen was measured using microfluidic flow chambers in order to determine the effects of VWF domains in regulating platelet adhesion in models of vascular injury (Fig. 3A, 3B). Here, fluorescent human washed platelets supplemented with erythrocytes at physiological hematocrit and 10μg/mL recombinant human VWF-variants were perfused over collagen at 1000/s (~40dyn/cm2). Here, compared to hV (wild-type) and hCV, hNV contributed to more extensive platelet thrombosis (Fig. 3A) and larger contiguous thrombi (Fig. 3B). Platelet deposition was negligible in controls lacking recombinant VWF. All VWF-variants bound collagen equally. Thus, the mutations do not affect VWF-collagen binding (Supplemental Fig. S3).
Figure 3. Thrombus formation and human platelet translocation.
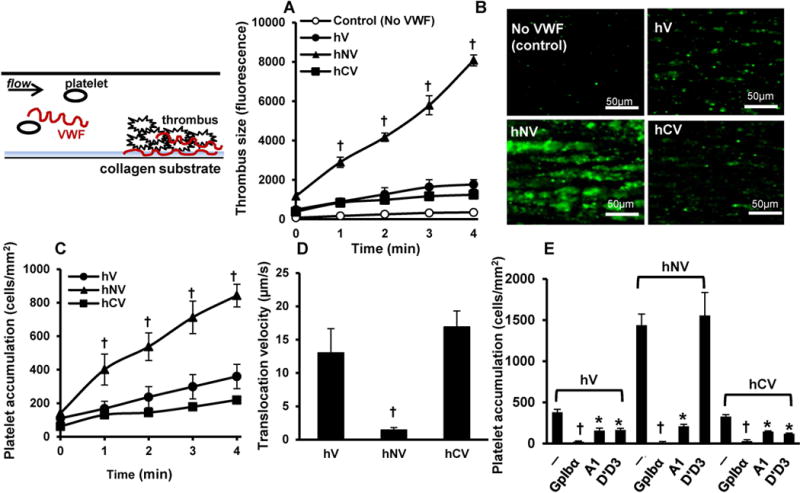
A-B. 108/mL BCECF-labeled fluorescent human platelets were suspended in plasma-free blood supplemented with 10 μg/ml recombinant human VWF variants. The mixture was perfused over Type I collagen substrates for 4-5 min at a wall shear rate of 1000 s−1 (Shear stress ~40 dyn/cm2). A. Thrombus formation quantified based on total fluorescence intensity in the field of view. Control runs were plasma-free without addition of VWF. B. Representative thrombus images at 4 min for various cases. C-E. 108 washed platelets/mL were perfused over substrates bearing recombinant VWF variants for 4 min at a wall shear stress of 3 dyn/cm2. Both platelet accumulation (C) and translocation velocity (D) were recorded. E. In some cases, blocking mAbs were added at 20μg/mL to inhibit platelet binding. Data are mean + SD for 4min time point. Statistical symbols used are same as in Fig. 1. hV displays greater tendency to form thrombus.
The platelet translocation assay measured washed human platelet accumulation (Fig. 3C) and translocation velocity (Fig. 3D) at a wall stress of 3 dyn/cm2, on substrates that had physisorbed hV, hCV or hNV. Equivalent amounts of VWF were immobilized in all cases. Here, significantly higher platelet accumulation (Fig. 3C) and lower translocation velocity (Fig. 3D) was observed on hNV compared to other constructs. Platelet adhesion was specifically blocked by mAbs against GpIbα and VWF-A1, with anti-D’D3 mAb DD3.1 only reducing adhesion on hV and hCV (Fig. 3E).
Similar observations as with hNV and hCV were made using constructs where the mucin insert was placed at aa1238 at the N-terminus (hNV[1238]) and aa1458 at the C-terminus (hCV[1458]) (Supplemental Fig. S4). Thus, all data support the notion that VWF-D’D3 is the dominant regulator of A1-GpIbα binding in both solution (Fig. 2) and substrate assays (Fig. 3).The A2-domain had a small effect, relative to VWF-D’D3.
N-terminal spacing augments chimeric-VWF GpIbα binding
Since murine GpIbα does not bind human VWF [26], chimeric VWF constructs were created for in vivo studies (Figure 4A). Here, h[mA1]V is identical to human VWF (hV), except that the human-A1 is replaced by murine-A1. Insertion of the CD43 spacer at the N- or C-terminus of mA1-domain resulted in h[mA1]NV and h[mA1]CV.
Figure 4. Human-murine chimeric VWF construct expression and function.
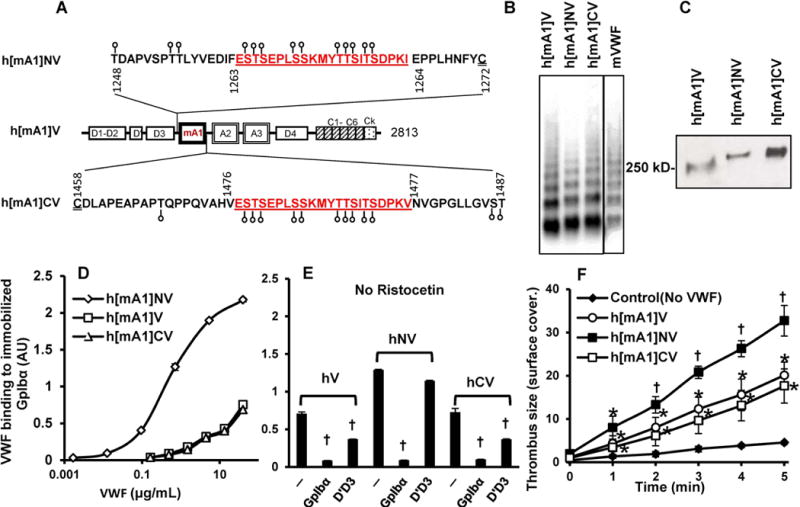
A. The human-murine VWF chimera ‘h[mA1]V’ contains full length human VWF with murine A1-domain replacing human-A1. Insertion of the 22 amino acid mucin sequence from human CD43 at the N-and C-terminus resulted in ‘h[mA1]NV’ and ‘h[mA1]CV’. B. Agarose gel electrophoresis-western blotting of non-reduced chimeric VWF variants and murine VWF (mVWF) expressed in HEK293T cells. C. 8% SDS-PAGE analysis of chimeric VWF variants showing higher molecular mass of h[mA1]NV and h[mA1]CV compared to h[mA1]V. D. Binding of different concentrations of chimeric VWF variants to 4μg/ml immobilized human GpIbα-Fc in microtiter plates. E. Binding of VWF variants to immobilized GpIbα-Fc in the presence or absence of 20 μg/mL blocking mAb. F. Mouse platelet adhesion and thrombus formation on fibrillar collagen in the presence of chimeric VWF. 107 VWF−/− washed mouse platelets/mL supplemented with 10μg/ml VWF chimeric proteins were perfused over collagen substrates at 1000 s−1. Thrombus formation quantified % substrate covered by platelets over time. Control runs did not contain supplemental VWF. All data are mean + SD from 3-4 independent runs. Error bars are too small to be visible in many cases. Statistical symbols are same as in Fig. 1. h[mA1]NV is more active compared to the other proteins. hCV is not different from hV.
h[mA1]V, h[mA1]NV and h[mA1]CV were functionally similar to hV, hNV and hCV, respectively (Fig. 4B-4F). All proteins expressed well in HEK293Ts, with h[mA1]NV displaying slightly smaller multimers (Fig. 4B). A ~12 kDa increase in monomer mass was noted for h[mA1]NV and h[mA1]CV with respect to h[mA1]V (Fig. 4C). In microtiter assays, h[mA1]NV binding to GpIbα exhibited an apparent KD of 0.31μg/mL, similar to hNV (Fig. 4D vs. Fig. 1E). This was higher than the binding affinity of GpIbα for either h[mA1]V or h[mA1]CV. The molecular recognition could be blocked by the anti-GpIbα mAb. Additionally, the anti-D’D3 mAb DD3.1 reduced h[mA1]V and h[mA1]CV, but not h[mA1]NV, binding to GpIbα (Fig. 4E). In the presence of ristocetin, all VWF variants displayed high binding to GpIbα, which could be blocked by anti-GpIbα mAb but not DD3.1 (Supplemental Fig. S5). In the thrombus formation assay also, N-terminal mucin insertion enhanced thrombus formation (Fig. 4F). h[mA1]CV displayed function similar to h[mA1]V. Thus, again, the D’D3-A1 spatial distance is a critical feature regulating VWF-GpIbα binding.
Hydrodynamic injection of h[mA1]NV into mouse causes thrombocytopenia resembling VWD Type 2B
Bleeding and thrombosis is complex in vivo since many different blood proteins interact with VWF and the hemodynamics is also heterogeneous [1]. To determine if domain-level structural features control VWF function in vivo, pLive vectors encoding for different chimeric proteins were hydrodynamically injected into VWF−/− mice (Figure 5). 48h post-injection, profound thrombocytopenia was evident in h[mA1]NV expressing mice with 70-90% reduction in platelet count (Fig. 5A). Average platelet counts in PBS and h[mA1]V injected mice was no different from uninjected VWF−/−. VWF expression in this model lasted 1-2 weeks, with platelet counts being depressed for 7-10 days following h[mA1]NV expression (Supplemental Fig. S6). Mice recovered normally thereafter.
Figure 5. Hydrodynamic injection.
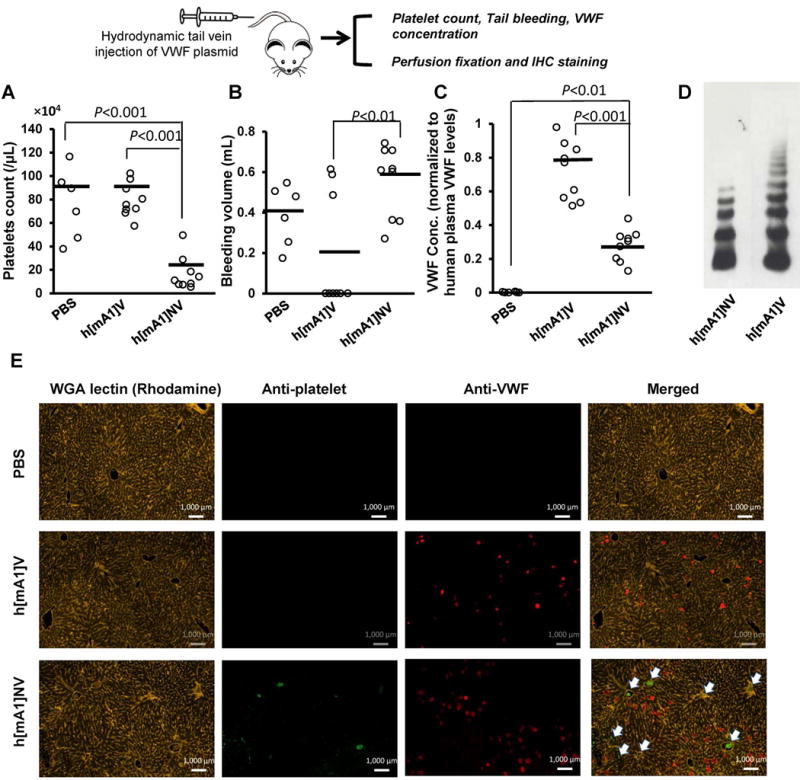
VWF variants were expressed in VWF−/− mice by hydrodynamic injection. Panels present the following data 48h post-injection: A. Platelet counts; B. Bleeding volumes after tail transection over 30 min. C. Chimeric VWF concentrations in mouse plasma measured with respect to human VWF standards prepared by pooling plasma from 5 healthy donors. D. Multimer distribution of h[mA1]V and h[mA1]NV in mouse plasma. E. Immunohistochemical staining of mouse liver sections. Platelet thrombi were detected using anti platelet DyLight-488 (green) and VWF using anti-VWF Alexa-647 (Dako, red). Both h[mA1]V and h[mA1]NV are robustly expressed in mice at 48h. Hepatic thrombi were found in most vessels upon h[mA1]NV expression (white arrow) and these animals also exhibited profound thrombocytopenia up to 10 days following hydrodynamic injection.
In tail bleed measurements, h[mA1]V corrected bleeding time and reduced bleeding volume in a majority of VWF−/− mice. However, bleeding continued unabated in all mice expressing h[mA1]NV (Fig. 5B, Supplementary Figure S7). The extent of bleeding was greater in the h[mA1]NV injected mice compared to PBS controls, though the difference did not reach statistical significance. While h[mA1]V expressing mice had plasma VWF concentrations at ~80% that of pooled human plasma, this was reduced to ~25% in the h[mA1]NV mice (Fig. 5C). The h[mA1]NV mice also lacked larger VWF multimers (Fig. 5D), consistent with the notion that N-terminal mucin insertion results in a hyper-adhesive VWF protein akin to VWD Type-2B mutants. Unlike h[mA1]NV, h[mA1]CV did not affect either platelet count (~106/μL) or bleeding time (~5 min) with plasma VWF levels remaining comparable to humans.
Immunohistochemical analysis revealed that VWF was expressed in hepatocytes at similar levels in both h[mA1]NV and h[mA1]V animals (Fig. 5E). The proteins were also expressed in the lung (Supplemental Fig. S8), but not other organs examined using immunohistochemistry (brain, spleen, kidney and heart). Platelet-rich thrombi were commonly observed in the portal veins and hepatic arteries of the h[mA1]NV mice (Fig. 5E, Supplemental Fig. S9), and this may account for the thrombocytopenia. Intravascular thrombi were not observed in other organs. Trichrome stained sections did not reveal differences in interstitial and perivascular collagen deposition in h[mA1]NV mice compared to controls (data not shown). Plasma bilirubin levels remained unchanged following h[mA1]NV expression (Fig. S6).
Overall, D’D3 critically regulates VWF adhesive function in vivo by tuning A1-GpIbα binding. Hydrodynamic injection of h[mA1]NV into VWF−/− mice results in VWD type-2B like symptoms with reduced plasma VWF expression, lower protein multimer distribution and profound thrombocytopenia [27]. Additionally, the introduction of this hyper-adhesive VWF form did not cause histologic abnormalities detectable under light microscopic examination.
Fluid shear and ristocetin may alter the structure of the D’D3-A1 interface
To determine if the physical spacing between VWF-A1 and -D’D3 may change under physiological shear, we screened a panel of anti-D’D3 IgG mAbs to identify entities that display greater binding to hNV compared to hV or hCV (Supplemental Figure S10). These studies identified mAb DD3.3 as a reagent displaying 1.5-2 fold greater binding to hNV compared to the other constructs. MAb DD3.5 served as a control as it did not bind differentially to the VWF proteins. When ristocetin was added in ELISA, DD3.3 binding was increased to all VWF constructs by 3-7 fold (Fig. 6A), with no change noted for DD3.5 (Fig. 6B). When wild-type VWF (hV) immobilized on anti-VWF Ab bearing beads was subjected to hydrodynamic shear in a cone-plate viscometer, also, mAb DD3.3 binding to beads increased progressively with applied shear rate >2400/s (Fig. 6C). No shear-dependent change in DD3.5 binding was observed (Fig. 6D). Thus, DD3.3 is a novel anti-D’D3 conformational sensitive mAb that preferentially binds the active form of VWF. Further, the data suggest that domain-level structural changes may occur at the VWF D’D3-A1 interface upon physiological fluid flow application and ristocetin addition.
Figure 6. VWF conformation change detected using anti-D’D3 mAb DD3.3.
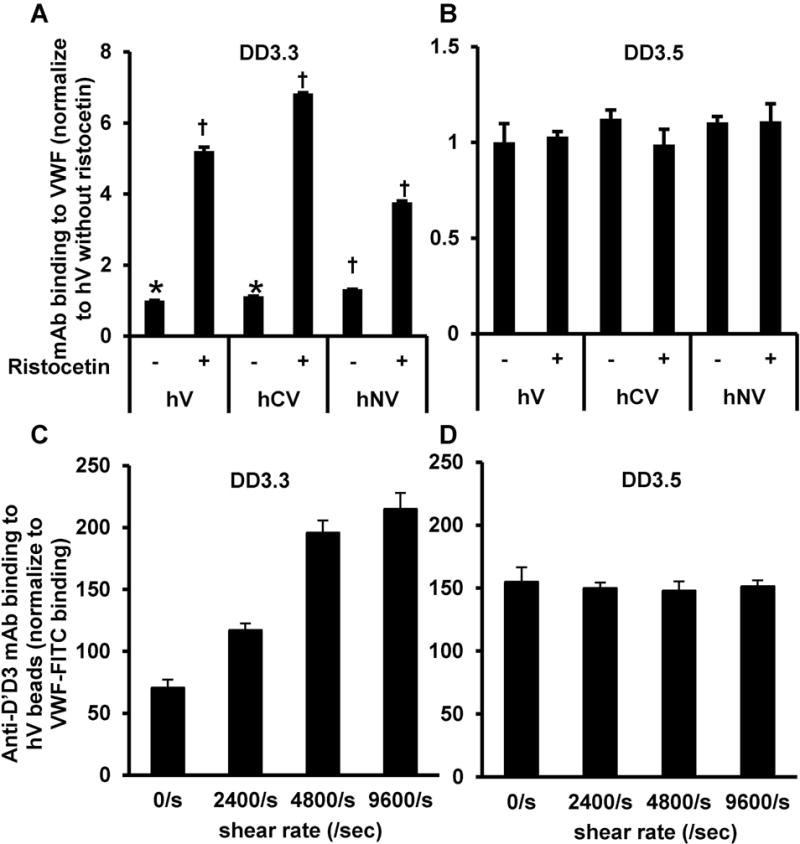
A-B. ELISA plates were coated overnight with 10μg/ml of the different VWF constructs. 20μg/ml DD3.3 (panel A) or DD3.5 (panel B) was then added to the wells in the presence or absence of risocetin. MAb binding was measured in ELISA using HRP conjugated rabbit anti-mouse Ab. DD3.3 displays greater binding to hNV compared to hV or hCV, with risocetin augmenting mAb binding to all constructs. DD3.5 binding to all VWF proteins was similar and this was not altered by ristocetin (panel B). C-D. 10μg/mL wild-type VWF (hV) was bound onto beads bearing anti-VWF capture Ab. These beads were washed and subjected to various shear rates for 5 min. in the presence of 20μg/mL mAbs against D’D3 (DD3.3 in panel C, DD3.5 in panel D) along with FITC conjugated polyclonal anti-VWF Ab (Dako). DD3.3/3.5 binding was normalized based on signal from FITC conjugated anti-VWF Ab. The binding of DD3.3, but not DD3.5, to immobilized VWF increased upon shear application.
DISCUSSION
This manuscript shows that mucin spacer insertion between the VWF D’D3- and A1-domains enhances VWF binding to platelet GpIbα in a variety of settings. This increased spacing augments platelet binding to VWF, and also thrombus formation on collagen under physiological flow. Platelet recruitment was also augmented on VWF substrates and platelet translocation velocity was decreased. We choose the O-glycan cluster to increase the physical separation between domains in this study, since this results in a stiff spacer unlike the classic Gly-Ser linker which is flexible [28]. Insertion of the mucin-spacer at either aa1263 (after the ‘N-terminal flanking peptide’ [7, 29, 30]) or aa1238 (before the ‘flanking peptide’) at the N-terminal of VWF-A1 had a similar biological function. Thus, the ‘flanking peptide’ may not contribute to A1-domain function in the context of multimeric VWF.
In contrast to the N-terminal insertion, similar changes at the C-terminus at the A1-A2 interface (at either aa1476 or aa1458) only marginally affected VWF function, with a slight decrease in ADAMTS13 proteolysis rates and a small effect on thrombus formation on collagen. Indeed, others have previously suggested that the A2-domain may bind VWF-A1 and reduce its ability to engage GpIbα [10]. While this may be true in some settings, based on our systematic side-by-side comparison of multimeric VWF, it is more likely that the physical spacing between D’D3 and A1 is the critical regulator of A1-GpIbα binding rates under physiological conditions.
To ensure that the conclusions of this work are not artifacts due to mucin insertion, we tested the effect of mucin insertion at multiple locations at the N- and C-terminus. We also screened a panel of mAbs against the VWF-D’D3 domain and applied them in studies with wild-type VWF. These studies revealed a non-function blocking mAb DD3.3 (Supplemental Fig. S11), whose binding to VWF was augmented in the presence of ristocetin and also hydrodynamic shear. In this regard, ristocetin binds aa1237-1266 and aa1458-1472 at the N- and C-terminus of VWF-A1 [31], and this binding interaction is not abolished in our mutants. Based on observations that DD3.3 binding is enhanced by ristocetin, the data suggest that ristocetin may promote structural rearrangements at the A1-D’D3 interface in addition to promoting VWF-GpIbα binding. Besides DD3.3, the function-blocking mAb DD3.1 inhibited VWF-GpIbα binding when D’D3 and A1 were proximal to each other both in native hV and h[mA1]V, but it was ineffective when the D’D3-A1 distance was increased using the spacer in hNV and h[mA1]NV. DD3.1 was also ineffective upon ristocetin addition. Together, the data using the two mAbs support the notion that the physical distance between D’D3 and A1 may be altered when VWF undergoes shear induced conformation changes (Fig. 7, [32]). The detailed mapping of the mAb binding sites on D’D3 is awaited as this may reveal the protein structure at the D’D3-A1 interface. Others have also demonstrated enhanced binding of single domain VWD type 2B mutants (e.g. I546V) to platelet GpIbα under shear [33]. More studies are needed to determine if structural changes at the D’D3-A1 interface accompany VWF binding to collagen, and the relative importance of this during VWD type 2B disease where single nucleotide polymorphisms located away from the A1-GpIbα binding interface augment VWF-platelet adhesion interactions [1, 2, 6].
Figure 7. Conceptual model.
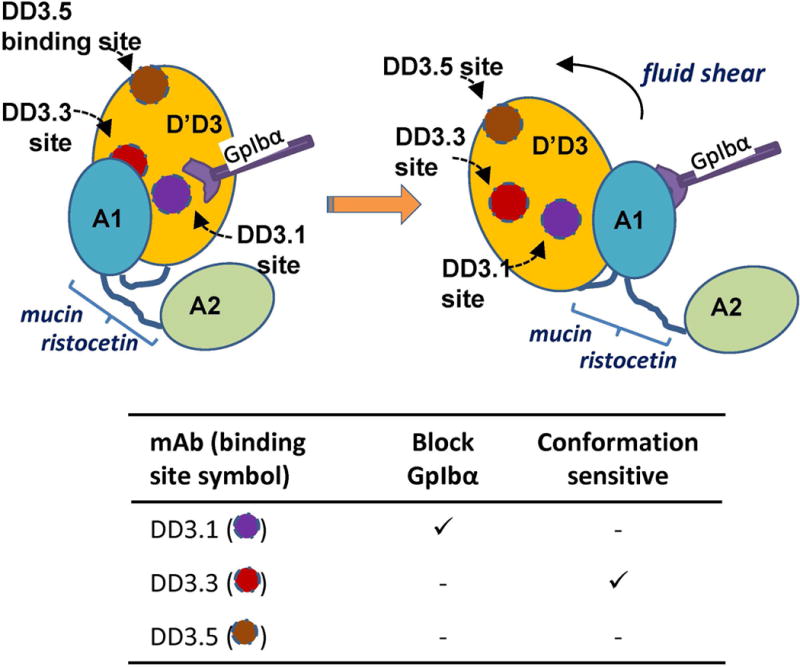
The data suggest that mAbs DD3.1 and DD3.3 may bind close to the D’D3-A1 interface. DD3.1 sterically hinders VWF-GpIbα binding interactions, while DD3.3 binds a partially cryptic epitope. mAb DD3.5 binds away from this interface. Application of shear, mucin insert or ristocetin to VWF causes structural changes at the interface, augmenting mAb DD3.3 binding and making DD3.1 an ineffective inhibitor of GpIbα binding. Note that the image is not drawn to scale, and circular regions used to mark mAb binding sites are hypothetical.
Besides basic science, the findings highlighted in this manuscript may be of translational significance. In this regard, the mucin insertion between VWF-D’D3 and -A1 in multimeric VWF significantly augmented platelet activation in solution based assays where VWF was sheared with platelets at 100 dyn/cm2 in a cone-plate viscometer. This structural change also decreased the apparent KD of VWF-GpIbα binding by >50-100-fold. Based on these dramatic effects, it is possible that the physical separation between D’D3 and A1 may control platelet function under high shear conditions as observed in stenosed arteries, and prosthetic devices like heart valves, ECMOs (extracorporeal membrane oxygenation) and LVADs (left ventricular assist devices) [34, 35].
In vivo, the chimeric VWF protein h[mA1]V largely restored the homeostatic defect in VWF−/− mice. VWF expression levels in these animals, 2-days following hydrodynamic injection, were comparable to that in humans and blood platelet counts were normal. Unlike h[mA1]V and h[mA1]CV, h[mA1]NV expression in VWF−/− mice consistently resulted in lower protein expression levels in circulation, reduced multimer distribution, thrombocytopenia and enhanced tail-vein bleeding. In this regard, the lower expression of h[mA1]NV may be due to its higher affinity binding to murine platelet GpIbα that results in faster protein clearance, especially for the higher multimers. The dramatic loss of platelets from circulation may also be due to the enhanced clearance of VWF-induced activated platelets and thrombi in the liver. These observations are similar to previous work that used hydrodynamic injection to create VWD-Type-2B in mouse [36, 37], though platelet depletion is more robust in this work. Additionally, due to the nature of the construct design, there is no need to wait for several weeks to allow VWF levels to stabilize when using our vectors as protein levels are already close to that of humans within 2-3 days [38].
In summary, the spatial proximity between VWF-D’D3 and A1 is likely a critical feature regulating VWF binding to GpIbα in shear based ex vivo studies and in vivo. This may be a key structural feature controlling thrombosis and hemostasis in man.
Supplementary Material
ESSENTIALS.
Role of von Willebrand Factor (VWF) domains in regulating platelet adhesion was studied in vivo.
Multimeric VWF with spacers at the N- and C-terminus of VWF-A1 were systematically tested.
N-terminal modified VWF avidly bound platelet GpIbα, causing VWD Type2B like phenotype in mice.
Novel anti-D’D3 mAbs suggest that changes at the D’D3-A1 interface may be biologically relevant.
Acknowledgments
We are grateful to Drs. Anil Chauhan and Denisa Wagner for providing VWF−/− mice. We also thank Dr. Jessy J. Alexander for technical advice. The work was supported by NIH grant HL77258.
Footnotes
AUTHOR CONTRIBUTIONS
C. Zhang designed and performed experiments, interpreted results and wrote the paper. A. Kelkar M. Nasirikenari and M. Sveinsson designed and performed experiments. J. T. Y. Lau designed experiments and interpreted results. U. C. Sharma and S. Pokharel analyzed results. S. Neelamegham conceptualized and directed the project, interpreted results and wrote the paper. All authors approved the final manuscript.
CONFLICTS OF INTEREST
None declared
References
- 1.Gogia S, Neelamegham S. Role of fluid shear stress in regulating VWF structure, function and related blood disorders. Biorheology. 2015;52:319–35. doi: 10.3233/BIR-15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005;56:173–91. doi: 10.1146/annurev.med.56.082103.104713. [DOI] [PubMed] [Google Scholar]
- 3.Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120:449–58. doi: 10.1182/blood-2012-01-405134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh I, Shankaran H, Beauharnois ME, Xiao Z, Alexandridis P, Neelamegham S. Solution structure of human von Willebrand factor studied using small angle neutron scattering. J Biol Chem. 2006;281:38266–75. doi: 10.1074/jbc.M607123200. [DOI] [PubMed] [Google Scholar]
- 5.Lof A, Muller JP, Brehm MA. A biophysical view on von Willebrand factor activation. J Cell Physiol. 2018;233:799–810. doi: 10.1002/jcp.25887. [DOI] [PubMed] [Google Scholar]
- 6.Ruggeri ZM. Von Willebrand factor: looking back and looking forward. Thromb Haemost. 2007;98:55–62. [PubMed] [Google Scholar]
- 7.Auton M, Sowa KE, Behymer M, Cruz MA. N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J Biol Chem. 2012;287:14579–85. doi: 10.1074/jbc.M112.348573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madabhushi SR, Zhang C, Kelkar A, Dayananda KM, Neelamegham S. Platelet GpIba binding to von Willebrand Factor under fluid shear:contributions of the D’D3-domain, A1-domain flanking peptide and O-linked glycans. J Am Heart Assoc. 2014;3:e001420. doi: 10.1161/JAHA.114.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulrichts H, Udvardy M, Lenting PJ, Pareyn I, Vandeputte N, Vanhoorelbeke K, Deckmyn H. Shielding of the A1 domain by the D’D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. 2006;281:4699–707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 10.Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J Thromb Haemost. 2007;5:1363–70. doi: 10.1111/j.1538-7836.2007.02536.x. [DOI] [PubMed] [Google Scholar]
- 11.Tischer A, Campbell JC, Machha VR, Moon-Tasson L, Benson LM, Sankaran B, Kim C, Auton M. Mutational Constraints on Local Unfolding Inhibit the Rheological Adaptation of von Willebrand Factor. J Biol Chem. 2016;291:3848–59. doi: 10.1074/jbc.M115.703850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumas JJ, Kumar R, McDonagh T, Sullivan F, Stahl ML, Somers WS, Mosyak L. Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibalpha complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J Biol Chem. 2004;279:23327–34. doi: 10.1074/jbc.M401659200. [DOI] [PubMed] [Google Scholar]
- 13.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–9. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 14.Madabhushi SR, Shang C, Dayananda KM, Rittenhouse-Olson K, Murphy M, Ryan TE, Montgomery RR, Neelamegham S. von Willebrand factor (VWF) propeptide binding to VWF D’D3 domain attenuates platelet activation and adhesion. Blood. 2012;119:4769–78. doi: 10.1182/blood-2011-10-387548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tischer A, Cruz MA, Auton M. The linker between the D3 and A1 domains of vWF suppresses A1-GPIbalpha catch bonds by site-specific binding to the A1 domain. Protein Sci. 2013;22:1049–59. doi: 10.1002/pro.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowak AA, Canis K, Riddell A, Laffan MA, McKinnon TA. O-linked glycosylation of von Willebrand factor modulates the interaction with platelet receptor glycoprotein Ib under static and shear stress conditions. Blood. 2012;120:214–22. doi: 10.1182/blood-2012-02-410050. [DOI] [PubMed] [Google Scholar]
- 17.Aponte-Santamaria C, Huck V, Posch S, Bronowska AK, Grassle S, Brehm MA, Obser T, Schneppenheim R, Hinterdorfer P, Schneider SW, Baldauf C, Grater F. Force-sensitive autoinhibition of the von Willebrand factor is mediated by interdomain interactions. Biophys J. 2015;108:2312–21. doi: 10.1016/j.bpj.2015.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jentoft N. Why are proteins O-glycosylated? Trends Biochem Sci. 1990;15:291–4. doi: 10.1016/0968-0004(90)90014-3. [DOI] [PubMed] [Google Scholar]
- 19.Shogren R, Gerken TA, Jentoft N. Role of glycosylation on the conformation and chain dimensions of O-linked glycoproteins: light-scattering studies of ovine submaxillary mucin. Biochemistry. 1989;28:5525–36. doi: 10.1021/bi00439a029. [DOI] [PubMed] [Google Scholar]
- 20.Dayananda KM, Singh I, Mondal N, Neelamegham S. von Willebrand factor self-association on platelet GpIbalpha under hydrodynamic shear: effect on shear-induced platelet activation. Blood. 2010;116:3990–8. doi: 10.1182/blood-2010-02-269266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shankaran H, Alexandridis P, Neelamegham S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood. 2003;101:2637–45. doi: 10.1182/blood-2002-05-1550. [DOI] [PubMed] [Google Scholar]
- 22.Gogia S, Kelkar A, Zhang CJ, Dayananda KM, Neelamegham S. Role of calcium in regulating the intra- and extracellular cleavage of von Willebrand factor by the protease ADAMTS13. Blood Advances. 2017;1:2063–74. doi: 10.1182/bloodadvances.2017009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marx I, Christophe OD, Lenting PJ, Rupin A, Vallez MO, Verbeuren TJ, Denis CV. Altered thrombus formation in von Willebrand factor-deficient mice expressing von Willebrand factor variants with defective binding to collagen or GPIIbIIIa. Blood. 2008;112:603–9. doi: 10.1182/blood-2008-02-142943. [DOI] [PubMed] [Google Scholar]
- 24.Denis C, Methia N, Frenette PS, Rayburn H, Ullman-Cullere M, Hynes RO, Wagner DD. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95:9524–9. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012 doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Tan K, Zhou H, Lo HF, Tronik-Le Roux D, Liddington RC, Diacovo TG. Modifying murine von Willebrand factor A1 domain for in vivo assessment of human platelet therapies. Nat Biotechnol. 2008;26:114–9. doi: 10.1038/nbt1373. [DOI] [PubMed] [Google Scholar]
- 27.Casari C, Du V, Wu YP, Kauskot A, de Groot PG, Christophe OD, Denis CV, de Laat B, Lenting PJ. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood. 2013;122:2893–902. doi: 10.1182/blood-2013-03-493312. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–69. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ju L, Dong JF, Cruz MA, Zhu C. The N-terminal flanking region of the A1 domain regulates the force-dependent binding of von Willebrand factor to platelet glycoprotein Ibalpha. J Biol Chem. 2013;288:32289–301. doi: 10.1074/jbc.M113.504001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng W, Wang Y, Druzak SA, Healey JF, Syed AK, Lollar P, Li R. A discontinuous autoinhibitory module masks the A1 domain of von Willebrand factor. J Thromb Haemost. 2017 doi: 10.1111/jth.13775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadler JE. Redeeming ristocetin. Blood. 2010;116:155–6. doi: 10.1182/blood-2010-04-276394. [DOI] [PubMed] [Google Scholar]
- 32.Singh I, Themistou E, Porcar L, Neelamegham S. Fluid shear induces conformation change in human blood protein von Willebrand factor in solution. Biophys J. 2009;96:2313–20. doi: 10.1016/j.bpj.2008.12.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doggett TA, Girdhar G, Lawshe A, Schmidtke DW, Laurenzi IJ, Diamond SL, Diacovo TG. Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow: the GPIb(alpha)-vWF tether bond. Biophys J. 2002;83:194–205. doi: 10.1016/S0006-3495(02)75161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marasco SF, Lukas G, McDonald M, McMillan J, Ihle B. Review of ECMO (extra corporeal membrane oxygenation) support in critically ill adult patients. Heart Lung Circ. 2008;17(Suppl 4):S41–7. doi: 10.1016/j.hlc.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood. 2016;127:3133–41. doi: 10.1182/blood-2015-10-636480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golder M, Pruss CM, Hegadorn C, Mewburn J, Laverty K, Sponagle K, Lillicrap D. Mutation-specific hemostatic variability in mice expressing common type 2B von Willebrand disease substitutions. Blood. 2010;115:4862–9. doi: 10.1182/blood-2009-11-253120. [DOI] [PubMed] [Google Scholar]
- 37.Rayes J, Hollestelle MJ, Legendre P, Marx I, de Groot PG, Christophe OD, Lenting PJ, Denis CV. Mutation and ADAMTS13-dependent modulation of disease severity in a mouse model for von Willebrand disease type 2B. Blood. 2010;115:4870–7. doi: 10.1182/blood-2009-11-254193. [DOI] [PubMed] [Google Scholar]
- 38.Morioka Y, Casari C, Wohner N, Cho S, Kurata S, Kitano A, Christophe OD, Lenting PJ, Li R, Denis CV, Prevost N. Expression of a structurally constrained von Willebrand factor variant triggers acute thrombotic thrombocytopenic purpura in mice. Blood. 2014;123:3344–53. doi: 10.1182/blood-2013-10-531392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.