

Abstract
Huntington’s disease (HD) is a complex neurodegenerative disorder that has no cure. Although treatments can often be given to relieve symptoms, the neuropathology associated with HD cannot be stopped or reversed. HD is characterized by degeneration of the striatum and associated pathways that leads to impairment in motor and cognitive functions as well as psychiatric disturbances. Although cell and rodent models for HD exist, longitudinal study in a transgenic HD nonhuman primate (NHP; i.e. rhesus macaque; HD monkeys) shows high similarity in its progression with human patients. Progressive brain atrophy and changes in white matter integrity examined by magnetic resonance imaging (MRI) are coherent with the decline in cognitive behaviors related to corticostriatal functions and neuropathology. HD monkeys also express higher anxiety and irritability/aggression similar to human HD patients that other model systems have not yet replicated. While a comparative model approach is critical for advancing our understanding of HD pathogenesis, HD monkeys could provide a unique platform for preclinical studies and long-term assessment of translatable outcome measures. This review summarizes the progress in the development of the transgenic HD monkey model and the opportunities for advancing HD preclinical research.
Keywords: Transgenic animal model, nonhuman primates, Huntington’s disease, comparative animal models
Introduction
Neurodegenerative disorders such as Parkinson’s disease (PD) and Huntington’s disease (HD) that severely impair basal ganglia functions are complex disorders that lead to motor and/or psychological impairments. Different models from cell culture to rodents to nonhuman primates (NHPs) have been developed to investigate disease mechanisms and develop cures for PD and HD. The key for clinical translation to assess effective diagnostics and the therapeutic efficacy of novel treatments has relied on the development of animal models with measurable clinical and pathological features consistent with those seen in human patients. In that regard, this review will highlight the progress made towards the development of a transgenic HD monkey model. Data from these animals will be compared with those gathered from various rodent HD models. The advantages and challenges in developing such an animal model will be discussed.
Brief Overview of Basal Ganglia
The basal ganglia play a key role in motor and non-motor functions affected in various brain disorders including PD and HD. The basal ganglia consist of the dorsal striatum that comprises the caudate nucleus and putamen, and the ventral striatum that comprises the nucleus accumbens and olfactory tubercle. In general, the caudate nucleus can be subdivided into the head, body and tail (Yeterian and Van Hoesen 1978). Although the caudate nucleus and putamen are separated by the internal capsule and often referred to collectively as the dorsal striatum in primates, they are functionally distinct with the putamen involved in sensorimotor processing while the caudate nucleus is involved in cognitive functions (Hewitt 1961; Smith et al. 2014). Basal ganglia nuclei also include the pallidum that is comprised of the internal and external segments of the globus pallidus (GP) in primates, which is referred to as GPi and GPe, respectively. The subthalamic nucleus (STN) is another important structure in the basal ganglia. Moreover, the substantia nigra is comprised of two sub-nuclei. The substantia nigra pars compacta (SNc) contains dopaminergic neurons and the substantia nigra reticulata (SNr) contains GABAergic projection neurons.
The striatum receives input from nearly all of the cerebral cortex. In the dorsal striatum, major cortical inputs originate from associative and sensorimotor cortices (Selemon and Goldman-Rakic 1985), while the ventral striatum receives its main cortical input from limbic cortices. Although relatively homogenous in cell density, acetylcholinesterase staining (and other neurochemical markers) reveal a patchwork pattern to the striatum (Graybiel and Ragsdale 1978), referred to as striosomes and matrix compartments. Dysregulation of this compartmentation may be involved in the development of repetitive motor behaviors (Canales and Graybiel 2000).
Striatal neurons can be categorized as projection neurons and interneurons. The main projection neurons are medium spiny neurons (MSNs). These GABAergic neurons are found in both the striosomes and matrix, accounting for over 90-95% of the total neuron population in the rat striatum (Fujiyama et al. 2011; Kawaguchi et al. 1989; Oorschot 1996; Penny et al. 1988). MSNs of the striosomes receive input from the corticolimbic cortex and amygdala, and project to the SNc (Fujiyama et al. 2011; Graybiel and Ragsdale 1978; Kawaguchi et al. 1989; Kincaid and Wilson 1996). Matrix MSNs mainly receive input from the sensorimotor cortex and nigrostriatal pathway and project to the GP and SNr (Fujiyama et al. 2011; Graybiel and Ragsdale 1978; Kawaguchi et al. 1989; Kincaid and Wilson 1996).
The direct and indirect pathway model has been widely used for the understanding of basal ganglia functions (DeLong 1990). The direct pathway is comprised of neurons that preferentially express D1 dopamine receptors, substance P and dynorphin, and project directly to the GPi and SNr via monosynaptic connections that link the striatum and basal ganglia output nuclei. Selective activation of this pathway is thought to facilitate locomotion, likely through disinhibition of thalamocortical neurons from their tonic GABAergic control by the basal ganglia output nuclei (Freeze et al. 2013; Roseberry et al., 2016). On the other hand, the indirect pathway is comprised of neurons that preferentially express D2 dopamine receptors and enkephalin and project to the GPe and the STN via polysynaptic pathways that link the striatum and GPi/SNr. The indirect pathway is considered as the NO-GO pathway because its selective activation decreases locomotion, most likely through increased basal ganglia inhibitory tone over thalamocortical neurons that reduces cortical outflow (Freeze et al. 2013; Lim et al. 2014; Roseberry et al., 2016, Sato and Parent 1998). The distinct functions of the two pathways allow bidirectional modulation of the basal ganglia network. Normal basal ganglia function relies on balanced activity between these two pathways (Lim et al. 2014).
The functions of the direct and indirect pathways are mediated through the GABAergic MSNs, the principal output cells whose activities are regulated by striatal interneurons (Hu et al. 2014; Lim et al. 2014). There are four main types of striatal interneurons based on their neurochemical content which include (1) choline acetyltransferase (ChAT), (2) parvalbumin (PV), (3) calretinin (CR) and (4) neuronal nitric oxide synthase (NOS), β-nicotinamide adenine dinucleotide phosphate-diaphorase (NADPH-d), somatostatin (SS) and neuropeptide Y (NPY) (Figueredo-Cardenas et al. 1996; Hu et al. 2014; Lanciego et al. 2012; Maurice et al. 2015; Petryszyn et al. 2014; Petryszyn et al. 2017; Smith et al. 2014; Wu and Parent 2000). Morphological heterogeneity among striatal interneurons are well described in rats, monkeys and humans (Petryszyn et al. 2014). Striatal interneurons constitute approximately 20%-25% of all striatal neurons in primates, but only 2–3 % in rodents (Graveland and DiFiglia 1985; Graveland et al. 1985; Tepper and Bolam 2004). Cholinergic (or ChAT) interneurons in the striatum are large and aspiny (Lim et al. 2014). Cholinergic interneurons are categorized as tonically active neurons (TANs) and play a key role in dopamine-dependent striatal plasticity, reward-related learning and motivated behaviors (Aosaki et al. 1994; Apicella 2007; Wang et al. 2006). Parvalbumin (PV) interneurons are also named GAGAergic fast-spiking parvalbumin-positive interneurons that are involved in feedback and feedforward inhibition of projection neurons activity (Hu et al. 2014; Lee et al. 2017). Recent study demonstrated that suppression or over-activation of PV interneurons activity can regulate MSN firing (Lee et al., 2017), which provides evidence for their key regulatory role in striatal microcircuits to enhance early stage learning (Lee et al. 2017; Xu et al. 2016). Calretinin (CR) interneurons are the most abundant striatal interneurons in nonhuman primates and humans (Petryszyn et al. 2014; Wu and Parent 2000). The rodent striatum comprises primarily medium-sized CR interneurons, while the monkey and human striatum contain both large and medium-sized CR interneurons that create an elaborated network (Petryszyn et al. 2017). Moreover, large-sized CR+/ChAT+ striatal interneurons are only found in monkeys and humans, but not in rodents, which further suggests their crucial role in striatal functions in primates (Petryszyn et al. 2017). The fourth population of striatal interneurons co-express NADPH-d/SS/NPY/NOS and are categorized as “persistent and low-threshold spike” neurons (Kawaguchi et al. 1995; Smith et al. 2014). However, variations in the quantity and combination of these chemicals were observed, suggesting unique functional roles, perhaps even related to neurodegeneration and excitotoxicity (Figueredo-Cardenas et al. 1996). Overall, the basal ganglia regulate activity of multiple brain regions in order to control motion function, motor learning, executive functions, and emotions (Lanciego et al. 2012). Additional details about the function and anatomy of the basal ganglia can be found in other reviews part of this Special Issue.
HD Symptoms
HD patients display a variety of symptoms that can be classified as motor, cognitive or psychiatric in nature. Although motor symptoms of the disease are often the most notable and the reason for the initial name “Huntington’s chorea,” a multidimensional approach assessing all three impacted areas may be more appropriate for diagnosis (Biglan et al. 2013; Rub et al. 2016). One of the earliest motor symptoms of HD is impairment of eye saccades (Rub et al. 2016; Tabrizi et al. 2012). Many juvenile-onset patients also experience rigidity and seizure (Cloud et al. 2012; Geevasinga et al. 2006; Nance and Myers 2001; van Dijk et al. 1986). Patients quickly deteriorate in motor ability (Roos 2010; Ross et al. 2014; Tabrizi et al. 2012; Tabrizi et al. 2013), most notably in bradykinesia and chorea (Biglan et al. 2013).
Other early symptoms include cognitive deficits such as speech delay in juvenile-onset HD (Solomon et al. 2007; Yoon et al. 2006). In fact, mild cognitive deficits may be noticed before formal diagnosis (Lawrence et al. 1998; Tabrizi et al. 2012; Vaccarino et al. 2011). In a study in which subjects were asymptomatic but genetically confirmed for the HD mutation, there were already signs of cognitive decline (Lawrence et al. 1998). These “preclinical” subjects showed deficits in attention set shifting with significantly more errors than non-carriers on a visual discrimination test involving an extradimensional shift. Cognitive symptoms progress as disease severity increases. In early stages of symptomatic HD, patients started showing “subcortical” dementia, with errors in executive function like planning and decision-making which is different from Alzheimer’s disease (Lawrence et al. 1996). These early-stage HD patients were also significantly worse at pattern and spatial recognition, exhibited shortened spatial span, used a less efficient search strategy during spatial working memory tasks, had slower thinking times and continued to show difficulty with attention set shifting. At advanced stages, patients experienced more widespread dementia that included temporal lobe and hippocampal cognition (Lange et al. 1995). Most patients have difficulty with memory recall (Zizak et al. 2005), specifically immediate memory recall (Ho et al. 2003). Primary deficits include decreased processing speed, decreased attention, difficulty with initiation, poor executive function independent of medication, motor impairment or altered psychological state (Bates et al. 2015; Beglinger et al. 2010; Duff et al. 2010; Ho et al. 2003; Paulsen et al. 2013; Peavy et al. 2010; Ross et al. 2014; Tabrizi et al. 2013). These studies suggest cognitive decline begins pre-symptomatically and progressively worsens along with the disease course in HD patients (Beglinger et al. 2010; Duff et al. 2010; Stout et al. 2011).
Psychiatric symptoms include aggression, affective disorders, irritability, obsession-like symptoms, behavioral disorders and personality disorders (Berrios et al. 2001). Perhaps the first psychiatric symptoms include apathy (Tabrizi et al. 2013) and difficulty recognizing emotion (Tabrizi et al. 2012). Depression is common and suicide is increased in HD patients compared to the general population (Di Maio et al. 1993). Some have suggested that the presence of psychiatric symptoms, such as affective disorders like depression, may be genetically linked to the family tree (Folstein et al. 1983). In fact, this study showed that in certain families, depression rates were as high as 41% and began as many as 20 years before HD onset. Suicide is a major concern in HD, even being noted as a feature of the illness in Huntington’s original description (Huntington 1872). There is not only an increase in suicide rates among HD patients, but also in patients who are suspected of having HD, but have not yet been diagnosed (Di Maio et al. 1993; Schoenfeld et al. 1984). This suggests that psychiatric symptoms may actually peak during early stages of the illness and not related to the diagnosis itself.
Many of these symptoms result from damage to the pathways connecting to and from the striatum or secondary brain regions affected by HD. Oftentimes rodent models are limited from replicating the HD phenotype seen in patients (Chan et al. 2015; Howland and Munoz-Sanjuan 2014; Morton and Howland 2013; Pouladi et al. 2013). No effective treatment has been developed based on successful outcomes in rodent models of HD which is largely due to inherent differences between the two species, but new rodent models are being developed with better representation of human conditions including neuropathology and molecular mechanisms (Alexandrov et al. 2016; Ament et al. 2017; Howland and Munoz-Sanjuan 2014; Pouladi et al. 2013). There is no doubt that rodent models will continue to be a relevant model for elucidating HD pathogenesis and assessing new therapies (Crook and Housman 2011); however, it is also clear that a large preclinical animal model is needed that could better capture a broader, if not full, spectrum of progressively evolving clinical symptoms in human HD patients (Chan et al. 2015; Howland and Munoz-Sanjuan 2014; Morton and Howland 2013; Pouladi et al. 2013).
Huntington Disease Overview-Genetics and Pathology
HD is a genetic disorder caused by an unstable CAG expansion at the N-terminus of the Huntingtin (HTT) gene (MacDonald et al. 1993; Snell et al. 1993). HD is characterized by neuronal aggregates and cell loss initially at the striatum (Alzheimer 1911; Jelgersma 1908) and cerebral cortex, which subsequently extend to other brain regions (Sapp et al. 1997). With CAG repeats above 40, individuals are at risk for developing HD (Gil and Rego 2008; Langbehn et al. 2010; Li and Li 2006; Ross et al. 2014). The CAG expansion in the precoding region for HTT causes misfolding of the HTT protein and the formation of HTT aggregates (Bates et al. 2015; Crook and Housman 2013; DiFiglia et al. 1997; Gupta et al. 2012; Sieradzan et al. 1999). These changes result in oligomerization and aggregation of HTT in neurons throughout the brain, disrupting cellular functions including transcriptional dysregulation of BDNF and impairment of proteostasis that have led to the loss or alteration of neural functions such as synaptic dysfunction, mitochondrial toxicity and impairing axonal transport, thus resulting in the elevation of glutamate, reduction of BDNF and neuronal death (Bates et al. 2015; Crook and Housman 2013; Sieradzan et al. 1999).
HD aggressively targets the GABAergic MSNs of the striatum (Crook and Housman 2013; Davies and Ramsden 2001; Gil and Rego 2008; Li and Li 2006; Ross et al. 2014), affecting both the direct and indirect pathway with earlier pathology in the indirect pathway neurons (Deng et al. 2004; Vonsattel and DiFiglia 1998). Mouse models for HD suggest that aggregates in the MSNs precede symptom onset (Laforet et al. 2001). The first areas of the striatum compromised include the caudal putamen, the tail of the caudate nucleus and the dorsomedial head of the caudate nucleus (Rub et al. 2016; Rub et al. 2015; Tabrizi et al. 2012; Vonsattel et al. 1985). During early stages of the disease, nuclear inclusions and cell loss increase and spread through the rest of the striatum that lead to degeneration of the GPe, SNr and SNc (Deng et al. 2004; Rub et al. 2016; Rub et al. 2015). Although there is significant loss of projection neurons such as MSNs, the interneurons are often spared in HD (Petryszyn et al. 2017). For example, the sparing of the CR interneurons has been suggested for their neuroprotection role in maintaining intracellular calcium homeostatsis via calcium binding property of calretinin (Petryszyn et al. 2017). Direct interaction between CR and mutant HTT (mHTT) has been demonstrated in vitro while over expression of CR can ameliorate mHTT induced cytotoxicity (Dong et al. 2012). In the cerebral cortex, isocortical layers III, V and VI show the most severe cell loss (Rub et al. 2015). As the disease progresses, secondary structures such as the motor and limbic pathways of the thalamus, cerebellum, oculomotor nuclei of the brainstem, pontine nucleus, inferior olive, reticulo-tegmental pons, raphe nucleus, superior olive and vestibular nuclei are also implicated (Rub et al. 2015). It has been theorized that HD is a multisystem degenerative disease in which all affected structures are connected to well-known targets, but spread far beyond the striatum and its cortical circuits (Lange and Aulich 1986; Rub et al. 2013; Vonsattel 2008). An fMRI study suggested that impairment is due not simply to atrophy of these structures, but to decreased connectivity (Dumas et al. 2013). Even presymptomatic HD patients showed signs of decreased connectivity of the left frontal, right parietal and bilateral visual cortices (Dumas et al. 2013). A recent report on changes in white matter of HD monkey further support the widespread progressive temporal-spatial microstructural changes of fiber bundles connecting cortical areas to the striatum as disease progress (Meng et al. 2017). Clearly the neural pathology of HD is a complex multisystem disorder and not limited to the striatum and cerebral cortex.
HD animal models
Mouse HD models
Many transgenic mouse models have been developed and used extensively in HD research trying to replicate human HD conditions to find a cure for HD. These include transgenic mice expressing the N-terminal of the HTT gene(Davies et al. 1997; Schilling et al. 1999), transgenic mice expressing full-length HTT (Slow et al. 2005) and CAG repeats knock-in mice (Ament et al. 2017; Holter et al. 2013; Kumar et al. 2016; Wheeler et al. 2000). Among the wealth of HD mouse models, R6/2 and N171-82Q are two of the most commonly used models in HD research. Both models were developed about 20 years ago and were very well characterized (Mangiarini et al. 1996; Schilling et al. 1999). The R6/2 mouse carries exon 1 with 115-156 CAG repeats and 262 base pairs of intron 1 under control of the human HTT promoter (Mangiarini et al. 1996). R6/2 mice show signs of motor deficit between five to six weeks, failure to gain weight at seven weeks, neurodegeneration at 10 weeks, and death between 12-14 weeks (Stack et al. 2005; Turmaine et al. 2000; Wade et al. 2008). N171-82Q mice carry a 171 amino acid fragment of human HTT with 82 CAG repeats under control of the mouse prion promoter (Schilling et al. 1999). N171-82Q mice fail to gain weight at eight to10 weeks, show motor deficit at 12 weeks, develop neurodegeneration at 16-20 weeks and die at 24-30 weeks (Li et al. 2003; Saydoff et al. 2006).
Monkey HD model
In addition to rodent models, the recent development of HD monkeys has led to a new interest in transgenic nonhuman primate modeling of human diseases (Jennings et al. 2016; Niu et al. 2015; Niu et al. 2014; Yang et al. 2008). Longitudinal assessment continues throughout the lifespan of HD monkeys and is ongoing. A current effort is underway to establish a self-sustainable colony and the production of HD monkeys for HD research community with emphasis in preclinical research. One key distinction between rhesus macaque and humans is the HTT gene in rhesus macaques carries only 10-11 CAG repeats while threshold for CAG repeats in human HD is ~35 (Bates et al. 2015; Putkhao et al. 2013). To date, data from eight HD monkeys have been reported, and five of them were part of a longitudinal study since they were one week old (Berrios et al. 2001; Chan et al. 2015; Chan et al. 2014; Kocerha et al. 2013; Meng et al. 2017; Yang et al. 2008). rHD1-5 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids. rHD1 carried a single copy of the transgene with 29 CAG repeats and rHD2 had two copies of the transgene with 83 CAG repeats (Chan et al. 2014; Yang et al. 2008). rHD3 expressed 84 CAG repeats with two copies, rHD4 expressed 27 CAG repeats with two copies and rHD5 expressed 88 CAG repeats with two copies (Yang et al. 2008). rHD3-5 were euthanized soon after birth due to severe motor impairment. rHD6, 7 and 8 carried exons 1-10 of the human HTT gene coding N-terminal 508 amino acids with 67-72 CAG repeats under control of the human HTT promoter (Chan et al. 2015; Kocerha et al. 2013; Meng et al. 2017; Raper et al. 2016).
Comparative neuropathological hallmarks of HD Patients to HD Mice and NHPs
As mentioned, neuropathology in HD patients begins in the striatum and cerebral cortex (Alzheimer 1911; Jelgersma 1908; Rub et al. 2016; Sapp et al. 1997). It then follows the circuitry of the striatum to affect widespread regions of the brain in later stages of the disease (Rub et al. 2015; Vonsattel et al. 1985). Both R6/2 and N171-82Q express high levels of the mutant HTT in the brain. Immunohistochemistry revealed nuclear aggregates of mutant HTT in the striatum of both mouse models. Striatal atrophy is found in the R6/2 model with up to 25% neuronal loss (Stack et al. 2005). Cortical and striatal atrophy is also found in the N171-82 Q model (Aggarwal et al. 2012). Ultimately, atrophy of the thalamus, hippocampus and piriform cortex occurred in the R6/2 model (Aggarwal et al. 2012). The N171-82Q mice also showed significant atrophy of the neocortex, hippocampus, piriform cortex and amygdala by the end of the disease at five to six months of age (Aggarwal et al. 2012). Furthermore, changes in neural activity of the GPe and decreased connectivity of the MSNs of the indirect pathway in R6/2 mice were also observed (Akopian et al. 2016; Cepeda et al. 2013).
Similar to HD rodent models, post-mortem brain tissues of infant HD monkeys (rHD4 and 5) revealed high levels of mutant HTT aggregate in the striatum, cerebral cortex, hippocampus and cerebellum (Fig 1a). Widespread nuclear inclusions and axonal aggregates of mutant HTT were also observed throughout the striatum and cerebral cortex (Fig 1b–f). Axons appeared dystrophic and fractured, indicating distress and degeneration (Wang et al. 2008). Recent studies on two adult HD monkeys (rHD1 and rHD7) at five years of age reveal the accumulation of mHTT aggregate, intranuclear inclusions and extensive neuronal loss in the striatum, but with a distinct pattern correlated with genotypes and expression levels of the mHTT transgene (Chan et al. 2015). rHD1 had significant loss of striatal mass while rHD7 retained comparable volume with control monkeys even with a significant decrease in neural cells, suggesting ongoing neurodegeneration in rHD7 (Chan et al. 2015). Together with progressive decline in cognitive and motor impairment in these animals, neuropathology supports the dysfunction in the fronto-striatal pathways and abnormal striatal outflow (Chan et al. 2015). Although neuropathological studies on HD monkeys are limited due to the number of available animals and resources, further in-depth neuropathological study on HD monkeys and comparison with rodents and humans will provide insightful mechanisms on HD pathogenesis.
Fig 1. Neuropathological changes in NHP model for HD.
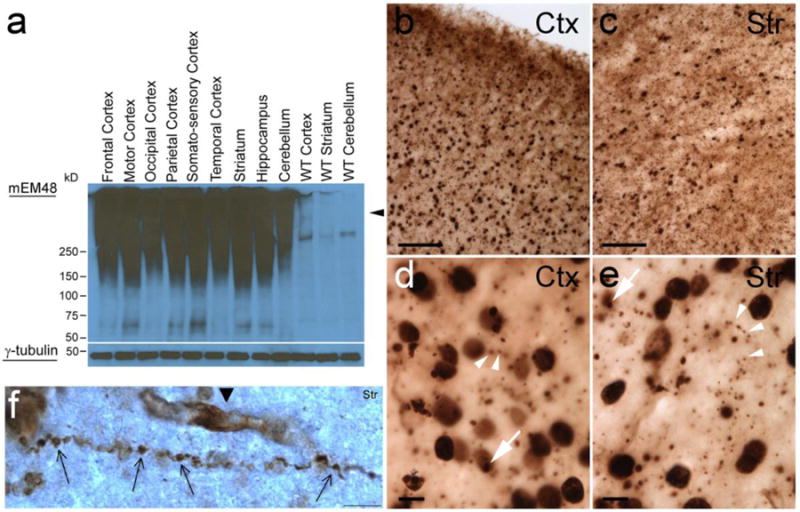
(a) Immunoblot for mEM48 shows high molecular mass oligomeric HTT (arrow head) and soluble HTT using γ-Tubulin as a control. (b–e) HTT inclusions in the cerebral cortex and striatum as shown by immunohistochemistry for mEM48. (b–c) Scale bars = 100mm (d–e) Scale bars = 10mm. (f) HTT aggregates causing disruption of the axon in a cortical neuron, scale bar = 5 μm (Yang et al. 2008; Wang et al 2008).
Using MRI, brain volume changes were observed in a small cohort of HD and wild-type control monkeys with respect to age. The rate of the change in brain volume was influenced by mutant HTT transgenes. rHD1 had the highest mutant HTT expression and heavily aggregated mutant HTT protein and exhibited a significantly faster reduction rate in striatal volume and enlargement of lateral ventricles compared to the controls (Fig 2a, b) and HD monkeys (rHD6-8s) with lower mutant HTT expression (Chan et al. 2015). These results suggest progressive striatal atrophy, similar to that seen in HD patients. In vivo proton magnetic resonance spectroscopy (MRS) for N-acetylaspartate (NAA) was used to assess neuronal survival (Chan et al. 2015). MRS data showed decreased NAA in the caudate nucleus and putamen of HD monkeys compared to the controls, suggesting the loss of striatal neurons (Fig 2c). Stereological counting of cells in the caudate nucleus and putamen of post-mortem brain sections indicated a decrease, not just in striatal volume, but in the number of neurons in both the caudate nucleus and putamen (Fig 2d, e). Interestingly, rHD1 and rHD7 carried two distinct mutant HTT transgenes in which rHD1 developed at a more rapid rate of striatal atrophy and cell loss. Although MRI did not reveal significant striatal atrophy in rHD7 compared to the controls, MRS and post-mortem neuropathology indicated significant reduction in neurons that further suggest a slower progression rate occurred in rHD7 compared to rHD1, while the loss of striatal neurons preceded the loss of striatal volume (Chan et al. 2015). These results indicate progressive death of striatal neurons, not simply shrinkage or glial cell loss as disease progress in HD monkeys (Chan et al. 2015). Recent study on white matter integrity of HD monkeys by longitudinal diffuse tensor imaging (DTI) reveals progressive changes in whole brain white matter as disease progress from infancy to adulthood (Meng et al. 2017). Thus the combination of noninvasive structural MRI and MRS is important to reveal HD status of the affected areas because compensatory response to neuronal cell loss might occur to maintain physical mass of the areas at early stages of the disease. Together with DTI, noninvasive MR imaging can be used as a powerful diagnostic tool for clinical assessment.
Fig 2. Decrease in striatal volume and cell number in HD NHPs.
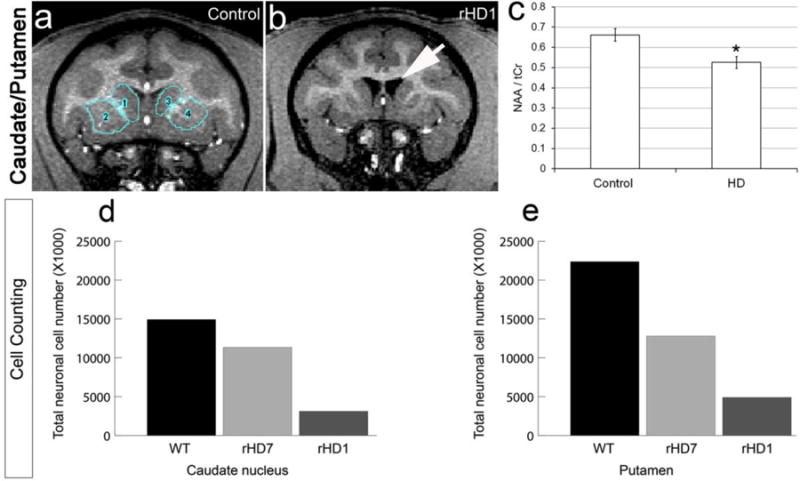
(a–b) Increase in lateral ventricles and decrease in striatal volume of HD transgenic NHP compared to control NHP shown by MRI. (c) Decrease in striatal NAA at 48 months of HD NHP compared to control NHP assessed by proton MRS. (d) Decrease in Nissl-positive cells by stereological counting in the caudate nucleus and (e) putamen of HD NHPs compared to control. rHD1 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids with 29 polyQ repeats. rHD7 carried exons 1-10 of the human HTT gene coding N-terminal 508 amino acids with approximately 67-72Q under the control of the human HTT promoter. (Chan et al. 2015).
While neuropathological assessment of HD monkeys is ongoing and very limited data is available for comprehensive comparison with rodent models and human patients, encouraging data including evidence of axonal aggregates, cell distress and neurodegeneration revealed in HD monkey brain suggest the resemblance between HD monkeys and humans patients. Even though neuropathology of HD monkeys may be similar to that seen in HD patients, in-depth analyses with additional HD monkeys in the future is critical to fuhrther confirm and support our findings, thus to better assess the potential of the HD monkey model in HD translational research.
Cognitive behavioral assessment in HD rodents and monkeys
Perhaps the most notable symptoms for HD are motor deficits, with the hallmark feature being a dance-like movement called chorea (Huntington 1872). Many patients also experience rigidity and bradykinesia (Biglan et al. 2013). Although not as obvious, oculomotor impairment is often one of the first symptoms to present (Tabrizi et al. 2012; Rub et al. 2015).
Several tests have been developed to evaluate motor deficits in mice. These tests include swimming, walking across a narrow beam and walking or running on a spinning rod. However, some mouse models such as the short-stop and C6R models never show motor deficit (Wang et al. 2008). R6/2 mice begin to display motor symptoms as early as five to six weeks and N171-82Q mice start showing deficits after 12 weeks (Pouladi et al. 2013; Wang et al. 2008). In the swimming tank test where mice swim to a platform to escape the water, R6/2 mice displayed abnormal front and hind limb kicking (Carter et al. 1999). This abnormal swimming stroke caused them to take longer to reach the platform than control mice. As animals age, R6/2 mice also showed decreased speed in beam walking, with some even falling off the beam during late stages of sickness (Carter et al. 1999). N171-82Q mice also showed decreased speed crossing the beam (Southwell et al. 2009). On the spinning rotarod test, both R6/2 and N171-82Q mice progressively worsened on maintaining balance on the spinning rotarod (Carter et al. 1999). However, N171-82Q mice did not exhibit difficulty until five months of age (Southwell et al. 2009). With age, R6/2 mice began to show gait abnormalities, most notably with reduced stride length and increased front/hind paw overlap (Carter et al. 1999), while N171-82Q mice showed abnormal gait characterized by hyperkinesia and rigidity (Preisig et al. 2016).
Compared to HD rodents, HD monkeys with severe impairment presented with dystonia, chorea, difficulty swallowing and difficulty breathing as early as at birth (Yang et al. 2008). In order to evaluate motor impairment over time, a rating scale, Huntington’s Disease Primate Motor Rating Scale (HDPMRS), similar to that used in patients was developed. Using a scale of 0-4, the test assesses ability, bradykinesia, rigidity, dystonia and chorea in various regions of the body. HD monkeys had an elevated score on the assessment compared to age-matched controls at two years of age (Chan et al. 2014). Lower limb dystonia was the most noticeable deficit. Motor impairment interfered with cognitive testing around 16 months of age and was measured by the object discrimination reversal (ODR) and detour reaching/barrier task. In this study, monkeys had to reach around a barrier to retrieve a reward such as M&M candy. The HD NHPs had significantly more motor-related problems with reward retrieval on both easy and difficult tasks (Fig 3) (Chan et al. 2015; Chan et al. 2014). These findings aligned with neuropathology of rHD1 and rHD6, 7 & 8 which suggest fronto-striatal dysfunction as discussed previously. rHD1 also presented with tonic-clonic seizures around 22 months of age. Seizure is commonly developed in a juvenile form HD patient rather than an adult form (Cloud et al. 2012; Geevasinga et al. 2006; van Dijk et al. 1986). Overall, motor impairment gradually increased with age in HD monkeys (Chan et al. 2015; Chan et al. 2014).
Fig 3. Motor difficulty in HD NHPs.
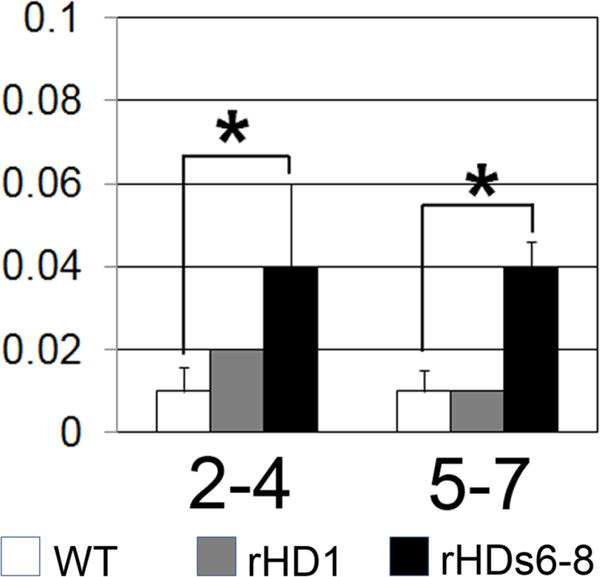
HD NHPs show increased motor problems on an object retrieval detour task at 16 months. rHD1 showed no impairment, but rHDs6-8 showed significant (p<0.05) impairment. rHD1 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids with 29 polyQ repeats. rHD6, 7, and 8 (rHDs6-8), on the other hand, carried exons 1-10 of the human HTT gene coding N-terminal 508 amino acids with approximately 67-72Q under the control of the human HTT promoter. rHD1 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids with 29 polyQ repeats. (Chan et al. 2015).
In addition to motor symptoms, HD patients also experience cognitive difficulties. Oftentimes, patients are already showing signs of cognitive impairment at the onset of motor symptoms (Duff et al. 2010; Paulsen et al. 2013; Peavy et al. 2010; Stout et al. 2012; Vonsattel and DiFiglia 1998). Speech and language delay, psychiatric and cognitive difficulties were observed in juvenile HD (Ribai et al. 2007; Yoon et al. 2006). Cognitive deficit in HD is very different from dementia in Alzheimer’s disease. Impairment in HD is most notable with attention shifting and executive function (Lawrence et al. 1996), with more global deficits in late stages of the disease (Lange et al. 1995).
In the spatial learning task called the Morris water maze, R6/2 mice acquired the task as easily as control mice (Lione et al. 1999). However, they failed to continue to improve on subsequent trials like control mice did starting at nine weeks of age (Fink et al. 2013). This difference was not due to motor impairment, as swimming speed was no different between the two groups until day 16 of testing when R6/2 mice began to swim more slowly (Lione et al. 1999). These findings indicate that the mice were not impaired at learning pre-symptomatically, but showed cognitive decline after onset of motor symptoms. Additionally, a two-choice tank swim test was used to test visual and reversal discrimination. Once HD mice became symptomatic, their ability to acquire this task severely decreased (Lione et al. 1999; Menalled and Brunner 2014; Oakeshott et al. 2013). The HD mice also failed to learn reversal discrimination at older ages using light as a visual stimulus. R6/2 mice were significantly worse than control mice at alternation tests in a T maze (Lione et al. 1999). These results show cognitive decline in R6/2 HD mice can be assessed.
Due to the much longer lifespan of monkeys, age-appropriate behavioral tests can be designed throughout life. Both control monkeys and rHD1 showed normal acquisition of cognitive tasks early in life (Chan et al. 2014). This finding showed normal cognitive development, which is consistent with that of HD patients. However, with age, rHD1 showed deterioration of cognitive skills. At eight months, there was a delay on a pattern discrimination test (Fig 4a). By nine months, rHD1 consistently failed to reach criterion for a discrimination task when all age-matched control NHPs were successful (Fig 4b) (Chan et al. 2014). These tests evaluate function of the striatocortical loop, meaning that the neuropathology seen in these monkeys was producing a cognitive deficit similar to that of humans. In an object discrimination task meant to test prefrontal cortical function, this same HD monkey was making almost twice the mistakes of control monkeys at 16 months. This difference was notable for barrier and perseveration reaches in both moderate and difficult version of the test (Fig 4c, d). rHD1 also showed impairment on a recognition memory task testing the function of the medial temporal lobe and hippocampus. Impairment was seen as early as four months and persisted through the 16-month testing (Fig 4e, f). In addition to rHD1, three additional HD monkeys (rHD6-8s) with expectation of slower disease progression also participated in similar cognitive testing (Chan et al. 2015). At eight months, they performed more barrier reaches and perseveration errors than age-matched control monkeys (Fig 5a, b). Interestingly, at 36 months, they showed increased latency on a visuomotor lifesaver task in which they had to remove candy lifesavers off a metal rod that could not be ascribed to motor deficits (Fig 5c, d). All monkeys acquired tasks normally, which indicate normal cognitive development. Based on the aforementioned reports, HD monkeys displayed cognitive deficit with increasing age (Chan et al. 2015).
Fig 4. Cognitive impairment in HD NHPs.
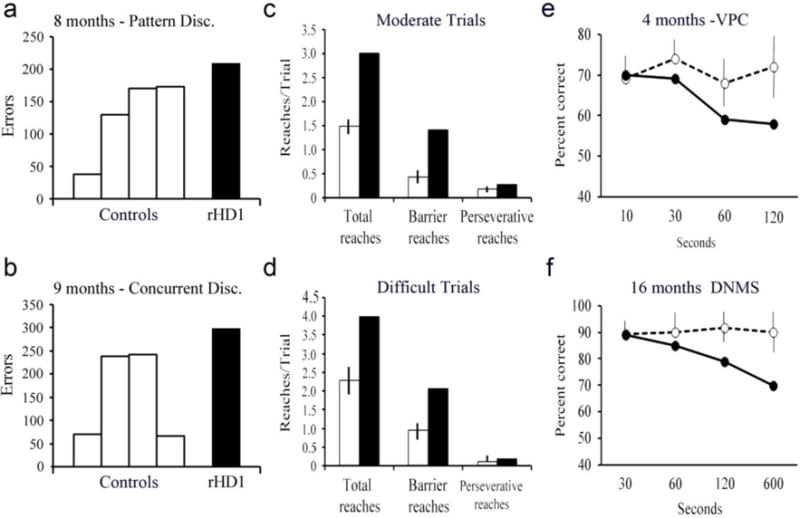
(a) HD NHPs show a slight increase in the number of errors made before reaching criteria on a pattern discrimination task at 8 months. (b) There is also a slight increase in errors at a concurrent discrimination task at 9 months. At 16 months, HD NHPs made almost twice as many barrier and preservative reaches as control NHPs on moderate (c) and difficult (d) trials. (e) Memory recognition test visual paired comparison at 4 months, scores are percent correct when looking at novel objects at a delay of 10, 30, 60, or 120 seconds. (f) Memory recognition test delayed non-matching to sample at 16 months, scores are percent correct at 30, 60, 120, and 600 seconds. rHD1 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids with 29 polyQ repeats. rHD7 carried exons 1-10 of the human HTT gene coding N-terminal 508 amino acids with approximately 67-72Q under the control of the human HTT promoter. (Chan et al. 2014).
Fig 5. Cognitive impairment in HD NHPs.
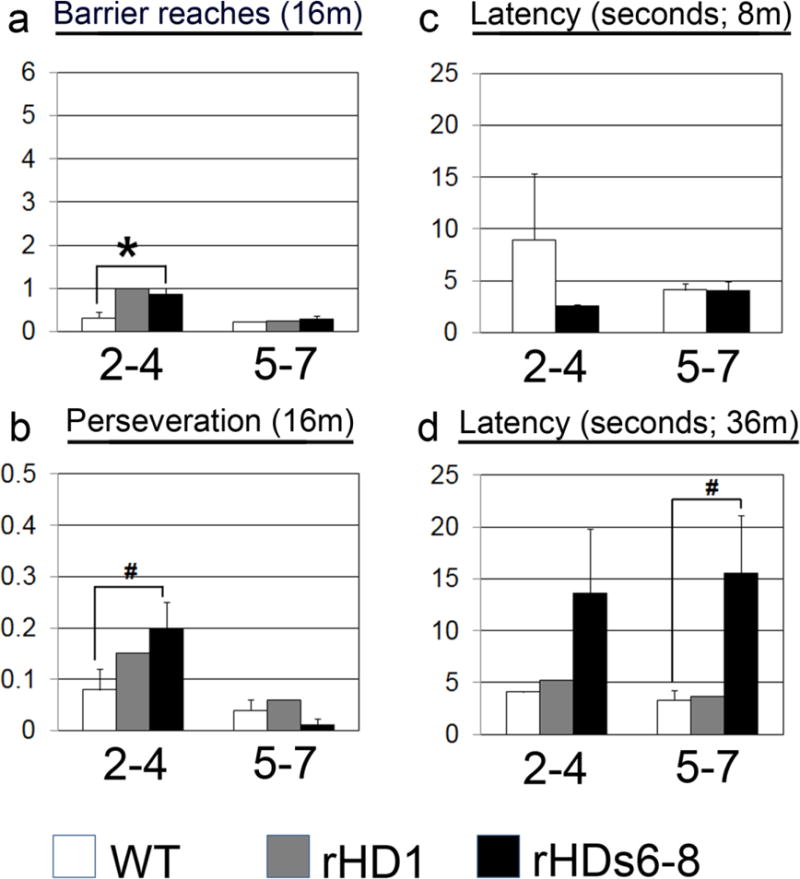
(a) At 16 months, HD NHPs showed more barrier reaches than control NHPs on an object retrieval detour task. (b) At 16 months, HD NHPs also showed an increase in the number of preservative reaches on the same task. (c-d) HD NHPs increased in latency on a visuomotor task at 36 months (d), but not at 8 months (c) compared to control NHPs. rHD1 carried exon 1 of the human HTT gene regulated by human polyubiquitin-C promoter, which expressed N-terminal 67 amino acids with 29 polyQ repeats. rHD7 carried exons 1-10 of the human HTT gene coding N-terminal 508 amino acids with approximately 67-72Q under the control of the human HTT promoter. (Chan et al. 2015).
HD monkeys displayed progressive cognitive deficits similar to those of HD patients that exhibit damage to subcortical processing as well as executive functions of the prefrontal cortex. It is important to note that an ongoing effort is to characterize the HD monkey model from infancy and throughout their lifespan in order to lay out the disease development timeline with milestone clinical events. Although this is an ongoing study, recent reports suggest the promise and potential of the HD monkey model in facilitating the finding of HD cures. Nonetheless, future study in a larger cohort of HD monkeys will help in compiling necessary data before cognitive therapies could be effectively tested using only the NHP model.
Psychiatric disturbance is one of the clinical areas that most HD patients also experience (Killoran and Biglan 2012; Raper et al. 2016; Ross 2004; Thompson et al. 2012). Psychiatric impairment may not be linked to other markers of disease course or symptoms, but rather they seem to develop independently (Zappacosta et al. 1996). Symptoms include aggression, irritability, affective disorders, behavioral disorders and personality disorders (Berrios et al. 2001; Bouwens et al. 2015; Dale and van Duijn 2015; Paulsen et al. 2005; Van den Stock et al. 2015). Depression is common (Paulsen et al. 2005), and suicide is increased in HD patients compared to the general population (Di Maio et al. 1993).
Studying the psychiatric status of mice is a challenging task with limitations that might be difficult to overcome. Open field-testing is one of the most common methods to study emotions such as fear. Depression in mice can be studied by using a forced swim test in which “depressed” mice float instead of swimming or trying to climb out. R6/2 mice show increased tendency of floating shortly after symptoms emerge when compared to control mice on a forced swim test, suggesting possible depression (Ciamei et al. 2015). Monitoring home cage behavior such as grooming, fighting and time spent in groups are also commonly used for evaluating mouse psychiatric conditions. However, quantitative measurement of psychiatric behavioral changes such as emotion and mood swing remains challenging and difficult in rodents due to limitation in quantitative tools for measuring progression of a broad spectrum of psychiatric symptoms observed in HD patients.
Assessment of psychiatric changes in NHPs is feasible and well established (Raper et al. 2016). Acute stressor has been successfully used to measure emotional and hormonal responses in nonhuman primates including HD monkeys (Kalin and Shelton 1998; Raper et al. 2016; Raper et al. 2013). HD monkeys were evaluated at five years of age, which is the equivalent to a young adult human. At this age, HD monkeys showed increased coo vocalizations (Fig 6a). Baby monkeys usually use this call to try to re-connect with their family. Using a coo vocalization at this age and in this way suggests increased anxiety (Hauser 1991; Pfefferle et al. 2014; Rowell and Hinde 1962). They also showed less freezing compared to age-matched control monkeys (Fig 6b), but increased hostility (Fig 6c). These two findings combined suggest that, although HD monkeys may exhibit decreased fear, they also have increased anger and aggression (Raper et al. 2016). In fact, one of the HD monkeys, rHD1, had to be stopped from visuospatial cognitive testing due to self-injurious behavior developed during testing (Chan et al. 2015). Study on emotional dysfunctions in adult HD monkeys strongly suggested the development of increased anxiety and irritability/aggression and are parallel with hyperactivity in innate immune response (Raper et al. 2016). Similar observation has also been reported in human HD patients (Bjorkqvist et al. 2008; Bouwens et al. 2015; Dale and van Duijn 2015; Forrest et al. 2010; Paulsen et al. 2005; Shannon and Fraint 2015; Van den Stock et al. 2015; Vassos et al. 2007; Wild and Tabrizi 2014). Due to the high homology in socio-emotional behavior between humans and NHPs (Watson and Platt 2012; Yue et al. 2014), findings in HD monkeys further suggest their potential in replicating progressive psychiatric disturbance in HD patients which is important in understanding HD pathogenesis and developing early diagnostic tools as well as novel cures for HD. One of the ongoing efforts is to develop a cohort of second generation HD monkeys and establish a small social group, thus progressive decline in psychiatric functions and change in social behavior can be closely monitored and possible biological markers can be identified to reveal disease progression and translation readiness.
Fig 6. Psychological symptoms in HD NHPs.
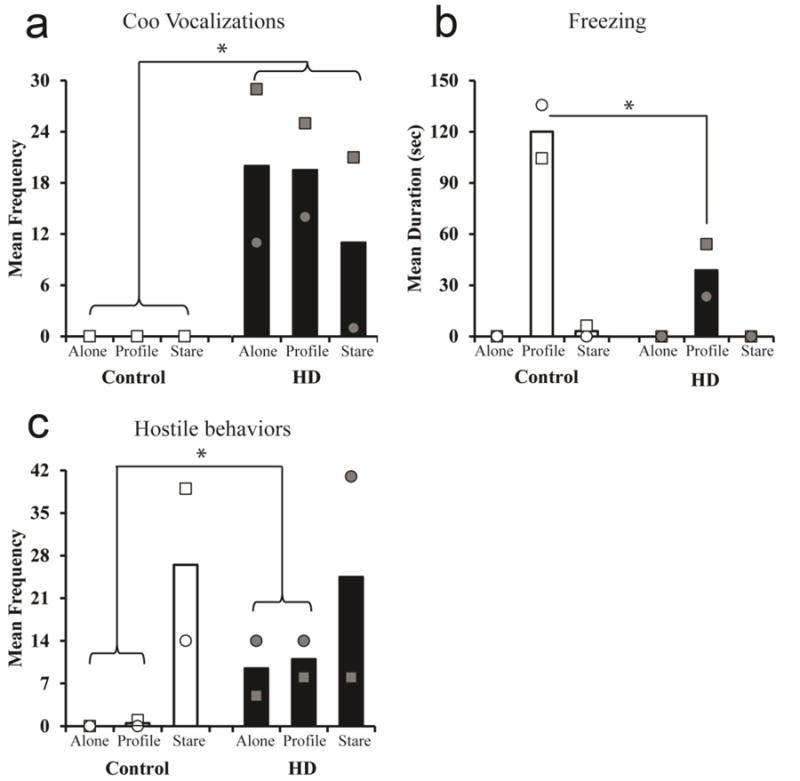
(a) Mature HD NHPs showed an increase in coo vocalizations when alone or introduced to an intruders profile or stare compared to control NHPs. (b) HD NHPs exhibited decreased freezing compared to control NHPs when presented with an intruder profile. (c) Overall, HD NHPs increased in hostile behavior compared to control NHPs when alone or introduced to an intruder’s profile (Raper et al. 2016).
Anxiety, aggression, irritability and self-injurious behavior have all been identified in HD patients and in HD monkeys (Dale and van Duijn 2015; Raper et al. 2016; Van den Stock et al. 2015; Vassos et al. 2007). The HD monkey model provides a unique opportunity and platform for understanding how HD impacts psychiatric behaviors. To date, no effective treatments have been developed using the currently used HD mouse models (Wild and Tabrizi 2014). With further characterization, HD monkeys can be a potential preclinical large animal model to study all three aspects of the disease; motor, cognitive and psychiatric. Thus novel therapeutics, or combinations of therapies, can be evaluated by assessing all symptoms simultaneously.
Discussion
Over the past several decades, NHPs have provided models for various diseases, many of which have resulted in successful therapies and vaccines. For example, deep brain stimulation, a commonly used and highly successful treatment for Parkinson’s disease, was first tested in a NHP model for the disease (Benazzouz et al. 1993) and later corroborated in patients (Limousin et al. 1995). Nonetheless, practicality and ethical boundaries should be cautiously assessed prior to considering the NHP model.
Although HD monkeys may hold great promise, there are limitations in NHP research. Unlike rodents, rhesus macaque has a long pubertal age and gestation time; thus, the breeding of HD monkeys is a relatively challenging, slow and expensive process. Neurodegenerative disease such as HD is a progressive inherited disorder that evolves throughout the lifespan of affected individuals. There is increasing evidence that suggests that motor deficits are preceded by cognitive behavioral dysfunctions and the development of early dysfunctional biomarkers has been the key interest in multiple longitudinal human studies and the development of therapeutic targets. While the development of HD in human patients is a relatively long process dictated by the size of CAG repeats, the benefit of a large animal model such as HD monkeys that is capable of capturing key clinical features with similar disease progression determined by using similar clinical assessment tools for humans may facilitate clinical translation (Howland and Munoz-Sanjuan 2014; Menalled and Brunner 2014; Morton and Howland 2013; Pouladi et al. 2013). Thus the longer developmental course in HD monkeys may allow key clinical development of HD that may not be possible to capture in short lived rodents. The recently established Transgenic Huntington’s Disease Monkey Resource (THDMR) sponsored by the NIH has a specific mission to produce and promote the preclinical applications of the HD monkey model (Chan et al. 2015; Meng et al. 2017; Moran et al. 2015; Raper et al. 2016). Besides the production of HD monkeys, a biological material bank was also established with samples including serum, plasma and cerebrospinal fluid (CSF) that were collected throughout the lifespan of HD monkeys from prodromal to symptomatic stages are available for the HD research community.
Increased interest in cell models for HD may be appropriate for studying the response of isolated neurons in terms of cell death, aggregates, or dystrophia (An et al. 2012; Carter et al. 2014; Kunkanjanawan et al. 2016). However, isolated cells cannot address the complex circuitry of the human brain or, obviously, the symptoms of the disease. Mouse models for the disease may be used to address the neuropathology and motor symptoms, but assessment of cognitive and psychiatric symptoms remains challenging and their translational value has yet to be determined. There is no perfect model of human diseases, as HD rodent and NHP models are equally important for the understanding of HD pathogenesis and the cures for HD. Translational value of rodents has drawn concerns after failure in clinical translation while the increasing interest in developing large preclinical animal models further suggest the potential of the HD monkeys (Menalled and Brunner 2014; Philips et al. 2014; Zeidler et al. 2015). In fact, our HD monkey is the first transgenic NHP model of human disease and is the prototype model of the species. Continued effort in longitudinal assessment and the development of social group study will greatly benefit our understanding on how cognitive behavioral dysregulation has evolved in HD. HD monkeys provide a unique opportunity to investigate some of the most important clinical aspects in HD that are largely unanswered. HD monkeys also allow the testing of therapeutics against the neuropathological, motor, cognitive and psychiatric changes associated with the disease and can be assessed simultaneously using similar human clinical tools. Therefore, one should consider it unethical not to consider the potential of HD monkeys in advancing preclinical research and facilitating clinical translation of novel therapeutics and treatments for HD patients.
Acknowledgments
We thank the Yerkes National Primate Research Center (YNPRC) veterinarian staff, primate enrichment team and animal care personnel for providing superior medical and daily care to HD monkeys as disease progressed. Editorial assistance provided by Ms. Leslee Sinclair. The Transgenic Huntington’s Disease Monkey Resource (THDMR) and this study are supported by a grant awarded by the ORIP/NIH (OD010930) to AWSC. YNPRC is supported by the National Center for Research Resources P51RR165 and is currently supported by the Office of Research and Infrastructure Program (ORIP)/OD P51OD11132.
References
- Aggarwal M, Duan W, Hou Z, Rakesh N, Peng Q, Ross CA, Miller MI, Mori S, Zhang J. Spatiotemporal mapping of brain atrophy in mouse models of Huntington's disease using longitudinal in vivo magnetic resonance imaging. Neuroimage. 2012;60(4):2086–2095. doi: 10.1016/j.neuroimage.2012.01.141. S1053-8119(12)00177-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian G, Barry J, Cepeda C, Levine MS. Altered membrane properties and firing patterns of external globus pallidus neurons in the R6/2 mouse model of Huntington's disease. J Neurosci Res. 2016;94(12):1400–1410. doi: 10.1002/jnr.23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov V, Brunner D, Menalled LB, Kudwa A, Watson-Johnson J, Mazzella M, Russell I, Ruiz MC, Torello J, Sabath E, Sanchez A, Gomez M, Filipov I, Cox K, Kwan M, Ghavami A, Ramboz S, Lager B, Wheeler VC, Aaronson J, Rosinski J, Gusella JF, MacDonald ME, Howland D, Kwak S. Large-scale phenome analysis defines a behavioral signature for Huntington's disease genotype in mice. Nat Biotechnol. 2016;34(8):838–844. doi: 10.1038/nbt.3587. nbt.3587 [pii] [DOI] [PubMed] [Google Scholar]
- Alzheimer A. Uber die anatomische Grundlage der Huntington’schen Chorea und der choreaischen Bewegung uberhaupt. Neurol Centralblatt. 1911;30:891–892. [Google Scholar]
- Ament SA, Pearl JR, Grindeland A, St Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, Kwak S, Howland D, Lee MY, Baxter D, Scherler K, Wang K, Geman D, Carroll JB, MacDonald ME, Carlson G, Wheeler VC, Price ND, Hood LE. High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington's disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet. 2017;26(5):913–922. doi: 10.1093/hmg/ddx006. 3045004 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM. Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11(2):253–263. doi: 10.1016/j.stem.2012.04.026. S1934-5909(12)00337-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994;14(6):3969–3984. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella P. Leading tonically active neurons of the striatum from reward detection to context recognition. Trends Neurosci. 2007;30(6):299–306. doi: 10.1016/j.tins.2007.03.011. doi:S0166-2236(07)00076-8 [pii] 10.1016/j.tins.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5. nrdp20155 [pii] [DOI] [PubMed] [Google Scholar]
- Beglinger LJ, Duff K, Allison J, Theriault D, O’Rourke JJ, Leserman A, Paulsen JS. Cognitive change in patients with Huntington disease on the Repeatable Battery for the Assessment of Neuropsychological Status. J Clin Exp Neuropsychol. 2010;32(6):573–578. doi: 10.1080/13803390903313564. 916351889 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benazzouz A, Gross C, Feger J, Boraud T, Bioulac B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci. 1993;5(4):382–389. doi: 10.1111/j.1460-9568.1993.tb00505.x. [DOI] [PubMed] [Google Scholar]
- Berrios GE, Wagle AC, Markova IS, Wagle SA, Ho LW, Rubinsztein DC, Whittaker J, Ffrench-Constant C, Kershaw A, Rosser A, Bak T, Hodges JR. Psychiatric symptoms and CAG repeats in neurologically asymptomatic Huntington's disease gene carriers. Psychiatry Res. 2001;102(3):217–225. doi: 10.1016/s0165-1781(01)00257-8. doi:S0165-1781(01)00257-8 [pii] [DOI] [PubMed] [Google Scholar]
- Biglan KM, Zhang Y, Long JD, Geschwind M, Kang GA, Killoran A, Lu W, McCusker E, Mills JA, Raymond LA, Testa C, Wojcieszek J, Paulsen JS. Refining the diagnosis of Huntington disease: the PREDICT-HD study. Front Aging Neurosci. 2013;5:12. doi: 10.3389/fnagi.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205(8):1869–1877. doi: 10.1084/jem.20080178. doi:10.1084/jem.20080178jem.20080178 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens JA, van Duijn E, van der Mast RC, Roos RA, Giltay EJ. Irritability in a Prospective Cohort of Huntington's Disease Mutation Carriers. J Neuropsychiatry Clin Neurosci. 2015;27(3):206–212. doi: 10.1176/appi.neuropsych.14030051. [DOI] [PubMed] [Google Scholar]
- Canales JJ, Graybiel AM. A measure of striatal function predicts motor stereotypy. Nat Neurosci. 2000;3(4):377–383. doi: 10.1038/73949. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19(8):3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RL, Chen Y, Kunkanjanawan T, Xu Y, Moran SP, Putkhao K, Yang J, Huang AH, Parnpai R, Chan AW. Reversal of cellular phenotypes in neural cells derived from Huntington's disease monkey-induced pluripotent stem cells. Stem Cell Reports. 2014;3(4):585–593. doi: 10.1016/j.stemcr.2014.07.011. S2213-6711(14)00241-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Galvan L, Holley SM, Rao SP, Andre VM, Botelho EP, Chen JY, Watson JB, Deisseroth K, Levine MS. Multiple sources of striatal inhibition are differentially affected in Huntington's disease mouse models. J Neurosci. 2013;33(17):7393–7406. doi: 10.1523/JNEUROSCI.2137-12.2013. 33/17/7393 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Jiang J, Chen Y, Li C, Prucha MS, Hu Y, Chi T, Moran S, Rahim T, Li S, Li X, Zola SM, Testa CM, Mao H, Villalba R, Smith Y, Zhang X, Bachevalier J. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS One. 2015;10(5):e0122335. doi: 10.1371/journal.pone.0122335. PONE-D-14-45291 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Xu Y, Jiang J, Rahim T, Zhao D, Kocerha J, Chi T, Moran S, Engelhardt H, Larkin K, Neumann A, Cheng H, Li C, Nelson K, Banta H, Zola SM, Villinger F, Yang J, Testa CM, Mao H, Zhang X, Bachevalier J. A two years longitudinal study of a transgenic Huntington disease monkey. BMC Neurosci. 2014;15:36. doi: 10.1186/1471-2202-15-36. doi:10.1186/1471-2202-15-361471-2202-15-36 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciamei A, Detloff PJ, Morton AJ. Progression of behavioural despair in R6/2 and Hdh knock-in mouse models recapitulates depression in Huntington's disease. Behav Brain Res. 2015;291:140–146. doi: 10.1016/j.bbr.2015.05.010. S0166-4328(15)00339-3 [pii] [DOI] [PubMed] [Google Scholar]
- Cloud LJ, Rosenblatt A, Margolis RL, Ross CA, Pillai JA, Corey-Bloom J, Tully HM, Bird T, Panegyres PK, Nichter CA, Higgins DS, Jr, Helmers SL, Factor SA, Jones R, Testa CM. Seizures in juvenile Huntington's disease: frequency and characterization in a multicenter cohort. Mov Disord. 2012;27(14):1797–1800. doi: 10.1002/mds.25237. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron. 2011;69(3):423–435. doi: 10.1016/j.neuron.2010.12.035. S0896-6273(10)01085-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook ZR, Housman DE. Surveying the landscape of Huntington's disease mechanisms, measurements, and medicines. J Huntingtons Dis. 2013;2(4):405–436. doi: 10.3233/JHD-130072. B5813326VH31603H [pii] [DOI] [PubMed] [Google Scholar]
- Dale M, van Duijn E. Anxiety in Huntington's Disease. J Neuropsychiatry Clin Neurosci. 2015;27(4):262–271. doi: 10.1176/appi.neuropsych.14100265. [DOI] [PubMed] [Google Scholar]
- Davies S, Ramsden DB. Huntington's disease. Mol Pathol. 2001;54(6):409–413. doi: 10.1136/mp.54.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. doi: 10.1016/s0092-8674(00)80513-9. doi:S0092-8674(00)80513-9 [pii] [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13(7):281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A. Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27(3):143–164. doi: 10.1016/j.jchemneu.2004.02.005. S0891061804000286 [pii] [DOI] [PubMed] [Google Scholar]
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington's disease. J Med Genet. 1993;30(4):293–295. doi: 10.1136/jmg.30.4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dong G, Gross K, Qiao F, Ferguson J, Callegari EA, Rezvani K, Zhang D, Gloeckner CJ, Ueffing M, Wang H. Calretinin interacts with huntingtin and reduces mutant huntingtin-caused cytotoxicity. J Neurochem. 2012;123(3):437–446. doi: 10.1111/j.1471-4159.2012.07919.x. [DOI] [PubMed] [Google Scholar]
- Duff K, Beglinger LJ, Theriault D, Allison J, Paulsen JS. Cognitive deficits in Huntington's disease on the Repeatable Battery for the Assessment of Neuropsychological Status. J Clin Exp Neuropsychol. 2010;32(3):231–238. doi: 10.1080/13803390902926184. 911813851 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas EM, van den Bogaard SJ, Hart EP, Soeter RP, van Buchem MA, van der Grond J, Rombouts SA, Roos RA. Reduced functional brain connectivity prior to and after disease onset in Huntington's disease. Neuroimage Clin. 2013;2:377–384. doi: 10.1016/j.nicl.2013.03.001. S2213-1582(13)00022-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueredo-Cardenas G, Morello M, Sancesario G, Bernardi G, Reiner A. Colocalization of somatostatin, neuropeptide Y, neuronal nitric oxide synthase and NADPH-diaphorase in striatal interneurons in rats. Brain Res. 1996;735(2):317–324. doi: 10.1016/0006-8993(96)00801-3. doi:0006-8993(96)00801-3 [pii] [DOI] [PubMed] [Google Scholar]
- Fink KD, Rossignol J, Crane AT, Davis KK, Bombard MC, Bavar AM, Clerc S, Lowrance SA, Song C, Lescaudron L, Dunbar GL. Transplantation of umbilical cord-derived mesenchymal stem cells into the striata of R6/2 mice: behavioral and neuropathological analysis. Stem Cell Res Ther. 2013;4(5):130. doi: 10.1186/scrt341. doi:10.1186/scrt341scrt341 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein S, Abbott MH, Chase GA, Jensen BA, Folstein MF. The association of affective disorder with Huntington's disease in a case series and in families. Psychol Med. 1983;13(3):537–542. doi: 10.1017/s0033291700047966. [DOI] [PubMed] [Google Scholar]
- Forrest CM, Mackay GM, Stoy N, Spiden SL, Taylor R, Stone TW, Darlington LG. Blood levels of kynurenines, interleukin-23 and soluble human leucocyte antigen-G at different stages of Huntington's disease. J Neurochem. 2010;112(1):112–122. doi: 10.1111/j.1471-4159.2009.06442.x. JNC6442 [pii] [DOI] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, Kreitzer Control of Basal Ganglia Output by Direct and Indirect Pathway Projection Neurons. J Neurosci. 2013;33(47):18531–18539. doi: 10.1523/JNEUROSCI.1278-13.2013. 33/47/18531 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F, Sohn J, Nakano T, Furuta T, Nakamura KC, Matsuda W, Kaneko T. Exclusive and common targets of neostriatofugal projections of rat striosome neurons: a single neuron-tracing study using a viral vector. Eur J Neurosci. 2011;33(4):668–677. doi: 10.1111/j.1460-9568.2010.07564.x. [DOI] [PubMed] [Google Scholar]
- Geevasinga N, Richards FH, Jones KJ, Ryan MM. Juvenile Huntington disease. J Paediatr Child Health. 2006;42(9):552–554. doi: 10.1111/j.1440-1754.2006.00921.x. doi:JPC921 [pii] 10.1111/j.1440-1754.2006.00921.x. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27(11):2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. EJN6310 [pii] [DOI] [PubMed] [Google Scholar]
- Graveland GA, DiFiglia M. The frequency and distribution of medium-sized neurons with indented nuclei in the primate and rodent neostriatum. Brain Res. 1985;327(1–2):307–311. doi: 10.1016/0006-8993(85)91524-0. doi:0006-8993(85)91524-0 [pii] [DOI] [PubMed] [Google Scholar]
- Graveland GA, Williams RS, DiFiglia M. A Golgi study of the human neostriatum: neurons and afferent fibers. J Comp Neurol. 1985;234(3):317–333. doi: 10.1002/cne.902340304. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Ragsdale CW., Jr Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci U S A. 1978;75(11):5723–5726. doi: 10.1073/pnas.75.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Jie S, Colby DW. Protein misfolding detected early in pathogenesis of transgenic mouse model of Huntington disease using amyloid seeding assay. J Biol Chem. 2012;287(13):9982–9989. doi: 10.1074/jbc.M111.305417. M111.305417 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Groenewegen HJ, Grove EA, Nauta WJ. Efferent connections of the ventral pallidum: evidence of a dual striato pallidofugal pathway. J Comp Neurol. 1985;235(3):322–335. doi: 10.1002/cne.902350304. [DOI] [PubMed] [Google Scholar]
- Hauser MD. Sources of acoustic variation in rhesus macaque (Macaca mulatta) vocalizations. Ethology. 1991;89:29–46. [Google Scholar]
- Hewitt W. The development of the human internal capsule and lentiform nucleus. J Anat. 1961;95:191–199. [PMC free article] [PubMed] [Google Scholar]
- Ho AK, Sahakian BJ, Brown RG, Barker RA, Hodges JR, Ane MN, Snowden J, Thompson J, Esmonde T, Gentry R, Moore JW, Bodner T. Profile of cognitive progression in early Huntington's disease. Neurology. 2003;61(12):1702–1706. doi: 10.1212/01.wnl.0000098878.47789.bd. [DOI] [PubMed] [Google Scholar]
- Holter SM, Stromberg M, Kovalenko M, Garrett L, Glasl L, Lopez E, Guide J, Gotz A, Hans W, Becker L, Rathkolb B, Rozman J, Schrewed A, Klingenspor M, Klopstock T, Schulz H, Wolf E, Wursta W, Gillis T, Wakimoto H, Seidman J, MacDonald ME, Cotman S, Gailus-Durner V, Fuchs H, de Angelis MH, Lee JM, Wheeler VC. A broad phenotypic screen identifies novel phenotypes driven by a single mutant allele in Huntington's disease CAG knock-in mice. PLoS One. 2013;8(11):e80923. doi: 10.1371/journal.pone.0080923. PONE-D-13-35381 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Munoz-Sanjuan I. Mind the gap: models in multiple species needed for therapeutic development in Huntington's disease. Mov Disord. 2014;29(11):1397–1403. doi: 10.1002/mds.26008. [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345(6196):1255263. doi: 10.1126/science.1255263. 1255263 [pii] 345/6196/1255263 [pii] [DOI] [PubMed] [Google Scholar]
- Huntington G. On chorea. Med Surg Reporter. 1872;26(317–321) [Google Scholar]
- Jelgersma G. Uber anatomische Befunde bei Paralyis agitans und bei chronischer Chorea. Neurol Contralblatt. 1908;(27):995–996. [Google Scholar]
- Jennings CG, Landman R, Zhou Y, Sharma J, Hyman J, Movshon JA, Qiu Z, Roberts AC, Roe AW, Wang X, Zhou H, Wang L, Zhang F, Desimone R, Feng G. Opportunities and challenges in modeling human brain disorders in transgenic primates. Nat Neurosci. 2016;19(9):1123–1130. doi: 10.1038/nn.4362. doi:10.1038/nn.4362nn.4362 [pii] [DOI] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE. Ontogeny and stability of separation and threat-induced defensive behaviors in rhesus monkeys during the first year of life. Am J Primatol. 1998;44(2):125–135. doi: 10.1002/(SICI)1098-2345(1998)44:2<125::AID-AJP3>3.0.CO;2-Y. doi:10.1002/(SICI)1098-2345(1998)44:2<125::AID-AJP3>3.0.CO;2-Y [pii]10.1002/(SICI)1098-2345(1998)44:2<125::AID-AJP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18(12):527–535. doi: 10.1016/0166-2236(95)98374-8. doi:0166-2236(95)98374-8 [pii] [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Emson PC. Intracellular recording of identified neostriatal patch and matrix spiny cells in a slice preparation preserving cortical inputs. J Neurophysiol. 1989;62(5):1052–1068. doi: 10.1152/jn.1989.62.5.1052. [DOI] [PubMed] [Google Scholar]
- Killoran A, Biglan KM. Therapeutics in Huntington's Disease. Curr Treat Options Neurol. 2012 doi: 10.1007/s11940-012-0165-x. [DOI] [PubMed] [Google Scholar]
- Kincaid AE, Wilson CJ. Corticostriatal innervation of the patch and matrix in the rat neostriatum. J Comp Neurol. 1996;374(4):578–592. doi: 10.1002/(SICI)1096-9861(19961028)374:4<578::AID-CNE7>3.0.CO;2-Z. doi:10.1002/(SICI)1096-9861(19961028)374:4<578::AID-CNE7>3.0.CO;2-Z [pii] 10.1002/(SICI)1096-9861(19961028)374:4<578::AID-CNE7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Kocerha J, Liu Y, Willoughby D, Chidamparam K, Benito J, Nelson K, Xu Y, Chi T, Engelhardt H, Moran S, Yang SH, Li SH, Li XJ, Larkin K, Neumann A, Banta H, Yang JJ, Chan AW. Longitudinal transcriptomic dysregulation in the peripheral blood of transgenic Huntington's disease monkeys. BMC Neurosci. 2013;14:88. doi: 10.1186/1471-2202-14-88. 1471-2202-14-88 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Tallaksen-Greene S, Crowley MR, Crossman DK, Morton AJ, Van Groen T, Kadish I, Albin RL, Lesort M, Detloff PJ. Allelic series of Huntington's disease knock-in mice reveals expression discorrelates. Hum Mol Genet. 2016;25(8):1619–1636. doi: 10.1093/hmg/ddw040. ddw040 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkanjanawan T, Carter RL, Prucha MS, Yang J, Parnpai R, Chan AW. miR-196a Ameliorates Cytotoxicity and Cellular Phenotype in Transgenic Huntington's Disease Monkey Neural Cells. PLoS One. 2016;11(9):e0162788. doi: 10.1371/journal.pone.0162788. PONE-D-15-49080 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21(23):9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. doi:21/23/9112 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciego JL, Luquin N, Obeso JA. Functional neuroanatomy of the basal ganglia. Cold Spring Harb Perspect Med. 2012;2(12):a009621. doi: 10.1101/cshperspect.a009621. doi:10.1101/cshperspect.a009621a009621 [pii] cshperspect.a009621 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langbehn DR, Hayden MR, Paulsen JS. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):397–408. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange HW, Aulich A. Die Huntingtonsche Krankheit. Hippokrates; Stuttgart: 1986. Die Hirnatrophie bei der Huntingtonschen Keankheit; pp. 25–41. [Google Scholar]
- Lange KW, Sahakian BJ, Quinn NP, Marsden CD, Robbins TW. Comparison of executive and visuospatial memory function in Huntington's disease and dementia of Alzheimer type matched for degree of dementia. J Neurol Neurosurg Psychiatry. 1995;58(5):598–606. doi: 10.1136/jnnp.58.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AD, Hodges JR, Rosser AE, Kershaw A, ffrench-Constant C, Rubinsztein DC, Robbins TW, Sahakian BJ. Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain. 1998;121(Pt 7):1329–1341. doi: 10.1093/brain/121.7.1329. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Hodges JR, Rosser AE, Lange KW, Robbins TW. Executive and mnemonic functions in early Huntington's disease. Brain. 1996;119(Pt 5):1633–1645. doi: 10.1093/brain/119.5.1633. [DOI] [PubMed] [Google Scholar]
- Lee K, Holley SM, Shobe JL, Chong NC, Cepeda C, Levine MS, Masmanidis SC. Parvalbumin Interneurons Modulate Striatal Output and Enhance Performance during Associative Learning. Neuron. 2017;93(6):1451–1463 e1454. doi: 10.1016/j.neuron.2017.02.033. doi:S0896-6273(17)30141-1 [pii] 10.1016/j.neuron.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li XJ. Multiple pathways contribute to the pathogenesis of Huntington disease. Mol Neurodegener. 2006;1:19. doi: 10.1186/1750-1326-1-19. doi:1750-1326-1-19 [pii] 10.1186/1750-1326-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Yu ZX, Li CL, Nguyen HP, Zhou YX, Deng C, Li XJ. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J Neurosci. 2003;23(17):6956–6964. doi: 10.1523/JNEUROSCI.23-17-06956.2003. doi:23/17/6956 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6:22. doi: 10.3389/fnsyn.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limousin P, Pollak P, Benazzouz A, Hoffmann D, Le Bas JF, Broussolle E, Perret JE, Benabid AL. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345(8942):91–95. doi: 10.1016/s0140-6736(95)90062-4. [DOI] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB. Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci. 1999;19(23):10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH, Gray J, Conneally PM, Young A, Penney J, et al. Gametic but not somatic instability of CAG repeat length in Huntington's disease. J Med Genet. 1993;30(12):982–986. doi: 10.1136/jmg.30.12.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/s0092-8674(00)81369-0. doi:S0092-8674(00)81369-0 [pii] [DOI] [PubMed] [Google Scholar]
- Maurice N, Liberge M, Jaouen F, Ztaou S, Hanini M, Camon J, Deisseroth K, Amalric M, Kerkerian-Le Goff L, Beurrier C. Striatal Cholinergic Interneurons Control Motor Behavior and Basal Ganglia Function in Experimental Parkinsonism. Cell Rep. 2015;13(4):657–666. doi: 10.1016/j.celrep.2015.09.034. S2211-1247(15)01045-1 [pii] [DOI] [PubMed] [Google Scholar]
- Menalled L, Brunner D. Animal models of Huntington's disease for translation to the clinic: best practices. Mov Disord. 2014;29(11):1375–1390. doi: 10.1002/mds.26006. [DOI] [PubMed] [Google Scholar]
- Meng Y, Jiang J, Bachevalier J, Zhang X, Chan AW. Developmental Whole Brain White Matter Alterations in Transgenic Huntington's Disease Monkey. Sci Rep. 2017;7(1):379. doi: 10.1038/s41598-017-00381-8. 10.1038/s41598-017-00381-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Chi T, Prucha MS, Ahn KS, Connor-Stroud F, Jean S, Gould K, Chan AW. Germline transmission in transgenic Huntington's disease monkeys. Theriogenology. 2015;84(2):277–285. doi: 10.1016/j.theriogenology.2015.03.016. S0093-691X(15)00143-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Howland DS. Large genetic animal models of Huntington's Disease. J Huntingtons Dis. 2013;2(1):3–19. doi: 10.3233/JHD-130050. L584238235002QV6 [pii] [DOI] [PubMed] [Google Scholar]
- Nance MA, Myers RH. Juvenile onset Huntington’s disease–clinical and research perspectives. Ment Retard Dev Disabil Res Rev. 2001;7(3):153–157. doi: 10.1002/mrdd.1022. doi:10.1002/mrdd.1022 [pii] 10.1002/mrdd.1022. [DOI] [PubMed] [Google Scholar]
- Niu Y, Guo X, Chen Y, Wang CE, Gao J, Yang W, Kang Y, Si W, Wang H, Yang SH, Li S, Ji W, Li XJ. Early Parkinson's disease symptoms in alpha-synuclein transgenic monkeys. Hum Mol Genet. 2015;24(8):2308–2317. doi: 10.1093/hmg/ddu748. ddu748 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F, Ji S, Zhou Q, Huang X, Ji W, Sha J. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell. 2014 doi: 10.1016/j.cell.2014.01.027. doi:S0092-8674(14)00079-8 [pii] 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- Oakeshott S, Farrar A, Port R, Cummins-Sutphen J, Berger J, Watson-Johnson J, Ramboz S, Howland D, Brunner D. Deficits in a Simple Visual Go/No-go Discrimination Task in Two Mouse Models of Huntington's Disease. PLoS Curr. 2013;5 doi: 10.1371/currents.hd.fe74c94bdd446a0470f6f905a30b5dd1ecurrents.hd.fe74c. 94bdd446a0470f6f905a30b5dd1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366(4):580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. doi:10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0 [pii]10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Nehl C, Hoth KF, Kanz JE, Benjamin M, Conybeare R, McDowell B, Turner B. Depression and stages of Huntington's disease. J Neuropsychiatry Clin Neurosci. 2005;17(4):496–502. doi: 10.1176/jnp.17.4.496. doi:17/4/496 [pii] 10.1176/jnp.17.4.496. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Smith MM, Long JD. Cognitive decline in prodromal Huntington Disease: implications for clinical trials. J Neurol Neurosurg Psychiatry. 2013;84(11):1233–1239. doi: 10.1136/jnnp-2013-305114. jnnp-2013-305114 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peavy GM, Jacobson MW, Goldstein JL, Hamilton JM, Kane A, Gamst AC, Lessig SL, Lee JC, Corey-Bloom J. Cognitive and functional decline in Huntington's disease: dementia criteria revisited. Mov Disord. 2010;25(9):1163–1169. doi: 10.1002/mds.22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny GR, Wilson CJ, Kitai ST. Relationship of the axonal and dendritic geometry of spiny projection neurons to the compartmental organization of the neostriatum. J Comp Neurol. 1988;269(2):275–289. doi: 10.1002/cne.902690211. [DOI] [PubMed] [Google Scholar]
- Petryszyn S, Beaulieu JM, Parent A, Parent M. Distribution and morphological characteristics of striatal interneurons expressing calretinin in mice: a comparison with human and nonhuman primates. J Chem Neuroanat. 2014;59–60:51–61. doi: 10.1016/j.jchemneu.2014.06.002. S0891-0618(14)00043-X [pii] [DOI] [PubMed] [Google Scholar]
- Petryszyn S, Parent A, Parent M. The calretinin interneurons of the striatum: comparisons between rodents and primates under normal and pathological conditions. J Neural Transm (Vienna) 2017 doi: 10.1007/s00702-017-1687-x. doi:10.1007/s00702-017-1687-x10.1007/s00702-017-1687-x [pii] [DOI] [PubMed] [Google Scholar]
- Pfefferle D, Kazem AJ, Brockhausen RR, Ruiz-Lambides AV, Widdig A. Monkeys spontaneously discriminate their unfamiliar paternal kin under natural conditions using facial cues. Curr Biol. 2014;24(15):1806–1810. doi: 10.1016/j.cub.2014.06.058. doi:10.1016/j.cub.2014.06.058S0960-9822(14)00774-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Rothstein JD, Pouladi MA. Preclinical models: needed in translation? A Pro/Con debate. Mov Disord. 2014;29(11):1391–1396. doi: 10.1002/mds.26010. [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013;14(10):708–721. doi: 10.1038/nrn3570. doi:10.1038/nrn3570nrn3570 [pii] [DOI] [PubMed] [Google Scholar]
- Preisig DF, Kulic L, Kruger M, Wirth F, McAfoose J, Spani C, Gantenbein P, Derungs R, Nitsch RM, Welt T. High-speed video gait analysis reveals early and characteristic locomotor phenotypes in mouse models of neurodegenerative movement disorders. Behav Brain Res. 2016;311:340–353. doi: 10.1016/j.bbr.2016.04.044. S0166-4328(16)30259-5 [pii] [DOI] [PubMed] [Google Scholar]
- Putkhao K, Kocerha J, Cho IK, Yang J, Parnpai R, Chan AW. Pathogenic cellular phenotypes are germline transmissible in a transgenic primate model of Huntington's disease. Stem Cells Dev. 2013;22(8):1198–1205. doi: 10.1089/scd.2012.0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper J, Bosinger S, Johnson Z, Tharp G, Moran SP, Chan AW. Increased irritability, anxiety, and immune reactivity in transgenic Huntington's disease monkeys. Brain Behav Immun. 2016;58:181–190. doi: 10.1016/j.bbi.2016.07.004. doi:S0889-1591(16)30198-2 [pii]10.1016/j.bbi.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper J, Wilson M, Sanchez M, Machado CJ, Bachevalier J. Pervasive alterations of emotional and neuroendocrine responses to an acute stressor after neonatal amygdala lesions in rhesus monkeys. Psychoneuroendocrinology. 2013;38(7):1021–1035. doi: 10.1016/j.psyneuen.2012.10.008. S0306-4530(12)00343-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribai P, Nguyen K, Hahn-Barma V, Gourfinkel-An I, Vidailhet M, Legout A, Dode C, Brice A, Durr A. Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch Neurol. 2007;64(6):813–819. doi: 10.1001/archneur.64.6.813. doi:64/6/813 [pii] 10.1001/archneur.64.6.813. [DOI] [PubMed] [Google Scholar]
- Roseberry TK, Lee AM, Lalive AL, Wilbrecht L, Bonci A, Kreitzer AC. Cell-Type-Specific Control of Brainstem Locomotor Circuits by Basal Ganglia. Cell. 2016;164(3):526–537. doi: 10.1016/j.cell.2015.12.037. doi: 0.1016/j.cell.2015.12.037S0092-8674(15)01701-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5(1):40. doi: 10.1186/1750-1172-5-40. doi:1750-1172-5-40 [pii] 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA. Huntington's disease: new paths to pathogenesis. Cell. 2004;118(1):4–7. doi: 10.1016/j.cell.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–216. doi: 10.1038/nrneurol.2014.24. doi:10.1038/nrneurol.2014.24nrneurol.2014.24 [pii] [DOI] [PubMed] [Google Scholar]
- Rowell TE, Hinde RA. Vocal communication by the rhesus monkey (Macaca mulatta) P Zoological Society of London. 1962;138:279–294. [Google Scholar]
- Rub U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D, Paulson HL, Bohl J, von Gall C, Vonsattel JP, Korf HW, den Dunnen WF. Degeneration of the cerebellum in Huntington's disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol. 2013;23(2):165–177. doi: 10.1111/j.1750-3639.2012.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Seidel K, Heinsen H, Vonsattel JP, den Dunnen WF, Korf HW. Huntington's disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016;26(6):726–740. doi: 10.1111/bpa.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Seidel K, Vonsattel JP, Lange HW, Eisenmenger W, Gotz M, Del Turco D, Bouzrou M, Korf HW, Heinsen H. Huntington's Disease (HD): Neurodegeneration of Brodmann's Primary Visual Area 17 (BA17) Brain Pathol. 2015;25(6):701–711. doi: 10.1111/bpa.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42(4):604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- Sato F, Parent A. Efferent projections of single neurons in the external segment of the globus pallidus in monkeys. Neuroscience Research. 1998;31(1):S173. [Google Scholar]
- Saydoff JA, Garcia RA, Browne SE, Liu L, Sheng J, Brenneman D, Hu Z, Cardin S, Gonzalez A, von Borstel RW, Gregorio J, Burr H, Beal MF. Oral uridine pro-drug PN401 is neuroprotective in the R6/2 and N171-82Q mouse models of Huntington's disease. Neurobiol Dis. 2006;24(3):455–465. doi: 10.1016/j.nbd.2006.08.011. doi:S0969-9961(06)00196-3 [pii]10.1016/j.nbd.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8(3):397–407. doi: 10.1093/hmg/8.3.397. doi:ddc057 [pii] [DOI] [PubMed] [Google Scholar]
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington's disease. J Neurol Neurosurg Psychiatry. 1984;47(12):1283–1287. doi: 10.1136/jnnp.47.12.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS. Longitudinal topography and interdigitation of corticostriatal projections in the rhesus monkey. J Neurosci. 1985;5(3):776–794. doi: 10.1523/JNEUROSCI.05-03-00776.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon KM, Fraint A. Therapeutic advances in Huntington's Disease. Mov Disord. 2015;30(11):1539–1546. doi: 10.1002/mds.26331. [DOI] [PubMed] [Google Scholar]
- Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol. 1999;156(1):92–99. doi: 10.1006/exnr.1998.7005. doi:S0014-4886(98)97005-4 [pii]10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102(32):11402–11407. doi: 10.1073/pnas.0503634102. doi:0503634102 [pii]10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Wichmann T, DeLong MR. Corticostriatal and mesocortical dopamine systems: do species differences matter? Nat Rev Neurosci. 2014;15(1):63. doi: 10.1038/nrn3469-c1. doi:10.1038/nrn3469-c1nrn3469-c1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat Genet. 1993;4(4):393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- Solomon AC, Stout JC, Johnson SA, Langbehn DR, Aylward EH, Brandt J, Ross CA, Beglinger L, Hayden MR, Kieburtz K, Kayson E, Julian-Baros E, Duff K, Guttman M, Nance M, Oakes D, Shoulson I, Penziner E, Paulsen JS. Verbal episodic memory declines prior to diagnosis in Huntington's disease. Neuropsychologia. 2007;45(8):1767–1776. doi: 10.1016/j.neuropsychologia.2006.12.015. doi:S0028-3932(07)00008-5 [pii]10.1016/j.neuropsychologia.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell AL, Ko J, Patterson PH. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington's disease. J Neurosci. 2009;29(43):13589–13602. doi: 10.1523/JNEUROSCI.4286-09.2009. 29/43/13589 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490(4):354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Stout JC, Jones R, Labuschagne I, O’Regan AM, Say MJ, Dumas EM, Queller S, Justo D, Santos RD, Coleman A, Hart EP, Durr A, Leavitt BR, Roos RA, Langbehn DR, Tabrizi SJ, Frost C. Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington's disease. J Neurol Neurosurg Psychiatry. 2012;83(7):687–694. doi: 10.1136/jnnp-2011-301940. doi:10.1136/jnnp-2011-301940jnnp-2011-301940 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout JC, Paulsen JS, Queller S, Solomon AC, Whitlock KB, Campbell JC, Carlozzi N, Duff K, Beglinger LJ, Langbehn DR, Johnson SA, Biglan KM, Aylward EH. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. 2011;25(1):1–14. doi: 10.1037/a0020937. 2010-20839-001 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, Kennard C, Landwehrmeyer B, Stout JC, Borowsky B, Scahill RI, Frost C, Langbehn DR. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11(1):42–53. doi: 10.1016/S1474-4422(11)70263-0. S1474-4422(11)70263-0 [pii] [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12(7):637–649. doi: 10.1016/S1474-4422(13)70088-7. S1474-4422(13)70088-7 [pii] [DOI] [PubMed] [Google Scholar]
- Tepper JM, Bolam JP. Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol. 2004;14(6):685–692. doi: 10.1016/j.conb.2004.10.003. doi:S0959-4388(04)00155-2 [pii]10.1016/j.conb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koos T, Ibanez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. doi: 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JC, Harris J, Sollom AC, Stopford CL, Howard E, Snowden JS, Craufurd D. Longitudinal evaluation of neuropsychiatric symptoms in Huntington's disease. J Neuropsychiatry Clin Neurosci. 2012;24(1):53–60. doi: 10.1176/appi.neuropsych.11030057. [DOI] [PubMed] [Google Scholar]
- Tulloch IF, Arbuthnott GW, Wright AK. Topographical organization of the striatonigral pathway revealed by anterograde and retrograde neuroanatomical tracing techniques. J Anat. 1978;127(Pt 2):425–441. [PMC free article] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2000;97(14):8093–8097. doi: 10.1073/pnas.110078997. 110078997 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino AL, Sills T, Anderson KE, Borowsky B, Craufurd D, Giuliano J, Goodman L, Guttman M, Kupchak P, Ho AK, Paulsen JS, J CS, van Kammen DP, Evans K. Assessment of cognitive symptoms in prodromal and early huntington disease. PLoS Curr. 2011;3:RRN1250. doi: 10.1371/currents.RRN1250. doi:10.1371/currents.RRN1250 k/-/-/19jerwgzmryar/16 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Stock J, De Winter FL, Ahmad R, Sunaert S, Van Laere K, Vandenberghe W, Vandenbulcke M. Functional brain changes underlying irritability in premanifest Huntington's disease. Hum Brain Mapp. 2015;36(7):2681–2690. doi: 10.1002/hbm.22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk JG, van der Velde EA, Roos RA, Bruyn GW. Juvenile Huntington disease. Human genetics. 1986;73(3):235–239. doi: 10.1007/BF00401235. [DOI] [PubMed] [Google Scholar]
- Vassos E, Panas M, Kladi A, Vassilopoulos D. Higher levels of extroverted hostility detected in gene carriers at risk for Huntington's disease. Biol Psychiatry. 2007;62(12):1347–1352. doi: 10.1016/j.biopsych.2006.12.016. doi:S0006-3223(06)01583-6 [pii]10.1016/j.biopsych.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):55–69. doi: 10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44(6):559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wade A, Jacobs P, Morton AJ. Atrophy and degeneration in sciatic nerve of presymptomatic mice carrying the Huntington's disease mutation. Brain Res. 2008;1188:61–68. doi: 10.1016/j.brainres.2007.06.059. doi:S0006-8993(07)01554-5 [pii] 10.1016/j.brainres.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet. 2008;17(17):2738–2751. doi: 10.1093/hmg/ddn175. ddn175 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50(3):443–452. doi: 10.1016/j.neuron.2006.04.010. doi:S0896-6273(06)00278-9 [pii] 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Watson KK, Platt ML. Of mice and monkeys: using non-human primate models to bridge mouse- and human-based investigations of autism spectrum disorders. J Neurodev Disord. 2012;4(1):21. doi: 10.1186/1866-1955-4-21. doi:1866-1955-4-21 [pii] 10.1186/1866-1955-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9(4):503–513. doi: 10.1093/hmg/9.4.503. doi:ddd068 [pii] [DOI] [PubMed] [Google Scholar]
- Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington's disease: what's in the pipeline? Mov Disord. 2014;29(11):1434–1445. doi: 10.1002/mds.26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Parent A. Striatal interneurons expressing calretinin, parvalbumin or NADPH-diaphorase: a comparative study in the rat, monkey and human. Brain Res. 2000;863(1–2):182–191. doi: 10.1016/s0006-8993(00)02135-1. doi:S0006-8993(00)02135-1 [pii] [DOI] [PubMed] [Google Scholar]
- Xu M, Li L, Pittenger C. Ablation of fast-spiking interneurons in the dorsal striatum, recapitulating abnormalities seen post-mortem in Tourette syndrome, produces anxiety and elevated grooming. Neuroscience. 2016;324:321–329. doi: 10.1016/j.neuroscience.2016.02.074. S0306-4522(16)00227-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Cheng PH, Banta H, Piotrowska-Nitsche K, Yang JJ, Cheng EC, Snyder B, Larkin K, Liu J, Orkin J, Fang ZH, Smith Y, Bachevalier J, Zola SM, Li SH, Li XJ, Chan AW. Towards a transgenic model of Huntington's disease in a nonhuman primate. Nature. 2008;453(7197):921–924. doi: 10.1038/nature06975. doi:10.1038/nature06975nature06975 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeterian EH, Van Hoesen GW. Cortico-striate projections in the rhesus monkey: the organization of certain cortico-caudate connections. Brain Res. 1978;139(1):43–63. doi: 10.1016/0006-8993(78)90059-8. doi:0006-8993(78)90059-8 [pii] [DOI] [PubMed] [Google Scholar]
- Yoon G, Kramer J, Zanko A, Guzijan M, Lin S, Foster-Barber A, Boxer AL. Speech and language delay are early manifestations of juvenile-onset Huntington disease. Neurology. 2006;67(7):1265–1267. doi: 10.1212/01.wnl.0000238390.86304.4e. doi:67/7/1265 [pii]10.1212/01.wnl.0000238390.86304.4e. [DOI] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, Shen Y, Pervouchine DD, Djebali S, Thurman RE, Kaul R, Rynes E, Kirilusha A, Marinov GK, Williams BA, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See LH, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K, Bender MA, Zhang M, Byron R, Groudine MT, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu YC, Rasmussen MD, Bansal MS, Kellis M, Keller CA, Morrissey CS, Mishra T, Jain D, Dogan N, Harris RS, Cayting P, Kawli T, Boyle AP, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi VS, Cline MS, Erickson DT, Kirkup VM, Learned K, Sloan CA, Rosenbloom KR, Lacerda de Sousa B, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent WJ, Ramalho Santos M, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo PJ, Wilken MS, Reh TA, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds AP, Neph S, Humbert R, Hansen RS, De Bruijn M, Selleri L, Rudensky A, Josefowicz S, Samstein R, Eichler EE, Orkin SH, Levasseur D, Papayannopoulou T, Chang KH, Skoultchi A, Gosh S, Disteche C, Treuting P, Wang Y, Weiss MJ, Blobel GA, Cao X, Zhong S, Wang T, Good PJ, Lowdon RF, Adams LB, Zhou XQ, Pazin MJ, Feingold EA, Wold B, Taylor J, Mortazavi A, Weissman SM, Stamatoyannopoulos JA, Snyder MP, Guigo R, Gingeras TR, Gilbert DM, Hardison RC, Beer MA, Ren B. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515(7527):355–364. doi: 10.1038/nature13992. nature13992 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappacosta B, Monza D, Meoni C, Austoni L, Soliveri P, Gellera C, Alberti R, Mantero M, Penati G, Caraceni T, Girotti F. Psychiatric symptoms do not correlate with cognitive decline, motor symptoms, or CAG repeat length in Huntington's disease. Arch Neurol. 1996;53(6):493–497. doi: 10.1001/archneur.1996.00550060035012. [DOI] [PubMed] [Google Scholar]
- Zeidler S, Hukema RK, Willemsen R. The quest for targeted therapy in fragile X syndrome. Expert Opin Ther Targets. 2015;19(10):1277–1281. doi: 10.1517/14728222.2015.1079176. [DOI] [PubMed] [Google Scholar]
- Zizak VS, Filoteo JV, Possin KL, Lucas JA, Rilling LM, Davis JD, Peavy G, Wong A, Salmon DP. The ubiquity of memory retrieval deficits in patients with frontal-striatal dysfunction. Cogn Behav Neurol. 2005;18(4):198–205. doi: 10.1097/01.wnn.0000192134.53616.39. doi:00146965-200512000-00003 [pii] [DOI] [PubMed] [Google Scholar]