

Abstract
Objective
Fibroblast‐like synoviocytes (FLS) produce key synovial fluid and tissue components to ensure joint integrity under healthy conditions, whereas they become cancer‐like and aggressively contribute to joint degeneration in inflammatory arthritis. The aim of this study was to determine whether the SOXC transcription factors SOX4 and SOX11, whose functions are critical in joint development and many cancer types, contribute to FLS activities under normal and inflammatory conditions.
Methods
We inactivated the SOXC genes in FLS from adult mice and studied the effect on joint homeostasis and tumor necrosis factor (TNF)–induced arthritis. We used primary cells and synovial biopsy specimens from arthritis patients to analyze the interactions between inflammatory signals and SOXC proteins.
Results
Postnatal inactivation of the SOXC genes had no major effect on joint integrity in otherwise healthy mice. However, it hampered synovial hyperplasia and joint degeneration in transgenic mice expressing human TNF. These effects were explained by the ability of SOX4/11 to amplify the pathogenic impact of TNF on FLS by increasing their survival and migration. SOXC RNA levels were not changed by TNF and other proinflammatory cytokines, but SOXC proteins were strongly stabilized and able to potentiate the TNF‐induced up‐regulation of genes involved in FLS transformation. Substantiating the relevance of these findings in human disease, SOXC protein levels, but not RNA levels, were significantly higher in inflamed synovium than in noninflamed synovium from arthritis patients.
Conclusion
SOXC proteins are targets and pivotal mediators of proinflammatory cytokines during FLS transformation in arthritic diseases. Targeting of these proteins could thus improve current strategies to treat arthritic diseases and possibly other inflammatory diseases.
The internal compartment of joint capsules, known as the synovium, is composed of synovial lining, which is rich in fibroblasts, and synovial intima, a loose connective tissue with interspersed fibroblasts and endothelial cells. The synovial lining and intima fibroblasts are collectively referred to as fibroblast‐like synoviocytes (FLS). Under healthy conditions, FLS produce major constituents of the synovial fluid and synovial tissue extracellular matrix, including lubricin (encoded by Prg4), hyaluronan, and heparan sulfate proteoglycans 1, 2. Apart from ensuring joint health, FLS are also known to be transformed into cells that actively contribute to the pathogenesis of chronic inflammatory joint diseases, including but not limited to rheumatoid arthritis (RA), psoriatic arthritis, and gout 3. They also contribute to the synovitis that worsens cartilage degeneration in osteoarthritis (OA) 4.
In all of these diseases, proinflammatory cytokines transform FLS into aggressive, joint‐destroying cells that increase in numbers, invade joint spaces and tissues, and produce enzymes that directly participate in the degradation of articular cartilage and subchondral bone. These cells also secrete cytokines and chemoattractants that recruit immune cells into the hypertrophied granulation synovial tissue (i.e., synovial pannus formation). In this transformed state, FLS strikingly resemble metastatic cancer cells 3, 4, 5, 6. To date, therapies directly aimed at preventing or reverting FLS transformation have not been developed. The lack of such therapies is largely attributable to insufficient understanding of the molecular mechanisms underlying FLS behavior under both healthy and transformed conditions.
SOX4, SOX11, and SOX12 form group C of the transcription factors containing a SOX DNA‐binding domain 7, 8. SOXC proteins share more identity with one another than with other SOX proteins, and they possess a unique type of transactivation domain. This domain is more potent in SOX4 and SOX11 than in SOX12 9. Sox4 and Sox11 are primarily coexpressed in various types of multipotent progenitor cells, and they were shown in mice to act largely in redundancy to determine the behavior and survival of these cells. Sox4‐null, Sox11‐null, and Sox4/11–double‐ conditional‐ null mutant mice therefore exhibit major defects in the development of organs such as the skeleton, heart, brain, and eyes 10, 11, 12, 13, 14, 15. Although Sox12 is often coexpressed with Sox4 and Sox11, it provides only a minor contribution to these functions, and Sox12‐null mice develop normally throughout life 10. SOX4 and SOX11 have been shown to be highly expressed in prostate, breast, leukemia, colorectal, and other forms of cancer in humans. SOX4 has been recognized as a master regulator of cell proliferation and metastasis in several cancer types, with SOX11 recognized as a poor prognosis marker in lymphoma and breast cancer subtypes 16, 17, 18, 19, 20. To date, however, the importance of SOXC proteins in other adult tissue in physiologic and pathologic processes remains largely unknown.
We previously showed that conditional inactivation of the SOXC genes in mouse embryos drastically affected the survival and fate determination of skeletogenic cells and resulted in a cartilaginous skeleton that failed to grow and ossify and that also completely lacked joints 12. We therefore speculated that the SOXC genes could also have critical roles in the adult skeleton. In the current study, focusing on FLS and using mouse and in vitro models as well as biopsy specimens from patients with arthritis, we demonstrated that SOX4 and SOX11 are direct targets and mediators of proinflammatory cytokines and are thereby required for the cancer‐like transformation of FLS that occurs in arthritic joints.
Materials and methods
Mice. Mice were used as approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Mice carrying Sox4– and Sox11–conditional null alleles and Sox12‐null alleles were previously described 11, 21. The Sox4– and Sox11–conditional null alleles contain loxP sites that flank the entire coding region of the genes. Thus, upon recombination by Cre recombinase, no protein is produced from these alleles. Mice expressing human tumor necrosis factor (TNF) carried 2 transgenes that enable tetracycline‐on (Tet–On)– mediated regulation of TNF expression. 22. The first transgene expresses reverse tetracycline trans activator 2S (rtTA2S) protein. The second transgene is designed to express human TNF protein only when rtTA2S protein is bound by tetracycline compounds. The mice received drinking water supplemented with 0.5 mg doxycycline (a tetracycline compound)/ml (Sigma) and 5% sucrose for 2 weeks. Prg4 CreERt2 mice have been previously described 23. Cre recombinase activity was induced by administering intraperitoneal injections of 1 mg tamoxifen (Sigma‐Aldrich) diluted in olive oil/10 gm body weight.
Mouse FLS cultures. FLS were isolated by digesting interphalangeal joint synovium with 1 mg/ml of collagenase IV (Sigma‐Aldrich) for 2 hours at 37°C 24. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum (FCS; Invitrogen) and used at passage 4. They were immunophenotyped using an LSRII flow cytometer (Becton Dickinson) and fluorescein isothiocyanate–labeled CD90.2 and allophycocyanin‐conjugated CD14 antibodies (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract).
Histology and RNA in situ hybridization. Paraffin‐embedded sections of mouse limbs were generated after tissue fixation in 4% paraformaldehyde for 48 hours, followed by demineralization in 0.5M EDTA (pH 7.5) for 2 weeks. The sections were deparaffinized with Histo‐Clear (National Diagnostics) and rehydrated in graded ethanol solutions. Staining with Lerner 3 Hematoxylin (Lerner Laboratories) and Eosin Y (Polysciences) was performed according to the instructions of the manufacturers. Safranin O (0.1% in water; Sigma‐Aldrich) was used to stain cartilage proteoglycans. RNA in situ hybridization was performed using digoxigenin‐labeled and BM‐Purple substrate (Roche) 25. Sox4, Sox11, and Prg4 antisense RNA probes were previously described 9, 26. Cell death was assessed with a TUNEL fluorescence imaging kit (Invitrogen). Cell proliferation was detected by EdU incorporation following intraperitoneal injection of EdU (5 mg/10 gm body weight; Invitrogen) and detected using a Click‐iT assay (Invitrogen). Slides were mounted using Permount (Thermo Fisher Scientific) or DAPI‐containing Vectashield (Vector). The slides were visualized with a Leica DM2500 microscope and captured using a digital camera (QImaging MicroPublisher 5.0 with Real‐Time Viewing) with a 10× objective lens. All images were acquired using QCapture Pro 6.0 (QImaging) and processed with Adobe Photoshop 7.0 software.
Human synovium and FLS. Synovium specimens were collected from OA patients undergoing total knee arthroplasty, with approval from the Cleveland Clinic Institutional Review Board (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). Inflamed synovium and noninflamed synovium were distinguished based on gross appearance and increased expression of interleukin‐6 (IL‐6). FLS were isolated by digesting tissue for 3 hours with 1 mg/ml of collagenase IV solution 24. The FLS were cultured in DMEM containing 10% FCS and used at passage 4. RA FLS were generated by digesting synovial biopsy specimens from patients with RA, which were obtained by arthroscopic biopsy performed at the Academic Medical Centre Amsterdam 27. The FLS were used in experiments at passage 6 or 7.
In vitro assays. Replication‐deficient adenoviral particles encoding FLAG‐SOX11, Escherichia coli β‐galactosidase (lacZ product), or Cre recombinase were generated at the University of Iowa Gene Transfer Vector Core 12. Cells were infected with 100 plaque‐forming units/cell. For transient transfection of plasmids encoding FLAG‐SOX4 and FLAG‐SOX11, 3 × 105 SW‐982 cells (human synovial fibroblasts; ATCC) were exposed to mixtures containing 1 μg of plasmid and 3 μl of FuGENE 6 (Roche) for up to 24 hours 9. Cells were treated with recombinant human TNF, IL‐1α, IL‐6, or IL‐10 (R&D Systems), MG‐132, and cycloheximide (Sigma‐Aldrich), as described in Results. Cell migration was assessed using a scratch assay. FLS were grown to confluence in 12‐well dishes in DMEM containing 10% FCS, followed by overnight culture in medium containing 0.5% FCS. Wounding was performed with a 200‐μl pipette tip. Cells were treated with recombinant proteins for 4 hours, fixed in 4% formaldehyde, and stained with trypan blue. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and semi‐dry Western blotting on nitrocellulose membranes (Li‐Cor) were performed under standard conditions (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). Signals were detected using ECL Prime Western Blotting Detection Reagent (Life Technologies).
Quantitative reverse transcription–polymerase chain reaction (RT‐PCR). Total RNA was prepared using TRIzol (Life Technologies) and an RNeasy Mini Kit (Qiagen). Complementary DNA was synthesized with a SuperScript IV First‐Strand Synthesis System (Life Technologies) and amplified with primers (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract) using SYBR Green PCR Master Mix (Applied Biosystems). PCR conditions were as follows: 1 cycle at 95°C for 10 seconds followed by 40 cycles at 95°C for 5 seconds and 60°C for 30 seconds. Relative messenger RNA (mRNA) levels were calculated using the ∆Ct method.
Statistical analysis. All data presented are representative of ≥2 independent experiments. Quantitative data are presented as the mean ± SD of at least triplicate biologic samples. Student's 2‐tailed t‐test was used to calculate the significance of sample differences in mouse and in vitro assays. A paired 2‐tailed t‐test was used to calculate the significance of differences between inflamed and noninflamed human synovial tissue. P values less than 0.05 were considered significant.
Results
Expression of Sox4 and Sox11 in adult mouse FLS. We began the study by analyzing the expression of Sox4 and Sox11 in the joints of 6‐week‐old mice. Quantitative RT‐PCR assays revealed that both genes were expressed in synovial tissue (Figure 1A). Their proteins were readily detected on Western blots, using a SOX4‐specific antibody and a pan–SOXC antibody, which binds SOX11 more efficiently than SOX4 (Figure 1B; see also Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). This pan–SOXC antibody did not permit distinguishing SOX11 from SOX4, because both proteins exhibited an apparent molecular weight of 72 kd on SDS‐PAGE. RNA in situ hybridization showed that the expression domains of Sox4 and Sox11 overlapped with that of Prg4 (encoding lubricin) in the synovial lining and also included the synovial intima and articular cartilage (Figure 1C).
Figure 1.
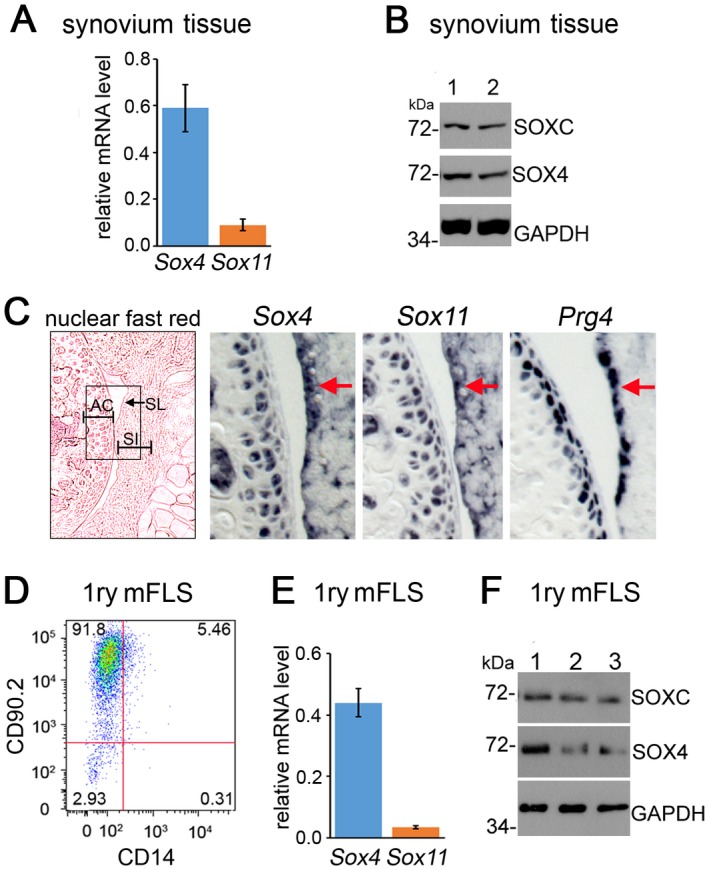
SOX4 and SOX11 are expressed in mouse fibroblast‐like synoviocytes (mFLS). A, Levels of Sox4 and Sox11 mRNA in synovial tissue from 6‐week‐old mice, as measured by quantitative reverse transcription–polymerase chain reaction and normalized to GAPDH mRNA levels. B, Expression of SOXC and SOX4 protein in whole synovium lysates from 2 wild‐type mice (mice 1 and 2). Western blots were hybridized with SOX4‐specific and pan–SOXC antibodies. GAPDH was used as the loading control. The molecular weights of protein standards running close to the proteins of interest are indicated. C, RNA in situ hybridization in adjacent knee sections from 6‐week‐old mice, using Sox4, Sox11, and Prg4 RNA probes. A nuclear fast red–stained section is shown in the left panel (original magnification × 5); the boxed area indicates the areas that are shown at higher magnification (original magnification × 20) in the middle and right panels. Arrows indicate synovial lining (SL). D, Flow cytometric phenotyping of mouse primary synovial cells at passage 4, using fluorescein isothiocyanate–conjugated CD90.2 and allophycocyanin‐conjugated CD14 antibodies. E, Levels of Sox4 and Sox11 mRNA in primary (1ry) mFLS similar to those in D. F, Detection of SOX4 and SOX11 proteins on Western blots of whole lysates from the immunophenotyped cells in D. Values in A and E are the mean ± SD. AC = articular cartilage; SI = synovial intima.
FLS account for the majority of cells present in healthy synovium 2. Thus, they likely generated most, if not all, of the signals detected for SOX4 and SOX11 in whole‐synovium samples. To support this conclusion, we established primary cultures of adherent synovial cells at passage 4. Flow cytometric analysis indicated that most cells were FLS, because 91.8% expressed the fibroblast cell surface marker CD90.2, and only 5.46% expressed the CD14 macrophage marker (Figure 1D). Sox4 and Sox11 mRNAs were detected in these cells (Figures 1E and F). We concluded that Sox4 and Sox11 are expressed in FLS and therefore could have important roles in FLS physiology in adult mice.
SOXC expression in the Prg4 + cell lineage may be dispensable for joint integrity in young adult mice. To test whether SOXC genes have a role in Prg4 + cells in the joints of juvenile and young adult mice, we generated mice harboring Sox4– and Sox11–conditional null alleles, Sox12‐null alleles, and a Prg4 allele with a GFPCreERt2 cassette inserted into its translation initiation codon. This Prg4 CreERt2 allele was previously shown to generate tamoxifen‐dependent Cre recombinase activity in the synovial lining, superficial articular chondrocytes, and tendons of adult mice 23. SOXC mutant mice and age/sex‐matched control mice were injected with tamoxifen at 3–4 weeks of age and analyzed for joint defects at 2 months of age (Figure 2A). RNA in situ hybridization showed that, as expected, the expression of Sox4 and Sox11 was down‐regulated in synovium and articular chondrocytes from Sox4 fl/fl 11 fl/fl 12 –/– Prg4 CreERt2 mouse knees (Figure 2B). As evaluated by quantitative RT‐PCR assays, the efficiency of Sox4 and Sox11 inactivation in the synovium was 65–75%, whereas Prg4 expression was not changed (Figure 2C). Moreover, no obvious histologic changes were seen in the synovial lining and articular cartilage (Figures 2D and E). Taken together, these data suggested that the SOXC genes in synovium and cartilage‐lining cells are dispensable for joint integrity in young adult mice.
Figure 2.
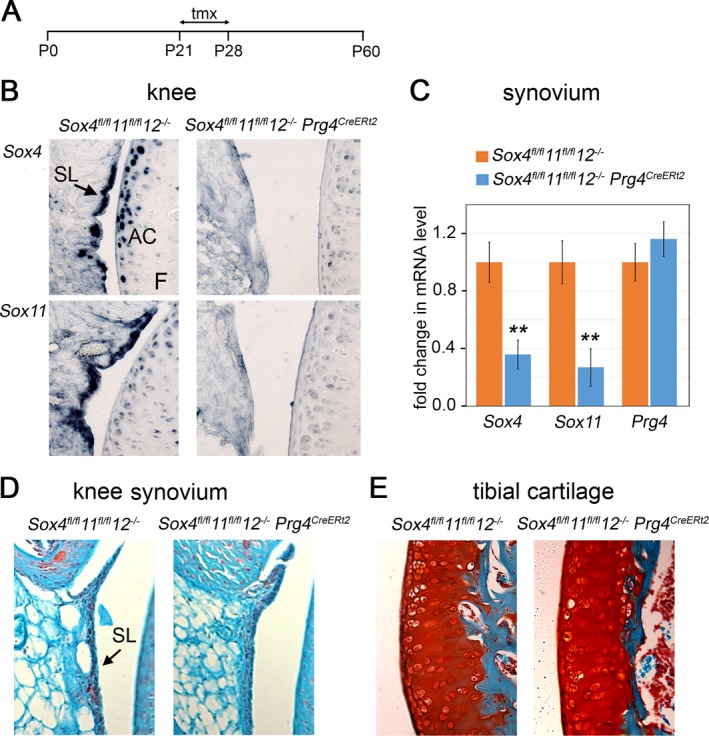
Inactivation of SOXC genes in the Prg4 + cell lineage does not affect joint integrity in young mice. A, Schematic representation of the timeline of the experiment. Tamoxifen (tmx) was injected on postnatal days 21 (P21) and 60. B, RNA in situ hybridization in adjacent mouse knee sections, using Sox4 and Sox11 RNA probes. Expression of Sox4 and Sox11 was down‐regulated in synovium and articular chondrocytes from Sox4 fl/fl 11 fl/fl 12 –/– Prg4 CreERt2 mouse knees. C, Fold changes in Sox4 and Sox11 mRNA levels in pooled knee and hip synovial tissue samples (n = 3 mice per genotype), as assessed by quantitative reverse transcription–polymerase chain reaction. Values are the mean ± SD. ** = P < 0.01. D and E, Safranin O–stained knee synovium (D), and fast green–counterstained tibial cartilage (E). F = femur (see Figure 1 for other definitions). Original magnification × 20 in B, D, and E.
Effect of SOXC inactivation on TNF‐induced arthritis in mice. To investigate the roles of the SOXC genes in joints under the stress of inflammation, we generated Sox4 fl/f 11 fl/fl 12 –/– Prg4 CreERt2 Tnf tg/0 rtTA tg/0 mice and compared them to Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 control mice (see Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). Mice expressing Tnf tg and rtTA tg were previously shown to widely express human TNF upon doxycycline administration and to develop inflammatory arthritis in interphalangeal joints and psoriasis‐like inflammation in nail beds 22. We injected the mice with tamoxifen at 3–4 weeks of age, treated them with doxycycline beginning at 5 weeks of age, and analyzed them at 7 weeks of age (Figure 3A). External analysis of the forepaws did not indicate any abnormality in SOXC‐deficient mice (Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 Prg4 CreERt2) and control mice (Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0) (Figure 3B). Histologic analysis of interphalangeal joints after Safranin O staining revealed no significant loss of proteoglycan in the articular cartilage of Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 Prg4 CreERt2 mice (Figures 3B and D). Hematoxylin and eosin staining also revealed no differences in synovial lining cellularity. Thus, these data, obtained for interphalangeal joints, corroborated the conclusion reached by analyzing the knee joints (Figure 2) that the SOXC genes are not critically needed for joint health in young adult mice.
Figure 3.

SOXC gene inactivation protects against tumor necrosis factor (TNF)–induced joint degeneration in mice. A, Schematic representation of the timeline of the experiment. Tamoxifen was injected at 3–4 weeks of age, and doxycycline (dox) treatment began at 5 weeks of age. B and C, Safranin O/fast green (B) and hematoxylin and eosin (H&E) (C) staining of sections from the distal interphalangeal joint of forepaw digit III. The boxed areas in the left panels (original magnification × 2.5) indicate the areas that are shown at higher magnification (original magnification × 20) in the middle and right panels. Arrowhead shows loss of Safranin O–stained cartilage‐specific proteoglycans. Arrows indicate invasion the joint cavity by synovial tissue. Asterisk indicates synovial hyperplasia. D, Fold change in the intensity of Safranin O staining in articular cartilage. E, Fold change in the size of the synovial pannus. Cartilage staining intensity and synovial pannus size were measured in 3 sections per mouse (30 μm apart), using ImageJ software. In D and E, symbols represent individual mice (n = 5 per group); bars show the mean ± SD. See Figure 2 for other definitions.
As expected, the interphalangeal joints of Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 rtTA tg/0 mice exhibited articular cartilage damage, increased cellularity of the synovium, and invasion of the synovial cells into the joint cavity (Figures 3C–E). In contrast, Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 rtTA tg/0 Prg4 CreERt2 mice had significantly reduced cartilage erosion and synovial hyperplasia compared to Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 rtTA tg/0 mice. These differences in synovium and cartilage disease status between control and SOXC‐deficient mice under the condition of TNF expression could not be attributed to differences in the serum levels of TNF in mice of both genotypes (see Supplementary Figure 2B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). Accordingly, the digits of the TNF‐expressing SOXC‐deficient mice (Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 rtTA tg/0 Prg4 CreERt2) were as swollen as those of the TNF‐expressing control mice (Sox4 fl/fl 11 fl/fl 12 –/– Tnf tg/0 rtTA tg/0). Moreover, the extent of hyperplasia under the forepaw nail beds, where Prg4 CreERt2 is not active, was similar in both mouse types (see Supplementary Figure 2C). Together, these data demonstrated that SOXC gene deletion in the Prg4 cell lineage in young adult mice hampered the ability of TNF to exert pathogenic actions in the joints.
Requirement of SOXC genes for induction of FLS aggressiveness. Increased proliferation, survival, and ability to migrate are defining pathologic features of FLS 3. We used an EdU incorporation assay to test whether SOXC genes increased the proliferation rate of synovial cells in response to TNF. Although EdU‐labeled cells could be readily detected in subchondral bone, hardly any were found in interphalangeal cartilage and synovial tissue in control and TNF‐expressing mice (see Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). An in vitro assay of cell cycle phases showed that primary FLS were equally proliferative when stimulated with TNF, when forced to overexpress SOX11, and when both treatments were applied together (see Supplementary Figure 3B). Thus, in our mouse model, neither TNF nor SOXC genes transformed FLS by increasing their proliferation.
We used TUNEL staining to assess cell death. In both control and SOXC‐deficient mice, the death rate of cells in the synovium was 1% at 7 weeks of age (Figures 4A and B). Interestingly, this death rate was 3.8% in TNF‐overexpressing mice and was twice as high (8%) in SOXC‐deficient TNF‐overexpressing mice. We then tested primary FLS from Sox4 fl/fl 11 fl/fl 12 –/– mice, using flow cytometric quantification of annexin V positivity (Figure 4C). These cell populations showed 3‐fold more apoptotic cells when infected with a Cre‐expressing adenovirus than when infected with a control, lacZ‐expressing adenovirus. Upon TNF treatment, the percentage of apoptotic cells doubled, but only in the SOXC‐deficient population. Collectively, these in vivo and in vitro data suggested that the SOXC genes promote synovial hyperplasia in response to TNF by, at least in part, reducing the FLS death rate.
Figure 4.
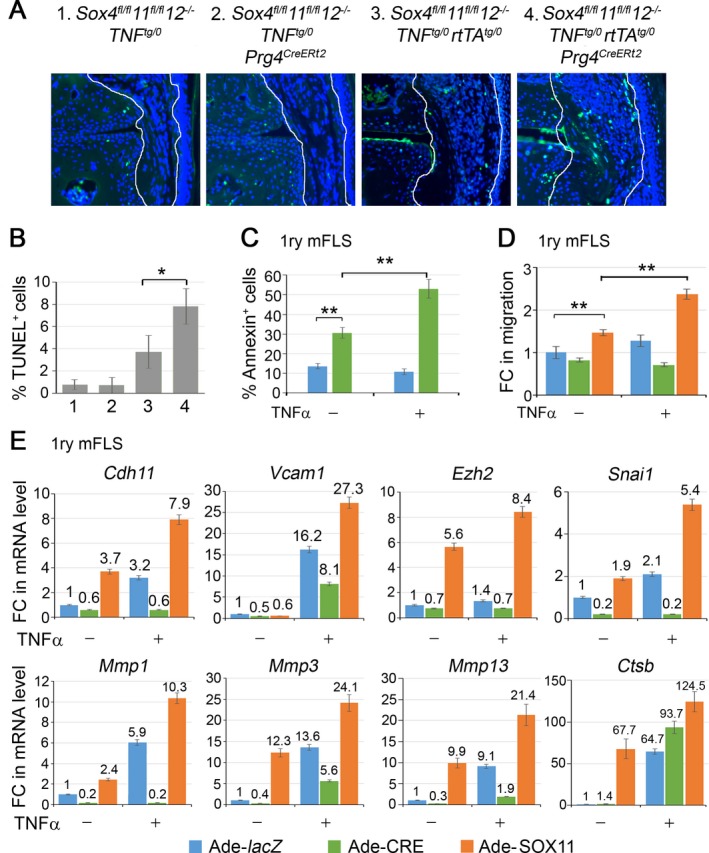
SOXC genes promote FLS transformation. A, TUNEL staining (green) and DAPI counterstaining (blue) of cell nuclei. Original magnification × 20. B, Death rate of cells in the synovium (n = 3 mice per genotype), as determined by TUNEL staining. C, Flow cytometric quantification of annexin V–positive cells in primary FLS from Sox4 fl/fl 11 fl/fl 12 –/– mice. Cells were infected with lacZ‐ or Cre‐expressing adenovirus for 16 hours and then treated with 5 ng/ml tumor necrosis factor (TNF) for 8 hours. D, Fold changes (FCs) in FLS migration to wounded area, as assessed by in vitro wound‐healing assay. Confluent cultures of FLS from Sox4 fl/fl 11 fl/fl 12 –/– mice were infected with lacZ‐, Cre‐, or SOX11‐expressing adenovirus (Ade) for 16 hours, a cell monolayer was scratched with a pipette tip, and the cultures were either treated with 5 ng/ml TNF for 4 hours or were left untreated. E, Fold changes in the levels of mRNA for genes required for efficient cell migration and FLS transformation. GAPDH mRNA levels were used for normalization. Values are the mean ± SD of triplicate cultures per condition. * = P < 0.05; ** = P < 0.01 by Student's 2‐tailed t‐test. See Figure 1 for other definitions.
Our in vivo experiments showed that SOXC inactivation precluded joint cavity invasion by synovial cells in TNF‐expressing mice. To evaluate whether the SOXC genes directly affect FLS migration, we used an in vitro wound‐healing assay, in which a cell monolayer was locally scratched, and cells migrating into the wounded area were counted. SOXC inactivation slightly impaired cell migration, whereas SOX11 overexpression significantly increased it (Figure 4D; see also Supplementary Figure 3C, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40386/abstract). Interestingly, TNF marginally increased the migration of control cells, but it was totally ineffective in SOXC‐deficient cells. The effect of TNF was potentiated by SOX11 overexpression. Because these effects were observed within 4 hours of TNF treatment, it is likely that they truly reflected increased cell migration, rather than increased cell survival or proliferation.
Finally, we tested whether SOXC proteins impacted the effect of TNF on the expression of genes required for efficient cell migration and FLS transformation. These genes included Cdh11 (cadherin 11) and Vcam1 (vascular cell adhesion protein 1), which encode cell surface proteins, Ezh2 (enhancer of zeste 3 polycomb repressive complex 2 subunit), Snai1 (Snail family transcriptional repressor 1), Mmp1, Mmp3, and Mmp13 (matrix metalloproteinase 1 [MMP‐1], MMP‐3, and MMP‐13, respectively), and Ctsb (cathepsin B). Sox4/11 loss blocked or reduced the ability of TNF to up‐regulate the expression of each of these genes except Ctsb. SOX11 overexpression up‐regulated the expression of Cdh11, Snai1, Mmp1, Mmp3, and Mmp13, and combined TNF treatment and SOX11 overexpression greatly increased the expression of all 8 genes (Figure 4E). Taken together, these data indicated that SOXC genes enhance the ability of TNF to transform FLS into aggressive, cancer‐like cells.
Stabilization of SOX4/11 proteins by inflammatory cytokines. Having discovered that the SOXC genes potentiate TNF signaling in FLS, we next sought to determine whether they are targets of this pathway. Treatment of mouse primary FLS with TNF did not change the levels of SOX4 and SOX11 mRNA, in spite of significantly increasing the level of mRNA for IL6, a TNF signaling target (Figure 5A). Interestingly, however, the same treatment up‐regulated the level of SOX4/11 protein by >5‐fold (Figure 5B). We then investigated whether other cytokines that are abundantly present in arthritic joints had effects comparable to those of TNF. We tested the effects of IL‐1α and IL‐6, which are also proinflammatory, and IL‐10, which is antiinflammatory, on human synovial sarcoma–derived FLS‐like SW‐982 cells. As in mouse primary FLS, SOX4 and SOX11 mRNAs were readily detected in SW‐982 cells (Figure 5C). None of the cytokines changed the levels of these mRNAs, even though TNF, IL‐1, and IL‐6 increased the level of mRNA for CCL2 (encoding a monocyte chemoattractant protein [MCP] known as CCL2 or MCP‐1) by >10‐fold, and IL‐10 increased it by almost 3‐fold. Remarkably, TNF, IL‐1, IL‐10, and IL‐6 all increased the levels of SOX4/11 protein with a robustness closely paralleling their effect on the CCL2 RNA level (Figure 5D). The SOX proteins were primarily located in the cell nucleus in all conditions, indicating that the cytokines did not affect their nuclear–cytoplasmic shuttling.
Figure 5.
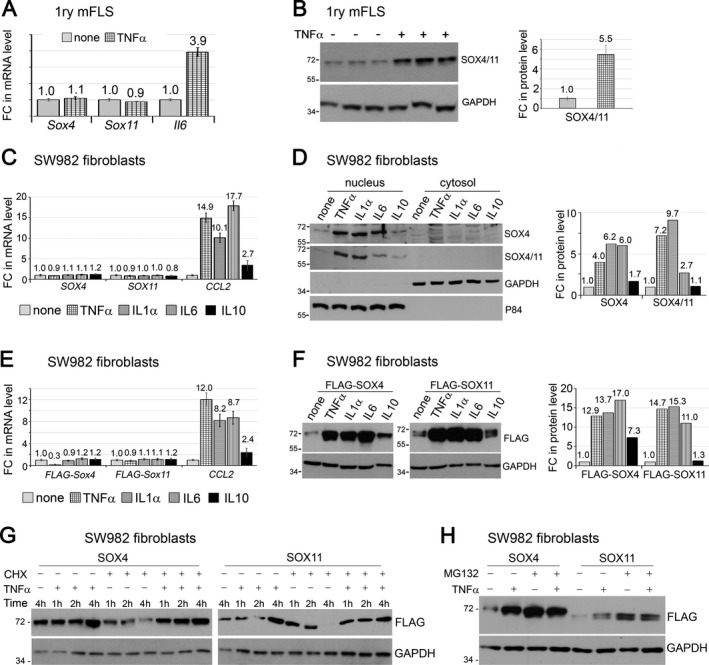
SOX4/11 proteins are stabilized by inflammatory cytokines. A and B, Fold changes (FCs) in Sox4, Sox11, and Il6 mRNA levels (A) and SOXC protein levels (B). Mouse FLS at passage 4 were treated with 5 ng/ml tumor necrosis factor (TNF) for 8 hours. Messenger RNA levels were measured by quantitative reverse transcription–polymerase chain reaction, and protein levels were determined by Western blotting of whole‐cell extracts from triplicate cultures. C and D, Fold changes in RNA (C) and protein (D) expression in SW‐982 cells treated with 5 ng/ml TNF, 10 ng/ml interleukin‐1α (IL‐1α), 10 ng/ml IL‐6, or 10 ng/ml IL‐10 for 8 hours. Nuclear protein P84 and GAPDH cytoplasmic protein were used as loading controls. E and F, Fold changes in FLAG‐SOX mRNA (E) and protein (F) levels in whole‐cell extracts. SW‐982 cells were transfected with expression plasmids encoding FLAG‐SOX4 and FLAG‐SOX11 for 16 hours and treated with cytokines, as described in C and D. G, SOX4 and SOX11 protein levels in whole‐cell extracts, as determined by Western blotting. SW‐982 cells were transfected with expression plasmids encoding FLAG‐SOX4 and FLAG‐SOX11 for 16 hours, after which time the medium was left unsupplemented or supplemented with 5 μg/ml cycloheximide (CHX) for 15 minutes, with or without subsequent supplementation with 5 ng/ml TNF for 1, 2, or 4 hours. H, FLAG‐SOX4 and FLAG‐SOX11 protein levels in SW‐982 cells treated with or without MG‐132 and/or TNF, as determined by Western blotting. Fold‐change values are the mean ± SD. See Figure 1 for other definitions.
The ability of the cytokines to increase SOX4/11 protein levels, but not RNA levels, implied involvement of a posttranscriptional mechanism. To investigate the nature of this mechanism, we first used expression plasmids encoding FLAG‐tagged mouse SOX4 and SOX11 proteins. The plasmids featured a heterologous cytomegalovirus promoter that, besides carrying the SOXC coding sequences, lacked any Sox4 or Sox11 regulatory sequences. As seen for the endogenous proteins, the inflammatory cytokines increased the levels of the FLAG‐SOX4 and FLAG‐SOX11 proteins without affecting their transcript levels (Figures 5E and F). This result consolidated the notion of a posttranscriptional effect and, since the expression plasmids lacked Sox4 and Sox11 5′‐ and 3′‐untranslated sequences, it suggested that the effect is posttranslational.
To investigate whether inflammatory cytokines affected SOX4 and SOX11 mRNA translation, we expressed FLAG‐SOX4 and FLAG‐SOX11 in SW‐982 cells and treated the cells with TNF. The protein levels robustly increased by 4 hours (Figure 5G). Blockade of new protein synthesis with the translation elongation inhibitor cycloheximide caused a progressive loss of FLAG‐SOX4 and FLAG‐SOX11 proteins in cells that were not treated with TNF but did not do so in TNF‐treated cells. These results thus provided evidence of a short life of the SOXC proteins in the absence, but not in the presence, of TNF.
To validate this novel notion, we tested the effect of blocking the proteasome protein degradation pathway with the MG‐132 compound. As anticipated, both TNF and MG‐132 increased the levels of FLAG‐SOX4 and FLAG‐SOX11 proteins, and combined treatment with TNF and MG‐132 did not further increase the protein levels (Figure 5H). Therefore, TNF signaling likely increased the SOX4 and SOX11 protein levels by inhibiting the proteasome pathway. Taken together, these data indicate that proinflammatory cytokines robustly increase SOXC protein stability in FLS.
Elevated SOX4/11 protein levels in inflamed synovium of arthritis patients. In order to establish whether SOXC protein stabilization also occurs in human synovium subjected to proinflammatory signals, we separately collected noninflamed and inflamed synovium biopsy specimens from OA patients who underwent knee arthroplasty. Inflamed synovium was recognized based on hyperplasia and erythema, and inflammation was confirmed by measuring the IL‐6 mRNA level (Figure 6A). Interestingly, although the levels of SOX4 and SOX11 mRNA showed no consistent pattern of inflammation‐dependent change in the 7 patients who were tested (Figure 6A), the SOX4/11 protein levels were higher in inflamed synovial tissue than in patient‐matched noninflamed tissue. The increase varied from <2‐fold to >9‐fold (Figure 6B). Taken together, these data support the novel assertion that the SOXC proteins are stabilized in the inflamed synovium of OA patients.
Figure 6.
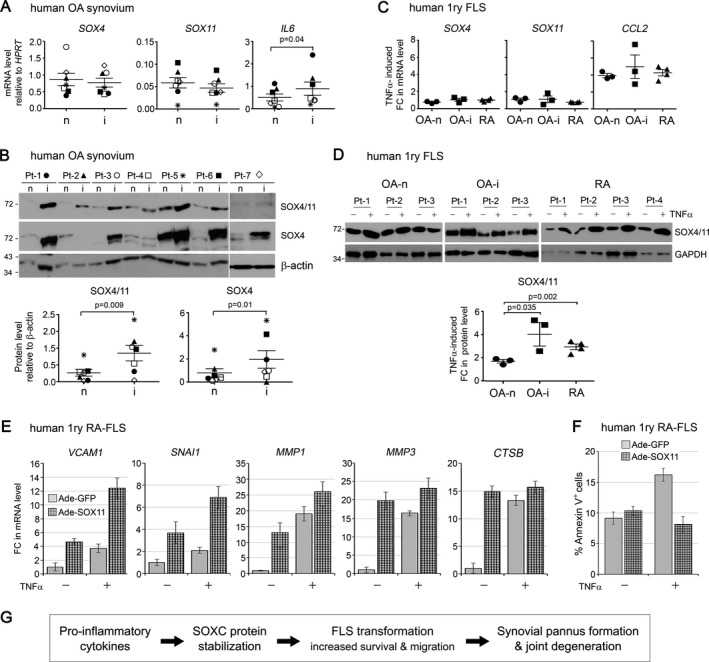
SOX4/11 protein levels are increased in inflamed synovium and FLS from patients with arthritic disease. A and B, Tumor necrosis factor (TNF)–induced fold changes (FCs) in levels of SOX4/11 mRNA (A) and protein (B) in noninflamed (n) and inflamed (i) synovial tissue biopsy specimens from 7 patients with osteoarthritis (OA) (Pt‐1 to Pt‐7) as assessed by quantitative reverse transcription‐polymerase chain reaction and Western blotting, respectively. C and D, TNF‐induced fold changes in levels of SOX4/11 mRNA (C) and protein (D) in noninflamed and inflamed synovial tissue biopsy specimens from patients with OA (Pt‐1 to Pt‐3) and patients with rheumatoid arthritis (RA) (Pt‐1 to Pt‐4). FLS were treated with 5 ng/ml TNF for 8 hours. E, Fold changes in levels of mRNA for the indicated genes in RA FLS. Cells were treated with adenoviruses (Ade) for 16 hours and then with or without 5 ng/ml TNF for 8 hours. HPRT mRNA levels were used for normalization. F, Percentage of annexin V–positive (apoptotic) cells among RA FLS treated with adenoviruses for 16 hours and then with or without TNF for 16 hours. G, Model of the inflammation/SOX4/11 axis identified in this study. Proinflammatory signals triggered by cytokines such as TNF, interleukin‐1 (IL‐1), and IL‐6 induce stabilization and accumulation of the SOX4/11 proteins in FLS. The SOXC transcription factors are thereby allowed to actively participate in the aggressive transformation of FLS by up‐regulating genes that promote cell survival and migration. Hence, SOXC proteins are targets and mediators of inflammatory pathways that cause accelerated joint degeneration in arthritic diseases. See Figure 1 for other definitions.
We next compared the effect of TNF on SOX4/11 mRNA and protein levels in primary FLS isolated from patients with OA and patients with RA. Similar to mouse FLS, FLS from patients with OA and patients with RA responded to TNF treatment with an increase in the CCL2 mRNA level without a significant change in SOX4 and SOX11 mRNA levels (Figure 6C). Interestingly, the level of SOX4/11 protein increased ≤2‐fold in the FLS isolated from noninflamed OA synovium and increased significantly more, up to 3‐fold or 4‐fold, in the FLS isolated from inflamed synovium from patients with OA and patients with RA (Figure 6D). This suggests that cells isolated from inflamed tissue have acquired increased responsiveness to TNF with regard to SOX4/11 protein stabilization.
Finally, we tested whether overexpression of SOXC proteins in human primary RA FLS impacted the ability of TNF to up‐regulate the expression of genes required for efficient cell migration and invasion. We observed that, similar to results obtained in mouse FLS, SOX11 overexpression alone was sufficient to increase the expression of VCAM1 and SNAI1, whose gene products are known to promote cell migration (Figure 6E). Genes encoding for the proteolytic enzymes MMP‐1, MMP‐3, and cathepsin B, which help FLS to become invasive, also had increased expression. The combination of SOX11 overexpression and the addition of TNF resulted in higher expression of VCAM1, SNAI1, and MMP1 than when both treatments were applied independently. Furthermore, the percentage of FLS undergoing apoptosis upon TNF treatment was reduced by SOX11 overexpression (Figure 6F). Together, these data strongly suggest that SOXC proteins contribute toward enhancing the aggressive phenotype of RA FLS.
Discussion
This study revealed that the SOXC proteins are pivotal targets of a proinflammatory cytokine–initiated molecular axis that leads to the development of arthritic lesions in synovial joints (Figure 6G). While suggesting that the SOXC proteins are dispensable for joint integrity in juvenile and young adult mice, it demonstrated that these proteins are required in the Prg4 + cell lineage for synovial pannus formation and articular cartilage degeneration in TNF‐induced arthritic disease. It was then shown that SOXC proteins are very unstable in FLS under basal conditions but robustly stabilized upon stimulation with TNF and other proinflammatory cytokines. Proinflammatory pathways thereby use these transcription factors to amplify the expression of genes that promote FLS survival and migration, which are critical events in inflammation‐induced FLS transformation.
Our observation that SOXC genes may be dispensable for joint integrity in juvenile and young adult mice was not predicted. Indeed, we were able to detect the proteins and their mRNA in the synovium and articular cartilage of adult mice, and we and others previously showed the indispensability of SOXC genes for joint formation in mouse embryos 11, 13, 14, 15, 28, 29, 30. A possible explanation is that the SOXC proteins are present at very low levels or in an inactive form in healthy (nonarthritic) adult synovium and therefore cannot exert significant functions. Although SOXC inactivation did not cause major changes in the joints of young adult mice, it is possible that it would cause problems in the long term and in particular upon aging. Testing this possibility will require further investigation. The finding that SOXC genes are necessary for FLS transformation in arthritic mice is the first reported evidence that these genes, and in fact, any member of the SOX family, are involved in positively mediating or exacerbating inflammatory pathways. Interestingly, SOXC mRNA levels were not changed in FLS treated with these cytokines, but the SOXC protein levels were quickly and dramatically increased. Thus, constitutive expression of the SOXC genes allows healthy FLS to quickly respond to proinflammatory signals.
Similar to SOX4 and SOX11, SOX9 (a group E SOX protein) is also a target of proinflammatory cytokines, but the underlying mechanisms are distinct for the 2 types of SOX proteins, and their consequences are opposite. Indeed, the SOXC proteins are stabilized in response to proinflammatory pathways, whereas the SOX9 mRNA and protein levels are quickly and sharply down‐regulated. Although SOXC inactivation was shown in this study to be protective for articular cartilage, SOX9 inactivation leads to chondrocyte de differentiation and hence to cartilage degeneration 31. These opposite mechanisms and consequences are not unexpected when one considers that SOX9 belongs to the SOXE group of the SOX family, that the SOXC and SOXE proteins share only ~50% identity in their DNA‐binding domain and none outside this domain, and that the 2 types of genes have very different patterns of expression and modes of regulation. Moreover, we previously showed that the SOXC proteins are not only very different from SOX9 proteins but also have antichondrogenic properties that prevent presumptive joint and perichondrial mesenchyme in mouse embryos from undergoing ectopic chondrogenesis 12. One of these properties was the ability to stabilize β‐catenin and thereby synergize with canonical Wnt signaling to efficiently repress the levels and the activity of SOX9. By extrapolation, we surmise that SOXC inactivation in the joints of the mice used in this study may have protected articular cartilage at least in part by increasing the levels of SOX9 in chondrocytes.
Our findings suggest that SOXC genes contribute to synovial hyperplasia in arthritic disease by increasing FLS survival but not by affecting cell proliferation. Similarly, we and other investigators previously showed that the SOXC genes promote survival of skeletal mesenchyme, neuronal progenitors, and other types of progenitor cells without affecting cell proliferation 11, 30. It is known that FLS are not highly proliferative in adulthood. Only a small proportion of the FLS is known to undergo an extremely slow rate of proliferation, similar to that of stem‐like cells 32, 33. Moreover, it has also been shown that cell proliferation does not play a significant role in TNF‐induced FLS transformation 3. Thus, our findings that SOXC genes do not affect cell proliferation in the synovium are consistent with these observations. Transformed FLS are known to resist TNF‐induced apoptosis in spite of expressing functional Fas (cell surface death receptors), TRAILR‐1 and TRAILR‐2, and TNF receptor 1 (TNFR1) and TNFR2 34. Here, we showed that the absence of the SOXC genes causes TNF‐exposed FLS to lose their apoptosis resistance.
We also showed that besides promoting cell survival, the SOXC genes allowed FLS invasion of joint cavities and migration over plastic surfaces. Studies of epithelial cancers showed that SOX4 and SOX11 promote metastasis. The epithelial mesenchymal transition transcription factor SNAI1 and histone methyltransferase EZH2 are direct transcriptional targets of SOX4 in cancer cells 19. Both genes are also TNF signaling targets with key roles in promoting FLS invasiveness 35, 36. Cadherin 11 is an FLS surface marker and a target of TNF 37. Its inactivation protects against development of TNF‐induced joint degeneration in mice. We have shown here that Ezh2, Cdh11, and Snai1 are targets of the SOXC/TNF molecular axis. It is thus likely that SOXC proteins directly up‐regulate the expression of the TNF target genes that support FLS transformation in arthritic disease.
In spite of years of research in to many processes, mechanisms underlying SOXC regulation remain largely unknown. SMAD proteins activated by transforming growth factor β were shown to directly up‐regulate SOX4 transcription in cancer cells and T cells 4, 38. Our study sheds new light on SOXC regulation by showing that protein stabilization is central downstream of inflammatory signals. We speculate that a similar mechanism is likely at play in specific developmental and physiologic processes. However, the signals that then stabilize the SOXC proteins have not yet been identified.
Direct targeting of proinflammatory cytokines with biologic compounds is a prevalent strategy in current clinical practice to reduce or delay permanent joint degeneration. This strategy, however, is neither ideal nor sufficient, because global interference with the immune system results in unwanted effects such as development of drug resistance, infections, and cancers 39, 40, 41, 42. We demonstrated that down‐regulating the expression of SOXC genes in the synovial lining and articular chondrocytes significantly reduced synovitis and articular cartilage erosion. We therefore propose the SOXC genes or proteins as potential alternative or complementary drug targets for treating inflammation‐induced joint pathology.
Targeting SOXC genes/proteins is especially tempting considering that these factors have thus far been found to be critical in disease development but not in physiologic processes governing the function and maintenance of tissue in adults. However, more studies are needed to validate their candidacy as drug targets. SOXC transcription factors are expressed in a broad range of tissues and cell types, including immune cells that accumulate in the synovial pannus 43. Further understanding of the mechanistic regulation of the interactions between SOXC proteins and inflammation in additional models of inflammation‐induced arthritis will be necessary. Although it has long been considered that developing drugs that target transcription factors is not feasible, a number of compounds directed toward preventing their interactions with DNA or protein partners have recently been successfully developed. They include small molecule inhibitors that target STAT‐3 dimerization, and p53–Mdm2, p300–HIF1α, and SOX18–RBP‐J interactions 44, 45, 46, 47. Therefore, targeting the SOXC proteins or their stabilization mechanism using systemic delivery of small molecule inhibitors is an option to be explored, because it could soon become a safe and effective therapy for arthritic diseases and other types of inflammatory diseases.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Bhattaram had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Bhattaram, Muschler, Lefebvre.
Acquisition of data
Bhattaram, Muschler, Wixler, Lefebvre.
Analysis and interpretation of data
Bhattaram, Lefebvre.
Supporting information
Acknowledgments
We thank T. Haqqi and all members of the Lefebvre, Muschler, and Haqqi laboratories for providing assistance during the study. We also thank M. Longworth and O. Wessely for their advice during manuscript preparation.
Supported by the Arthritis National Research Foundation (grant ANRF1405PB to Dr. Bhattaram), the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR‐070736 to Dr. Bhattaram, grant AR‐063733 to Dr. Muschler, and grants AR‐046249 and AR‐068308 to Dr. Lefebvre), and the Rheumatology Research Foundation (grant to Dr. Lefebvre).
References
- 1. Iwanaga T, Shikichi M, Kitamura H, Yanase H, Nozawa‐Inoue K. Morphology and functional roles of synoviocytes in the joint. Arch Histol Cytol 2000;63:17–31. [DOI] [PubMed] [Google Scholar]
- 2. Smith MD. The normal synovium. Open Rheumatol J 2011;5:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bottini N, Firestein GS. Duality of fibroblast‐like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 2013;9:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhattaram P, Chandrasekharan U. The joint synovium: a critical determinant of articular cartilage fate in inflammatory joint diseases. Semin Cell Dev Biol 2017;62:86–93. [DOI] [PubMed] [Google Scholar]
- 5. De Bari C. Are mesenchymal stem cells in rheumatoid arthritis the good or bad guys? Arthritis Res Ther 2015;17:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther 2017;19:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development 2013;140:4129–44. [DOI] [PubMed] [Google Scholar]
- 8. Lefebvre V, Dumitriu B, Penzo‐Mendez A, Han Y, Pallavi B. Control of cell fate and differentiation by Sry‐related high‐mobility‐group box (Sox) transcription factors. Int J Biochem Cell Biol 2007;39:2195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dy P, Penzo‐Mendez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. The three SoxC proteins—Sox4, Sox11 and Sox12—exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res 2008;36:3101–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoser M, Potzner MR, Koch JM, Bosl MR, Wegner M, Sock E. Sox12 deletion in the mouse reveals nonreciprocal redundancy with the related Sox4 and Sox11 transcription factors. Mol Cell Biol 2008;28:4675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhattaram P, Penzo‐Mendez A, Sock E, Colmenares C, Kaneko KJ, Vassilev A, et al. Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat Commun 2010;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhattaram P, Penzo‐Mendez A, Kato K, Bandyopadhyay K, Gadi A, Taketo MM, et al. SOXC proteins amplify canonical WNT signaling to secure nonchondrocytic fates in skeletogenesis. J Cell Biol 2014;207:657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang KC, Hertz J, Zhang X, Jin XL, Shaw P, Derosa BA, et al. Novel regulatory mechanisms for the SoxC transcriptional network required for visual pathway development. J Neurosci 2017;37:4967–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paul MH, Harvey RP, Wegner M, Sock E. Cardiac outflow tract development relies on the complex function of Sox4 and Sox11 in multiple cell types. Cell Mol Life Sci 2014;71:2931–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potzner MR, Tsarovina K, Binder E, Penzo‐Mendez A, Lefebvre V, Rohrer H, et al. Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development 2010;137:775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bilir B, Osunkoya AO, Wiles WG IV, Sannigrahi S, Lefebvre V, Metzger D, et al. SOX4 is essential for prostate tumorigenesis initiated by PTEN ablation. Cancer Res 2016;76:1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roisman A, Stanganelli C, Nagore VP, Richardson GV, Scassa ME, Bezares RF, et al. SOX11 expression in chronic lymphocytic leukemia correlates with adverse prognostic markers. Tumour Biol 2015;36:4433–40. [DOI] [PubMed] [Google Scholar]
- 18. Vervoort SJ, van Boxtel R, Coffer PJ. The role of SRY‐related HMG box transcription factor 4 (SOX4) in tumorigenesis and metastasis: friend or foe? Oncogene 2013;32:3397–409. [DOI] [PubMed] [Google Scholar]
- 19. Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial‐mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013;23:768–83. [DOI] [PubMed] [Google Scholar]
- 20. Shepherd JH, Uray IP, Mazumdar A, Tsimelzon A, Savage M, Hilsenbeck SG, et al. The SOX11 transcription factor is a critical regulator of basal‐like breast cancer growth, invasion, and basal‐like gene expression. Oncotarget 2016;7:13106–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Penzo‐Mendez A, Dy P, Pallavi B, Lefebvre V. Generation of mice harboring a Sox4 conditional null allele. Genesis 2007;45:776–80. [DOI] [PubMed] [Google Scholar]
- 22. Retser E, Schied T, Skryabin BV, Vogl T, Kanczler JM, Hamann N, et al. Doxycycline‐induced expression of transgenic human tumor necrosis factor α in adult mice results in psoriasis‐like arthritis. Arthritis Rheum 2013;65:2290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozhemyakina E, Zhang M, Ionescu A, Ayturk UM, Ono N, Kobayashi A, et al. Identification of a Prg4‐expressing articular cartilage progenitor cell population in mice. Arthritis Rheumatol 2015;67:1261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ntougkos E, Chouvardas P, Roumelioti F, Ospelt C, Frank‐Bertoncelj M, Filer A, et al. Genomic responses of mouse synovial fibroblasts during tumor necrosis factor–driven arthritogenesis greatly mimic those in human rheumatoid arthritis. Arthritis Rheumatol 2017;69:1588–600. [DOI] [PubMed] [Google Scholar]
- 25. Chen D, Jarrell A, Guo C, Lang R, Atit R. Dermal β‐catenin activity in response to epidermal Wnt ligands is required for fibroblast proliferation and hair follicle initiation. Development 2012;139:1522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rhee DK, Marcelino J, Baker M, Gong Y, Smits P, Lefebvre V, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest 2005;115:622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wixler V, Cromme C, Retser E, Meyer LH, Smyth N, Muhlenberg K, et al. FHL2 regulates the resolution of tissue damage in chronic inflammatory arthritis. Ann Rheum Dis 2015;74:2216–23. [DOI] [PubMed] [Google Scholar]
- 28. Gnedeva K, Hudspeth AJ. SoxC transcription factors are essential for the development of the inner ear. Proc Natl Acad Sci U S A 2015;112:14066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schilham MW, Clevers H. HMG box containing transcription factors in lymphocyte differentiation. Semin Immunol 1998;10:127–32. [DOI] [PubMed] [Google Scholar]
- 30. Thein DC, Thalhammer JM, Hartwig AC, Crenshaw EB III, Lefebvre V, Wegner M, et al. The closely related transcription factors Sox4 and Sox11 function as survival factors during spinal cord development. J Neurochem 2010;115:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murakami S, Lefebvre V, de Crombrugghe B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin‐1 and tumor necrosis factor‐α. J Biol Chem 2000;275:3687–92. [DOI] [PubMed] [Google Scholar]
- 32. Roelofs AJ, Zupan J, Riemen AH, Kania K, Ansboro S, White N, et al. Joint morphogenetic cells in the adult mammalian synovium. Nat Commun 2017;8:15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kurth TB, Dell'Accio F, Crouch V, Augello A, Sharpe PT, de Bari C. Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Rheum 2011;63:1289–300. [DOI] [PubMed] [Google Scholar]
- 34. Orosa B, González A, Mera A, Gómez‐Reino JJ, Conde C. Lysophosphatidic acid receptor 1 suppression sensitizes rheumatoid fibroblast‐like synoviocytes to tumor necrosis factor–induced apoptosis. Arthritis Rheum 2012;64:2460–70. [DOI] [PubMed] [Google Scholar]
- 35. Lauzier A, Lavoie RR, Charbonneau M, Gouin‐Boisvert B, Harper K, Dubois CM. Snail is a critical mediator of invadosome formation and joint degradation in arthritis. Am J Pathol 2016;186:359–74. [DOI] [PubMed] [Google Scholar]
- 36. Trenkmann M, Brock M, Gay RE, Kolling C, Speich R, Michel BA, et al. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Ann Rheum Dis 2011;70:1482–8. [DOI] [PubMed] [Google Scholar]
- 37. Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, et al. Cadherin‐11 in synovial lining formation and pathology in arthritis. Science 2007;315:1006–10. [DOI] [PubMed] [Google Scholar]
- 38. Kuwahara M, Yamashita M, Shinoda K, Tofukuji S, Onodera A, Shinnakasu R, et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF‐β and suppresses TH2 differentiation. Nat Immunol 2012;13:778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schaeverbeke T, Truchetet ME, Kostine M, Barnetche T, Bannwarth B, Richez C. Immunogenicity of biologic agents in rheumatoid arthritis patients: lessons for clinical practice. Rheumatology (Oxford) 2016;55:210–20. [DOI] [PubMed] [Google Scholar]
- 40. Tragiannidis A, Kyriakidis I, Zundorf I, Groll AH. Invasive fungal infections in pediatric patients treated with tumor necrosis α (TNF‐α) inhibitors. Mycoses 2017;60:222–9. [DOI] [PubMed] [Google Scholar]
- 41. Atzeni F, Gianturco L, Talotta R, Varisco V, Ditto MC, Turiel M, et al. Investigating the potential side effects of anti‐TNF therapy for rheumatoid arthritis: cause for concern? Immunotherapy 2015;7:353–61. [DOI] [PubMed] [Google Scholar]
- 42. Ehrenstein MR, Wing C. The BAFFling effects of rituximab in lupus: danger ahead? Nat Rev Rheumatol 2016;12:367–72. [DOI] [PubMed] [Google Scholar]
- 43. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh‐hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 2014;20:62–8. [DOI] [PubMed] [Google Scholar]
- 44. Miyoshi K, Takaishi M, Nakajima K, Ikeda M, Kanda T, Tarutani M, et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA‐21, a Stat3 inhibitor. J Invest Dermatol 2011;131:108–17. [DOI] [PubMed] [Google Scholar]
- 45. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, et al. Small molecule blockade of transcriptional coactivation of the hypoxia‐inducible factor pathway. Cancer Cell 2004;6:33–43. [DOI] [PubMed] [Google Scholar]
- 46. Chene P. Inhibiting the p53‐MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer 2003;3:102–9. [DOI] [PubMed] [Google Scholar]
- 47. Fontaine F, Overman J, Moustaqil M, Mamidyala S, Salim A, Narasimhan K, et al. Small‐molecule inhibitors of the SOX18 transcription factor. Cell Chem Biol 2017;24:346–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials