

Abstract
Ligand activation of the angiotensin II type 1 receptor (AT1R), a member of the G protein-coupled receptor (GPCR) family, stimulates intracellular signaling to mediate a variety of physiological responses. The AT1R is also known to be a mechanical sensor. When activated by mechanical stretch, the AT1R can signal via the multifunctional adaptor protein β-arrestin, rather than through classical heterotrimeric G protein pathways. To date, the AT1R conformation induced by membrane stretch in the absence of ligand was thought to be the same as that induced by β-arrestin-biased agonists, which selectively engage β-arrestin thereby preventing G protein coupling. Here, we show that in contrast to the β-arrestin-biased agonists TRV120023 and TRV120026, membrane stretch uniquely promotes the coupling of the inhibitory G protein (Gαi) to the AT1R to transduce signaling. Stretch-triggered AT1R-Gαi coupling is required for the recruitment of β-arrestin2 and activation of downstream signaling pathways, such as EGFR transactivation and ERK phosphorylation. Our findings demonstrate additional complexity in the mechanism of receptor bias in which the recruitment of Gαi is required for allosteric mechanoactivation of the AT1R-induced β-arrestin-biased signaling.
Keywords: β-arrestin, angiotensin II, G protein, mechanotransduction
1 | INTRODUCTION
The angiotensin II type 1 receptor (AT1R), a member of the G protein-coupled receptor (GPCR) superfamily, is a well-known regulator of blood pressure and cardiac function.1 In response to the balanced agonist angiotensin II (AngII), AT1Rs couple to the heterotrimeric G-protein triggering the dissociation of Gαq from Gβ, which subsequently activates the signaling effector phospholipase C (PLC) to promote generation of the second messengers inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG).1 This G protein-mediated signaling is rapidly turned off by the receptor desensitization, a process that involves phosphorylation of the C-terminal tail of the AT1R mediated by G protein-coupled receptor kinases (GRKs) and recruitment of the multifunctional scaffold protein β-arrestin that impairs further G protein coupling.2–4 It is now known that β-arrestins also function as signal transducers in their own right, regulating a large network of cellular responses.5 Several AT1R ligands, such as [Sar1, Ile4, Ile8]-AngII (SII) and TRV120023, have been shown to selectively activate β-arrestin-mediated pathways and therefore termed as β-arrestin-biased agonists.6,7
Our current understanding for ligand stimulated AT1R signaling is based on a mechanism whereby ligand binding stabilizes distinct receptor conformations allowing them to engage signal transducers such as G proteins or β-arrestin to affect activation of an array of signaling pathways, a concept known as functional selectivity.1,6,8–12 Interestingly, activation of AT1Rs can be mechanosensitive, whereby membrane stretch allosterically stabilizes a distinct β-arrestin-biased AT1R conformation triggering ligand-independent β-arrestin-mediated signaling.13,14 The precise molecular mechanism by which mechanical stress activates AT1R signaling is not clearly understood.
It is now well appreciated that AT1Rs may couple to multiple signaling transducers with different efficacy and kinetics depending on cellular systems and stimuli.15,16 For example, in proximal tubule cells, the AngII-activated AT1R recruits Gαi leading to the inhibition of adenylate cyclase activity.17 In cardiac fibroblasts, AngII activates the AT1R-mediated ERK signaling through the Gβγ subunits of Gαi18 and the weak β-arrestin-biased agonist [Sar1, Ile4, Ile8]-angiotensin II (SII) can promote AT1R coupling to both Gαq and Gαi.15
We, therefore, set out to determine whether Gαi plays a role as a signal transducer for the AT1R under conditions of mechanoactivation. Indeed, we show that membrane stretch promotes the recruitment of Gαi to the AT1R, which is required for the activation of β-arrestin-biased signaling as measured by epidermal growth factor receptor (EGFR) internalization and extracellular signal-regulated kinases (ERK) phosphorylation. In contrast, Gαi is not required for signaling induced by the balanced agonist AngII or the β-arrestin-biased agonist TRV120023 and TRV120026. Notably, our findings demonstrate mechanistic heterogeneity for AT1R-mediated β-arrestin-biased signaling induced by mechanoactivation versus biased ligands and support a concept that membrane stretch may stabilize a biased AT1R conformation that is distinct from that stabilized by a β-arrestin-biased ligand.
2 | MATERIALS AND METHODS
2.1 | Cell culture
HEK 293 cells stably expressing hemagglutinin (HA)-tagged AT1Rs were cultured in minimum essential medium (MEM) supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (Thermofisher Scientific, Waltham, MA), and 100 μg/mL of zeocin (Invitrogen, Carlsbad, CA) at 37°C in a humidified environment with 5% CO2 as previously described.13 HEK 293 cells stably expressing cyan fluorescent protein (CFP)-tagged AT1R and yellow fluorescent protein (YFP)-tagged β-arrestin2 were cultured in MEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, 300 μg/mL of zeocin, and 150 μg/mL of Geneticin (Thermofisher Scientific).
2.2 | Generation of Gαi knockout cell line with CRISPR-Cas9 gene editing
Plasmids carrying S. pyogenes Cas9 (SpCas9) next to a cloning site for guide RNA (gRNA) with EGFP (pSpCas9 (BB)-2A-GFP, Addgene 48138) were obtained from Addgene (deposited by the laboratory of Dr. F. Zhang19). Designing of the guide RNAs for Gαi and cloning the guide RNAs into the Cas9 plasmids were performed as previously described.19 Gαi1 was targeted using guide sequence oligos (top: CACCGCGCCGTCCTCACGGA GGTTG; bottom: AAACCAACCTCCGTGAGGACGGCGC), Gαi2 was targeted using guide sequence oligos (top: CACCGAG ACAACCGCCCGGTACTGC, bottom: AAACGCAGTACCG GGCGGTTGTCTC), and Gαi3 was targeted using guide sequence oligos (top: CACCGGGACGGCTAAAGATTGA CTT; bottom: AAACAAGTCAATCTTTAGCCGTCCC). The guide sequence oligos were cloned into pSpCas9 (BB)-2A-GFP. Plasmids targeting thethree Gαi subtypes were co-transfectedinto HEK293 cells. GFP positive cells were selected by fluorescence-activated cell sorting, diluted for growth and single cell colonies were obtained. The Gαi knockout were confirmed by Western blot.
2.3 | Proximity ligation assay
Proximity ligation assay (PLA) was performed following manufacture’s protocol (Sigma-Aldrich, St. Louis, MO) and previously published paper.20 Briefly, HEK293 cells stably expressing HA-tagged AT1R were plated on collagen-coated glass bottom cell culture dishes. After overnight incubation in serum-free medium with vehicle or 200 ng/mL pertussis toxin (List Biological Labs, Campbell, CA), cells were stimulated with 1 μM AngII (Sigma-Aldrich), 1 μM TRV120023 (synthesized by GenScript, Inc., Piscataway, NJ), or osmotic stretch (by adding same volume of double-distilled H2O to the medium as previously described14) for 10 min at 37°C in a humidified environment with 5% CO2, and fixed with 4% PFA. Fixed cells were permeabilized with 0.2% Triton-X100 for 10 min, blocked with manufacture’s blocking solution for 30 min, and incubated overnight with anti-Gαi mouse monoclonal antibody (NewEast Biosciences, King of Prussia, PA) and anti-HA rabbit antibody (Sigma-Aldrich) at 4°C. The next day, cells were subsequently incubated with Duolink In Situ PLA Probe Anti-Mouse MINUS and Anti-Rabbit PLUS for 1 h, ligation buffer for 30 min, amplification buffer (Duolink In Situ Detection Reagents Green) for 100 min, and mounted with Duolink In Situ Mounting Medium with DAPI. Images were recorded using the Zeiss Axio Observer Z1 confocal microscope. Quantification of PLA signal representing the AT1R-Gαi interaction was performed by averaging the intensity of the green fluorescence per cell and subtracting the non-specific background. Background signal was determined as green fluorescence intensity per cell in wildtype HEK293 cells without HA-tagged AT1R overexpression.
2.4 | Co-immunoprecipitation
HEK293 cells stably expressing HA-tagged AT1Rs were transfected with Gαi2 for the detection of AT1R-Gαi co-IP, and not transfected for the AT1R-Gαq co-IP experiments. Cells were incubated in serum-free medium with vehicle or 200 ng/mL PTX for overnight, and then stimulated with 1 μM AngII, 10 μM TRV120023 or osmotic stretch for 10 min. After stimulation, cells were lysed in 0.5% MNG lysis buffer (20 mM HEPES, 150 mM NaCl, 0.5% Lauryl Maltose Neopentyl Glycol, 2 mM sodium orthovanadate, 1 mM PMSF, 10 mM sodium fluoride, 10 μg/ml aprotinin, 5 μg/mL leupeptin and phosphatase inhibitors) with gentle agitation at 4°C for 1 h, and then centrifuged for 15 min at 13 200 rpm. 0.5–1 mg of protein was incubated with 30 μL of anti-HA magnetic beads (Pierce, Waltham, MA) for overnight. Immunoprecipitates were then separated by SDS-PAGE, transferred to PVDF membrane (Bio-Rad, Hercules, CA) and subjected to immunoblotting with various primary antibodies. Immunoblots were detected using enhanced chemiluminiscence (Thermo Fisher Scientific) and analyzed with ImageJ software.
2.5 | Confocal microscopy
In the β-arrestin translocation assay, HEK 293 cells stably expressing CFP-tagged AT1R and YFP-tagged β-arrestin2 were transiently transfected with GRK5 using Fugene 6 (Roche, Basel, Switzerland) and then plated in collagen-coated glass bottom cell culture dishes. After overnight serum-starvation with vehicle or 200 ng/mL PTX, cells were stimulated with 1 μM AngII, 1 μM or 10 μM TRV120023, 1 μM or 10 μM TRV120026, or osmotic stretch for 10 min, and then fixed with 4% PFA. In the EGFR internalization assay, HEK293 cells stably expressing HA-tagged AT1R were transiently transfected with green fluorescent protein (GFP)-tagged EGFR and GRK5 plasmids, and stimulated and fixed as described above. Cells were visualized using a Zeiss Axio Observer Z1 confocal microscope. Quantification of β-arrestin translocation was performed by counting the percentage of the cells showing intracellular aggregates of β-arrestin. Similarly, quantification of EGFR internalization was performed by counting the percentage of cells showing EGFR internalization.
2.6 | ERK phosphorylation assay
We performed immunoblotting as previously described.13 In brief, HEK293 cells stably expressing HA-tagged AT1Rs were plated on collagen-coated six-well dishes at a confluence of 60–70%. Cells were serum-starved for overnight and pretreated with vehicle or inhibitors (PTX: 200 ng/mL for overnight; Ro318425 (Sigma-Aldrich): 1 μM for 30 min; U73122 (Sigma-Aldrich): 10 μM for 30 min; AG1478 (Sigma-Aldrich): 1 μM for 30 min), and then stimulated with 1 μM AngII, 1 μM or 10 μM TRV120023, 1μM or 10 μM TRV120026, or osmotic stretch for 10 min. After stimulation, cells were lysed in 1% NP-40 lysis buffer (20 mM Tris, pH7.4, 137 mM NaCl, 20% glycerol, 1% Nonidet P-40, 2 mM sodium orthovanadate, 1 mM PMSF, 10 mM sodium fluoride, 10 μg/mL aprotinin, 5 μg/mL leupeptin and phosphatase inhibitors) with gentle agitation for 1 h at 4°C and centrifuged for 15 min at 13 200 rpm. Protein concentrations were measured with Bio-Rad protein assay reagent. Proteins were separated by SDS-PAGE, and transferred to PVDF membrane and blocked with 5% skim-milk. Membranes were incubated with phospho ERK1/2 (1:1000 Cell Signaling, Danvers, MA) or total ERK1/2 (1:2000, EMD Millipore, Billerica, MA) antibody overnight with gentle agitation at 4°C, and then Horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Protein bands were visualized by ECL with the Syngene Imager and quantified by densitometry using GeneSnap (Syngene, Cambridge, UK) or ImageJ software. In left ventricle (LV) of the mice, tissues were homogenized in NP-40 lysis buffer. Immunoblotting was performed using the protocol described above.
2.7 | Ex-vivo LV balloon stretch
Animal studies were carried out according to approved protocols and animal welfare regulations of Duke University Medical Center’s Institutional Review Boards. We performed ex-vivo LV balloon stretch experiment as previously described.13 Briefly, 8–12 weeks old, gender matched wild type mice (purchased from Jackson laboratory, Bar Harbor, ME) were pretreated with 25 μg/kg of PTX or vehicle through intraperitoneal (IP) injection at 48 h before experiment. LV balloon was made by stretching a polypropylene membrane into the shape of the LV cavity. Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (2.5 mg/kg), and heparinized by IP injection. After 10 min, chest was opened and heart was excised. Aorta was cannulated to the needle and balloon was inserted to LV through mitral valve. Heart was retrograde perfused with buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, 0.5 mM Na-EDTA, and 5.5 mM glucose with O2 bubbling through Langendorff apparatus (Hugo Sachs Harvard Apparatus, Holliston, MA) set at 37°C. LV balloon was connected to a Statham P23 Db pressure transducer (Gould Statham Instruments, Westminister, CA) through Polyethylene-50 (PE-50) for pressure monitoring. Intraballoon pressure was recorded continuously with a pressure-recording system (MacLab, Millar Instruments, Houston, TX). Balloon inflation was started after 10 min perfusion, and diastolic intraballoon pressure was kept within 30–50 mmHg with minor adjustment. Control was defined as perfusion without balloon inflation for identical periods of time. After 10 min balloon inflation, heart was removed from the system, and snap frozen in liquid nitrogen after removing atrium and right ventricle. If intraballoon pulse pressure was less than 20 mmHg or heart rate was less than 100 bpm continuously, the heart was excluded from this study.
2.8 | Statistics
Data are expressed as mean ± SEM. Statistical significance was determined with a one- or two-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons using GraphPad Prism software. A P-value of <0.05 was considered significant.
3 | RESULTS
3.1 | Membrane stretch promotes the recruitment of Gαi to AT1Rs
Previous work has shown that ligand stimulated AT1Rs may transduce signaling through multiple G proteins such as Gαq and Gαi, as well as the multifunctional adaptor protein β-arrestin.15 It is now appreciated that AT1Rs, in response to biomechanical membrane stretch, also activate signaling but in a Gαq-independent β-arrestin-dependent manner.13,14 To determine whether Gαi is involved in this stretch-induced AT1R signaling, we investigated the effect of membrane stretch on AT1R-Gαi coupling. HEK293 cells stably expressing HA-tagged AT1R were stimulated with the balanced agonist AngII, the β-arrestin-biased agonist TRV120023, or hypotonicity-induced membrane stretch, respectively. The recruitment of Gαi to AT1R was then measured with an in situ proximity ligation assay (PLA), in which protein-protein interactions can be directly visualized and quantified by the fluorescence signal. We found that osmotic stretch significantly increased the amount of Gαi recruited to AT1R, shown as the green PLA signal (Figure 1). Pretreatment with pertussis toxin (PTX), a Gαi inhibitor that catalyzes the ADP-ribosylation of Gαi and prevents its coupling to receptors, blocked the stretch-induced Gαi recruitment to AT1Rs. Notably, the balanced agonist AngII and the β-arrestin biased agonist TRV120023 did not alter the PLA signal.
FIGURE 1.
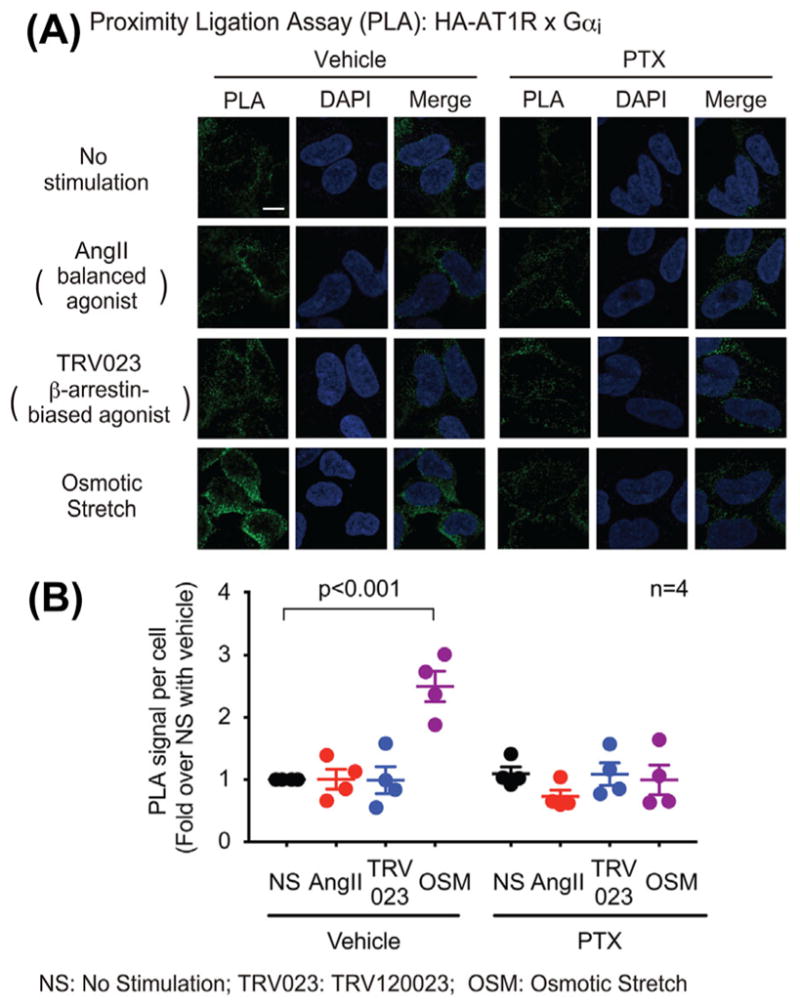
Osmotic stretch promoted Gαi recruitment to AT1R, while the balanced agonist AngII and the β-arrestin-biased agonist TRV120023 did not have significant effect. HEK293 cells stably expressing HA-tagged AT1Rs were pretreated with vehicle or 200 ng/mL PTX for overnight, then stimulated with 1 μM AngII, 1 μM TRV120023 or osmotic stretch for 10 min. In proximity ligation assay (PLA), cells were immuno-stained with Gαi antibody raised in mouse and HA antibody raised in rabbit. The recruitment of Gαi to AT1R is indicated by the green fluorescence signal. A: Representative image showings osmotic stretch-induced Gαi recruitment to AT1R. Scale bar = 10 μM. B: The PLA signal was quantified as the average intensity of the green fluorescence per cell, and normalized by subtracting the non-specific background determined as the green fluorescence intensity per cell in wildtype HEK293 cells without HA-AT1R overexpression. Data represent the mean ± SEM for four independent experiments. Statistical significance versus unstimulated cells was assessed using one-way ANOVA with Bonferroni correction
To further demonstrate recruitment of Gαi to the stretch-activated AT1R, we performed co-immunoprecipitation experiments. Consistent with the PLA results, the level of Gαi bound to AT1R was increased by osmotic stretch, but not affected by AngII and TRV120023 (Figures 2A and 2B). In contrast, AngII promoted the recruitment of Gαq to AT1R, while neither TRV120023 nor stretch had any effect (Figures 2C and 2D), which is consistent with previous studies suggesting that TRV120023- and stretch-induced signaling is independent of Gαq. Thus, in contrast to the balanced agonist AngII and the β-arrestin-biased agonist TRV120023, Gαi is only recruited to the mechano activated AT1R.
FIGURE 2.
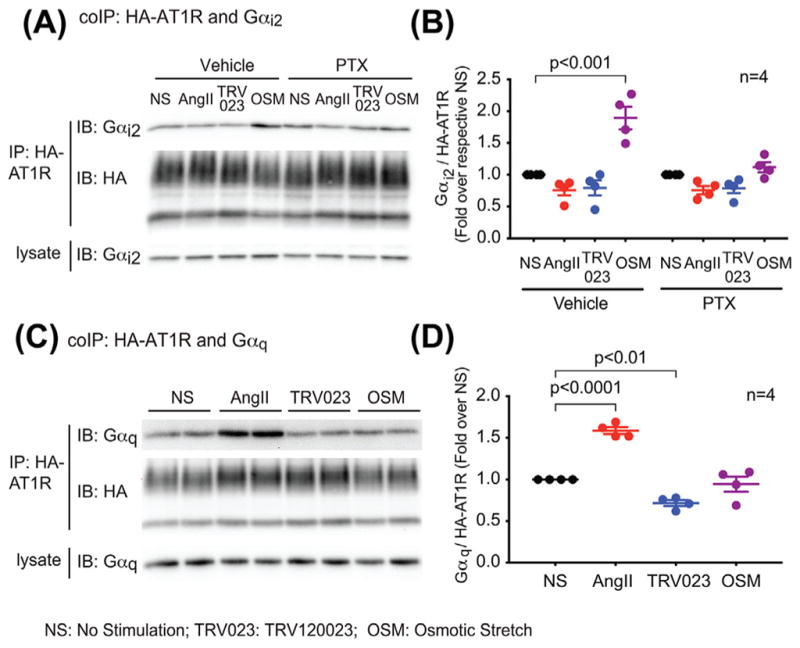
Osmotic stretch specifically induced AT1R coupling to Gαi, but not Gαq. In contrast, AngII promoted AT1R-Gαq coupling, while TRV120023 and osmotic stretch did not have significant effect. A and B: Osmotic stretch increases the amount of Gαi bound to AT1R, which was blocked by the PTX pretreatment. HEK293 cells stably expressing HA-tagged AT1Rs were transfected with Gαi2 48 h before experiment, and pretreated with vehicle or 200 ng/mL PTX in serum-free medium for overnight. Cell were stimulated with 1 μM AngII, 10 μM TRV120023, or osmotic stretch for 10 min. Gαi recruitment was determined with co-immunoprecipitation assay. HA-AT1Rs were immunoprecipitated with anti-HA magnetic beads, and bound Gαi2 was detected with western blot. C and D: AngII specifically induced Gαq recruitment to AT1R. AT1R stable cells were serum-starved for overnight, then stimulated with 1 μM AngII, 10 μM TRV120023 or osmotic stretch for 10 min. The AT1R-Gαq coupling was determined with co-immunoprecipitation. Signals were quantified by densitometry, normalized to the immunoprecipitated AT1R level, and expressed as fold change over the unstimulated cells. Data represent the mean ± SEM for n independent experiments as marked on the figure. Statistical significance versus unstimulated cells was assessed using one-way ANOVA with Bonferroni correction
3.2 | Gαi is required for membrane stretch-induced β-arrestin2 recruitment to AT1Rs
To dissect the role of Gαi in stretch-induced AT1R signaling, we tested the effect of the Gαi inhibitor PTX on β-arrestin2 recruitment using HEK293 cells stably expressing both CFP-tagged AT1R and YFP-tagged β-arrestin2. Due to low abundance in HEK293 cells of G protein-coupled receptor kinase 5 (GRK5), a kinase that mediates AT1R phosphorylation required for osmotic stretch-induced β-arrestin recruitment,13 cells were transfected with GRK5. In the absence of stimulation, AT1R localizes on the plasma membrane and β-arrestin2 is evenly distributed in cytoplasm (Figure 3A). When cells were stimulated with AngII, TRV120023, or osmotic stretch, β-arrestin2 translocated from cytoplasm to endocytic vesicles where it co-localized with the internalized AT1R (AngII: 87.0 ± 1.7%, 1 μM TRV120023: 73.8 ± 2.1%, 10 μM TRV120023: 73.3 ± 6.3%, osmotic stretch: 36.9 ± 2.1% of the cells showed β-arrestin2 translocation, respectively, all P < 0.0001 compared with unstimulated cells), indicating the recruitment of β-arrestin2 to AT1R. Pretreatment with PTX significantly blocked the osmotic stretch-induced β-arrestin2 recruitment (8.3 ± 1.4%, P < 0.0001 compared with vehicle-pretreated cells); while without any effect on the AngII- and TRV120023-induced response (with PTX, AngII: 84.5 ± 2.0%, 1 μM TRV120023: 75.8 ± 2.0%, 10 μM TRV120023: 74.7 ± 6.8%, P = n.s. compared with vehicle) (Figure 3B).
FIGURE 3.

Gαi is required for the osmotic stretch-induced β-arrestin2 recruitment to the AT1R. HEK 293 cells stably expressing CFP-tagged AT1R and YFP-tagged β-arrestin2 were transiently transfected with GRK5. After pretreatment with vehicle or 200 ng/mL PTX for overnight, cells were stimulated with 1 μM AngII, 1 μM, or 10 μM TRV120023, or osmotic stretch for 10 min. A: In response to AngII, TRV120023, or osmotic stretch, AT1R redistributed from the plasma membrane to intracellular puncta, and the cytosolic evenly distributed β-arrestin2 translocated and co-localized with the internalized AT1Rs, indicating the β-arrestin2 recruitment to AT1Rs. Pretreatment with PTX significantly inhibited osmotic stretch-induced β-arrestin2 recruitment, whereas has no effect to AngII- or TRV120023-induced response. Scale bar = 10 μM. B: Quantification of β-arrestin2 translocation was expressed as the percentage of cells showing intracellular aggregates of β-arrestin. Data represent the mean ± SEM for n independent experiments as marked on the figure. Statistical significance versus unstimulated cells, or between the vehicle and PTX pretreated group, was assessed using two-way ANOVA with Bonferroni correction
We previously showed that the mechanoactivated AT1R has specificity for β-arrestin2.13 To test whether PTX would influence β-arrestin1 translocation to the AT1R, we stably expressed both CFP-tagged AT1Rs and YFP-tagged β-arrestin1 in HEK293 cells and performed recruitment assays. While the recruitment of β-arrestin1 to AT1R was robustly induced by AngII and TRV120023, and was not affected by PTX pretreatment, β-arrestin1 translocation was not observed in cells treated with hypotonic stretch (Supplementary Figure S1).
Taken together, these results indicate a requirement of Gαi for osmotic stretch-induced β-arrestin2 recruitment to the AT1R, and indicates important heterogeneity in AT1R signaling induced by a β-arrestin-biased agonist compared to that by membrane stretch.
3.3 | Mechanoactivated-AT1R induces EGFR transactivation and ERK1/2 phosphorylation in a Gαi-dependent manner
Both ligand stimulation and mechanoactivation of AT1R promotes the shedding of membrane-bound EGF and subsequent transactivation of EGFR in a β-arrestin-dependent manner.13,21 To dissect the mechanism of stretch-induced Gαi-dependent AT1R signaling, we tested the effect of PTX on AT1R-mediated EGFR transactivation. We transfected HEK293 cells stably expressing AT1Rs with GFP-tagged EGFRs and monitored EGFR activation by GFP internalization with confocal microscopy. In the absence of stimulation, EGFRs were located on the plasma membrane. Stimulation of AT1R induced EGFR internalization as shown by redistribution into cellular aggregates (AngII: 44.4 ± 1.3%, TRV120023: 12.0 ± 1.7%, or osmotic stretch: 25.6 ± 2.6% of the cells showed EGFR internalization, AngII: P < 0.0001, TRV120023: P < 0.01, osmotic stretch: P < 0.0001 compared with unstimulated cells) (Figures 4A and 4B). Pretreatment with PTX significantly inhibited EGFR internalization induced by osmotic stretch (with PTX, 9.6 ± 1.6%, P < 0.0001 compared with vehicle-pretreated cells), whereas have no effect on AngII or TRV120023 (with PTX, AngII: 42.8 ± 1.9%, TRV120023: 11.2 ± 1.4%, P = n.s. compared with vehicle) (Figures 4A and 4B). These data indicate that Gαi is required for AT1R-mediated EGFR transactivation promoted by osmotic stretch, but not for the response induced by AngII or the β-arrestin-biased ligand TRV120023.
FIGURE 4.

Mechanoactivated AT1R signaling is dependent on Gαi. A and B: PTX selectively blocked AT1R-mediated EGFR internalization induced by osmotic stretch. HEK 293 cells stably expressing AT1R were transiently transfected with GFP-tagged EGFR and GRK5. Cells were pretreated with vehicle or 200 ng/mL PTX for overnight, and then stimulated with 1 μM AngII, 1 μM TRV120023, or osmotic stretch for 10 min. Quantification of EGFR internalization was performed by counting the percentage of the cells showing intracellular aggregates of GFP-EGFR. All three stimulations promoted EGFR internalization, whereas only the osmotic stretch-induced response is PTX-sensitive. Scale bar = 10 μM. C and D: Gαi is required for osmotic stretch-induced ERK1/2 phosphorylation in HEK293 cells. AT1R stable cells pretreated with vehicle or PTX were stimulated with 1 μM AngII, 1 μM TRV120023 or osmotic stretch for 10 min. Cell lysates were analyzed for phosphorylated ERK1/2 (pERK1/2) and total ERK1/2 (tERK1/2) with Western blot. The pERK1/2 signals were quantified by densitometry, normalized to tERK1/2, and expressed as fold change over unstimulated cells. ERK1/2 phosphorylation was significantly increased in response to AngII or osmotic stretch. PTX pretreatment diminished osmotic stretch-induced ERK1/2 phosphorylation, whereas has no effect on AngII-induced response. E and F: PTX blocked mechanical stretch-induced ERK1/2 phosphorylation in mice heart. In ex vivo perfused wild type heart, LV balloon stretch significantly increased ERK1/2 phosphorylation. Pretreatment with PTX (25 μg/kg) significantly inhibited balloon stretch-induced ERK1/2 phosphorylation. Quantification of pERK1/2 signal was performed as mentioned above. Data represent the mean ± SEM for n independent experiments or animals as marked on the figure. Statistical significance was assessed using two-way ANOVA with Bonferroni correction
We next determined the effect of PTX on AT1R-mediated ERK1/2 phosphorylation. In HEK293 cells stably expressing AT1Rs, both AngII and osmotic stretch significantly increased ERK1/2 phosphorylation, whereas TRV120023 showed a mild increase (Figures 4C and 4D). Similar to that observed for EGFR transactivation, PTX blocked the osmotic stretch-induced ERK1/2 signaling (P < 0.001 compared to vehicle), but did not alter the AngII response (Figures 4C and 4D).
To determine whether a similar signaling mechanism is involved in vivo, we next tested whether Gαi is necessary to activate mechanical stretch-induced AT1R signaling in mouse heart tissue. A polypropylene membrane balloon was placed in the left ventricle (LV) of a Langendorff-perfused mouse heart and then inflated to apply a mechanical stretch on the heart. We have previously shown that stretch of the heart induces ERK1/2 phosphorylation in a AT1R- and β-arrestin2-dependent manner.13 While LV balloon stretch of hearts significantly increased the phosphorylation of ERK1/2 (P < 0.001 compared with control), pretreatment with PTX abolished this response (P < 0.01 compared with the vehicle-pretreated hearts), indicating a requirement of Gαi for stretch-induced ERK1/2 signaling in vivo (Figures 4E and 4F).
To determine the relative contribution of Gαq and EGFR transactivation on AT1R signaling, we tested the effect of the protein kinase c (PKC) inhibitor Ro318425, phospholipase C inhibitor U73122 and the EGFR inhibitor AG1478 on ERK1/2 phosphorylation. Both Ro318425 and U73122 blocked AngII-induced ERK1/2 signaling (for both inhibitors, P < 0.0001 compared with vehicle), while neither had effect on the osmotic stretch response (Figure 5A–D). In contrast, AG1478 blocked ERK1/2 phosphorylation induced by either AngII (P < 0.0001 compared with vehicle) or osmotic stretch (P < 0.01 compared with vehicle) (Figures 5E and 5F).
FIGURE 5.

Osmotic stretch-induced ERK1/2 phosphorylation is independent of Gαq, whereas dependent on EGFR transactivation. A and B: Osmotic stretch-induced ERK1/2 phosphorylation was not blocked by Ro318425, suggesting Gαq is not required for this response. AT1R stable cells were pretreated with 1 μM PKC inhibitor Ro318425 for 30 min, then stimulated with 1 μM AngII, 1 μM TRV120023, or osmotic stretch for 10 min. C and D: The PLC inhibitor U73122 blocked the AngII-induced ERK1/2 phosphorylation, whereas had no effect on the osmotic stretch response. Cells were pretreated with 10 μM U73122. E and F: AG1478 blocked ERK1/2 phosphorylation induced by AngII or osmotic stretch, suggesting the ERK1/2 activation is mediated by the transactivated EGFRs. AT1R stable cells were pretreated with 1 μM EGFR inhibitor AG1478 for 30 min before stimulation. The pERK1/2 signals were quantified by densitometry, normalized to tERK1/2, and expressed as fold change over unstimulated cells. Data represent the mean ± SEM for n independent experiments or animals as marked on the figure. Statistical significance was assessed using two-way ANOVA with Bonferroni correction
Collectively, these data support a concept that the balanced ligand AngII stimulates AT1R ERK1/2 signaling through both the Gαq and β-arrestin transducers, while mechanoactivated AT1R signaling is mediated through a Gαi-dependent, β-arrestin-dependent mechanism.
3.4 | TRV120026, another β-arrestin-biased agonist, induces β-arrestin-biased AT1R signaling independent of Gαi
To determine whether the Gαi dependency for the β-arrestin-biased AT1R signaling is specifically induced by mechanical stress, we tested the effect of the Gαi inhibitor PTX on the AT1R signaling induced by TRV120026, another β-arrestin-biased agonist with very similar potency of β-arrestin recruitment to AT1R.7,22 In HEK293 cells stably expressing the CFP-tagged AT1R and YFP-tagged β-arrestin2, TRV120026 significantly induced β-arrestin2 translocation, to the same extent of TRV120023 response (1 μM TRV120026: 62.0 ± 2.0%, 10 μM TRV120026: 72.7 ± 2.4%, 10 μM TRV120023: 72.0 ± 2.3% of cells showed β-arrestin2 translocation, all P < 0.0001 compared with unstimulated cells) (Figures 6A and 6B). Pretreatment of PTX have no effect on TRV120026-induced response (Figures 6A and 6B), suggesting that Gαi is not required for TRV120026-induced β-arrestin2 translocation. Similar to TRV120023, TRV120026 induced mild increase of ERK1/2 phosphorylation, which was not affected by PTX pretreatment (Figures 6C and 6D). Taken together, these data suggest that the β-arrestin-biased agonists TRV120023 and TRV120026 both induce AT1R signaling in a Gαi-independent manner, which is distinct from that induced by osmotic stretch.
FIGURE 6.

TRV120026 induces the β-arrestin-biased AT1R signaling in a Gαi-independent manner, consistent with the TRV120023 response. A and B: PTX had no effect on the TRV120026-induced β-arrestin2 recruitment to AT1R. HEK 293 cells stably expressing CFP-tagged AT1R and YFP-tagged β-arrestin2 were transiently transfected with GRK5. After pretreatment with vehicle or 200 ng/mL PTX for overnight, cells were stimulated with 1 μM AngII, 10 μM TRV120023, 1 μM or 10 μM TRV120026, or osmotic stretch for 10 min. Scale bar = 10 μM. Quantification of β-arrestin2 translocation was expressed as the percentage of cells showing intracellular aggregates of β-arrestin. C and D: similar to the effect of TRV120023, TRV120026 induced a mild increase of ERK1/2 phosphorylation, which was not PTX-sensitive. HEK293 cells stably expressing AT1R were pretreated and stimulated as mentioned above. The pERK signals were quantified by densitometry, normalized to tERK, and expressed as fold change over unstimulated cells. Data represent the mean ± SEM for n independent experiments as marked on the figure. Statistical significance was assessed using two-way ANOVA with Bonferroni correction
3.5 | Depletion of Gαi by CRISPR-Cas9 gene editing blocks osmotic stretch-induced β-arrestin2 recruitment to AT1Rs
To more robustly determine the role of Gαi in osmotic stretch-induced AT1R signaling, we measured β-arrestin2 translocation in a Gαi knockout cell line23 that is completely deficient of all three subtypes of Gαi generated by CRISPR/Cas9 gene editing (Figure 7A). In wild type HEK293 cells transfected with CFP-tagged AT1R and YFP-tagged β-arrestin2 (together with GRK5), stimulation with AngII, TRV120023, TRV120026, and osmotic stretch induced the translocation of β-arrestin2. In the Gαi knockout cells, the osmotic stretch-induced β-arrestin2 translocation was blocked, whereas the responses induced by other stimuli were not affected (Figures 7B and 7C). These data are consistent with our PTX experiments and robustly demonstrate that Gαi is required for stretch-induced β-arrestin2 recruitment to the AT1R.
FIGURE 7.

Osmotic stretch-induced β-arrestin2 recruitment to AT1R was blocked in Gαi knockout cells. A: Western blot showing the knockout of Gαi in HEK293 cells by CRISPR-Cas9 gene editing. B: AngII, TRV120023, and TRV120026 induced β-arrestin2 translocation in Gαi knockout cells to the same extent as that in wild type cells. In contrast, the osmotic stretch-induced response was blocked in Gαi knockout cells. Gαi knockout cells were transfected with CFP-tagged AT1R, YFP-tagged β-arrestin2, and GRK5. Cells were stimulated with 1 μM AngII, 10 μM TRV120023, 10 μM TRV120026, or osmotic stretch for 10 min. B: Quantification of β-arrestin2 translocation was expressed as the percentage of cells showing intracellular aggregates of β-arrestin. Data represent the mean ± SEM for n independent experiments as marked on the figure. Statistical significance was assessed using two-way ANOVA with Bonferroni correction
4 | DISCUSSION
In this study, we identify a new molecular mechanism for β-arrestin-biased AT1R signaling. Membrane stretch, which has been shown to activate β-arrestin-biased AT1R signaling without activating Gαq, specifically promotes the recruitment of another subtype of G proteins, Gαi, to the AT1R (Figure 8). AT1R-Gαi coupling is required for the recruitment of β-arrestin to the receptor and the induction of subsequent β-arrestin-dependent signaling as measured by EGFR internalization and ERK1/2 phosphorylation. In contrast, the Gαi recruitment and the dependence of Gαi for β-arrestin-mediated signaling was not observed for the balanced agonist AngII, or, remarkably, the β-arrestin-biased agonists TRV120023 and TRV120026 (Figure 8). These results indicate mechanistic divergence of AT1R-mediated β-arrestin-biased signaling triggered by the different modes of receptor activation, membrane stretch and β-arrestin-biased ligands in this case.
FIGURE 8.

Schematic model of AT1R signaling induced by different modes of receptor activation. Different stimulations stabilize the receptors into distinct active conformational states, allowing the recruitment of unique subsets of transducers that subsequently activates divergent arrays of downstream signaling. When induced by the balanced agonist AngII, the AT1Rs transduce signaling through both Gαq and β-arrestins, whereas the biased agonists TRV120023 and TRV120026 preferentially induce β-arrestin signaling without activating G proteins. In contrast, another β-arrestin-biased receptor stimulation, mechanical stress, specifically promotes the recruitment of Gαi to the receptors and activates β-arrestin signaling through a mechanism distinct from that induced by the biased ligands
It is well established that GPCR can engage to multiple G protein subtypes, and the efficacy and kinetics of distinct G protein coupling are regulated by the specificity of ligands or allosteric modulators.16 Although in our study we did not observe Gαi recruitment to AngII-stimulated AT1Rs, previous studies have shown that AngII may promote AT1R-Gαi coupling to inhibit adenylyl cyclase and to regulate Ca2+ channels in certain tissue or cell types.24–26 In addition, a recent study reported that SII, an AT1R ligand previously described as a β-arrestin-biased agonist, can also weakly activates Gαq and Gαi.15 Despite the variation of the effect of AngII on Gαi activation, which might due to the difference in experimental systems or assay sensitivity, it is clear that in response to different stimuli AT1Rs can engage distinct transducers to induce cellular signaling.
A full understanding of the diversity of GPCR signaling through distinct transducers activated by different stimuli remains to be elucidated. Besides their role in receptor internalization and desensitization, β-arrestins are also critical transducers for GPCR signaling that involved in a wide diversity of physiological and pathological processes. Interestingly, the G protein signaling and β-arrestin signaling are not mutually exclusive. The signaling mediated by many Gαi-coupled receptors requires both Gαi and β-arrestin.27 Upon the previous understanding, the β-arrestin-mediated signaling of most Gαs or Gαq receptors appears to be independent of G proteins.27 However, our study suggests that certain modes of stimulation, mechanoactivation in this case, may switch the primarily Gαq-coupled receptors to coupling to Gαi. The now Gαi-coupled receptors then activate downstream β-arrestin signaling.
Considerable structural information is now available for GPCRs in complex with ligands or signal transducers and support the concept that receptors adopt distinct conformations to selectively regulate different arrays of downstream signaling.28,29 Biophysical analysis of mechanical stretch-activated AT1R suggest that the AT1R undergoes anticlockwise rotation and a shift of transmembrane domain 7 into the ligand-binding pocket, whereas AT1R blocker candesartan stabilized the receptor in the inactive conformation and prevented stretch induced conformational change.30 Our previous work suggested that membrane stretch induces a β-arrestin-biased conformational state of AT1Rs and an active β-arrestin conformation indistinguishable from that induced by the biased agonist TRV120023.13,14 However, we now show that membrane stretch induces a distinct AT1R-β-arrestin signaling complex through its recruitment of Gαi suggesting the existence of heterogeneity in biased-receptor conformations.
Given the importance of GRK-mediated receptor phosphorylation on the recruitment of β-arrestins to the receptor, the concept of a bar code of receptor phosphorylation has emerged as a potential mechanism for how ligand-specific receptor conformations selectively recruit specific transducers.12,31,32 According to this hypothesis, the receptor adopts distinct conformational states when bound to specific ligands, and each then recruits unique subsets of GRKs favoring distinct regions of the receptor, leading to phosphorylation of distinct amino acid residues on the c-terminal tail of the receptor, generating a phosphorylation “bar code.” These distinct GRK-mediated phosphorylation patterns direct the recruitment of signal transducers, such as β-arrestins, stabilizing them into unique active conformations and activating diversity in signaling profiles. This hypothesis is compatible with previous findings for the AT1R signaling whereby GRK5 and GRK6 are required for β-arrestin-mediated MAPK activation,13 whereas GRK2 and GRK3 mediate receptor internalization.3 Therefore, in our conceptual model, we speculate that membrane stretch induces a unique AT1R conformation that exposes an allosteric binding sites for Gαi, and the bound Gαi in turn stabilizes a unique receptor conformation allowing receptor phosphorylation at specific residues on the c-terminal tail of the receptor by a unique subset of GRKs. The stretch-induced GRK-mediated distinctive phosphorylation pattern of the AT1R then induces a unique β-arrestin-biased receptor conformational state, leading to the activation of subsequent biased signaling. Future phosphoproteomic studies will be needed to determine the specific AT1R c-terminus phosphorylation pattern induced by balanced agonist, β-arrestin-biased agonist or membrane stretch.
AT1R plays vital roles in the regulation of cardiovascular function, and is one of the major targets for the therapeutic treatment of cardiovascular diseases such as congestive heart failure and hypertension.1,9,33 As Gαq-mediated AT1R signaling has been shown to mediate cardiomyocyte hypertrophy whereas β-arrestin-mediated signaling is thought to promote cardioprotection,34–36 the β-arrestin-biased agonist may provide novel clinical implications on promoting cardiac contractility and simultaneously blocking cardiac dysfunction.22,37–39 With our data now showing that two different β-arrestin-biased stimuli, membrane stretch and the biased ligands likely engage distinct AT1R molecular states to activate β-arrestin signaling, the potential to screen for unique biased ligands that can distinguish these different pathways may be possible.
In conclusion, we show that membrane stretch specifically promotes AT1R coupling to Gαi, triggering activation of β-arrestin-biased signaling. These new insights for mechanoactivation of the AT1R reveal additional regulatory complexity for biased agonism.
Supplementary Material
Acknowledgments
Funding information: National Institutes of Health, Grant numbers: HL56687, HL75443; American Heart Association, Grant number: 14PRE20480352
This work was supported by National Institutes of Health grants HL56687 and P01 HL75443 to HAR and a AHA Predoctoral Fellowship 14PRE20480352 to JW.
Footnotes
Additional Supporting Information may be found online in the supporting information tab for this article.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 2.Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 3.Kim J, Ahn S, Ren XR, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A. 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 5.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strachan RT, Sun JP, Rominger DH, et al. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR) J Biol Chem. 2014;289:14211–14224. doi: 10.1074/jbc.M114.548131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 9.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. Beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem. 2014;289:28271–28283. doi: 10.1074/jbc.M114.585067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauliere A, Bellot M, Paris H, et al. Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol. 2012;8:622–630. doi: 10.1038/nchembio.961. [DOI] [PubMed] [Google Scholar]
- 16.Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, Martemyanov KA. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal. 2015;8:ra123. doi: 10.1126/scisignal.aab4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crajoinas RO, Polidoro JZ, Carneiro de Morais CP, Castelo-Branco RC, Girardi AC. Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am J Physiol Cell Physiol. 2016;311:C768–C776. doi: 10.1152/ajpcell.00191.2016. [DOI] [PubMed] [Google Scholar]
- 18.Zou Y, Komuro I, Yamazaki T, et al. Cell type-specific angiotensin II-evoked signal transduction pathways: critical roles of Gbetagamma subunit, Src family, and Ras in cardiac fibroblasts. Circ Res. 1998;82:337–345. doi: 10.1161/01.res.82.3.337. [DOI] [PubMed] [Google Scholar]
- 19.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choo YY, Hagen T. Mechanism of cullin3 E3 ubiquitin ligase dimerization. PLoS ONE. 2012;7:e41350. doi: 10.1371/journal.pone.0041350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent beta-arrestin2 and Gq/protein kinase Czeta pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J Biol Chem. 2009;284:11953–11962. doi: 10.1074/jbc.M808176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Violin JD, DeWire SM, Yamashita D, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Hanada K, Staus DP, et al. Galphai is required for carvedilol-induced beta1 adrenergic receptor beta-arrestin biased signaling. Nat Commun. 2017;8:1706. doi: 10.1038/s41467-017-01855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hescheler J, Rosenthal W, Hinsch KD, Wulfern M, Trautwein W, Schultz G. Angiotensin II-induced stimulation of voltage-dependent Ca2+ currents in an adrenal cortical cell line. EMBO J. 1988;7:619–624. doi: 10.1002/j.1460-2075.1988.tb02855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maturana AD, Casal AJ, Demaurex N, Vallotton MB, Capponi AM, Rossier MF. Angiotensin II negatively modulates L-type calcium channels through a pertussis toxin-sensitive G protein in adrenal glomerulosa cells. J Biol Chem. 1999;274:19943–19948. doi: 10.1074/jbc.274.28.19943. [DOI] [PubMed] [Google Scholar]
- 26.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 27.Lefkowitz RJ. Ar restins come of age: a personal historical perspective. Prog Mol Biol Transl Sci. 2013;118:3–18. doi: 10.1016/B978-0-12-394440-5.00001-2. [DOI] [PubMed] [Google Scholar]
- 28.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaidehi N, Kenakin T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr Opin Pharmacol. 2010;10:775–781. doi: 10.1016/j.coph.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Yasuda N, Miura S, Akazawa H, et al. Conformational switch of angiotensin II type 1 receptor underlying mechanical stress-induced activation. EMBO Rep. 2008;9:179–186. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nobles KN, Xiao K, Ahn S, et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou XE, He Y, de Waal PW, et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell. 2017;170:457–469. e413. doi: 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–766. doi: 10.1016/s0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- 34.Fan G, Jiang YP, Lu Z, et al. A transgenic mouse model of heart failure using inducible Galpha q. J Biol Chem. 2005;280:40337–40346. doi: 10.1074/jbc.M506810200. [DOI] [PubMed] [Google Scholar]
- 35.Benigni A, Corna D, Zoja C, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajagopal K, Whalen EJ, Violin JD, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. {beta}-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem. 2009;284:8855–8865. doi: 10.1074/jbc.M808463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC., Jr Cardiorenal actions of TRV120027, a novel ss-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4:770–778. doi: 10.1161/CIRCHEARTFAILURE.111.962571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.