

Abstract
Cold-inducible RNA-binding protein (CIRP) is a novel inflammatory mediator that stimulates the release of proinflammatory cytokines from macrophages in sepsis. Given the immune dysregulation that characterizes sepsis, the effect of CIRP on other immune cells is an area of increasing interest that has not yet been studied. In the present study, we hypothesized that extracellular CIRP promotes activation of T lymphocytes in the spleen during sepsis. We observed that mice subjected to sepsis by cecal ligation and puncture showed significantly higher expression of the early activation markers CD69 and CD25 at 20 h on CD4+ splenic T cells, and significantly higher CD69 expression on CD8+ splenic T cells compared with sham-operated controls. Furthermore, at 20 h after receiving intravenous injection of recombinant murine CIRP (rmCIRP, 5 mg/kg body weight (BW)) or PBS (vehicle), those mice receiving rmCIRP showed significantly increased expression of CD69 and CD25 on both CD4+ and CD8+ splenic T cells. This effect, however, was not seen in TLR4-deficient mice after rmCIRP injection. In addition, treatment with CIRP predisposed CD4+ T cells to a Th1 hyperinflammatory response profile, and influenced CD8+ T cells toward a cytotoxic profile. Taken together, our findings indicate that CIRP is a proinflammatory mediator that plays an important role in T-cell dysregulation during sepsis in a TLR4-dependent manner.
Keywords: CIRP, immune response, lymphocytes, sepsis, T-cell activation
INTRODUCTION
After several decades of evolving definitions attempting to effectively define sepsis, the most recent consensus guidelines characterize it as a dysregulated host response to infection that results in life-threatening organ dysfunction.1 It is a serious clinical entity that can ultimately lead to shock and death, with recent global mortality estimates as high as 5.3 million lives per year.2, 3, 4 In the United States, sepsis is a major public health problem accounting for over 1 million hospitalizations and over $24 billion in treatment costs annually.5, 6
The immune response to sepsis is multifactorial, and is often described as consisting of two stages, the timing of which is likely complex and overlapping. The first component is a state of hyperinflammation, also referred to as the ‘cytokine storm’.7 This is typically thought to predominate early on in the course of the disorder and is generally attributed to the actions of the innate immune system, including macrophages, neutrophils and natural killer (NK) cells, as well as a subset of the T lymphocyte population, natural killer T (NKT) cells. The second component of the immune response is a state of immunosuppression in which patients are particularly susceptible to secondary infection, a major cause of morbidity and mortality in sepsis.7, 8 This aspect of the immune response can largely be attributed to dysfunction of the adaptive immune system, including B and T lymphocytes.
One component of the immune response to sepsis is inflammation caused by pathogen-associated molecular patterns (PAMPs), which are elements of invading organisms that trigger cytokine release on detection by pathogen recognition receptors (PRRs) present on immune cells. A second component of the immune response to sepsis is caused by damage-associated molecular patterns (DAMPs). DAMPs are endogenous molecules that, under normal conditions, remain hidden from recognition by the immune system. However, in times of cellular stress (for example, hypoxia, ischemia, mechanical injury), these molecules are released and trigger inflammation.9
Cold-inducible RNA-binding protein (CIRP) is a 172-amino acid nuclear protein that has recently been identified as a DAMP.10 It is a cold-shock protein, originally found to impair cell growth in the setting of hypothermia.11 CIRP can bind to RNAs and modulate them at the post-transcriptional level,12 and is involved in regulating diverse cellular physiological processes, including cell growth, senescence and apoptosis.13 More recently it has also been found at increased levels in humans with sepsis and hemorrhagic shock. In addition, in animal models neutralization with anti-CIRP antibody prolonged survival and attenuated organ injury in sepsis and hemorrhagic shock.10 Furthermore, in vitro work demonstrated that CIRP is able to stimulate the release of the proinflammatory cytokines TNF-α and HMGB1 from macrophages.10 Given the immune dysregulation that characterizes sepsis, and the role of CIRP in sepsis and shock, the interaction of CIRP with other cells of the immune system is an area of particular interest. Specifically, its effect on T lymphocytes is worth exploring, because T cells have a special role in the immune response, being key components of the adaptive system while also contributing to the innate immune response.
Many aspects of T-cell function can be studied, including antigen presentation and recognition, T-cell activation, proliferation, differentiation and effector functions. In the present study, we examined T-cell activation by looking at the expression of two surface molecules known to be upregulated on activation: CD69 and CD25. CD69 is well-established as an early activation antigen of immune cells.14, 15 It has been used as a marker of T-cell activation in septic animals and humans, as well as in children with pneumonia.16, 17, 18 CD25 is also known as the alpha subunit of the interleukin 2 (IL-2) receptor, and IL-2 is a cytokine implicated in lymphocyte proliferation. CD25 is upregulated slightly later in the course of T-cell activation, and has also been used as a marker of T-cell activation in animals and humans.15, 18, 19
In this study, we hypothesized that extracellular CIRP would have an important role in the activation of T cells and contribute to the immune dysregulation seen in sepsis. In order to examine this, we subjected mice to polymicrobial sepsis by cecal ligation and puncture (CLP), followed by flow cytometric analysis of splenic T lymphocytes. We then looked at changes in T-cell profiles in mice injected with recombinant murine CIRP (rmCIRP). We further explored the role of toll-like receptor 4 (TLR4) in this setting by injecting TLR4-deficient (Tlr4−/−) mice with rmCIRP. Lastly, we explored the downstream effects of CIRP on T-cell effector functions.
MATERIALS AND METHODS
Experimental animals
Adult male C57BL/6 mice (20-25 g) were purchased from Charles River Laboratories (Wilmington, MA, USA). On acquisition, mice were allowed to acclimate to the environment in our facility for a period of 7 days. Tlr4−/− mice were donated by the lab of Dr Kevin Tracey at the Feinstein Institute for Medical Research (Manhasset, NY, USA). All mice were housed in a temperature-controlled room with a 12-h light-dark cycle, fed a standard laboratory diet, and allowed water ad libitum. All experiments were performed in accordance with the guidelines for the use of experimental animals by NIH (Bethesda, MD, USA) and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Feinstein Institute for Medical Research. For all in vivo experiments, anesthesia was induced and maintained using inhaled isoflurane.
Polymicrobial sepsis model
Sepsis was induced by CLP, as previously described by Wisnoski et al.20 with minor modifications. Mice were randomly assigned to undergo CLP or sham operation. After anesthesia induction, mice were placed supine and their abdomens shaved and prepped with betadine solution. A midline incision was performed through the skin and peritoneum. The cecum was identified and extracorporalized. In sham-operated controls, the cecum was then returned to the abdomen and the incision was closed with non-absorbable suture. In CLP animals, 50% of the cecum was ligated with non-absorbable suture. The distal aspect of the cecum was then punctured twice with a 22-gauge needle, and a small amount of feces was extruded. The cecum was then returned to the abdominal cavity and the abdomen was closed with non-absorbable suture. In all cases animals received 0.5 ml of normal saline resuscitation subcutaneously, were observed to recover from anesthesia, and were then returned to their cages. At 20 h after operation, all mice were killed by CO2 asphyxiation and organs were collected for processing.
Purification of rmCIRP and removal of lipopolysaccharide
Transformed E. coli carrying the rat His-CIRP expression plasmid were inoculated in Luria–Bertani medium containing kanamycin overnight. They were subsequently induced with 1 mm isopropyl β-d-1-thiogalactopyranoside for an additional 6 h. Following this, the bacteria were collected by centrifugation and washed once with 20 mm Tris–HCl, pH 7.9. The pellet was resuspended in 20 mm Tris–HCl, pH 7.9, 500 mm NaCl and 5 mM imidazole. It was then lysed by sonication at 4 °C. The soluble extract was clarified by centrifugation at 20 000g for 1 h at 4 °C. Clear lysate was then loaded onto a nickel-nitrilotriacetic acid column (Novagen, Madison, WI, USA). Bound protein was washed with 20 mM Tris–HCl, pH 7.9, 500 mm NaCl and 100 mm imidazole, and was eluted in the same buffer supplemented with 1 m imidazole. All proteins were dialyzed with phosphate-buffered solution (PBS) and stored at −80 °C before use. Triton X-114 (Sigma-Aldrich, St Louis, MO, USA) was added to the purified rmCIRP protein at a final concentration of 5%. It was then rotated for 15 min at room temperature to ensure homogeneity. It was subsequently centrifuged at 14 000g for 12 min, and the upper aqueous phase (lipopolysaccharide (LPS)-free rmCIRP) was removed. The level of LPS was measured by a limulus amebocyte lysate (LAL) assay (Cambrex, East Rutherford, NJ, USA), and was <10 pg LPS/μg rmCIRP.
Administration of rmCIRP
Mice were randomly assigned to receive 5 mg/kg body weight rmCIRP or equivalent volume PBS (vehicle). After induction of anesthesia, mice were placed supine and the area over the right jugular vein was shaved and prepped with betadine. An incision was made and dissection carried out to the level of the jugular vein. This was then isolated, and rmCIRP or PBS injected. The vein was then ligated with non-absorbable suture and the incision closed. Animals were observed to recover from anesthesia and were then returned to their cages. At 20 h after injection, all mice were killed by CO2 asphyxiation and organs were collected for processing.
Western blot analysis
Spleen samples (100 mg) were lysed and homogenized in 250 μl lysis buffer (10 mm Tris-buffered saline pH 7.5, 1% Triton X-100, 1 mm ethylenediaminetetraacetic acid, 1 mm ethylene glycol tetraacetic acid) containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA) by sonication. After 30-min incubation at 4 °C, samples were centrifuged at 13,200 r.p.m. for 15 min. Supernatant was then collected, and sample protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Somerset, NJ, USA). Once protein concentration was determined, 50 μg samples were separated on 4–12% Bis–Tris gels and transferred to nitrocellulose membranes. Membranes were then incubated with primary antibody against CIRP or β-actin (SantaCruz Biotechnologies, Santa Cruz, CA, USA). Protein bands were detected by immunofluorescence using species-specific fluorescent secondary antibodies and the LI-COR Odyssey Fc Imager (LI-COR, Lincoln, NE, USA). Band intensity was measured using the NIH Image J densitometric software program (Bethesda, MD, USA).
Lymphocyte preparation from spleen and T-cell purification
Spleens were disrupted and homogenized by crushing and filtering through a sterile 70 μm nylon filter in complete RPMI medium (with 10% FBS, 1% Penn-Strep, 10 mm HEPES, 2 mm l-glutamine and 50 μm β-mercaptoethanol). Erythrocytes were eliminated using ACK Lysing Buffer (Quality Biological, Gaithersburg, MD, USA) and the cells were then washed and resuspended in complete RPMI medium. CD4+ and CD8+ T cells were purified from splenic lymphocytes by immunomagnetic negative selection using EasySep CD4+ T cell and CD8+ T Cell Isolation Kits (STEMCELL Technologies, Vancouver, BC, Canada) per manufacturer instructions.
Flow cytometry
Cells were stained with the following fluorescent antibodies in flow cytometry buffer (PBS, 2% fetal bovine serum, 0.1% sodium azide): APC-CD8, PerCP/Cy5.5-CD4, PE-CD69, PE/Cy7-CD25 (Biolegend, San Diego, CA, USA) and analyzed using a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA, USA). Flow cytometry data was analyzed using FlowJo 7.6.5 software (FlowJo LLC, Ashland, OR, USA).
CD4+ T cell Th1/Th2 PCR array and CD8+ T cell real-time polymerase chain reaction (qPCR)
Purified CD4+ and CD8+ T cells from the spleens of healthy mice were stimulated (in complete RPMI media on α-CD3/α-CD28-coated 24-well plates) with or without rmCIRP (1 μg/ml) for 20 h at 37 °C. Cells were then collected and total RNA was extracted using TRIzol reagent (Invitrogren, Carlsbad, CA, USA).
Total CD4+ T-cell RNA (0.5 μg) then underwent reverse transcription using the RT2 First Strand Kit (QIAGEN, Germantown, MD, USA) per manufacturer instructions. qPCR was then carried out using the RT2 Profiler PCR Array for Mouse Th1 & Th2 Responses (QIAGEN) per manufacturer instructions. An ABI 7300 real-time PCR machine (Applied Biosystems, Foster City, CA, USA) was used for amplification using absolute quantification under the thermal profile of 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Data was then analyzed using the manufacturer’s web-based data analysis suite (QIAGEN). A fold regulation threshold of ±2 with a P-value of 0.05 was used for detecting changes in gene expression between groups.
Total CD8+ T-cell RNA (2 μg) underwent reverse transcription using murine leukemia virus reverse transcriptase (Applied Biosystems). PCR was carried out in a 25 μl final volume containing 0.5 μl each of forward and reverse primers, 5 μl cDNA, 6.5 μl H2O and 12.5 μl Power SYBR Green PCR Master Mix (Applied Biosystems). An ABI 7300 real-time PCR machine was used for amplification under the thermal profile of 50 °C for 2 min and 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 sec and 60 °C for 1 min. Mouse β-actin mRNA levels were used for normalization. Relative expression of mRNA was calculated by the 2−ΔΔCt method, and results are expressed as fold change in comparison to control group. The sequences for the primers used in this study are as follows: mouse regulated on activation, normal T cell expressed and secreted (RANTES) 5′-GCCCACGTCAAGGAGTATTT-3′ (forward) and 5′-AGCAAGCAATGACAGGGAAG-3′ (reverse); mouse granzyme B 5′-ACCCAAAGACCAAACGTGCT-3′ (forward) and 5′-AGCAGGATCCATGTTGCTTC-3′ (reverse); mouse interferon gamma (IFN-γ) 5′-GGCTTTGCAGCTCTTCCTC-3′ (forward) and 5′-CCAGTTCCTCCAGATATCCAA-3′ (reverse); and mouse β-actin, 5′-CGTGAAAAGATGACCCAGATCA-3′ (forward) and 5′-TGGTACGACCAGAGGCATACAG-3′ (reverse).
Statistical analysis
All data are expressed as mean±s.e.m. and compared by two-tailed Student’s t-test for pair comparisons. Differences in values are considered significant if P<0.05.
RESULTS
Sepsis induces activation of splenic CD4+ and CD8+ T cells
We first examined the activation status of T cells in the spleen, a lymphoid organ and source of abundant T lymphocytes, in sepsis. At 20 h after sepsis induction by CLP, spleens were collected and processed to isolate lymphocytes for flow cytometric analysis. After acquisition, total cells were gated for the lymphocyte population based on forward and side scatter (FSC-A and SCC-A) characteristics; from this, the population staining positively for PerCP/Cy5.5-CD4 was gated (Figure 1a). This CD4+ T-cell population was subsequently gated on PE-CD69-positive and PE/Cy7-CD25-positive populations (Figure 1a). Sepsis resulted in a significant increase in the expression of activation markers CD69 and CD25 on CD4+ T cells compared to sham-operated controls (Figure 1b). This was true for the percentages of CD69+ (11.3 vs 18.9%, P<0.05) and CD25+ (12.9 vs 16.5%, P<0.05) cells, as well as for the expression levels of CD69 and CD25 per cell, indicated by the mean fluorescence intensity (MFI; CD69 MFI: 110 vs 147; CD25 MFI: 104 vs 126; P<0.05). For the CD8+ T cells, total splenocytes were again first gated for the lymphocyte population based on FSC-A and SCC-A characteristics (Figure 2a); from this, the population staining positively for APC-CD8 was gated (Figure 2a). This CD8+ population was subsequently gated on PE-CD69-positive and PE/Cy7-CD25-positive populations (Figure 2a). Sepsis resulted in a significant increase in the expression of CD69 on CD8+ T cells compared to sham-operated controls (Figure 2b). This was true for the percentages of CD69+ cells (4.8 vs 10.7%, P<0.05), as well as for the expression level of CD69 per cell, indicated by the MFI (98 vs 121, P<0.05).
Figure 1.

Activation of CD4+ splenic T cells after CLP. Mice underwent CLP or sham operation; spleens were collected 20 h later and processed to isolate lymphocytes. (a) A representative sample showing flow cytometry gating strategy. Total splenocytes were gated for the live lymphocyte population based on FSC and SSC characteristics. Lymphocytes were subsequently gated for the PerCP/Cy5.5-CD4-positive cell population and further gated into those staining positively for PE-CD69 and PE/Cy7-CD25. (b) Flow cytometric analysis of surface CD69 and CD25 expression on the gated splenic CD4+ T cells. Percentages of CD69+ and CD25+ cells and mean fluorescence intensity (MFI) of CD69 and CD25 expression are shown. Data expressed as mean±s.e.m. (n=5 per group) and compared by Student’s t-test. *P<0.05 vs Sham. CLP, cecal ligation and puncture; FSC, forward scatter; SSC, side scatter.
Figure 2.

Activation of CD8+ splenic T cells after CLP. Mice underwent CLP or sham operation; spleens were collected 20 h later and processed to isolate lymphocytes. (a) A representative sample showing flow cytometry gating strategy. Total splenocytes were gated for the live lymphocyte population based on FSC and SSC characteristics. Lymphocytes were subsequently gated for the APC-CD8-positive cell population and further gated into those staining positively for PE-CD69 and PE/Cy7-CD25. (b) Flow cytometric analysis of surface CD69 and CD25 expression on the gated splenic CD8+ T cells. Percentages of CD69+ and CD25+ cells and MFI of CD69 and CD25 expression are shown. Data expressed as mean±s.e.m. (n=5 per group) and compared by Student’s t-test. *P<0.05 vs Sham. CLP, cecal ligation and puncture; FSC, forward scatter; SSC, side scatter.
CIRP protein levels in the spleen are increased after CLP
After identifying the activation of splenic T cells in sepsis, we sought to determine the protein expression of CIRP in septic mice by western blotting. CIRP protein levels in the spleen were increased 2-, 3- and 4-fold compared with sham-operated controls at 5, 10 and 20 h after CLP, respectively (Figure 3). This result demonstrates an association between CIRP elevation and T-cell activation in sepsis.
Figure 3.

Protein levels of CIRP in the spleen after CLP. Mice underwent CLP or sham operation; spleens were collected at 5, 10 and 20 h after CLP and CIRP protein levels were determined by western blot. A representative blot is shown along with a graph representing densitometry analysis. Data expressed as mean±s.e.m. (n=2 mice in sham group and 3 mice per CLP group). *P<0.05 vs Sham by Student’s t-test. CLP, cecal ligation and puncture; CIRP, cold-inducible RNA-binding protein.
Administration of rmCIRP induces activation of splenic CD4+ and CD8+ T cells in mice
To further examine the effect of CIRP on the activation of T cells, rmCIRP was injected intravenously into healthy mice. Twenty hours after injection, spleens were collected and processed to isolate lymphocytes for flow cytometric analysis. rmCIRP administration resulted in significant increases in the expression of activation markers CD69 and CD25 on both CD4+ (Figure 4a) and CD8+ (Figure 4b) T cells compared to PBS administration. This was demonstrated by the percentages of CD69+ (CD4+CD69+: 13.8 vs 28.2%, CD8+CD69+: 5.4 vs 19.1%, P<0.05) and CD25+ (CD4+CD25+: 10.1 vs 14.8%, CD8+CD25+: 1.1 vs 1.4, P<0.05) cells, as well as by the expression levels of CD69 and CD25 per cell indicated by the MFI (CD69 MFI on CD4+T: 150 vs 285; CD69 MFI on CD8+T: 114 vs 174; CD25 MFI on CD4+T: 119 vs 162; CD25 MFI on CD8T: 41.7 vs 48.0; P<0.05).
Figure 4.

Activation of CD4+ and CD8+ splenic T cells by rmCIRP. rmCIRP (5 mg/kg BW) or PBS was intravenously injected in mice; spleens were collected 20 h later and processed to isolate lymphocytes. Flow cytometric analysis of surface CD69 and CD25 expression on the gated splenic CD4+ T cells (a) and CD8+ T cells (b) from rmCIRP- and PBS-injected mice. Percentages of CD69+ and CD25+ cells and mean fluorescence intensity (MFI) of CD69 and CD25 expression are shown. Data expressed as mean±s.e.m. (n=4–5 per group) and compared by Student’s t-test. *P<0.05 vs PBS. BW, body weight; rmCIRP, recombinant murine CIRP; PBS, phosphate-buffered solution.
TLR4 is required for CIRP-induced T-cell activation
It has been demonstrated that CIRP can directly bind to TLR4.10 Therefore, we further determined whether TLR4 was involved in CIRP-induced T-cell activation. Tlr4−/−mice were injected intravenously with rmCIRP. Twenty hours after injection, spleens were collected and processed to isolate lymphocytes for flow cytometric analysis. In Tlr4−/−mice, rmCIRP administration did not result in any increase in expression of CD69 or CD25 on CD4+ T cells compared to PBS administration (Figure 5), indicating that CIRP-induced T-cell activation is mediated in a TLR4-dependent manner.
Figure 5.

Mediation of splenic CD4+ T-cell activation by TLR4. rmCIRP (5 mg/kg BW) or PBS was intravenously injected in Tlr4−/− mice; spleens were collected 20 h later and processed to isolate lymphocytes. Flow cytometric analysis of surface CD69 and CD25 expression on the gated splenic CD4+ T cells from PBS- and rmCIRP-injected Tlr4−/− mice. Percentages of CD69+ (a) and CD25+ (c) cells and MFI of CD69 (b) and CD25 (d) expression on gated CD4+ T cells are shown. Data expressed as mean±s.e.m. (n=4–5 per group) and compared by Student’s t-test. BW, body weight; rmCIRP, recombinant murine CIRP; PBS, phosphate-buffered solution; MFI, mean fluorescence intensity; NS, no significance.
rmCIRP induces a hyperinflammatory Th1 response profile in CD4+ T cells
Activation is an initial step in the cascade of T-cell functions following antigen presentation and recognition. We next examined the downstream effects of T-cell activation induced by CIRP. We focused on the differentiation of CD4+ T cells to Th1 and Th2 helper cells, and measured the change in expression levels of 84 genes involved in Th1 and Th2 responses by PCR array. Those CD4+ T cells treated with rmCIRP showed significant upregulation of seven genes involved in T-helper cell immune responses (Figure 6). Two of these genes were CD4+ T-cell markers, one was a Th2 marker, and the remaining four were Th1 markers and related genes. Specifically, the Th1-associated genes interferon-γ (Ifng), interleukin 12 receptor beta 2 (Il12rb2), T-bet (Tbx21) and colony stimulating factor 2 (Csf2) were increased 7-, 5-, 4- and 2-fold, respectively, compared to control (P<0.05). Suppressor of cytokine signaling 3 (Socs3) and interleukin 6 (Il6), two CD4+ T-cell markers, were increased 3- and 4-fold, respectively (P<0.05). The only Th2-associated gene was interleukin 13 (Il13), which was increased 4-fold in CIRP-treated cells (P<0.05). No genes were downregulated. A complete list of examined genes and their fold regulation after CIRP treatment can be found in Supplementary Table 1.
Figure 6.
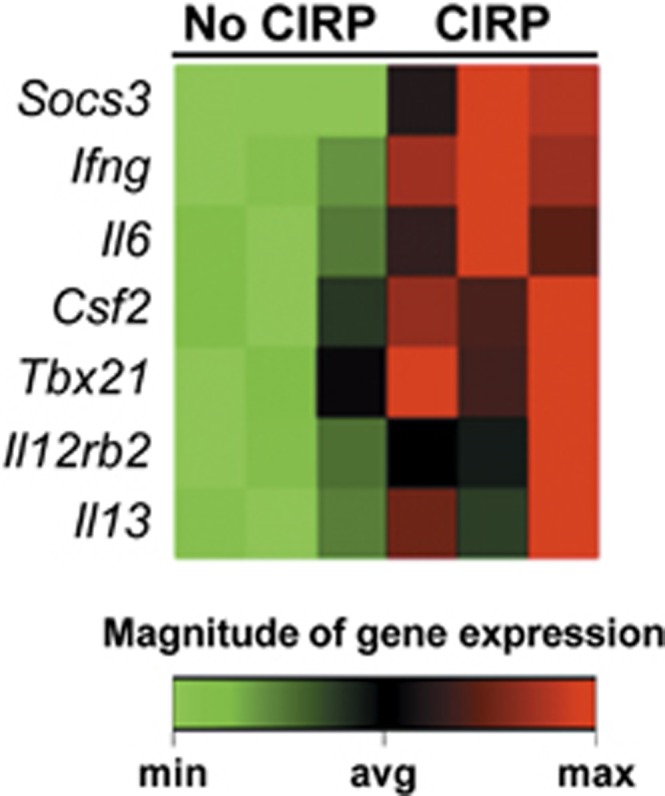
Clustergram of genes upregulated in CD4+ T cells after treatment with rmCIRP. Purified splenic CD4+ T cells (1 × 106/ml/well) were treated with rmCIRP (1 μg/ml) or PBS for 20 h during plate-bound anti-CD3/CD28 (1 μg/ml each) stimulation. An 84-gene PCR array was used to examine Th1 vs Th2 response profiles for CD4+ T cells. A clustergram featuring the seven genes that were significantly upregulated is shown (n=3 per group, P<0.05 by Student’s t-test). Of these, four are Th1-related genes (Ifng, Csf2, Tbx21 and Il12rb2), two are CD4 T-cell markers (Socs3 and Il6), and one is a Th2-related cytokine (Il13). No genes were downregulated. rmCIRP, recombinant murine CIRP; PCR, polymerase chain reaction.
rmCIRP induces a cytotoxic response profile in CD8+ T cells
We also examined the response of CD8+ T cells to rmCIRP at the transcriptional level. Granzyme B (Gzmb), a serine protease contained in the granules of cytotoxic lymphocytes, was upregulated 5-fold in rmCIRP-treated cells compared with untreated cells (P<0.05, Figure 7a). Two additional genes important for cytotoxic T-cell function, RANTES (Ccl5) and IFN-γ (Ifng), were upregulated 8- and 7-fold, respectively, although these increases did not reach statistical significance (Figures 7b and c). These findings suggest that CIRP induces a cytotoxic profile to promote the inflammatory response in sepsis.
Figure 7.

Increased gene expression in CD8+ T cells after treatment with rmCIRP. Purified splenic CD8+ T cells (1 × 106/ml/well) were treated with rmCIRP (1 μg/ml) or PBS for 20 h during plate-bound anti-CD3/CD28 (1 μg/ml each) stimulation. Fold increase in expression of granzyme B (a), RANTES (b) and IFN-γ (c) mRNA is shown relative to cells treated with PBS. β-actin was used as the reference gene. Data expressed as mean±s.e.m. (n=4 per group) and compared by Student’s t-test. *P<0.05 vs PBS. rmCIRP, recombinant murine CIRP; PBS, phosphate-buffered solution.
DISCUSSION
We have shown an increase in levels of CIRP protein in the spleen in an animal model of sepsis. This is consistent with previous research demonstrating elevated CIRP protein levels in the liver and serum of rats at 20 h after CLP.10 Likewise, previous studies have shown that CIRP is released into the serum in both animal models of sepsis and ICU patients with sepsis.10, 21 The spleen was our primary organ of interest for this investigation because it is the largest secondary lymphoid organ, containing ~5% of the body’s lymphocytes.22 In addition, in male C57BL/6 mice, just over 20% of splenocytes are comprised of CD4+ and CD8+ T lymphocytes.23
We have demonstrated an upregulation of the early lymphocyte activation markers CD69 and CD25 on CD4+ T cells, and shown that CD8+ T cells show upregulation of CD69, but not CD25, at the relatively early time point of 20 h after sepsis onset. Previous studies have shown increased expression of CD69 on stimulated human T cells in vitro and in septic ICU patients, as well as in various models of animal sepsis.15, 16, 17, 19 For example, in murine E. coli sepsis, those mice that received intravenous inoculation with E. coli demonstrated an increase in CD69 expression on both CD4+ and CD8+ T cells within 24 h, compared to PBS-injected mice.16 Interestingly, recent work has identified CD69’s importance as a regulator of immune responses, beyond its role in T-cell activation.24 In multiple chronic inflammatory conditions, including asthma,25 arthritis26 and inflammatory bowel disease,27 the absence of CD69 resulted in exacerbation of disease severity. Thus, CD69’s upregulation in sepsis may have an important part in regulating the inflammatory response, in addition to serving as a marker for lymphocyte activation. CD25 has additionally been used as a marker of T-cell activation in septic models, usually increasing later on in the course of sepsis.19 Our finding of an increased frequency of CD4+CD25+ lymphocytes in the spleen is consistent with previous study in murine sepsis.28
We have shown that the administration of rmCIRP in mice induces CD69 and CD25 upregulation in both CD4+ and CD8+ T cells. This extends our understanding of CIRP’s role in the immune response to sepsis as a molecule capable of initiating adaptive immune responses in addition to its documented role in stimulating cytokine release from macrophages.10 This is consistent with similar observations of other DAMPs in the immune response to critical illness. High-mobility group protein B1 (HMGB1), for example, was discovered to be released from macrophages after treatment with endotoxin, TNF-α or IL-1,29 and further that it can activate neutrophils and increase their expression of proinflammatory cytokines.30 Later study showed an association of HMGB1 with immunosuppression and T-cell dysfunction in the context of severe burn injury.31 These findings highlight the importance of DAMPs in both proinflammatory stress responses and post-injury T-cell dysfunction. Our current findings expand on this to include induction of T-cell activation as an important adaptive function of DAMPs. However, unlike after rmCIRP injection, we have not observed an increase in the CD8+CD25+ T-cell population in septic mice induced by CLP. In this polymicrobial sepsis model, many antigens and inflammatory mediators are released and elevated in the peritoneal cavity and circulation, which may counteract CIRP’s effect on CD25 expression in splenic CD8+ T cells. Detailed kinetic analysis of the timing of CD25 expression and elevation of inflammatory mediators after CLP may further facilitate the understanding of such differential responses.
T-cell anergy has been well-documented in the late stages of sepsis;32, 33, 34 given the recent work elucidating CD69’s immunoregulatory function discussed above, this molecule may serve an important link between T-cell activation in the initial response to sepsis and later T-cell anergy. CIRP’s ability to upregulate CD69 in the absence of sepsis lends further support to CIRP’s importance in immunoregulation. In addition, we have shown that CIRP induces an increase in CD4+CD25+ T lymphocytes, or regulatory T cells (Treg). This is consistent with previous findings in sepsis, in which the Treg population has been documented to increase, and this increase has been found to be in part responsible for the depletion of other T-cell populations in the late stages of sepsis.20, 35, 36, 37 This highlights yet another way in which CIRP can link the early, innate immune response to sepsis with late-stage anergy. Although we did not examine it in this study, another subset of regulatory T cells, CD4+CD69+CD25- T cells, has been shown to be involved in regulating immunosuppression in solid tumor mouse models and in patients with leukemia relapse after allogeneic hematopoietic cell transplantation.38, 39 These T cells do not express Foxp3, but express membrane-bound TGF-β1, which mediates suppression of T-cell proliferation.38 Whether CD4+CD69+CD25- T cells play a role in sepsis needs further investigation.
In this study, we have identified that CIRP activates T cells through a TLR4-dependent mechanism. TLR4 is a cell-surface receptor capable of identifying a diverse array of pathogenic ligands; its activation leads to a complex signal transduction pathway that results in the induction of expression of proinflammatory cytokines.40 Our findings are consistent with previously reported data on the role of TLR4 in CIRP-induced cytokine release from macrophages.10 The presence of TLR4 on T cells is well-established,41, 42, 43 and CIRP may exert its effect on these cells directly or through other TLR4-expressing cells. We have also shown an increase of CIRP levels in the spleen after CLP. CIRP is widely expressed in various cell types. Whether extracellular CIRP is released from the spleen or infiltrates from sites of infection to stimulate splenic T cells in sepsis requires further study.
In addition to identifying CIRP’s effects on T-cell activation, we further determined the role of CIRP in CD4+ and CD8+ T-cell polarization. In CIRP-treated CD4+ T cells, there was an increase in the expression of seven out of 84 genes in a Th1/Th2 PCR array. Four of these genes, including Tbx21, Ifng, Il12rb2 and Csf2, were associated with the Th1, or hyperinflammatory, response profile. Tbx21 is the gene encoding T-box transcription factor 21 or T-bet, which is a Th1-specific transcription factor that controls expression of the classic Th1 cytokine interferon-gamma (IFN-γ).44 Ifng is the gene that encodes IFN-γ itself; its main sources are NK and NKT cells, in addition to Th1 CD4+ T cells and cytotoxic CD8+ T cells.45 Il12rb2 codes for a subunit of the interleukin 12 receptor, a type I transmembrane protein whose inducible expression is driven by T-bet. IL-12 signaling through this receptor leads to the phosophorylation of STAT4, which acts synergistically with T-bet to stimulate IFN-γ expression and promote Th1 differentiation and proper function.46 Csf2 encodes colony stimulating factor 2, also known as granulocyte macrophage-colony stimulating factor, which has been shown to be required in order for mice to mount a Th1 response to infection.47 These results demonstrate a propensity for CD4+ T cells to differentiate toward a hyperinflammatory Th1 response profile in the presence of CIRP. Previous studies have demonstrated the important role of TLR4 in skewing immune responses toward a Th1 profile.48, 49 This is consistent with our findings demonstrating the role of TLR4 in mediating CIRP-induced T-cell activation, and the skewing of CIRP-treated cells towards a Th1 profile.
CIRP-treated CD8+ T cells demonstrated increased expression of three genes involved in cytotoxic T-cell responses: Gzmb, Ccl5 and Ifng. Gzmb encodes granzyme B, a serine protease that mimics the caspases in its ability to cleave target cell proteins after selected aspartate residues, making it the most powerful proapoptotic granzyme.50 It acts in concert with perforin to enter the cytosol of target cells and induce apoptosis. Its upregulation in CIRP-treated CD8+ T cells is suggestive of a cytotoxic profile in these cells. Ccl5 encodes RANTES, a proinflammatory chemokine degranulated from activated CD8+ T cells, which can lead to further activation of T cells as well as monocytes and neutrophils.51 IFN-γ, as discussed above, is an important inflammatory cytokine primarily produced from NK, NKT, Th1 CD4+ T cells and cytotoxic CD8+ T cells. RANTES and IFN-γ were upregulated in CD8+ T cells after CIRP treatment, although not to a significant degree, in our sample. Due to the variation of data among the tested samples, the fold change in expression of these two genes does not demonstrate statistical significance by the Student’s t-test. This suggests that subpopulations within the isolated splenic CD8+ T cells may have differential responses to rmCIRP.
SUMMARY
CIRP, a cold-shock protein recently discovered to act as a DAMP in sepsis, promotes splenic CD4+ and CD8+ T-cell activation in mice. CIRP-induced T-cell activation is mediated by TLR4 and leads to upregulation of proinflammatory gene expression in both CD4+ and CD8+ T cells. Our findings demonstrate an important role of CIRP in mediating T-cell dysfunction during sepsis.
Acknowledgments
We thank Dr Kevin Tracey for providing the Tlr4−/− mice and the members of the Flow Cytometry Core Facility at the Feinstein Institute for Medical Research for their input and technical assistance. This study was supported by the National Institutes of Health (NIH) Grants HL076179 and GM053008 (PW).
Footnotes
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website (http://www.nature.com/cmi)
The authors declare no conflict of interest.
Supplementary Material
References
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016; 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 2013; 369: 2063. [DOI] [PubMed] [Google Scholar]
- Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193: 259–272. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Delano MJ, Kelly KM, O'Malley KA, Efron PA, McAuliffe PF et al. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol. 2006; 177: 7943–7949. [DOI] [PubMed] [Google Scholar]
- Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther 2012; 10: 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med 2012; 40: 754–761. [DOI] [PubMed] [Google Scholar]
- Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol 2013; 93: 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013; 13: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med 2013; 19: 1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama H, Itoh K, Kaneko Y, Kishishita M, Yoshida O, Fujita J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol 1997; 137: 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F et al. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science 2012; 338: 379–383. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bührer C, Wellmann S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell Mol Life Sci 2016. e-pub ahead of print 4 May 2016 doi:10.1007/s00018-016-2253-7. [DOI] [PMC free article] [PubMed]
- González-Amaro R, Cortés JR, Sánchez-Madrid F, Martín P. Is CD69 an effective brake to control inflammatory diseases? Trends Mol Med 2013; 19: 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso A, Licenziati S, Corulli M, Canaris AD, De Francesco MA, Fiorentini S et al. Flow cytometric analysis of activation markers on stimulated T cells and their correlation with cell proliferation. Cytometry 1997; 27: 71–76. [DOI] [PubMed] [Google Scholar]
- van Schaik SM, Abbas AK. Role of T cells in a murine model of Escherichia coli sepsis. Eur J Immunol 2007; 37: 3101–3110. [DOI] [PubMed] [Google Scholar]
- Roger PM, Hyvernat H, Ticchioni M, Kumar G, Dellamonica J, Bernardin G. The early phase of human sepsis is characterized by a combination of apoptosis and proliferation of T cells. J Crit Care 2012; 27: 384–393. [DOI] [PubMed] [Google Scholar]
- Guo L, Liu F, Lu MP, Zheng Q, Chen ZM. Increased T cell activation in BALF from children with Mycoplasma pneumoniae pneumonia. Pediatr Pulmonol 2015; 50: 814–819. [DOI] [PubMed] [Google Scholar]
- Schmoeckel K, Traffehn S, Eger C, Potschke C, Broker BM. Full activation of CD4+ T cells early during sepsis requires specific antigen. Shock 2015; 43: 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnoski N, Chung CS, Chen Y, Huang X, Ayala A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock 2007; 27: 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J. The cold-inducible RNA–binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PLoS One 2015; 10: e0137721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol 2006; 34: 455–465. [DOI] [PubMed] [Google Scholar]
- The Jackson LaboratoryFlow-Cytometric Analysis of 11 Strains of Mice. MPD:Jaxpheno6. Mouse Phenome Database Website. The Jackson Laboratory: Bar Harbor, Maine: USA. Available at: http://phenome.jax.org. (accessed on 6 March 2016). [Google Scholar]
- Radulovic K, Niess JH. CD69 is the crucial regulator of intestinal inflammation: a new target molecule for IBD treatment? J Immunol Res 2015; 2015: 497056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Gomez M, Lamana A, Matesanz Marin A, Cortes JR, Ramirez-Huesca M et al. The leukocyte activation antigen CD69 limits allergic asthma and skin contact hypersensitivity. J Allergy Clin Immunol 2010; 126: 355–365; 65.e1-3. [DOI] [PubMed] [Google Scholar]
- Sancho D, Gomez M, Viedma F, Esplugues E, Gordon-Alonso M, Garcia-Lopez MA et al. CD69 downregulates autoimmune reactivity through active transforming growth factor-beta production in collagen-induced arthritis. J Clin Invest 2003; 112: 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic K, Manta C, Rossini V, Holzmann K, Kestler HA, Wegenka UM et al. CD69 regulates type I IFN-induced tolerogenic signals to mucosal CD4 T cells that attenuate their colitogenic potential. J Immunol 2012; 188: 2001–2013. [DOI] [PubMed] [Google Scholar]
- Sharma A, Yang WL, Matsuo S, Wang P. Differential alterations of tissue T-cell subsets after sepsis. Immunol Lett 2015; 168: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999; 285: 248–251. [DOI] [PubMed] [Google Scholar]
- Park JS, Arcaroli J, Yum HK, Yang H, Wang H, Yang KY et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol 2003; 284: C870–C879. [DOI] [PubMed] [Google Scholar]
- Zhang LT, Yao YM, Dong YQ, Dong N, Yu Y, Sheng ZY. Relationship between high-mobility group box 1 protein release and T-cell suppression in rats after thermal injury. Shock 2008; 30: 449–455. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 1999; 27: 1230–1251. [DOI] [PubMed] [Google Scholar]
- Felmet KA, Hall MW, Clark RS, Jaffe R, Carcillo JA. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J Immunol 2005; 174: 3765–3772. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Hui JJ, Chang KC et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 2001; 166: 6952–6963. [DOI] [PubMed] [Google Scholar]
- Cao C, Ma T, Chai YF, Shou ST. The role of regulatory T cells in immune dysfunction during sepsis. World J Emerg Med 2015; 6: 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med 2004; 32: 2329–2331. [DOI] [PubMed] [Google Scholar]
- Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F et al. Increased circulating regulatory T cells (CD4+CD25+CD127−) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med 2009; 35: 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Guo Q, Zhang M, Chen Z, Cao X. CD69+ CD4+ CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol 2009; 182: 111–120. [DOI] [PubMed] [Google Scholar]
- Zhao XS, Wang XH, Zhao XY, Chang YJ, Xu LP, Zhang XH et al. Non-traditional CD4+CD25-CD69+ regulatory T cells are correlated to leukemia relapse after allogeneic hematopoietic stem cell transplantation. J Transl Med 2014; 12: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Karmakar S, Babu SP. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis 2016; 20: 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner-Fischer M, Lider O et al. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol 2007; 179: 41–44. [DOI] [PubMed] [Google Scholar]
- Zanin-Zhorov A, Cohen IR. Signaling via TLR2 and TLR4 directly down-regulates T cell effector functions: the regulatory face of danger signals. Front Immunol 2013; 4: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci USA 2012; 109: 13064–13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000; 100: 655–669. [DOI] [PubMed] [Google Scholar]
- Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 2007; 96: 41–101. [DOI] [PubMed] [Google Scholar]
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity 2012; 37: 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC et al. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol 2005; 77: 914–922. [DOI] [PubMed] [Google Scholar]
- Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2001; 2: 947–950. [DOI] [PubMed] [Google Scholar]
- Netea MG, Van der Meer JW, Sutmuller RP, Adema GJ, Kullberg BJ. From the Th1/Th2 paradigm towards a Toll-like receptor/T-helper bias. Antimicrob Agents Chemother 2005; 49: 3991–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015; 15: 388–400. [DOI] [PubMed] [Google Scholar]
- Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol 2001; 22: 83–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.