

ABSTRACT
Tuberous sclerosis complex is an autosomal dominant disorder characterized by benign tumors arising from the abnormal activation of mTOR signaling in cells lacking TSC1 (hamartin) or TSC2 (tuberin) activity. To expand the genetic framework surrounding this group of growth regulators, we utilized the model eukaryote Schizosaccharomyces pombe to uncover and characterize genes that buffer the phenotypic effects of mutations in the orthologous tsc1 or tsc2 loci. Our study identified two genes: fft3 (encoding a DNA helicase) and ypa1 (encoding a peptidyle-prolyl cis/trans isomerase). While the deletion of fft3 or ypa1 has little effect in wild-type fission yeast cells, their loss in tsc1Δ or tsc2Δ backgrounds results in severe growth inhibition. These data suggest that the inhibition of Ypa1p or Fft3p might represent an ‘Achilles’ heel’ of cells defective in hamartin/tuberin function. Furthermore, we demonstrate that the interaction between tsc1/tsc2 and ypa1 can be rescued through treatment with the mTOR inhibitor, torin-1, and that ypa1Δ cells are resistant to the glycolytic inhibitor, 2-deoxyglucose. This identifies ypa1 as a novel upstream regulator of mTOR and suggests that the effects of ypa1 loss, together with mTOR activation, combine to result in a cellular maladaptation in energy metabolism that is profoundly inhibitory to growth.
KEY WORDS: Tuberous sclerosis complex, Schizosaccharomyces pombe, mTOR, Genetic buffering
Summary: We exploit the BioGRID gene interaction database to identify and characterize susceptibilities in fission yeast cells lacking tsc1 or tsc2, orthologs of the human TSC1 and TSC2 tumor suppressor genes.
INTRODUCTION
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by the development of benign tumors in tissues that include the heart, lung, kidney and brain. In addition, neurological phenotypes (e.g. seizures, learning disabilities, autism, hyperactivity, dementia and ataxia) are also commonly associated with the disorder. TSC affects as many as 40,000 individuals in the USA and between 1 to 2 million individuals worldwide. It has an estimated prevalence of one in 6000-8000 newborns and is seen in all races and in both sexes (Henske et al., 2016; Randle, 2017; Rosset et al., 2017). There is no cure.
TSC is caused by loss-of-function mutations in either TSC1 (encoding the protein, hamartin) or TSC2 (encoding the protein, tuberin). These genes function, at least in part, to inhibit the mechanistic target of rapamycin (mTOR) signaling pathway, which serves as a key regulator of cell proliferation, metabolism, and cell survival (Henske et al., 2016; Saxton and Sabatini, 2017). Orthologs of the TSC1 and TSC2 genes exist in a wide range of organisms, including the commonly used and genetically tractable model eukaryote, Schizosaccharomyces pombe (also known as fission yeast) (Hoffman et al., 2015).
While the inhibition of mTOR signaling through rapamycin treatment has shown some therapeutic promise, rapamycin-independent pathways related to ‘non-canonical’ TSC1 and TSC2 functions are also likely to play a role with respect to the manifestation of the disorder (Neuman and Henske, 2011). In addition, while rapamycin treatment alone has shown some efficacy in limiting the growth of tumor cells, combination therapies involving rapamycin and secondary drugs have proven even more useful in treating certain cancers (Li et al., 2014). Lastly, it is also clear that rapamycin treatment is purely cytostatic. Thus, the identification of alternate ‘drugable’ targets (the inhibition of which might result in a cytocidal response) may provide more effective options for treatment (Medvetz et al., 2015; Neuman and Henske, 2011; Switon et al., 2016).
Fortunately, the relatively recent development of genetic interaction analysis has provided a novel and unbiased avenue with which to pursue this goal. In such assays the phenotypic effects of a ‘query’ mutation – both alone and in combination with a panel of mutations representing the remaining genes in the genome – are assayed to uncover both negative and positive genetic interactions on a genome-wide scale (Baryshnikova et al., 2013; Dixon et al., 2009b; Kuzmin et al., 2016). In this way, functional relationships between the query gene and all other genes in the genome can be characterized in a systematic and unbiased manner. Such strategies have been used successfully in a wide variety of model systems including Saccharomyces cerevisiae, S. pombe, Escherichia coli, Caenorhabditis elegans, Drosophila melanogaster, and more recently in mammalian cell lines (Butland et al., 2008; Byrne et al., 2007; Costanzo et al., 2016; Housden et al., 2015, 2017; Ryan et al., 2012; Shen et al., 2017).
Of particular relevance to TSC are genes that negatively interact with both TSC1 and TSC2. Given the fact that tumor formation arises from loss of heterozygosity, this characteristic identifies such genes as potential therapeutic targets. This is to say, drugs inhibiting a negative interactor would presumably suppress only the growth of tumor cells (which bear two mutant copies of the affected TSC gene: the inherited mutant germline copy, and the copy affected by the ‘second-hit’) while leaving phenotypically normal cells (carrying only the mutant germline copy) unaffected. In addition, the fact that these interacting genes ‘buffer’ the phenotypic effects of hamartin and tuberin loss-of-function mutations, suggests that their detailed molecular/genetic analysis might provide novel insight into the molecular pathology of TSC and its many diverse associated phenotypes.
Due to the paucity of genome-wide genetic interaction data in human cell lines, we have explored the genetic framework surrounding the fission yeast orthologs of human TSC1 and TSC2 in the hope of identifying susceptibilities generally conserved in cells deficient in hamartin or tuberin function. While limited data exist, current research does indeed indicate some conservation in genetic interaction networks between organisms separated by significant evolutionary distances (Dixon et al., 2008, 2009a; Tosti et al., 2014).
In this report, we present the results of this preliminary exploration and identify fft3 (encoding a SMARCAD1 family ATP-dependent DNA helicase) and ypa1 (encoding a peptidyl-prolyl cis-trans isomerase) as strong negative interactors. While deletion of either gene has little phenotypic effect in normal cells, their loss in either tsc1 or tsc2 gene deletion mutants profoundly inhibits growth. Furthermore, we show that the targeted loss of Fft3p ATPase activity confers a similar growth defect in tsc1Δ or tsc2Δ mutant backgrounds. These data suggest that the inhibition of either Ypa1p or Fft3p (through targeting the ATPase domain) might represent an ‘Achilles’ heel’ of cells defective in hamartin or tuberin function. In addition, we identify Ypa1p as a novel upstream regulator of mTOR and present evidence suggesting that the phenotypic effects of ypa1 loss, together with ectopic mTOR activation, combine to induce a cellular maladaptation in energy metabolism that is profoundly inhibitory to growth.
RESULTS
Inspection of the S. pombe genetic interaction network reveals a group of nine interactors common to both tsc1 and tsc2
Genes that negatively interact with both TSC1 and TSC2 represent potential therapeutic targets (i.e. drugs inhibiting a negative interactor would be predicted to suppress the growth of tumor cells while leaving phenotypically normal cells unaffected). Due to the paucity of genetic interaction data in human cell lines, together with the abundance of such data in S. pombe, we initiated an exploratory study examining the genetic interaction network encompassing the fission yeast orthologs of human TSC1 and TSC2.
We began by definitively identifying the orthologous S. pombe genes using the DRSC Integrative Ortholog Prediction Tool. This revealed two open reading frames, SPAC22F3.13, and SPAC630.13c, as the sole orthologs of TSC1 and TSC2, respectively (Supplementary Files S1 and S2). Aptly named tsc1 and tsc2, these genes were previously identified as sharing significant similarity with human TSC1 and TSC2 and have been extensively characterized with respect to nutrient uptake and their roles in regulating the fission yeast Tor1p and Tor2p proteins (Aspuria and Tamanoi, 2008; Matsumoto et al., 2002; Nakase et al., 2013; van Slegtenhorst et al., 2004, 2005; Weisman et al., 2005, 2007). Fission yeast Tsc1p and Tsc2p interact physically, just like their human counterparts (Matsumoto et al., 2002), and share a similar overall domain structure with their respective human orthologs (Fig. S1).
Next, we mined the publicly available BioGRID database to identify genes exhibiting interactions with either tsc1 or tsc2. BioGRID is an open access database that catalogs protein, genetic and chemical interactions in all commonly used model organisms as well as in humans. Annotated from over 48,000 publications, this resource provides the most comprehensive archive of interactions currently available (Chatr-Aryamontri et al., 2017; Stark et al., 2006).
This analysis revealed a total of 99 tsc1-specific, and 143 tsc2-specific genetic interactions. These data were filtered by removing interactions that were supported by only a single observation (i.e. the interactions of the filtered set are supported by two or more experimental results). The remaining 17 tsc1-specific and 28 tsc2-specific interactions were then filtered again by removing genes that interact with tsc1, but not tsc2, as well as genes that interact with tsc2, but not tsc1. Of the remaining nine genes, two (fft3 and ypa1) were chosen for further detailed analysis as they display strong negative interactivity with tsc1 and tsc2, and they themselves possess clear human orthologs (Fig. 1). The fft3 gene encodes a SMARCAD1 family ATP-dependent DNA helicase that is known to suppress nucleosome turnover and that is involved in controlling nuclear organization and chromatin structure (Lee et al., 2017; Steglich et al., 2015; Strålfors et al., 2011; Taneja et al., 2017). The ypa1 gene encodes a peptidyl-prolyl cis/trans-isomerase involved in the regulation of protein phosphatase 2A (PP2A) and PP2A-like enzymes (Goyal and Simanis, 2012; Guo et al., 2014; Jordens et al., 2006; Leulliot et al., 2006). Neither has been previously characterized with respect to their relationship to TSC or mTOR signaling in any system.
Fig. 1.

Network diagram displaying fission yeast genes that genetically interact with tsc1 or tsc2. The diagram was created using the embedded BioGRID webtool powered by CytoscapeJS (http://thebiogrid.org/). Genes common to both tsc1 (A) and tsc2 (B) are shown in light blue. The ypa1 and fft3 genes are encircled in red.
Validation and quantitation of the identified negative genetic interactions
We began the next phase of the study by first confirming and carefully quantitating the severity of the observed negative genetic interactions. To this end, we first created tsc1 and tsc2 knockout strains in which the ura4+ deletion cassette was flanked by loxP and loxM3 sites. These sites were added so that we could later introduce alternate tsc1/tsc2 alleles using a technique based on recombinase mediated cassette exchange (see Materials and Methods). To confirm that the constructed knockout strains were true loss-of-function variants, we grew the mutants on EMM plates supplemented with 60 µg/ml canavanine (a toxic arginine analog). As expected, the knockout strains, in contrast to a wild-type control, were able to proliferate on canavanine media, reflecting the inability of tsc1 or tsc2 mutants to properly transport amino acids from the extracellular environment into the cell (van Slegtenhorst et al., 2004) (Fig. S2).
Having validated that the knockout strains were true loss-of-function mutants, we individually crossed the tsc1 and tsc2 gene deletion strains to fft3Δ or ypa1Δ mutants (obtained as part of version 4 of the Bioneer S. pombe gene deletion collection) (Kim et al., 2010). Double mutants were recovered and assayed for growth alongside a wild-type control and the respective single mutant strains. Assays were performed by monitoring colony size in rich media at 30°C over 5 days (see Materials and Methods). To avoid the possible confounding effects of background auxotrophic mutations, all strains examined in this way were prototrophic. As shown in Fig. 2, the colony sizes of fft3Δ tsc1Δ, fft3Δ tsc2Δ, ypa1Δ tsc1Δ and ypa1Δ tsc2Δ double mutants were significantly reduced (P<0.05) in comparison to both the wild-type control and the respective single mutants.
Fig. 2.
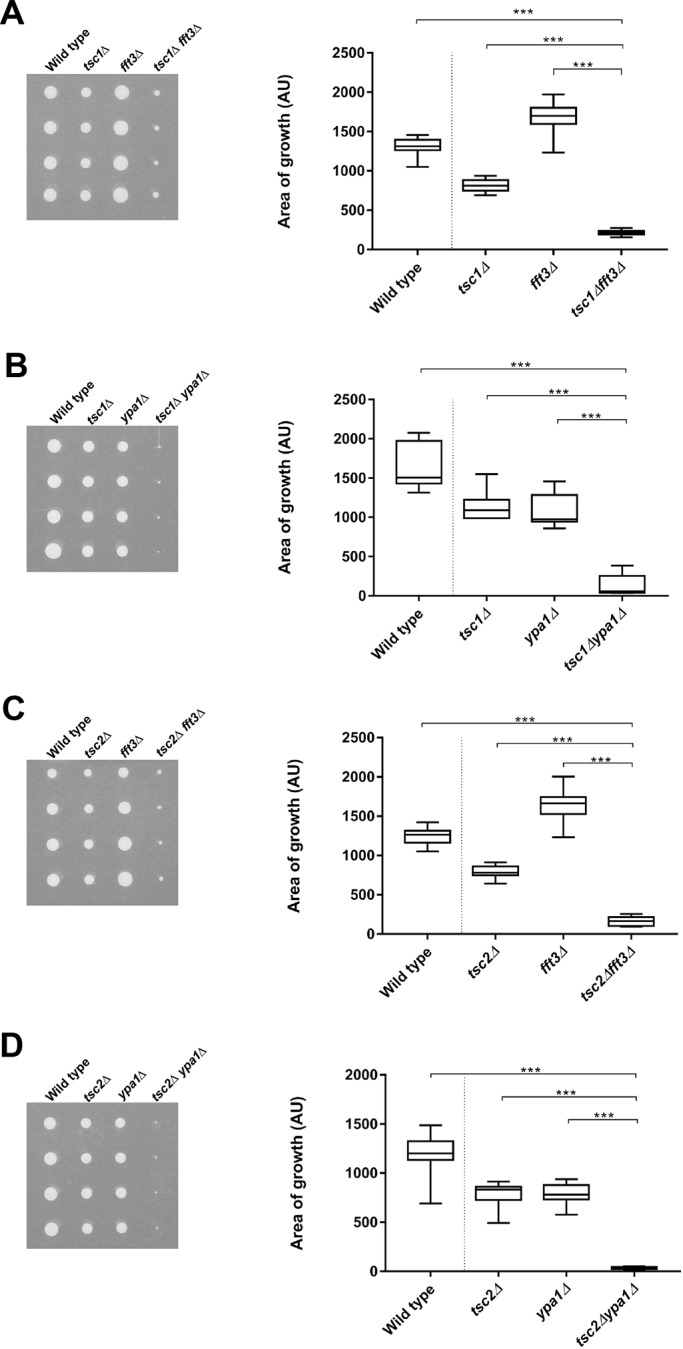
Cells bearing gene deletions in either fft3 or ypa1 are profoundly inhibited for growth in tsc1/tsc2 mutant backgrounds. (A,C) fft3. (B,D) ypa1. Individual cells of the indicated genotype were seeded (in quadruplicate) onto YES plates. Photographs (left) show colony size after 3 days' growth. Box and whisker plots (right) describe colony size (arbitrary units) after 3 days' growth on YES medium at 30°C. ***P<0.001.
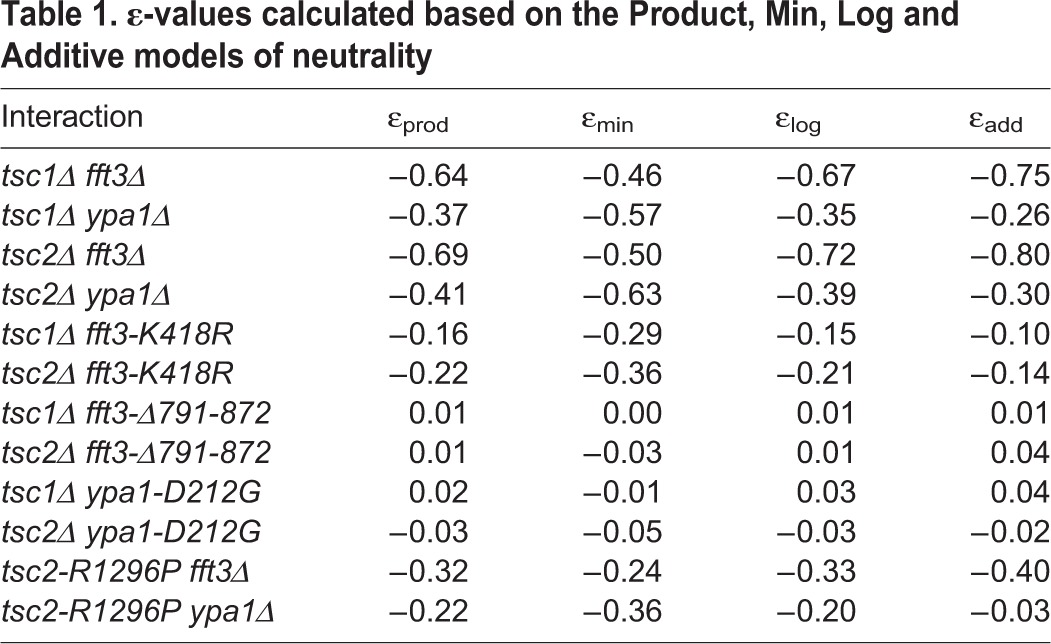
In order to quantitate the severity of the interactions, we next calculated ε-values (for each double mutant) corresponding to each of the four commonly used models of neutrality: the Product, Min, Log, and Additive models (Mani et al., 2008). These values provide a measure of the deviation of the observed double mutant fitness from the fitness expected if there was a ‘neutral’ interaction (see Materials and Methods). As shown in Table 1, the calculated ε-values were less than zero for each of the neutrality models, indicating that the double mutant phenotypes were indeed more severe than expected based on the phenotypes of the individual single mutants.
Table 1.
ε-values calculated based on the Product, Min, Log and Additive models of neutrality
Neighboring gene effects
Importantly, recent work in S. cerevisiae has shown that negative interactions can be falsely annotated due to neighboring gene effects (NGEs). In such cases, the deletion of a given gene affects the proper functioning of an adjacent gene leading to improper annotation of negative interactions (Atias et al., 2016; Baryshnikova and Andrews, 2012; Ben-Shitrit et al., 2012). While the mechanism of these effects remains unknown, it is possible that the deletion of a given locus alters chromatin structure and/or the regulatory characteristics of adjacent loci (Baryshnikova and Andrews, 2012). NGEs are relatively extensive (with measured rates ranging from 7% to 15%) and can distort large-scale genetic interactomes (Ben-Shitrit et al., 2012).
To eliminate the possibility that the loss-of-function of a gene adjacent to fft3 or ypa1 was in fact responsible for the observed negative interactions we examined the effects of atg14, spac25a8.03c and lip2 gene deletions. atg14 and spac25a8.03c are immediately adjacent to fft3 while lip2 is immediately downstream of ypa1 (Fig. S3). The gene immediately upstream of ypa1, trm7, is essential and thus could not be tested. The data obtained revealed that the tsc1Δ atg14Δ, tsc1Δ spac25a8.03cΔ, tsc1Δ lip2Δ, tsc2Δ atg14Δ, tsc2Δ spac25a8.03cΔ and tsc2Δ lip2Δ double mutants did not exhibit significant growth defects relative to the relevant single mutants (data not shown). While difficult to rule out NGEs resulting from the combined perturbation of both the gene of interest and a neighboring gene, these results at minimum demonstrate that the synthetic interactions characterized in this study are not influenced by NGEs resulting from the simple loss-of-function of adjacent genes.
fft3 mutants defective in ATPase activity negatively interact with both tsc1Δ and tsc2Δ
We were next interested in identifying specific domains in Fft3p or Ypa1p that might be pharmacologically targeted (in tsc1 or tsc2 mutant backgrounds) to recapitulate a negative interaction. Since Fft3p exhibits both helicase and ATPase activity, it was unclear if one or both biochemical functions needed to be abrogated in order to confer a negative interaction. We thus created two strains: one expressing an fft3 allele bearing a K418R substitution (ATPase dead) and one where the helicase domain (residues 791-872) was completely deleted (Fig. S4) (Costelloe et al., 2012). These strains were individually crossed to tsc1Δ and tsc2Δ knockouts and the double mutants isolated. Interestingly, a negative interaction was observed in fft3-K418R tsc1Δ and fft3-K418R tsc2Δ double mutants, but not in fft3-Δ791-872 tsc1Δ or fft3-Δ791-872 tsc2Δ mutants (Fig. 3A-D, Table 1). This suggests that the pharmacological inhibition of the ATPase domain of Fft3p might be specifically inhibitory to the growth of cells deficient in hamartin or tuberin function.
Fig. 3.

Cells bearing the fft3-K418R mutation are inhibited for growth in tsc1/tsc2 mutant backgrounds, while cells bearing the fft3-Δ791-872 mutation, or the ypa1-D212G mutation, show no synthetic growth defects with tsc1/tsc2 gene deletions. (A,C) fft3-K418R mutation. (B,D) fft3-Δ791-872 mutation. (E,F) ypa1-D212G mutation. Individual cells of the indicated genotype were seeded (in quadruplicate) onto YES plates. Box and whisker plots describe colony size (arbitrary units) after 3 days' growth on YES medium at 30°C. **P<0.01; ***P<0.001; ns, not significant.
Using a similar line of reasoning, we also constructed single and double mutant strains expressing a ypa1 allele (ypa1-D212G) that was reported to abrogate prolyl isomerase (PPIase) activity (Fig. S5) (Leulliot et al., 2006). Interestingly, in this case no negative interaction was observed (Fig. 3E,F, Table 1). This indicates that the loss of a yet unidentified molecular function is in fact responsible for the negative genetic interactions observed between ypa1Δ and tsc1Δ/tsc2Δ.
Using recombinase mediated cassette exchange to examine alternate tsc1 or tsc2 alleles
We were next interested in establishing an S. pombe platform where we could quickly and easily construct strains expressing wild-type or mutant tsc1 or tsc2 alleles under the control of their native promoters. To this end, we employed a previously described system based on recombinase-mediated cassette exchange (RMCE) (Watson et al., 2008). As we had previously introduced loxP and loxM3 sites into the respective deletion cassettes, we could now replace the ura4+ selectable marker present in the tsc1::ura4+/tsc2::ura4+ knockout strains (hereafter referred to as the ‘base’ strains) by simply transforming the respective knockouts with a vector that expresses the Cre recombinase and that contains tsc1 or tsc2 sequence variants flanked by the identical loxP and loxM3 sites (Fig. S6).
To test the RMCE system, we first exchanged the ura4+ selectable markers present in the tsc1::ura4+ and tsc2::ura4+ base strains with wild-type S. pombe tsc1 or tsc2 (creating the tsc1::tsc1Sp and tsc2::tsc2Sp strains). As expected, cells of these genotypes were unable to proliferate on canavanine plates indicating that they were indeed expressing functional Tsc1p and Tsc2p (Fig. S2). In an attempt to create a system where we could study the human hamartin and tuberin proteins in yeast, we also asked if human TSC1 or TSC2 could substitute for the S. pombe versions of the genes. We thus used RMCE to exchange the ura4+ selectable markers of the knockout strains with human TSC1 or TSC2 creating the tsc1::TSC1Hs and tsc2::TSC2Hs strains. Unfortunately, both strains (as well as tsc1::TSC1Hs tsc2::TSC2Hs cells co-expressing both human TSC1 and TSC2) were able to proliferate on canavanine media. Thus, the human hamartin and tuberin proteins are unable to substitute for their fission yeast counterparts in vivo (Fig. S2).
Up to this point in the study, all of the genetic interaction analyses had been performed using complete deletions of tsc1 or tsc2. We thus asked if the observed negative interactions were allele specific or if they might be observed more generally with all loss-of-function tsc1 or tsc2 alleles. To this end, we constructed Cre-recombinase exchange plasmids carrying tsc1 or tsc2 alleles bearing point mutations (tsc1-L346H, tsc1-G432R, tsc2-G296E, tsc2-N1191K, tsc2-N1199S, tsc2-R1296P) orthologous to those found clinically (Supplementary Files S1 and S2). These plasmids were transformed into the tsc1::ura4+ or tsc2::ura4+ base strains and cells expressing the respective tsc1 or tsc2 alleles were recovered (see Materials and Methods). To assess if the mutations did indeed result in loss-of-function, we assayed the growth of the tsc1 or tsc2 point mutant strains on canavanine media and observed that only two (tsc2-N1191K, tsc2-R1296P) were capable of growth (indicating that they alone represented true loss of function mutants) (data not shown). The two mutant alleles were then individually crossed into fft3Δ or ypa1Δ backgrounds to assess the growth rates of double mutant cells.
While both of the mutations clearly resulted in loss of tsc2 function (based on the ability of the respective mutants to grow on canavanine plates) only one of the two alleles (tsc2-R1296P) displayed negative interactivity with both fft3 and ypa1 (Fig. 4, Table 1). In contrast, the tsc2-N1191K double mutants did not grow significantly slower than the respective single mutants (data not shown). Thus, the observed interactions between tsc2 and ypa1 or fft3 are allele specific. This demonstrates that the loss of fft3 or ypa1 activity is not necessarily detrimental to growth in all cells exhibiting deficiencies in hamartin or tuberin function. Instead the results imply that genetic interaction might depend on the subtleties of Tsc1p/Tsc2p molecular function in an allele-specific manner (see Discussion).
Fig. 4.

Cells bearing the tsc2-R1296P mutation are inhibited for growth in fft3 or ypa1 mutant backgrounds. (A) fft3. (B) ypa1. Individual cells of the indicated genotype were seeded (in quadruplicate) onto YES plates. Box and whisker plots describe colony size (arbitrary units) after 3 days' growth on YES medium at 30°C. *P<0.05; **P<0.01; ***P<0.001.
Treatment with the mTOR inhibitor, torin-1, rescues the negative genetic interaction observed between ypa1 and tsc1/tsc2, but not between fft3 and tsc1/tsc2
We were next interested in better understanding the mechanism(s) behind the negative interactions observed between ypa1/fft3 and tsc1/tsc2. Since loss of tsc1/tsc2 leads to abnormal hyper-activation of the mTOR pathway we reasoned that Ypa1p or Fft3p function might be specifically required when cells were ‘locked’ in a state of high mTOR activity. To test this hypothesis, we examined the growth of ypa1Δ tsc1Δ, ypa1Δ tsc2Δ, fft3Δ tsc1Δ and fft3Δ tsc2Δ double mutants on media containing the mTOR inhibitor torin-1. Remarkably, we found that torin-1 treatment rescued the synthetic growth defect observed in ypa1Δ tsc1Δ and ypa1Δ tsc2Δ double mutants (Fig. 5). Thus, the ypa1-specific negative interaction results from defects in the canonical functions of tsc1 and tsc2 (i.e. their role in regulating mTOR signaling). Since torin-1 treatment is epistatic to the effects of the ypa1 deletion in tsc1Δ/tsc2Δ backgrounds, this result also strongly suggests that Ypa1p functions upstream of mTOR.
Fig. 5.
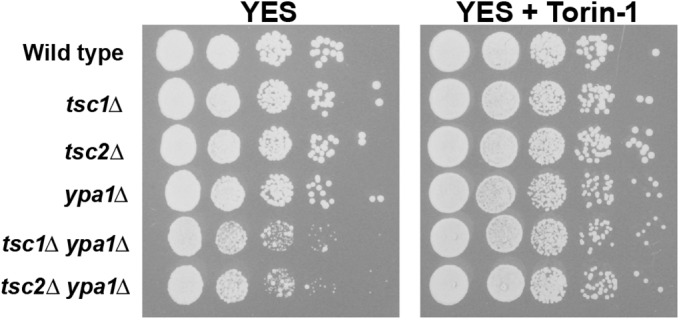
Treatment with the mTOR inhibitor, torin-1, rescues the synthetic growth defect observed in ypa1Δ tsc1/tsc2Δ mutants. Ten-fold serial dilutions of the indicated yeast cultures were plated on YES medium, or YES medium containing 750 nM torin-1, and incubated for 3 days at 30°C.
In contrast, torin-1 treatment had no effect on the negative interactions observed between fft3 and tsc1/tsc2 (data not shown). This suggests that the fft3-specific negative interactions result from defects in a non-canonical function of the Tsc1p/Tsc2p complex (i.e. a function independent of its role in modulating mTOR signaling).
ypa1Δ cells are resistant to the glycolytic inhibitor 2-deoxyglucose
The previous results suggested that the high metabolic activity conferred by hyperactive mTOR could not be tolerated in ypa1Δ mutant backgrounds. We thus examined whether or not ypa1 itself affected metabolism. We began by growing ypa1Δ cells in the presence of the glycolytic inhibitor 2-deoxyglucose (2-DG). 2-DG is a glucose analog in which the hydroxyl group at C-2 is replaced with hydrogen. It is actively imported by hexose transporters and can be phosphorylated to produce 2-DG-6-phosphate. Unlike glucose-6-phosphate, this form cannot be further metabolized and accumulates within the cell, inhibiting glycolytic flux (possibly through product inhibition of hexokinase) and reducing cellular ATP pools (Vishwanatha and D'Souza, 2017).
Remarkably, we found that ypa1Δ cells, as well as ypa1Δ tsc1Δ and ypa1Δ tsc2Δ double mutants, were highly resistant to 2-DG (Fig. 6). The observation that ypa1Δ mutants are resistant to the drug suggests that these cells have reduced glycolytic rates (i.e. they rely less on glycolysis to generate ATP). We suggest that this altered physiology, in conjunction with high mTOR levels brought about by the inactivation of Tsc1p/Tsc2p, leads to a conflicted and metabolically unbalanced state that profoundly inhibits growth (see Discussion).
Fig. 6.
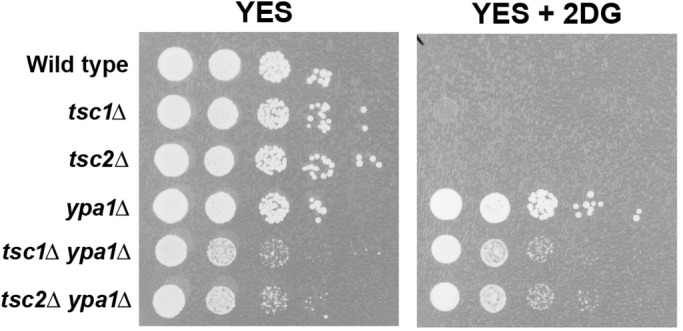
ypa1Δ cells are resistant to 2-DG. Ten-fold serial dilutions of the indicated yeast cultures were plated on YES medium, or YES medium containing 1 mg/ml 2-DG, and incubated for 3 days at 30°C.
DISCUSSION
An understanding of any biological process (including disease processes) goes well beyond elucidating the function of genes in isolation (Hartman et al., 2001; Hartwell, 2004). In fact, research of the past decade has made it abundantly clear that complex genetic interactions (i.e. buffering relationships between genes) play a crucial role in modulating the genotype-phenotype relationship (Baryshnikova et al., 2013; Butland et al., 2008; Byrne et al., 2007; Costanzo et al., 2016; Housden et al., 2017). Fortunately, methodologies capable of characterizing genetic interactions at the genome-wide level have been developed to aid researchers in grappling with this complexity (Dixon et al., 2009b; Kuzmin et al., 2016). The present study sought to exploit such data to more fully elucidate the molecular pathology of tsc1/tsc2 defects and to identify susceptibilities in cells lacking hamartin or tuberin function. This approach identified two S. pombe genes (fft3 and ypa1), the individual loss of which confer growth defects in hamartin/tuberin-deficient cells.
The molecular functions of fft3
The fft3 gene encodes a Swi/Snf2 family ATP-dependent chromatin remodeler that is highly conserved in eukaryotes. Previous work has shown that Fft3p is essential in maintaining heterochromatic structure in centromeric and sub-telomeric regions and furthermore that it suppresses the turnover of histones at heterochromatic loci to facilitate the epigenetic transmission of heterochromatin (Steglich et al., 2015; Strålfors et al., 2011; Taneja et al., 2017; Taneja and Grewal, 2017). Deletion of fft3 in S. pombe results in euchromatin formation, leading to incorrect histone modifications and the misregulation of gene expression (Strålfors et al., 2011).
Interestingly, Fft3p orthologs – Fun30 in S. cerevisiae and SMARCAD1 in humans – are known to aid in DNA end resection of double-stranded breaks (DSBs) via homologous recombination (Chen et al., 2012; Costelloe et al., 2012; Eapen et al., 2012). The inability to successfully repair DSBs compromises genome integrity and leads to prolonged G1/G2 checkpoint arrest, which often results in apoptosis (Massagué, 2004). Budding yeast Fun30 can increase the rate of 5′-to-3′ DNA resection and deactivate DNA damage checkpoint arrest (Eapen et al., 2012). This is done at the chromatin level, where Fun30 (as well as the human homolog SMARCAD1) can relax the tight histone-DNA interactions in nucleosomes adjacent to DSBs (Chen et al., 2012; Costelloe et al., 2012).
While Fft3p orthologs clearly play important roles in key cellular processes (transcription, DNA repair and DNA replication), the molecular mechanism by which the loss of fft3 inhibits growth in tsc1Δ or tsc2Δ backgrounds in fission yeast remains unknown. The fact that the growth defect is not rescued by torin-1 treatment suggests that a non-canonical function of the Tsc1p/Tsc2p complex (i.e. a function independent of its role in modulating mTOR signaling) is involved. The synthetic negative interaction also implies one of two scenarios: (1) that fft3 and tsc1/tsc2 function in parallel pathways that impinge on a shared (and as yet unknown) function affecting growth; or (2) that the interaction reflects ‘unidirectional compensation’, a situation in which one pathway normally prevents a potentially harmful cellular event that can be corrected by another pathway (Boone et al., 2007).
The molecular functions of ypa1
In contrast to fft3, much more is known with respect to ypa1 and its potential molecular role in modulating TSC signaling. In fact, based on both the published literature and our own experimental results, we have developed a working model with respect to the role of the Ypa1p protein (and its orthologs) in regulating TSC and mTOR signaling. Several key observations have led to this model, which posits the existence of a novel signaling axis extending from the cellular sensing of glucose to downstream mTOR activation. These findings are discussed in detail below.
The first series of observations relate to the known role of budding yeast Ypa1p (also known as Rrd1p) in regulating protein phosphatase 2A (PP2A) activity in response to glucose. Briefly, work in the budding yeast has shown that the addition of glucose to glucose-starved cells triggers the post-translational activation of PP2A, that Ypa1p physically interacts with PP2A-like phosphatases, and that full glucose-mediated activation of PP2A is dependent on Ypa1p (Castermans et al., 2012; Fellner et al., 2003; Van Hoof et al., 2005). It is thus clearly established that Ypa1p is required for the glucose-induced activation of PP2A, at least in budding yeast.
The second series of observations relates to the role of PP2A in inhibiting the activation of AMP-activated protein kinase (AMPK). In S. pombe, just as in other more developmentally complex eukaryotes, AMPK plays a key role in the regulation of cellular energy homeostasis (through positively regulating energy-producing pathways and inhibiting energy-consuming processes in response to low ATP levels). S. pombe AMPK is composed of three subunits: an alpha subunit (Ssp2p), a beta subunit (Amk2p), and a gamma subunit (Cbs2p). It is clear that nutrient starvation (glucose or nitrogen) triggers Thr-189 phosphorylation of the Ssp2p subunit and the activation of AMPK (Valbuena and Moreno, 2010). Interestingly, it has also been shown that human PP2A negatively regulates AMPK by dephosphorylating Thr-172 (equivalent to Thr-189 in fission yeast). These researchers also showed (through co-immunoprecipitation and colocalization studies) that PP2A directly interacts with AMPK (Joseph et al., 2015). While not directly shown experimentally in fission yeast, these results raise the possibility that fission yeast PP2A might also regulate AMPK through modulating Thr-189 phosphorylation.
The last series of observations relates to the regulation of Tsc1p/Tsc2p and mTOR by AMPK. In human HEK293 cells, it is clear that AMPK phosphorylates Tsc2p (on Thr-1227 and Ser-1345) in response to energy starvation and that these phosphorylation events enhance the activity of Tsc2p with respect to inhibiting mTOR (Inoki et al., 2003). Similarly, in S. pombe, nutrient stress has been shown to activate the Ssp2p alpha subunit of AMPK and reduce mTOR activity in a manner that is dependent on both Tsc1p and Tsc2p (Davie et al., 2015). It thus appears that AMPK, Tsc1/Tsc2 and mTOR function as a conserved signaling module that is sensitive to cellular energy status.
Bringing these three lines of reasoning together, we propose the existence of a signaling axis leading from the sensing of glucose to the activation of mTOR (through the intermediaries, Ypa1p, PP2A, AMPK and Tsc1p/Tsc2p) (Fig. 7A). It is important to note that in this model, Tsc1p/Tsc2p acts as a critical go-between in relaying information between AMPK and mTOR. Since AMPK positively regulates Tsc1p/Tsc2p and Tsc1p/Tsc2p negatively regulates mTOR, this regulatory design ensures that AMPK activity and mTOR activity are inverted with respect to each other (i.e. high AMPK leads to low mTOR, and low AMPK leads to high mTOR). We suggest that the role of Tsc1p/Tsc2p in balancing these two activities is crucial in ensuring that the cell maintains the proper balance between the nutrient-induced and starvation-induced activities of AMPK and mTOR. This is to say, in the presence of Tsc1p/Tsc2p, high catabolic rates (high AMPK) correlate with a starvation response (low mTOR). In contrast, high anabolic rates (low AMPK) correlate with the promotion of cell growth/proliferation (high mTOR). Finally, moderate catabolic rates (intermediate AMPK) correlate with a moderate starvation response (intermediate mTOR) (Fig. 7B).
Fig. 7.

Model describing the putative role of Ypa1p in mTOR signaling in fission yeast. (A) Hypothesized signaling axis extending from the sensing of glucose levels via Ypa1p to the activation of mTOR. Components listed in black type are considered fully active. Components listed in grey type are considered minimally active. (B) Abstract model describing the effects of mutations in ypa1 and tsc1/tsc2, both singly and in combination. (i) In the presence of glucose mTOR activity is high and AMPK activity is low. However, the system is free to shift in either direction as a function of glucose concentration. (ii) In ypa1Δ mutants, AMPK is locked in the ON position. The cell is maladapted, but the metabolic signals being output are consistent. (iii) In tsc1Δ/tsc2Δ mutants mTOR is locked in the ON position. The cell is maladapted, but again the metabolic signals being output are consistent. (iv) In ypa1Δ tsc1Δ/tsc2Δ mutants both mTOR and AMPK are locked in the ON position. The metabolic signals being output are contradictory, resulting in a synthetic growth defect. (v) Torin-1 treatment rescues the growth defect by turning mTOR OFF and restoring the balance.
Importantly, while mutants lacking either Ypa1p or Tsc1p/Tsc2p activity would indeed be maladapted, such cells would still maintain a ‘balance’ between AMPK and mTOR activity. For instance, in ypa1Δ mutants grown in nutrient rich conditions, AMPK would be abnormally locked in the ON position and mTOR in the OFF position. Likewise, in tsc1Δ/tsc2Δ mutants grown in nutrient poor conditions, AMPK would be abnormally locked in the OFF position and mTOR in the ON position. While maladapted, both cell types are consistent with respect to the metabolic signals being output to the cell (Fig. 7B).
Finally, consider the scenario in which cells lacks both Ypa1p and Tsc1p/Tsc2p activity (resulting in both AMPK and mTOR being locked in the ON position). In this case, the signals emanating from AMPK and mTOR are in direct contradiction; with AMPK promoting catabolic activity at the same time mTOR is promoting cell growth and proliferation (Fig. 7B). We hypothesize that the ‘conflicted and metabolically unbalanced’ cellular state characterized by the loss of both Ypa1p and Tsc1p/Tsc2p activity is the root cause of the synthetic growth defect observed in cells bearing loss-of-function mutations in both ypa1 and tsc1/tsc2. Importantly, it should be noted that the model predicts that the observed synthetic growth defect would be rescued by mTOR inhibition (by placing the cell back in a ‘balanced’ state) (Fig. 7B). As previously discussed, this is indeed the case as torin-1 treatment was seen to dramatically improve the growth of double mutant cells (Fig. 5). Lastly, and perhaps most intriguingly, these results also imply that cells devoid of hamartin/tuberin function would be especially vulnerable to treatments that directly promote AMPK activity.
Given this last point, it is interesting to speculate as to the potential conservation of the observed fft3- or ypa1-specific negative interactions in more developmentally complex eukaryotes. This is to say, it would be interesting to determine whether RNAi knockdown of either SMARCAD1 (human fft3 ortholog) or PPP2R4 (human ypa1 ortholog) in TSC1- or TSC2-deficient human cell lines would also result in reduced growth rates. If indeed conserved, then this raises the question of whether it might be possible to inhibit the growth of hamartin/tuberin deficient tumor cells by recapitulating the phenotypic effects of SMARCAD1 or PPP2R4 loss of function. In any event, it is hoped that a detailed understanding of ypa1 or fft3 function in fission yeast might reveal foundational knowledge that could be leveraged to reveal a previously unrecognized Achilles' heel exhibited by cells lacking hamartin/tuberin activity.
MATERIALS AND METHODS
Strains, media and growth conditions
S. pombe cells were cultured in YES media or EMM media supplemented as needed with adenine, histidine, leucine and/or uracil (Forsburg and Rhind, 2006). Liquid cultures were grown with shaking (200 rpm) at 30°C. All fission yeast strains used in this study were either derived from the Karagiannis laboratory collection, constructed during the course of this work (described below), or purchased from Bioneer Corporation as part of version 4 of the haploid gene deletion mutant library (Kim et al., 2010). All genetic crosses and general yeast techniques were performed using standard methods (Forsburg and Rhind, 2006).
Bioinformatics
Ortholog prediction and alignment was performed using the DRSC Integrative Ortholog Prediction Tool Version 5.5 (http://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl) using human TSC1 or human TSC2 as the input species and S. pombe as the output species. Scores less than one were excluded. Details of the tool can be found in Hu et al. (2011).
Genetic interaction network analysis was performed using the BioGRID webtool (http://thebiogrid.org/) using S. pombe tsc1 or tsc2 as input. Data were filtered by excluding both physical and chemical interactions, and by excluding genetic interactions supported by only one experimental result. Network diagrams were created using the embedded BioGRID webtool powered by CytoscapeJS using the Arbor layout. Details of the tool can be found in Chatr-Aryamontri et al. (2017).
Colony size assays
A Axioskop 40 micromanipulator (Zeiss, Oberkochen, Germany) was used to array single cells representing each genotype (in quadruplicate) upon YES agar plates. Plates were incubated at 30°C for 5 days. Digital images were taken at 24 h intervals. Colony size was determined by measuring the area of each colony from the images using the ‘measure’ tool of ImageJ (http://imagej.nih.gov/ij/). Statistically significant differences in colony size among the genotypes were determined using the non-parametric Wilcoxon matched pairs signed rank test (two-tailed). Three biological replicates were performed for each tested interaction.
ε-value calculations
Using colony size as a proxy for fitness, ε-values were calculated by subtracting the expected double mutant fitness from the observed double mutant fitness, ε=Wab−E(Wab). The expected fitness was calculated using one of four commonly used neutrality models. In the Product model, E(Wab)=Wa Wb. In the Min model, E(Wab)=min(Wa, Wb). In the Log model, E(Wab)=log2[(2Wa−1) (2Wb−1)+1]. In the Additive model, E(Wab)=Wa+Wb−1.
Spot assays
Strains of the indicated genotype were grown overnight at 30°C in liquid YES medium to an optical density of 0.5. Five microliters of undiluted culture, as well as four 10-fold serial dilutions (made in liquid YES), were then spotted onto YES-agar plates, or YES-agar plates containing 60 µg/ml canavanine (C-1625, Sigma-Aldrich), 750 nM torin-1 (A11587, AdooQ Bioscience, Irvine, USA), or 1 mg/ml 2-deoxyglucose (D-6134, Sigma-Aldrich). Growth was assayed visually after the plates had been incubated for 3 days at 30°C.
Construction of tsc1 and tsc2 gene deletion strains
Gene deletion cassettes for both the tsc1 and tsc2 genes were created by polymerase chain reaction (PCR) amplifying the ura4+ selectable marker (together with the loxP and loxM3 recombination sites) from the pAW1 vector using primers tsc11 (5′-TTA TCA ATG CTG CCA AGA CTT GCT ATC AGT ATA ATG TCG CAT AGT TGT ATA TCA ACG TTG ACT TTG CCA ACT TTG TAC GAC GGA TCC CCG GGT TAA TTA A-3′) and tsc12 (5′-AAT TAT TTT ATA TGG AAT GAG CAA GTA TGT TTT ATC ATA ATT GAC CAG TTC ATT TCA AGG ACC TTC AAA AAT ATA CCT ACG AAT TCG AGC TCG TTT AAA C-3′), or tsc13 (5′-TTA AGA GTT CAG ATT TGC TTT ATG TGG TTA TTC TGC TGA AGG TCC TAA TTT ATT GAC GTT GAA AAA TAA AGG CCA CAT AGC GGA TCC CCG GGT TAA TTA A-3′) and tsc14 (5′-ATA AAA ATT AAT TAA TGA TGG CAA GGC ACA ATC GTA ATC AAT CTT TTA ATT TAG GAC TTT TTA TAT GCC CTT ATG GCG AAT TCG AGC TCG TTT AAA C-3′), respectively. Strain ED666 (ura4-D18 leu1-32 ade6-210 h+) was then transformed with either the tsc1-specific or tsc2-specific cassette. Ura+ transformants were isolated and the respective gene deletions confirmed by colony PCR using primers tsc15 (5′-ATG TGG CAG ACT ACG CTA TCC T-3′) and tsc17 (5′-ATG CTT CCC CTA ATT CAT AGC A-3′) for the tsc1 deletion, or tsc16 (5′-AGC AAC CTA CGA GAG GAA GAT G-3′) and tsc18 (5′-GCG CAT AAC CCT TTC TAC ATT C-3′) for the tsc2 deletion.
Construction of fission yeast strains bearing site-directed mutations in fft3 or ypa1
C-terminal fragments of the fft3 and ypa1 genes bearing the desired mutations were synthesized by GenScript and cloned into the S. pombe integration vector, pJK210, using the EcoRI and SmaI sites, or the XhoI and SmaI sites, respectively. The pJK210-fft3-K418R and pJK210-fft3-Δ791-872 constructs were linearized within the fft3 sequence (upstream of the relevant mutations) using BglII and then transformed into a ura4-D18 strain using the lithium acetate protocol (Forsburg and Rhind, 2006). The pJK210-ypa1-D212G construct was similarly linearized with BstBI before being transformed into a ura4-D18 strain using the lithium acetate method. Ura4+ transformants were then assessed for integration at the fft3 or ypa1 loci through colony PCR. Proper integration at the fft3 locus was assessed using primers AR2F (5′-TCC CTC ATT TAC TTC CTC TGC TAA-3′) and AR2R (5′-TCC TAT GTT GTG TGG AAT TGT GAG-3′). Proper integration at the ypa1 locus was assessed using primers AR1F (5′-TAC AGC GTC TCA ATA TTG CAT CTG-3′) and AR1R (5′-TCC TAT GTT GTG TGG AAT TGT GAG-3′).
Recombinase-mediated cassette exchange of human TSC1 and TSC2 sequences at the S. pombe tsc1 or tsc2 loci
The full-length human TSC1 or TSC2 sequences were synthesized by Genscript and cloned into the XhoI and SpeI sites of the pAW8X vector (Watson et al., 2008). The tsc1::ura4+/tsc2::ura4+ base strains were then transformed with the pAW8X-TSC1/pAW8X-TSC2 vectors using the lithium acetate protocol (Forsburg and Rhind, 2006). As controls, the base strains were also transformed with the pAW8X-tsc1 and pAW8X-tsc2 vectors containing the fission yeast tsc1 and tsc2 genes. Leu+ transformants were then grown in YES media to allow the plasmid to be lost. Leu− cells in which the ura4+ gene was exchanged with TSC1/TSC2/tsc1/tsc2 were selected by growth on media containing 5-fluoroorotic acid (a drug counter-selectable to Ura+ cells). This created strains in which human TSC1 or TSC2, or fission yeast tsc1 or tsc2, were expressed from the endogenous fission yeast tsc1/tsc2 promoters at the native tsc1/tsc2 loci.
Construction of fission yeast strains bearing site-directed mutations in tsc1 or tsc2
The full-length S. pombe tsc1 or tsc2 genes (flanked by XhoI and SpeI sites) were synthesized by Genscript and cloned into the EcoRV site of the pUC57-vector. A Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, USA) was then used according to the manufacturer's protocol to generate the pUC57-tsc1-L346H, pUC57-tsc1-G432R, pUC57-tsc2-G296E, pUC57-tsc2-N1191K, pUC57-tsc2-N1199S and pUC57-tsc2-R1296P vectors. Mutagenic primers were designed using the NEBaseChanger webtool (http://nebasechanger.neb.com). DNA sequence analysis confirmed the incorporation of the desired changes. The respective tsc1 or tsc2 sequences were then digested from the pUC57 parent vector and cloned into the XhoI and SpeI sites of the pAW8X exchange vector. The tsc1::ura4+/tsc2::ura4+ base strains were then individually transformed with the pAW8X-tsc1-G432R, pAW8X-tsc2-G296E, pAW8X-tsc2-N1191K, pAW8X-tsc2-N1199S and pAW8X-tsc2-R1296P vectors using the lithium acetate protocol (Forsburg and Rhind, 2006). Leu+ transformants were then grown in YES medium to allow the plasmid to be lost. Leu− cells in which the ura4+ gene was exchanged with the tsc1/tsc2 mutants were selected by growth on medium containing 5-fluoroorotic acid (a drug counter-selectable to Ura+ cells). This created strains in which the respective tsc1 or tsc2 mutants were expressed from the endogenous fission yeast tsc1/tsc2 promoters at the native tsc1/tsc2 loci.
Supplementary Material
Acknowledgements
We thank Alexander Timoshenko for helpful discussions and expert technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.K.; Methodology: J.K.; Formal analysis: A.R., J.K.; Investigation: A.R., A.F., R.C., A.M., J.K.; Writing - original draft: J.K.; Writing - review & editing: A.R., A.F., R.C., A.M., J.K.; Supervision: J.K.; Project administration: J.K.; Funding acquisition: J.K.
Funding
This work was supported by the U.S. Department of Defense, under an award from the Tuberous Sclerosis Complex Research Program (W81XWH-14-1-0169). The opinions, interpretations, conclusions, and/or recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Supplementary information
Supplementary information available online at http://bio.biologists.org/lookup/doi/10.1242/bio.031302.supplemental
References
- Aspuria P.-J. and Tamanoi F. (2008). The Tsc/Rheb signaling pathway controls basic amino acid uptake via the Cat1 permease in fission yeast. Mol. Genet. Genomics 279, 441-450. 10.1007/s00438-008-0320-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atias N., Kupiec M. and Sharan R. (2016). Systematic identification and correction of annotation errors in the genetic interaction map of Saccharomyces cerevisiae. Nucleic Acids Res. 44, e50 10.1093/nar/gkv1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A. and Andrews B. (2012). Neighboring-gene effect: a genetic uncertainty principle. Nat. Methods 9, 341-343. 10.1038/nmeth.1936 [DOI] [PubMed] [Google Scholar]
- Baryshnikova A., Costanzo M., Myers C. L., Andrews B. and Boone C. (2013). Genetic interaction networks: toward an understanding of heritability. Annu. Rev. Genomics Hum. Genet. 14, 111-133. 10.1146/annurev-genom-082509-141730 [DOI] [PubMed] [Google Scholar]
- Ben-Shitrit T., Yosef N., Shemesh K., Sharan R., Ruppin E. and Kupiec M. (2012). Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat. Methods 9, 373-378. 10.1038/nmeth.1890 [DOI] [PubMed] [Google Scholar]
- Boone C., Bussey H. and Andrews B. J. (2007). Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 8, 437-449. 10.1038/nrg2085 [DOI] [PubMed] [Google Scholar]
- Butland G., Babu M., Díaz-Mejía J. J., Bohdana F., Phanse S., Gold B., Yang W., Li J., Gagarinova A. G., Pogoutse O. et al. (2008). eSGA: E. coli synthetic genetic array analysis. Nat. Methods 5, 789-795. 10.1038/nmeth.1239 [DOI] [PubMed] [Google Scholar]
- Byrne A. B., Weirauch M. T., Wong V., Koeva M., Dixon S. J., Stuart J. M. and Roy P. J. (2007). A global analysis of genetic interactions in Caenorhabditis elegans. J. Biol. 6, 8 10.1186/jbiol58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castermans D., Somers I., Kriel J., Louwet W., Wera S., Versele M., Janssens V. and Thevelein J. M. (2012). Glucose-induced posttranslational activation of protein phosphatases PP2A and PP1 in yeast. Cell Res. 22, 1058-1077. 10.1038/cr.2012.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N. K., O'Donnell L., Oster S., Theesfeld C., Sellam A. et al. (2017). The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369-D379. 10.1093/nar/gkw1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Cui D., Papusha A., Zhang X., Chu C.-D., Tang J., Chen K., Pan X. and Ira G. (2012). The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489, 576-580. 10.1038/nature11355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E. N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S. D. et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 10.1126/science.aaf1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costelloe T., Louge R., Tomimatsu N., Mukherjee B., Martini E., Khadaroo B., Dubois K., Wiegant W. W., Thierry A., Burma S. et al. (2012). The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581-584. 10.1038/nature11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie E., Forte G. M. A. and Petersen J. (2015). Nitrogen regulates AMPK to control TORC1 signaling. Curr. Biol. 25, 445-454. 10.1016/j.cub.2014.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J., Fedyshyn Y., Koh J. L. Y., Prasad T. S. K., Chahwan C., Chua G., Toufighi K., Baryshnikova A., Hayles J., Hoe K.-L. et al. (2008). Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. USA 105, 16653-16658. 10.1073/pnas.0806261105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J., Andrews B. J. and Boone C. (2009a). Exploring the conservation of synthetic lethal genetic interaction networks. Commun. Integr. Biol. 2, 78-81. 10.4161/cib.7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J., Costanzo M., Baryshnikova A., Andrews B. and Boone C. (2009b). Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 43, 601-625. 10.1146/annurev.genet.39.073003.114751 [DOI] [PubMed] [Google Scholar]
- Eapen V. V., Sugawara N., Tsabar M., Wu W.-H. and Haber J. E. (2012). The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 32, 4727-4740. 10.1128/MCB.00566-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner T., Lackner D. H., Hombauer H., Piribauer P., Mudrak I., Zaragoza K., Juno C. and Ogris E. (2003). A novel and essential mechanism determining specificity and activity of protein phosphatase 2A (PP2A) in vivo. Genes Dev. 17, 2138-2150. 10.1101/gad.259903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg S. L. and Rhind N. (2006). Basic methods for fission yeast. Yeast 23, 173-183. 10.1002/yea.1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A. and Simanis V. (2012). Characterization of ypa1 and ypa2, the Schizosaccharomyces pombe orthologs of the peptidyl proyl isomerases that activate PP2A, reveals a role for Ypa2p in the regulation of cytokinesis. Genetics 190, 1235-1250. 10.1534/genetics.111.138040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F., Stanevich V., Wlodarchak N., Sengupta R., Jiang L., Satyshur K. A. and Xing Y. (2014). Structural basis of PP2A activation by PTPA, an ATP-dependent activation chaperone. Cell Res. 24, 190-203. 10.1038/cr.2013.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J. L. IV, Garvik B. and Hartwell L. (2001). Principles for the buffering of genetic variation. Science 291, 1001-1004. 10.1126/science.291.5506.1001 [DOI] [PubMed] [Google Scholar]
- Hartwell L. (2004). GENETICS: robust interactions. Science 303, 774-775. 10.1126/science.1094731 [DOI] [PubMed] [Google Scholar]
- Henske E. P., Jóźwiak S., Kingswood J. C., Sampson J. R. and Thiele E. A. (2016). Tuberous sclerosis complex. Nat. Rev. Dis. Primer. 2, 16035 10.1038/nrdp.2016.35 [DOI] [PubMed] [Google Scholar]
- Hoffman C. S., Wood V. and Fantes P. A. (2015). An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics 201, 403-423. 10.1534/genetics.115.181503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden B. E., Valvezan A. J., Kelley C., Sopko R., Hu Y., Roesel C., Lin S., Buckner M., Tao R., Yilmazel B. et al. (2015). Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci. Signal. 8, rs9 10.1126/scisignal.aab3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden B. E., Nicholson H. E. and Perrimon N. (2017). Synthetic lethality screens using RNAi in combination with CRISPR-based knockout in Drosophila cells. Bio-Protoc. 7, e2119 10.21769/BioProtoc.2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N. and Mohr S. E. (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357 10.1186/1471-2105-12-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Li Y., Xu T. and Guan K.-L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829-1834. 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens J., Janssens V., Longin S., Stevens I., Martens E., Bultynck G., Engelborghs Y., Lescrinier E., Waelkens E., Goris J. et al. (2006). The protein phosphatase 2A phosphatase activator is a novel peptidyl-prolyl cis/trans-isomerase. J. Biol. Chem. 281, 6349-6357. 10.1074/jbc.M507760200 [DOI] [PubMed] [Google Scholar]
- Joseph B. K., Liu H.-Y., Francisco J., Pandya D., Donigan M., Gallo-Ebert C., Giordano C., Bata A. and Nickels J. T. (2015). Inhibition of AMP kinase by the protein phosphatase 2A heterotrimer, PP2APpp2r2d. J. Biol. Chem. 290, 10588-10598. 10.1074/jbc.M114.626259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.-U., Hayles J., Kim D., Wood V., Park H.-O., Won M., Yoo H.-S., Duhig T., Nam M., Palmer G. et al. (2010). Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617-623. 10.1038/nbt.1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin E., Costanzo M., Andrews B. and Boone C. (2016). Synthetic genetic array analysis. Cold Spring Harb. Protoc. 2016, pdb.prot088807 10.1101/pdb.prot088807 [DOI] [PubMed] [Google Scholar]
- Lee J., Choi E. S., Seo H. D., Kang K., Gilmore J. M., Florens L., Washburn M. P., Choe J., Workman J. L. and Lee D. (2017). Chromatin remodeller Fun30Fft3 induces nucleosome disassembly to facilitate RNA polymerase II elongation. Nat. Commun. 8, 14527 10.1038/ncomms14527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N., Vicentini G., Jordens J., Quevillon-Cheruel S., Schiltz M., Barford D., van Tilbeurgh H. and Goris J. (2006). Crystal structure of the PP2A phosphatase activator: implications for its PP2A-specific PPIase activity. Mol. Cell 23, 413-424. 10.1016/j.molcel.2006.07.008 [DOI] [PubMed] [Google Scholar]
- Li J., Kim S. G. and Blenis J. (2014). Rapamycin: one drug, many effects. Cell Metab. 19, 373-379. 10.1016/j.cmet.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R., St Onge R. P., Hartman J. L., Giaever G. and Roth F. P. (2008). Defining genetic interaction. Proc. Natl. Acad. Sci. USA 105, 3461-3466. 10.1073/pnas.0712255105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. (2004). G1 cell-cycle control and cancer. Nature 432, 298-306. 10.1038/nature03094 [DOI] [PubMed] [Google Scholar]
- Matsumoto S., Bandyopadhyay A., Kwiatkowski D. J., Maitra U. and Matsumoto T. (2002). Role of the Tsc1-Tsc2 complex in signaling and transport across the cell membrane in the fission yeast Schizosaccharomyces pombe. Genetics 161, 1053-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvetz D., Priolo C. and Henske E. P. (2015). Therapeutic targeting of cellular metabolism in cells with hyperactive mTORC1: a paradigm shift. Mol. Cancer Res. 13, 3-8. 10.1158/1541-7786.MCR-14-0343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase Y., Nakase M., Kashiwazaki J., Murai T., Otsubo Y., Mabuchi I., Yamamoto M., Takegawa K. and Matsumoto T. (2013). The fission yeast β-arrestin-like protein Any1 is involved in TSC-Rheb signaling and the regulation of amino acid transporters. J. Cell Sci. 126, 3972-3981. 10.1242/jcs.128355 [DOI] [PubMed] [Google Scholar]
- Neuman N. A. and Henske E. P. (2011). Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol. Med. 3, 189-200. 10.1002/emmm.201100131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle S. C. (2017). Tuberous sclerosis complex: a review. Pediatr. Ann. 46, e166-e171. 10.3928/19382359-20170320-01 [DOI] [PubMed] [Google Scholar]
- Rosset C., Netto C. B. O. and Ashton-Prolla P. (2017). TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet. Mol. Biol. 40, 69-79. 10.1590/1678-4685-gmb-2015-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C. J., Roguev A., Patrick K., Xu J., Jahari H., Tong Z., Beltrao P., Shales M., Qu H., Collins S. R. et al. (2012). Hierarchical modularity and the evolution of genetic interactomes across species. Mol. Cell 46, 691-704. 10.1016/j.molcel.2012.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R. A. and Sabatini D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 169, 361-371. 10.1016/j.cell.2017.03.035 [DOI] [PubMed] [Google Scholar]
- Shen J. P., Zhao D., Sasik R., Luebeck J., Birmingham A., Bojorquez-Gomez A., Licon K., Klepper K., Pekin D., Beckett A. N. et al. (2017). Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573-576. 10.1038/nmeth.4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A. and Tyers M. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535-D539. 10.1093/nar/gkj109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steglich B., Strålfors A., Khorosjutina O., Persson J., Smialowska A., Javerzat J.-P. and Ekwall K. (2015). The Fun30 chromatin remodeler Fft3 controls nuclear organization and chromatin structure of insulators and subtelomeres in fission yeast. PLoS Genet. 11, e1005101 10.1371/journal.pgen.1005101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strålfors A., Walfridsson J., Bhuiyan H. and Ekwall K. (2011). The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet. 7, e1001334 10.1371/journal.pgen.1001334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K., Kotulska K., Janusz-Kaminska A., Zmorzynska J. and Jaworski J. (2016). Tuberous sclerosis complex: from molecular biology to novel therapeutic approaches. IUBMB Life 68, 955-962. 10.1002/iub.1579 [DOI] [PubMed] [Google Scholar]
- Taneja N. and Grewal S. I. S. (2017). Shushing histone turnover: it's FUN protecting epigenome-genome. Cell Cycle 16, 1731-1732. 10.1080/15384101.2017.1360651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N., Zofall M., Balachandran V., Thillainadesan G., Sugiyama T., Wheeler D., Zhou M. and Grewal S. I. S. (2017). SNF2 family protein Fft3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 66, 50-62.e6. 10.1016/j.molcel.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosti E., Katakowski J. A., Schaetzlein S., Kim H.-S., Ryan C. J., Shales M., Roguev A., Krogan N. J., Palliser D., Keogh M.-C. et al. (2014). Evolutionarily conserved genetic interactions with budding and fission yeast MutS identify orthologous relationships in mismatch repair-deficient cancer cells. Genome Med. 6, 68 10.1186/s13073-014-0068-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valbuena N. and Moreno S. (2010). TOR and PKA pathways synergize at the level of the Ste11 transcription factor to prevent mating and meiosis in fission yeast. PloS ONE 5, e11514 10.1371/journal.pone.0011514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoof C., Martens E., Longin S., Jordens J., Stevens I., Janssens V. and Goris J. (2005). Specific interactions of PP2A and PP2A-like phosphatases with the yeast PTPA homologues, Ypa1 and Ypa2. Biochem. J. 386, 93-102. 10.1042/BJ20040887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst M., Carr E., Stoyanova R., Kruger W. D. and Henske E. P. (2004). tsc1+ and tsc2+ regulate arginine uptake and metabolism in Schizosaccharomyces pombe. J. Biol. Chem. 279, 12706-12713. 10.1074/jbc.M313874200 [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M., Mustafa A. and Henske E. P. (2005). Pas1, a G1 cyclin, regulates amino acid uptake and rescues a delay in G1 arrest in Tsc1 and Tsc2 mutants in Schizosaccharomyces pombe. Hum. Mol. Genet. 14, 2851-2858. 10.1093/hmg/ddi317 [DOI] [PubMed] [Google Scholar]
- Vishwanatha A. and D'Souza C. J. M. (2017). Multifaceted effects of antimetabolite and anticancer drug, 2-deoxyglucose on eukaryotic cancer models budding and fission yeast. IUBMB Life 69, 137-147. 10.1002/iub.1599 [DOI] [PubMed] [Google Scholar]
- Watson A. T., Garcia V., Bone N., Carr A. M. and Armstrong J. (2008). Gene tagging and gene replacement using recombinase-mediated cassette exchange in Schizosaccharomyces pombe. Gene 407, 63-74. 10.1016/j.gene.2007.09.024 [DOI] [PubMed] [Google Scholar]
- Weisman R., Roitburg I., Nahari T. and Kupiec M. (2005). Regulation of leucine uptake by tor1+ in Schizosaccharomyces pombe is sensitive to rapamycin. Genetics 169, 539-550. 10.1534/genetics.104.034983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman R., Roitburg I., Schonbrun M., Harari R. and Kupiec M. (2007). Opposite effects of tor1 and tor2 on nitrogen starvation responses in fission yeast. Genetics 175, 1153-1162. 10.1534/genetics.106.064170 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.