

ABSTRACT
The neutralizing antibody (nAb) response against the influenza virus hemagglutinin (HA) fusion glycoprotein is important for preventing viral infection, but we lack a comprehensive understanding of the mechanisms by which these antibodies act. Here we investigated the effect of nAb binding and the role of IgG bivalency in the inhibition of HA function for nAbs targeting distinct HA epitopes. HC19 targets the receptor binding pocket at the distal end of HA, while FI6v3 binds primarily to the HA2 fusion subunit toward the base of the stalk. Surprisingly, HC19 inhibited the ability of HA to induce lipid mixing by preventing the structural rearrangement of HA under fusion-activating conditions. These results suggest that nAbs such as HC19 not only act by blocking receptor binding but also inhibit key late-stage HA conformational changes required for fusion. Intact HC19 IgG was also shown to cross-link separate virus particles, burying large proportions of HA within aggregates where they are blocked from interacting with target membranes; Fabs yielded no such aggregation and displayed weaker neutralization than IgG, emphasizing the impact of bivalency on the ability to neutralize virus. In contrast, the stem-targeting nAb FI6v3 did not aggregate particles. The Fab fragment was significantly less effective than IgG in preventing both membrane disruption and fusion. We infer that interspike cross-linking within a given particle by FI6v3 IgG may be critical to its potent neutralization, as no significant neutralization occurred with Fabs. These results demonstrate that IgG bivalency enhances HA inhibition through functionally important modes not evident in pared-down Fab-soluble HA structures.
IMPORTANCE The influenza virus hemagglutinin (HA) fusion glycoprotein mediates entry into target cells and is the primary antigenic target of neutralizing antibodies (nAbs). Our current structural understanding of mechanisms of antibody (Ab)-mediated neutralization largely relies on the high-resolution characterization of antigen binding (Fab) fragments in complex with soluble, isolated antigen constructs by cryo-electron microscopy (EM) single-particle reconstruction or X-ray crystallography. Interactions between full-length IgG and whole virions have not been well characterized, and a gap remains in our understanding of how intact Abs neutralize virus and prevent infection. Using structural and biophysical approaches, we observed that Ab-mediated inhibition of HA function and neutralization of virus infectivity occur by multiple coexisting mechanisms, are largely dependent on the specific epitope that is targeted, and are highly dependent on the bivalent nature of IgG molecules.
KEYWORDS: antibody, bivalency, influenza, neutralization, electron microscopy, enveloped viruses
INTRODUCTION
Neutralizing antibodies (nAbs) play a key role in the immune response against viral pathogens by binding to viral surface proteins and preventing them from mediating cell entry and infection. The influenza virus hemagglutinin (HA) fusion glycoprotein is the dominant surface antigen and primary target of nAbs, with an estimated abundance of ∼300 to 500 HA glycoproteins per virion (1–3). HA is expressed as a single polypeptide chain, HA0, which trimerizes to form a 225-kDa spike on the virus surface (4). Each HA0 chain is cleaved by host proteases into the receptor binding domain, HA1, and the fusion subunit, HA2 (Fig. 1). HA mediates cell entry by binding to sialic acid on cellular glycoproteins and glycolipids, initiating internalization through endocytosis (4). As the endosomes mature, their lumens acidify, triggering HA to undergo a large-scale conformational change that drives the fusion of viral and endosomal membranes and generates a fusion pore through which the viral genetic material is delivered to the cytosol (4–7).
FIG 1.
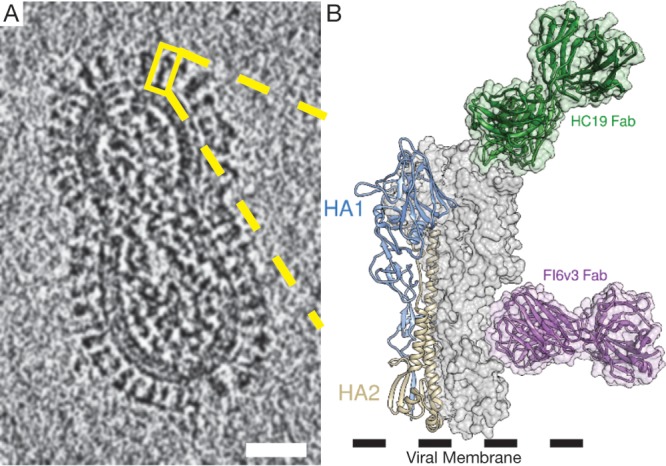
Cryo-electron tomography used to visualize the high density of HA on the viral surface. (A) An 8.0-nm computational slice through X31 influenza virus imaged by cryo-ET, with an individual HA trimer highlighted (yellow box). (B) Structural features of HA modeled in an expanded view of the highlighted trimer in panel A, with a single HA monomer consisting of the receptor binding domain (HA1) and the fusion domain (HA2) shown in a ribbon diagram (PDB accession number 4FNK). Epitopes for HC19 (PDB accession number 2VIR) and FI6v3 (PDB accession number 3ZTJ) are highlighted using the respective crystal structures. Bar = 25 nm.
Much of our understanding of the mechanisms of antibody (Ab)-mediated neutralization is based upon high-resolution structural studies of antigen binding (Fab) fragments in complex with soluble, isolated antigen constructs (8). Structures for several neutralizing antibodies against HA have been reported (9–13). Based upon high-resolution structures of Fab-HA complexes, it is usually inferred that the blockage of receptor binding sites or inhibition of conformational changes required for membrane fusion is responsible for neutralization. The static structures reveal antibody-antigen recognition in detail but do not necessarily enable the functional consequences of these interactions to be assessed. Recent structural and biophysical studies have begun to reveal the nature of the structural calisthenics performed by the HA fusion protein as well as the sequence of HA-induced membrane remodeling that lead to membrane fusion (5–7, 14, 15). With these new insights into fusion machinery function, it is possible to analyze mechanisms of the antibody-mediated disruption of these dynamic events that are essential for virus entry into cells. In addition, relatively few studies have characterized the interactions between full-length IgG and whole virions, where these modes of interaction are distinct from the picture provided by isolated Fab fragments due to the bivalency of IgG molecules and the multivalent display of antigens on the surface of most virus particles. Although the binding of IgG to antigens presented on the surface of the virus has been observed in some previous studies (16, 17), we are left with an incomplete picture of the relationship between IgG bivalency and mechanisms of inhibition of virus function. Interactions between intact IgG and proteins on the surface of the virus may increase binding avidity as well as expand the modes of neutralization through the cross-linking of antigenic sites on the same viral particle or on separate particles that lead to aggregation. These additional modes of action may be responsible for observations that high levels of virus neutralization are often observed even when saturation of binding sites by antibodies is not achieved (18–20).
Here we examined the interaction of two nAbs targeting different sites on the HA fusion protein presented on intact influenza virus. HC19 targets the receptor binding domain (RBD) on membrane-distal HA1, while FI6v3 targets the HA2 fusion subunit on the HA stalk (10, 13). We hypothesized that due to the location of the HC19 epitope on the highly exposed end of HA, the bivalency of IgG could enhance its mechanism of action by aggregating virus particles and by cross-linking adjacent HA trimers on a virion. In contrast, the epitope for FI6v3 is located further down along the HA spike, where it may be unable to reach across to HA on a separate particle (Fig. 1B) (13). More specifically, we sought to investigate the effect of antibodies targeting these distinct epitopes on the ability of HA to undergo the conformational changes required to drive membrane fusion to completion. In addition, we examined the impact of bivalency on the ability of IgG to arrest infectivity by comparing the neutralization of virus by intact IgG and monovalent Fab domains. We report that the inhibition of specific stages of HA-mediated fusion, starting with membrane disruption caused by fusion peptide insertion and followed by lipid mixing leading to pore formation as well as virus infectivity, showed dramatic differences for Fabs and intact IgG. These results suggest a pivotal and complex role for bivalency in effecting antibody neutralization of virus entry.
RESULTS
Binding affinities of IgG and Fab for soluble bromelain-released HA.
The affinity of bromelain-released HA (BHA) for HC19 and FI6v3 (IgG and the respective Fab) was determined by using biolayer interferometry (BLI) (Fig. 2). For BLI data collection, assays using anti-human Fab-CH1 biosensors were used to capture both IgG and isolated Fabs in our study. Anti-human Fc biosensors capturing IgG were also tested, yielding rates of association (kon) similar to those for isolated Fab; however, rates of dissociation (kdis) for IgG could not be accurately determined. We chose to use the anti-human Fab-CH1 biosensors as the common means of capturing both Fab and IgG in order to make the experiments and observed binding kinetics as comparable as possible.
FIG 2.

Affinities of IgG and Fab measured by using biolayer interferometry. (A) Binding sensorgrams show binding of BHA to immobilized HC19 IgG (blue), HC19 Fab (green), FI6v3 IgG (red), and FI6v3 Fab (orange). (B) Association and dissociation constants yield similar KDs between HC19 IgG and Fab and between FI6v3 IgG and Fab. Values shown are the averages and standard deviations of data from experiments repeated in triplicates.
BHA binds HC19 IgG and isolated Fab with nanomolar affinities (3.25 nM and 1.13 nM, respectively). The variance in the affinities between HC19 IgG and Fab is attributed to an accelerated kdis for IgG (Fig. 2B). kon values were similar for both HC19 IgG and Fab, and no avidity effects were detected for captured IgG when loading densities of 3 μg/ml, 5 μg/ml, and 10 μg/ml were compared. For FI6v3, the kon and kdis of BHA were similar and yielded only a modest difference in the KD (equilibrium dissociation constant) values (Fig. 2B). Overall, BLI shows that digestion of IgG into monovalent Fab does not impact the antibody paratope. The Fabs bind with affinities similar to those of full-length IgG, suggesting that any differences between IgG and Fab in subsequent experiments are due to the effects of bivalent Fab presentation by IgG and not a change in the Fab domain's capacity to bind HA.
Inhibition of HA-mediated membrane disruption and fusion activities by IgG and Fab.
Fluorescence spectroscopy was used to monitor HA-mediated fusion between influenza virus and liposomes and to compare the inhibitory effects of IgG and Fab fragments targeting two distinct epitopes on HA. DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indodicarbocyanine,4-chlorobenzenesulfonate salt)-labeled virus was preincubated with IgG or Fab at the desired Ab/HA ratios prior to the addition of sulforhodamine B (SRB)-encapsulating liposomes. Following the mixing of these components, the solution was acidified to trigger HA activation. The fluorescence assay enables us to monitor two stages of HA activity: (i) fusion peptide insertion into the target liposomal membrane, which leads to leakage and fluorescence dequenching of the encapsulated SRB dye, and (ii) lipid mixing of the merged membranes that occurs during fusion pore formation, reported by DiD dequenching (5, 6).
Increasing molar fractions of IgG and Fab relative to a constant amount of HA led to the increased inhibition of HA activities. Comparison of fusion peptide disruption of target membranes, monitored by SRB dequenching for HA1 head domain-targeting HC19 IgG and Fab, demonstrated that bivalent IgGs were more potent in their ability to reduce apparent HA activity than were monovalent Fabs, where levels of activity were only modestly diminished relative to those of the control (Fig. 3A). At the highest concentration of full-length IgG or Fab, SRB leakage was reduced by ∼75% for IgG, compared to only ∼20% inhibition for Fabs. A substantially smaller difference between IgG- and Fab-mediated inhibition was observed for the lipid-mixing stage of HA-mediated membrane fusion, which was inhibited to similar extents for both intact IgG and Fab (∼85% and 74% for IgG and Fab, respectively) (Fig. 3B). Thus, it appears that intact HC19 IgG could effectively inhibit both an early stage of fusion peptide-induced membrane disruption and the later stage of lipid mixing of HA-mediated membrane fusion, while the Fab fragment was effective only in inhibiting lipid mixing while permitting significant fusion peptide interactions with and disruption of membranes.
FIG 3.

Fluorescence fusion assay monitors antibody-mediated inhibition of HA function by tracking dequenching of the fluorescent dyes SRB, indicating membrane disruption, and DiD, indicating lipid mixing. Plotted are the inhibitory effects of IgG and Fab. The dashed line is a normalized version of the Fab inhibition data and represents inhibition at an equivalent number of Fab arms compared to IgG (1 Fab = 0.5 IgG). (A) HC19 inhibition of membrane disruption is enhanced through bivalent interactions between IgG and HA on X31 virus, even at subsaturating concentrations, where aggregation was observed by DLS. Fab binding has little impact on membrane disruption inhibition. (B) Extents of inhibition of lipid mixing by IgG and Fab are similar, suggesting that bivalency may not play a large role in fusion inhibition for HC19. (C) Membrane disruption is more potently inhibited by FI6v3 IgG than by monovalent Fab. (D) Inhibition of lipid mixing by FI6v3 IgG is greater than that by Fab, suggesting that cross-linking of neighboring trimers is important for HA inhibition.
For the stem-targeting broadly neutralizing antibody (bnAb) FI6v3, the Fab fragment showed a significantly reduced ability to inhibit fusion peptide-induced membrane disruption, monitored by SRB dequenching, compared to IgG, even at concentrations approaching saturation, ∼40% and ∼70%, respectively (Fig. 3C). Late-stage lipid mixing, as indicated by DiD dequenching, was potently inhibited by intact FI6v3 IgG, while surprisingly, FI6v3 Fab was far less effective in inhibiting membrane fusion, even at concentrations approaching equivalence with the binding sites on HA, 90% versus 50%, respectively (Fig. 3D). The presentation of the Fab domains in the context of intact, bivalent IgG thus appears to play a prominent role in the stem-targeted antibody's ability to arrest HA-mediated membrane fusion activity.
Neutralization of influenza virus by bivalent IgG versus monovalent Fab.
The inhibitory activity of HC19 and FI6v3 (both IgG and Fab) on influenza virus infection was evaluated by measuring the 50% tissue culture infective dose (TCID50) of X31 influenza virus. We hypothesized that neutralization would follow a profile similar to that for fluorescence fusion assays. A molar ratio of 1:1 (IgG/HA) was chosen, where HA would not be completely inhibited and a degree of fusion activity would remain. In the case of Fabs, the concentration was selected to include the equivalent number of Fab copies as present in IgG (two Fab copies per IgG copy); thus, the molar concentration of Fab was 2-fold higher than that of IgG.
Both HC19 and FI6v3 intact IgGs displayed strong neutralizing activity against X31 influenza virus, where 70% and >95% neutralizations were achieved, respectively (Fig. 4). FI6v3 was more efficient at inhibiting virus infectivity, even at an IgG/HA trimer ratio of only 1:1, suggesting that stem-directed antibodies require an occupancy of only a fraction of the binding sites to achieve potent neutralization. Antibodies such as HC19 that target the RBD may require a higher occupancy of binding sites to achieve levels of neutralization similar to those of FI6v3.
FIG 4.

Neutralization assay measuring TCID50. Antibody-mediated neutralization of influenza virus was evaluated by measuring the TCID50 of influenza virus. IgGs for both FI6v3 and HC19 displayed stronger neutralization than did the respective Fabs. While HC19 Fab could neutralize virus to some degree, FI6v3 Fab did not exhibit a significant ability to neutralize the virus (*, P > 0.05; **, P < 0.01).
HC19 IgG and Fab exhibited differences in the extents of neutralization that paralleled the trends in the inhibition of HA function observed in fluorescence fusion assays. HC19 IgG neutralized 70% of virus infectivity, compared to only 40% neutralization by an equivalent number of monovalent Fabs. Even larger differences in neutralization between IgG and Fab were observed for the stem-directed antibody FI6v3. More than 95% neutralization efficacy against X31 influenza virus was observed for FI6v3 IgG; however, no significant neutralization by Fab was detected. Overall, bivalent interactions by IgG appear to play a pivotal role in the antibody's ability to effectively neutralize influenza virus and prevent infection of cells.
Imaging of Fab:BHA complexes under neutral and acidic pH using negative-stain EM.
Negative-stain transmission electron microscopy (EM) imaging was performed on Fab:BHA complexes at pH 7.5 and 5.25 to assess the impact that Fab binding may have on the ability of BHA to undergo the acidic-pH-triggered conformational changes that are required to mediate fusion. Class averages of ∼8,000 BHA trimers at pH 7.5 exhibit the characteristic prefusion “peanut-shaped” structure that is observed on the surface of virus particles (Fig. 5A). After adjusting the pH to 5.25 for 10 min, BHA aggregates into rosettes commonly observed in solubilized fusion glycoproteins due to the formation of the stable postfusion helical bundle with tightly clustered fusion peptides (21–23).
FIG 5.
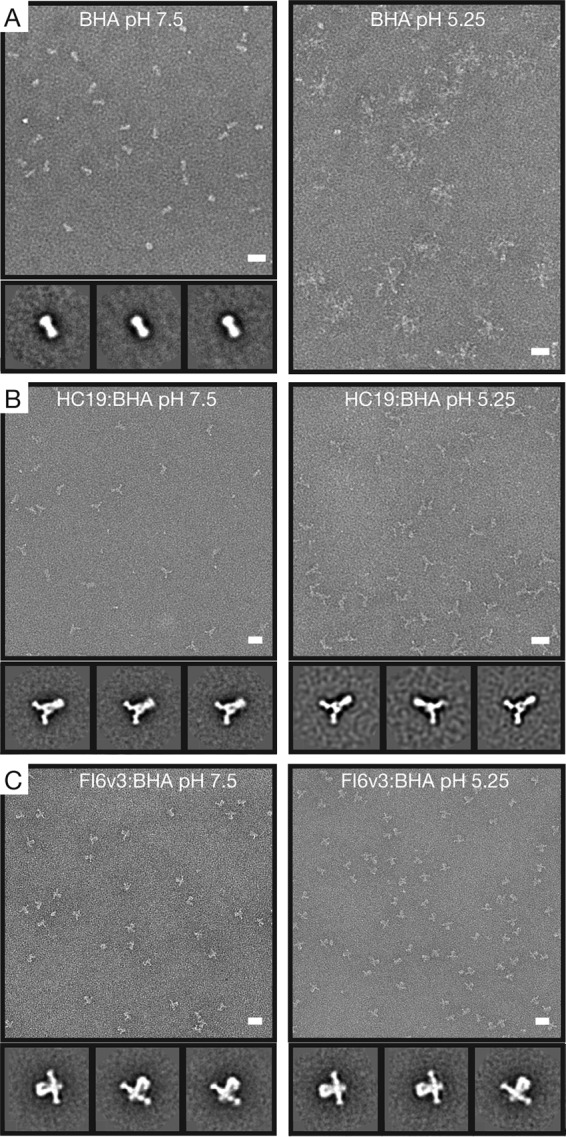
Negative-stain EM and 2D classification of Fab:BHA complexes under neutral and acidic conditions reveal structural similarities. (A) Electron micrographs and representative class averages of BHA at pH 7.5 and BHA that has been acidified for 10 min at pH 5.25. In response to acidic pH, BHA refolds into a postfusion conformation, forming small aggregates (rosettes) that are not amenable to 2D classification. (B) BHA bound by HC19 Fab at pH 7.5 maintains the prefusion form of the BHA trimer. After acidification for 10 min at pH 5.25, HC19 Fab appears to stabilize the BHA timer in a structure similar to that at neutral pH. (C) Both BHA bound by FI6v3 Fab at pH 7.5 and the Fab:BHA complex acidified for 10 min at pH 5.25 stabilize the BHA trimer and prevent refolding into a postfusion form. Bar = 20 nm.
Imaging of HC19 Fab bound to BHA showed classes of particles where Fab density could be observed projecting from the receptor binding domain on HA1 (Fig. 5B). The HC19 Fab:BHA complex at pH 7.5 displays a prefusion organization for the spike that was similar to that observed with BHA alone. After HC19 Fab and BHA were complexed for 1 h, the pH was adjusted to pH 5.25 for 10 min prior to grid preparation and imaging. Classification of particles shows that the acidified complex retains an organization similar to that of the neutral-pH complex, indicating that the binding of HC19 to BHA stabilizes the trimer in a state that is similar to that of the prefusion form, in which the HA spike remains largely intact (Fig. 5B). A modest narrowing of the base of the BHA stem is observed under acidic conditions with HC19 bound, which would be consistent with previous low-resolution imaging of HA-intermediate states on the surface of intact influenza virus virions, which suggested that the fusion peptide-associated regions were released from the main stem body and otherwise maintained an intact prefusion-like HA spike under activation conditions (24).
For FI6v3 Fab, density in the negative-stain EM images could be observed projecting from the HA2 fusion domain, consistent with the reported Fab-HA structure (13). Under neutral conditions, the BHA trimer remained in its prefusion form. The proposed mechanism of action for stem-directed antibodies is to prevent HA from undergoing critical changes necessary for fusion, and indeed, after exposure to acidic pH, the Fab:BHA complex, at the level of resolution afforded by negative-stain EM, appeared to maintain the prefusion organization (Fig. 5C).
Assessment of the ability of nAb to mediate particle aggregation by dynamic light scattering and negative-stain EM.
We hypothesized that an epitope's position on the HA spike could impact the ability of IgG to mediate interparticle aggregation. To test for interparticle aggregation, we used dynamic light scattering (DLS) to measure the time-dependent aggregation of influenza virus across various IgG/HA trimer molar ratios. We first examined if antibodies bound to the HC19 epitope at the RBD on the membrane-distal end of HA could mediate the cross-linking of HA on separate virus particles. DLS detected HC19-mediated aggregate formation at IgG/HA ratios of >1:3 (Fig. 6A). Over the final 30 min, at this relatively low IgG/HA ratio, a modest 11% ± 1% increase in the average particle radius was detected. This indicates that only a fraction of sites needs to be bound for aggregate formation to start to occur. Increasing the concentration of HC19 further promoted aggregation, with an increase in the average measured particle radius of 88% ± 6% relative to the control at IgG/HA ratios of 2:1. Aggregates showed a high degree of polydispersity, with a particle distribution ranging from 100 nm to 1 μm. Negative-stain imaging further illustrated the IgG-mediated aggregation of virus with increasing concentrations of HC19 (Fig. 6B). Interestingly, the particle size appeared to slightly decrease when approaching a 3:1 ratio (averaged radius of ∼170 nm). Such a reduction in aggregation may be due to a further coating of the viral surface where Ab competes for available HA molecules (25).
FIG 6.
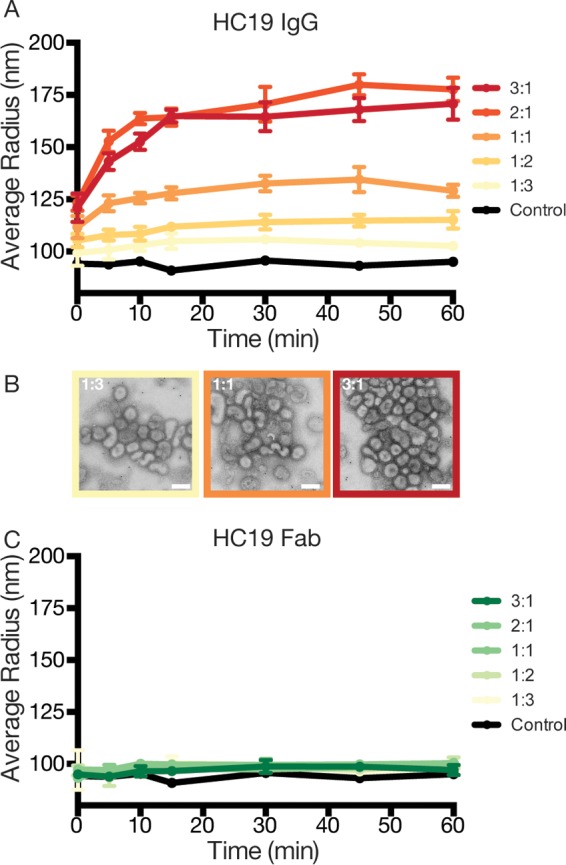
Dynamic light scattering monitors the time- and concentration-dependent aggregation of influenza virus X31 by HC19 IgG. (A) As the ratio of IgG/HA increases, HC19 aggregates virus particles, with peak aggregation occurring at 2:1 ratios. (B) Negative-stain micrographs further illustrate the extent of aggregation as the ratio of IgG/HA increases. (C) Digestion of IgG into monovalent Fab arms fails to aggregate influenza virus X31, emphasizing the complex binding that occurs between bivalent IgG and HA presented on the virus surface. Bar = 200 nm.
To investigate if aggregation was due to IgG bivalency, parallel experiments using HC19 digested into monovalent Fab fragments were carried out. HC19 Fab binding to HA on the surface of the virus had a negligible impact on the size of scattering populations, with only a 4% ± 2% increase across all ratios, which is likely the result of the bound Fab extending from the tops of HA trimers (Fig. 6C). The HC19 Fab data support the conclusion that aggregation by IgG is attributed to bivalent interactions in the intact HC19 antibody.
To test the dependency of the epitope location on the ability of IgG to aggregate the virus, we performed parallel experiments using FI6v3, a stem-directed bnAb. Both FI6v3 IgG and FI6v3 Fab failed to produce an increase in the particle radius as measured by DLS (Fig. 7A and C). Negative-stain EM micrographs of FI6v3 bound to X31 influenza virus particles appear monodispersed across a range of IgG concentrations (Fig. 7B). These data suggest that the aggregation of influenza virus is dependent not only on bivalent interactions but also on the epitope targeted by IgG.
FIG 7.
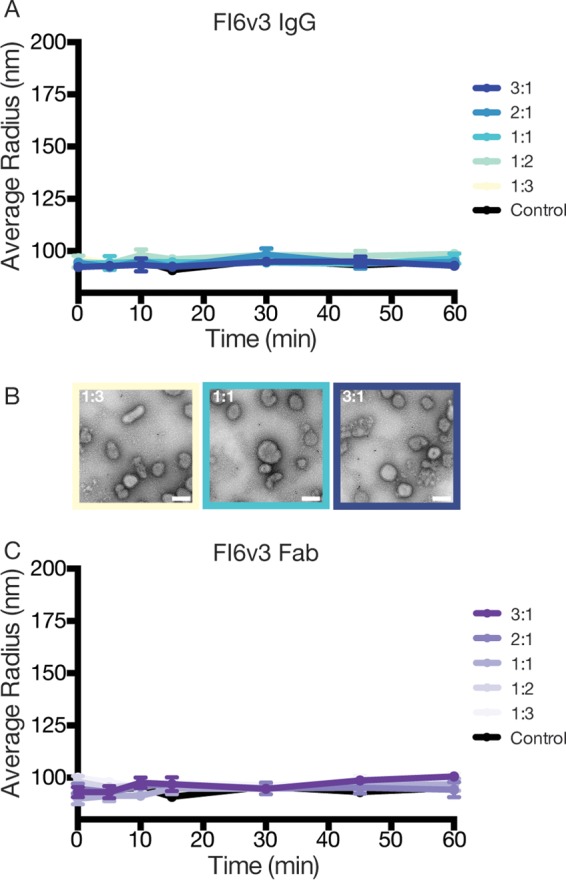
Dynamic light scattering does not detect aggregate formation of influenza virus X31 in the presence of IgG or Fab from stem-directed antibody FI6v3. (A) IgG incubated with influenza virus X31 at various ratios had no effect on particle radius in solution. (B) Negative-stain micrographs of influenza virus across various ratios of IgG/HA appear monodispersed, demonstrating the inability of FI6v3 to form bivalent interactions across multiple virus particles. (C) Similar to IgG, FI6v3 Fab fails to aggregate influenza virus X31. Bar = 200 nm.
HC19 IgG cross-linking of HA spikes imaged by cryo-ET.
From the negative-stain EM data, we observed an extensive aggregation of influenza virus by HC19 but were unable to resolve the 3-dimensional organization of IgG bound to HA trimers. To overcome this limitation, we used cryo-electron tomography (cryo-ET) to characterize IgG-virus interactions under more native conditions. Cryo-ET has been instrumental in the visualization of enveloped viruses that exhibit a pleomorphic structure and are unsuitable for traditional single-particle techniques, with the resolution being sufficient to distinguish different forms of viral surface glycoproteins alone and in complex with antibody Fabs (3, 5, 6, 26–28).
Tomographic analysis of HC19 associated with X31 virus demonstrated the antibody's ability to cross-link HA spikes within a given particle as well as to aggregate multiple particles by cross-linking HA spikes on separate viruses. Analysis of ∼20 tomograms revealed that the cross-linking of adjacent HA on a single virion appeared to be the dominant binding mode at lower concentrations of HC19 (IgG/HA ratio of 1:6), and the observed densities linking HA spikes were consistent with our negative-stain two-dimensional (2D) classification of HC19 bound to BHA (Fig. 5 and 8A and B). Both the cross-linking of adjacent HA molecules and aggregation of the virus were observed in ∼30 tomograms of samples collected at IgG/HA ratios of 1:3. Density for lipid and protein components across the interface of two viruses can be assigned from a radial density plot in which the complete HA density measures ∼14 nm across and is consistent with known crystal structures (Fig. 8C) (29). The density between the two particles shown in Fig. 8C is consistent with 2 Fab layers with a thickness of ∼7 nm, with the Fc fragment likely being localized in an adjacent x-y plane at a different z height.
FIG 8.

HC19 IgG-mediated cross-linking of HA on the surface of intact X31 influenza virus occurs between adjacent antigens on single particles and between antigens on separate particles, causing aggregation to occur. (A and B) A 4.4-nm-thick computational slice through HC19 IgG-complexed virus at a ratio of 1:6 (IgG/HA). Black arrowheads point toward IgG-cross-linked HA trimers on the surface of individual particles, and expanded views of these interactions are shown in the insets. (C) A 4.4-nm-thick computational slice through HC19 IgG-complexed virus at a ratio of 1:3 (IgG/HA). The radial density average at the interface between two virus particles was measured so that the distribution of lipid and protein components could be assigned. The blue box indicates the measured area used for the radial density plot. (D and E) Computational slices (8.0 nm thick) through HC19 IgG-complexed virus at a ratio of 1:2 (IgG/HA) illustrating extensive aggregation that occurs. Bar = 25 nm.
Further increasing the HC19/HA ratio to 1:2 (∼15 tomograms) led to extensive aggregation and coating of exposed HA by HC19 (Fig. 8D and E). At this HC19/HA ratio, clear imaging of virus bound by saturating amounts of IgG was not possible; due to the larger aggregates, the ice layer was too thick to provide sufficient beam penetration, resulting in poor image contrast.
In contrast to the high levels of HC19-induced influenza virus aggregation that were observed, FI6v3-bound viruses imaged by cryo-ET were largely monodispersed, as sampled across ∼10 tomograms. This is consistent with the observations by negative-stain EM and provides support for the hypothesis that the Ab-mediated aggregation of virus is heavily dependent upon the epitope location. The positioning of lobes of extra density that we attribute to antibody bound to an HA spike by the Fab domain is consistent with known cocrystal structures of FI6v3 Fabs bound to isolated HA trimers (13). Imaging of FI6v3-complexed virus at an IgG/HA ratio of 1:2 revealed regions where the organization of HA appears disrupted, with dense arrays of HA spikes that are normally observed on the viral surface no longer being apparent. It appears that the binding of IgG to the HA stem may cause this reorganization of HA by displacing the trimer spikes (Fig. 9).
FIG 9.

FI6v3 bound to X31 may disrupt the dense network of HA on the surface of the virus. Shown is a 6.4-nm-thick computational slice of FI6v3 bound to X31 at a ratio of 1:2 (IgG/HA). Black arrowheads point toward FI6v3 IgG-bound HA. Additional density ∼6 nm above the viral membrane corresponds to FI6v3 bound to the stem region of HA2. FI6v3 fails to aggregate X31 due to the epitope's location further down the HA stem. The IgG-virus mixture was incubated for 30 min prior to vitrification. Bar = 25 nm.
DISCUSSION
HA-mediated virus entry begins with receptor binding and ends with the merging of virus and host membranes that is driven by the fusion protein carrying out a sequence of dynamic conformational changes. For at least some influenza virus strains, including X31 H3N2 Aichi, this sequence of HA reorganization commences with fusion peptide release, only later to be followed by HA1 receptor binding subunit dissociation and large-scale HA2 fusion subunit refolding to the postfusion state (14, 24, 30). Binding of nAbs to HA is thought to interrupt various steps in the sequence of events that HA must traverse to carry out membrane fusion. From recent studies of whole viruses undergoing fusion, it has been possible to determine that as HA refolds, the viral and host membranes are drawn into tightly localized close apposition through the action of a set of several HA spikes (5, 6, 31, 32). These localized dimples transition to encompass increasingly large areas of virus and host membranes that are in direct contact (6). The formation of these tightly docked virus-target membrane intermediate states requires HA spikes to exhibit a degree of lateral mobility, as the fusion proteins are displaced from the membrane contact zones and instead are observed in partially refolded hairpin configurations at the periphery of membrane contact zones. Crystal structures of Fab bound to soluble antigen have been used to infer the stage at which the antibody is inhibiting the activity of HA; however, to understand the mechanism of inhibition, it is necessary to characterize the direct functional consequences of binding in the context of intact IgG, which can exhibit bivalent interactions and cross-linking of antigen copies on the virus surface. Here we sought to examine how intact, neutralizing antibodies targeting distinct epitopes on HA in the context of whole virus particles could interfere with the fusion protein's function and sequence of events that lead to virus entry.
The receptor binding subunit HA1 is highly immunodominant and often elicits subtype-specific Abs; however, neutralizing Abs with cross-reactivity against various HA subtypes, such as S139/1 and 5J8, have been detected (33, 34). Anti-HA1 Abs often block receptor binding and can prevent virus release from infected cells (35). A correlation between binding affinity and neutralization potency is commonly observed for Abs targeting the RBD, where bivalent interactions are thought to enhance antibody avidity (33, 34, 36). In instances where individual monovalent binding by Fab to HA across subtypes is weak, it has been proposed that bivalent binding by IgG may result in increased avidity, leading to broader antibody specificities and enhanced cross-subtype neutralization (33). The first nAb that we examined, HC19, has an epitope that overlaps the receptor binding site of HA1. The intraparticle spike cross-linking that we observed by cryo-ET, particularly at lower IgG/HA ratios, is consistent with this antibody exhibiting avid, bivalent binding. The 50% inhibitory concentration (IC50) for HC19 IgG (∼10 nM) in previously reported plaque reduction assays was 3 orders of magnitude lower than that observed for the respective Fab fragment, underscoring the greater potency of bivalent IgG (11). While we did not observe as dramatic an effect, we found that HC19 Fab was indeed significantly less effective than IgG in neutralizing virus, and data from fluorescence assays of the inhibition of HA-mediated fusion were in excellent agreement with the infectivity results. These results support the role of antibody bivalency in enhancing the activity of HC19.
Bivalent interactions of such antibodies may also result in the cross-linking of antigenic sites on separate particles, potentially expanding the modes of Ab-mediated neutralization by aggregating virions together. The angle at which the Fab arm binds to the HA spike, as seen in a previously reported crystal structure, is consistent with the Fab arms in intact IgG being able to reach adjacent trimers within a virus particle as well as bridge separate particles (10, 11). The binding angle and the specific epitope position on HA, even for antibodies targeting the HA1 subunit, are key determining factors of an antibody's ability to induce virus aggregation. It has been shown, for example, that HA1-directed nAbs 3F5 and 7B2 also form clusters of aggregated influenza virus particles (16), while nAbs targeting the hinge region of HA1, which is lower on the spike, orient the IgG molecule more in plane with the virus surface and have activity that is less dependent on antibody bivalency and spike cross-linking that could produce virus aggregation (37, 38).
When antibodies such as HC19 that target the receptor binding site produce large particle aggregates, numerous HAs that are not necessarily bound by an antibody are likely inaccessible due to their burial in the aggregates. In our DLS and EM studies, we observed that the degree of aggregation was largely dependent on the IgG concentration and that aggregates began to form rapidly even before the first measurement time point. Aggregates were detected at lower concentrations of HC19 IgG (1:3 ratio of IgG/HA), and the particle radius continued to increase until the Ab approached saturation of binding sites. At a 3:1 IgG/HA ratio, the average particle radius began to decrease, perhaps due to the saturation of the epitopes on the surface of individual virus particles. These data indicate that, in agreement with data from previous studies, aggregation occurs over a relatively narrow range of IgG/HA ratios (39–41).
Once the virus is aggregated into a large bolus, questions regarding the effect on infectivity remain. One possible outcome is the internalization of aggregates through an alternate, clathrin-independent pathway, giving rise to the delivery of a large quantity of virus into cells (42, 43). Previous studies on influenza virus suggest that aggregation may in some instances enhance infection rather than neutralize the virus through the delivery of a higher viral load to permissive cells (44, 45). Alternatively, aggregated virus could be degraded through phagocytosis by the use of Fc effector functions (46, 47). Indeed, the role of Ab-mediated aggregation further complicates mechanisms of neutralization of virus and may depend on the viral system being studied. For example, the IgG-induced aggregation of nonenveloped, icosahedral picornaviruses has been shown to contribute to Ab-mediated neutralization (48, 49). Aggregation of HIV-1 by dimeric IgA (tetravalent Fab presentation) is thought to inhibit movement across mucosal surfaces (50). In the present study, we observed enhanced neutralization by IgG compared to that by Fab at concentrations of IgG where extensive aggregation of influenza virus by the HA1-targeting HC19 nAb occurred. This suggests that the ability of IgG to aggregate virus can increase the efficacy of antibodies in inhibiting influenza virus infection. From our comparison of HC19 IgG and Fab activities with the complementary assays that we employed, we find that particle aggregation and interspike cross-linking modestly enhanced the activity of this nAb relative to the Fab fragment.
Additionally, in a previously unanticipated role, we found that HC19 and likely similar antibodies that bind to the HA1 receptor binding subunit also appear to be able to inhibit later stages of HA's conformational change that are required for lipid mixing. The fusion inhibition data shown in Fig. 3 suggest that HC19 may bind to the HA trimer in such a fashion that the fusion peptides can still be released and then bind to and perturb the target membrane; however, lipid mixing was dramatically inhibited, even by Fab alone. We note that these data are consistent with an HA fusion activation pathway where the fusion peptide can be released prior to HA1 dissociation without requiring full HA spike reorganization (14, 30, 51). Another HA1-targeted nAb, HC63, was previously shown to inhibit HA activation; however, in that case, the antibody itself bridges two HA1 subunits of the same trimer, preventing their dissociation (9, 12). For HC19, Fab interacts with a single protomer (10), and hence, we infer that it acts by stabilizing the subunit's conformation and, indirectly, the HA1-HA1 “cage” that prevents the full refolding of the HA2 subunit. In support of this model, negative-stain EM analysis of the soluble bromelain-released HA trimer bound by HC19 Fab and exposed to acidic pH indicated that the trimeric spike overall remained intact. It thus appears that the gross refolding of HA, required for the merging of the two membranes, is prevented when HC19 Fab was bound, although the fusion peptide appears to be deployed and able to disrupt membranes (Fig. 3 and 5). Alternatively, it is conceivable that with Fab bound to the HC19 epitope at the apex of the spike, the fusion machinery may be sterically blocked from drawing the two membranes into close enough proximity to fuse, despite allowing fusion peptide insertion into the target membrane. In total, the multiple mechanisms by which HC19 perturbs HA function, including blocking receptor binding, cross-linking HA spikes, aggregating particles, and inhibiting the conformational changes leading to membrane merging, are likely responsible for the reduction in infectivity observed in our neutralization assays (Fig. 4).
Whereas the effect of antibody bivalency in HC19 was measurable but relatively modest, in the case of FI6v3, IgG and Fab exhibited dramatic differences in their abilities to arrest the fusion process as well as neutralize virus and inhibit infection. We observed that fusion peptide-induced SRB dequenching and lipid mixing were far more potently inhibited by intact FI6v3 IgG than by Fabs. While it was tempting to speculate that differences in binding affinities could be the cause of this variation, BLI measurements of IgG and Fab bound to BHA, in the cases of both HC19 and FI6v3, yielded similar KDs, suggesting that these differences are instead due to the ability of IgG to form bivalent interactions. The highest levels of FI6v3 inhibition were observed for a range of IgG/HA ratios similar to those reported previously for other stem-directed antibodies, CR8020 and CR6261 (19). Our data are consistent with results from a single-particle fluorescence microscopy study of fusion inhibition, which suggested that bivalent IgG targeting the stems can disrupt networks of HA spikes that must work together for productive fusion to occur. Previous work showed that fusion requires the engagement of HA with a minimum of 3 to 5 neighboring spikes (5, 31, 32). In addition, in that fluorescence study, the authors deduced that ∼60% occupancy by stem-directed antibodies was required to inhibit fusion (19). That level of bound antibody is roughly consistent with ratios of IgG/HA spike (between 0.5:1 and 1:1) where we observed both potent inhibition of HA function and neutralization of virus infectivity. If bivalent IgG is cross-linking adjacent HA spikes on a virion, this would result in tightly bound networks of HA that have constrained lateral mobility. IgG displayed potent neutralizing activity with a 95% reduction in infectivity when bound at an IgG/HA ratio of 1:1. At this same ratio, Fab was unable to inhibit the virus, further suggesting that the ability of IgG to bind bivalently to HA is critical for FI6v3 and likely other stem-directed antibodies. Indeed, a significant decrease in fusion activity, as measured by fluorescence spectroscopy, materialized only once the Fab/HA ratio was 3:1. We postulate that complete saturation of the available binding sites on the trimeric HA spike may be necessary for Fab to have an inhibitory effect on the fusion protein's function. This may suggest that the three HA2 subunits per trimer are not cooperatively coupled to each other. In terms of using stem-targeting nAbs or engineered binders as entry-inhibiting antiviral agents, the relatively low potency of FI6v3 Fab may hint that high concentrations of inhibitors will be necessary for the compounds to be effective.
Why was FI6v3 IgG so effective when Fab was not? Our recent cryo-electron tomography studies depicting the interplay of HA and membrane remodeling during fusion indicate that shortly after initial contact and HA-induced dimple formation on the target membrane, large spans of the target and virus membranes are drawn into direct apposition (6). HA becomes excluded from these regions and must be laterally displaced to the periphery of the membrane contact zone. IgG bound to two HA spikes and, on a broader scale, cross-linking many spikes into a network, would prevent such displacement from occurring. Additionally, a cross-linked network of HA may act at an even earlier stage to prevent sufficient numbers of activated HA trimers from being recruited to the fusion site in order to produce the proper sequence of membrane docking and remodeling that can lead to fusion.
While bivalent interspike cross-linking by FI6v3 IgG within a given virus particle appears to be critical for its activity, in contrast to HC19, neither FI6v3 IgG nor Fab was able to aggregate virus. Stem-directed Abs such as FI6v3 must diffuse through a dense array of HA spikes on the surface of influenza virus, and the location of this epitope further down the HA stem prevents the Ab from being able to reach across to bridge two epitope sites on separate particles. Previous studies examined the HA distribution on the virus and inferred the accessibility of the HA2 stem epitope to antibodies (52) but did not actually examine whole virus complexed with antibody. Another cryo-electron tomography study previously examined a stem-targeting bnAb, C179, bound to H1N1 influenza virus virions but focused on the averaged structure of the trimer-antibody complex, which primarily revealed the orientation of one Fab domain projecting from the prefusion trimer (17). In that study, Harris et al. also observed that the distribution of trimers on virions was able to accommodate up to 75% antibody occupancy of HA. We note that for FI6v3, consistent with the findings of the study on C179, we also did not observe significant density for the Fc domain in an orientation where it would be accessible or extend beyond the HA arrays. We speculate that this may indicate that the Fc-mediated effects associated with stem-targeting nAbs such as FI6v3 may be targeting HA on the surface of infected cells, where the HA density may be lower, rather than on free virus (46, 47).
In conclusion, we observed that the Ab-mediated inhibition of HA function occurs by multiple complementary mechanisms and is largely dependent on the specific epitope that is targeted and on the bivalent nature of IgG molecules. We have shown that the ability of nAbs to aggregate influenza virus particles enhances the inhibition of HA at an early stage of fusion peptide-induced membrane disruption through the occlusion of infectious virions, even at concentrations well below antigen saturation. Surprisingly, for HC19, binding to HA also inhibited HA function at the lipid-mixing stage of the fusion pathway by preventing the refolding of HA through the stabilization of the trimer under fusion-activating conditions. Epitopes further down the HA stem do not exhibit cross-linking across separate particles, but the findings here support a model of neutralization where bivalent binding to a sufficient amount of antigen is needed to disrupt the cooperative network of HA required for fusion. The epitope that is targeted influences the type of bivalent interactions that can take place between HA and the virus and should be considered when developing a general model of Ab-mediated neutralization.
MATERIALS AND METHODS
IgG purification and Fab preparation.
nAb HC19 was transiently expressed by using the FreeStyle 293F expression system (Life Technologies), while FI6v3 was constitutively expressed from a stable HEK293F cell line. Plasmids encoding HC19 IgG and the stable cell line expressing FI6v3 IgG were kindly provided by Jesse Bloom at Fred Hutchinson Cancer Research Center (Seattle, WA). After production in HEK293F cells, secreted IgGs were isolated by centrifugation and further purified from the cell culture supernatant by gravity flow over protein A-agarose resin. IgGs were eluted from protein A by using 100 mM glycine buffer (pH 2.0). Fractions were analyzed by SDS-PAGE, pooled, and concentrated by using Vivaspin 2 centrifugal concentrators. Purified IgG was further digested into Fab domains by papain digestion (Pierce Fab preparation kit) and purified from Fc fragments and undigested IgG by gravity flow over protein A resin. Fractions were pooled and analyzed by SDS-PAGE to assess purity.
Influenza virus purification.
X31 (H3N2) influenza A virus grown in allantoic fluid of embryonated chicken eggs and purified by density gradient centrifugation was purchased from Charles River Laboratories. Virus stocks (2 mg/ml) were centrifuged at 2,320 relative centrifugal force (rcf) at 4°C for 5 min to remove residual egg protein contaminants. Virus in the supernatant was pelleted by centrifugation at 25,000 rcf for 60 min at 4°C and resuspended in HEPES-buffered saline (HBS) (pH 7.5) (150 mM NaCl, 50 mM citrate, 10 mM HEPES, 0.02% NaN3). For influenza virus used in fluorescence assays, the supernatant containing the virus was mixed with lipophilic tracer dye Vybrant DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indodicarbocyanine,4-chlorobenzenesulfonate salt) cell-labeling solution (Invitrogen), at a final dye concentration of 2.5% (vol/vol) for 60 min at 37°C prior to the final centrifugation.
HA quantitation.
The concentration of trimeric HA in influenza virus stocks was determined by Western blot analysis using BHA as a protein standard. X31 virus and known quantities of BHA (quantified by the A280) were run on NuPAGE 4% to 12% Bis-Tris gels and subsequently transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore). After transfer, membranes were blocked with 5% nonfat dry milk buffered in Tris-buffered saline (TBS) (pH 7.2)–150 mM NaCl–0.1% Tween 20 (TBST). Anti-HA-tagged antibody (Millipore) was diluted in blocking buffer at a 1:2,000 dilution and incubated with membranes for 1 h at room temperature. After incubation with primary antibody, membranes were washed with TBST. After washing, horseradish peroxidase (HRP)-conjugated protein G was diluted in blocking buffer at a 1:5,000 dilution and incubated with membranes for 1 h. Membranes were washed with TBS and developed by using enhanced chemiluminescence (ECL) detection methods. The band density for HA was measured and the concentration was calculated by using ImageJ software for gel analysis (53).
Antibody-mediated neutralization of virus measured by a TCID50 assay.
X31 (H3N2) influenza A virus mixed with either IgG at a 1:1 molar ratio or Fab at a 2:1 molar ratio to the HA trimer presented on the surface of the virus was incubated for 10 min at 37°C. The use of a 2:1 (Fab/HA) ratio offers a better comparison in our assay by accounting for the bivalent presentation of Fab on IgG and the number of potential interactions with the antigen. Samples of influenza virus without antibody were treated identically as a control. Neutralization was measured by a TCID50 assay in a 96-well-plate format. In the initial well of a column, 10 μl of the sample was added to 90 μl of influenza growth medium (IGM) (20 ml bovine serum albumin [BSA], 5 ml penicillin-streptomycin, 500 μl of 100 mg/ml CaCl2, 50 μl fetal bovine serum [FBS], and up to 500 ml of Opti-MEM). The total amount of HA from the virus in each initial well was 14.4 μg. The sample was then serially diluted 1:10 down the column of the plate. Fifty microliters of 105 MDCK-SIAT1-TMPRSS2 cells/ml (referred to as “MDCK cells”) was added to each well. Four columns were used per sample in each replicate. Plates were incubated at 37°C and scored for cytopathic effects at 72 h postinfection. Viral titers were calculated by Reed-Muench analysis implemented by the Python script at https://github.com/jbloom/reedmuenchcalculator. A total of 4 to 5 replicates for each sample was used for the statistical calculation of P values between groups by analysis of variance (ANOVA).
Dynamic light scattering.
Dynamic light scattering measurements were performed by using a Dynapro Nanostar instrument (Wyatt Technologies, Santa Barbara, CA). IgG or Fab was titrated into purified X31 influenza virus at the desired molar ratios of Ab to HA prior to addition to a low-volume quartz cuvette. For DLS measurements, the HA concentration was kept constant at 100 μg/ml. Concentrations of polyclonal Ab in serum in response to infection are estimated to be on the order of 1 mg/ml; however, concentrations of monoclonal Ab are not easily quantified. Therefore, concentrations of IgG ranging from 0.1 to 100 μg/ml, consistent with quantities used in previous neutralization assays, were examined (37, 54). Measurements were taken over the course of an hour at 25°C, and the mean estimated radius at each time point was calculated from 10 individual measurements.
Liposome preparation.
Liposomes composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipid and 25% cholesterol (molar ratio to DOPC) and decorated with 5% ganglioside (molar ratio), aNeu5Ac(2-3)bDGalp(1-3)bDGalNAc(1-4)[aNeu5Ac(2-3)]bDGalp(1-4)bDGlcp(1-1)Cer (gD1a), serving as a sialic acid receptor for the HA glycoprotein, were prepared by lipid extrusion according to a previously described protocol (5). DOPC, cholesterol, and gD1a were combined from chloroform stock solutions and dried under nitrogen gas. The lipid films were resuspended in buffer containing 25 mM SRB dye, 150 mM NaCl, 10 mM HEPES, 50 mM sodium citrate (pH 7.5), and 0.02% NaN3. After 10 freeze-thaw cycles in liquid nitrogen, liposomes were extruded 29 times through a 100-nm polycarbonate membrane (Avanti Polar Lipids). Extruded liposomes were passed over a PD-10 desalting column (GE Healthcare) and stored in pH 7.5 HEPES-citrate buffer. DLS was used to measure the average diameter and polydispersity of the liposomes at room temperature. The liposome diameter ranged from 165 nm to 182 nm, with low polydispersity.
Biolayer interferometry.
Binding kinetics of HC19 and FI6v3 IgGs and Fabs with trimeric BHA were determined by using BLI on an Octet Red system (FortéBio). Hydrated anti-human Fab-CH1 biosensors were immobilized for 4 min with purified IgG or Fab diluted to 10 μg/ml in phosphate-buffered saline (PBS) (pH 7.4) supplemented with 1% BSA, 0.03% Tween 20, and 0.02% NaN3. After reaching a stable baseline, antibody-immobilized tips were moved to wells containing a 2-fold dilution series of trimeric BHA to monitor association for 4 min. Tips were moved back to wells containing buffer to monitor dissociation for 8 min. Kinetic data were analyzed by using FortéBio Data Analysis 7.0 software. Average measurements from reference wells were subtracted, and data were processed by Savitzky-Golay filtering prior to fitting using a 1:1 binding model. Reported values are averages of data from experiments repeated in triplicate.
Fluorescence spectroscopy.
For fluorescence-monitored membrane fusion assays, DiD-labeled virus was incubated with IgG or Fab at 22°C for 30 min to allow complexes to form. The virus-antibody mixture was then added to an excess volume of SRB-labeled liposomes and transferred to a 40-μl fluorescence microcuvette. The reaction mixture was incubated at neutral pH at 37°C for 10 min to allow liposomes and the virus to reach a stable baseline. We have observed from our recently reported studies that pH 5.25 irreversibly triggers HA and provides optimal dequenching profiles in fluorescence fusion assays; therefore, fusion of the virus and liposomes was initiated by decreasing the pH to 5.25 by the addition of buffer containing 150 mM NaCl, 10 mM HEPES, 50 mM sodium citrate (pH 3.0), and 0.02% NaN3 (5, 6, 14). Measurements were collected on a Varian Cary Eclipse spectrophotometer using excitation/emission wavelength pairings of 644/655 nm for DiD and 565/586 nm for SRB, with 2.5-nm slit widths, at 37°C.
Once fusion of the liposome and viral membrane occurs, leakage of SRB from the hydrophilic compartment of the liposome relieves fluorescence quenching. Lipid mixing occurs as the virus and liposome fuse, and DiD dye diffuses through a larger membrane, where fluorescence dequenching is monitored. Fluorescence dequenching of SRB and DiD was normalized by using the formula [F(t) − F(0)]/[F(max) − F(0)], where F(0) is the fluorescence reading prior to acidification and F(max) is the fluorescence reading after complete dequenching by the addition of Triton X-100 detergent to a final concentration of 1%.
Negative-stain electron microscopy of influenza virus.
The HA concentration in spin-purified X31 influenza virus was determined by Western blot analysis. X31 influenza virus at 100 μg/ml (HA) was adsorbed onto glow-discharged C-Flat, 300-mesh, Cu grids (Electron Microscopy Sciences) and stained by using methylamine tungstate (Nano-W; Nanoprobes). Grids were imaged on a Tecnai T12 electron microscope operating at 120 keV and equipped with a 4k-by-4k charge-coupled-device (CCD) camera at a magnification of ×42,000, corresponding to a pixel size of 2.60 Å per pixel.
Negative-stain electron microscopy of the Fab:BHA complex.
The ability of Fab to stabilize prefusion BHA under fusion-activating conditions was assessed by performing reference-free 2D classification of the Fab:BHA complex under neutral and acidic pH conditions using the EMAN 2.1 image processing suite (55). In short, a 3-μl aliquot of the Fab:BHA complex at pH 7.4, or with the pH adjusted to 5.25 for 10 min, was applied to glow-discharged C-Flat, 300-mesh, Cu grids (Electron Microscopy Sciences) and stained by using Nano-W. Data were collected by using a Tecnai T12 transmission electron microscope operating at 120 keV. Images were taken by using a Gatan 4k-by-4k CCD camera at a magnification of either ×67,000 or ×52,000 and a defocus range of −3 to −1 μm, corresponding to a pixel size of 1.60 Å or 2.07 Å per pixel, respectively. Particles were selected by using interactive particle picking from ∼100 micrographs. Particle stacks of ∼8,000 particles were created and subjected to reference-free 2D classification and clustering to generate 50 2D classes. Representative class averages for each sample were chosen for clarity.
Cryo-EM and tomography processing.
Purified virus, 10-nm colloidal gold beads (Electron Microscopy Sciences), and purified IgG were combined at the desired ratios of Ab to HA. The combined solution was incubated at room temperature for 5 min prior to addition to glow-discharged holey carbon-coated grids (C-flat, 200 mesh; Electron Microscopy Sciences) and plunge frozen in liquid ethane by using a Vitrobot Mark IV specimen preparation unit (FEI Co.).
Vitrified grids were imaged at 200 keV in an FEI Tecnai F20 transmission electron microscope equipped with a 4k-by-4k CCD camera at a nominal magnification of ×50,000 (and binned by a factor of 2, yielding a pixel size of 4.4 Å per pixel) or a Gatan K2 Summit direct detector in counting mode at a calibrated magnification of ×11,500, corresponding to a pixel size of 3.2 Å per pixel. Images were acquired at a 3-μm defocus, and specimens were tilted in 2° increments from 58° to −58° by using the Leginon software package (56). The total electron dose for the entire tilt series was estimated to be ∼80 electrons/Å2. The K2 image frames were aligned prior to tomographic reconstruction (57). Tomograms were reconstructed by using the weighted back-projection method in the IMOD software package (58) and denoised as previously described (5).
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 GM099989.
We thank Jesse Bloom (Fred Hutchinson Cancer Research Center) for providing the plasmid encoding HC19 IgG and the stable cell line expressing FI6v3 IgG that were used in this study. We also thank members of the Lee laboratory and Thaddeus Davenport for helpful discussions regarding this study.
REFERENCES
- 1.Inglis SC, Carroll AR, Lamb RA, Mahy BW. 1976. Polypeptides specified by the influenza virus genome. I. Evidence for eight distinct gene products specified by fowl plague virus. Virology 74:489–503. doi: 10.1016/0042-6822(76)90355-X. [DOI] [PubMed] [Google Scholar]
- 2.Ruigrok RW, Andree PJ, Hooft van Huysduynen RA, Mellema JE. 1984. Characterization of three highly purified influenza virus strains by electron microscopy. J Gen Virol 65(Part 4):799–802. doi: 10.1099/0022-1317-65-4-799. [DOI] [PubMed] [Google Scholar]
- 3.Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. 2006. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A 103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 5.Lee KK. 2010. Architecture of a nascent viral fusion pore. EMBO J 29:1299–1311. doi: 10.1038/emboj.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gui L, Ebner JL, Mileant A, Williams JA, Lee KK. 2016. Visualization and sequencing of membrane remodeling leading to influenza virus fusion. J Virol 90:6948–6962. doi: 10.1128/JVI.00240-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chlanda P, Zimmerberg J. 2016. Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett 590:1940–1954. doi: 10.1002/1873-3468.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee PS, Wilson IA. 2015. Structural characterization of viral epitopes recognized by broadly cross-reactive antibodies. Curr Top Microbiol Immunol 386:323–341. doi: 10.1007/82_2014_413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. 2002. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 10.Bizebard T, Daniels R, Kahn R, Golinelli-Pimpaneau B, Skehel JJ, Knossow M. 1994. Refined three-dimensional structure of the Fab fragment of a murine IgG1, lambda antibody. Acta Crystallogr D Biol Crystallogr 50:768–777. doi: 10.1107/S0907444994001903. [DOI] [PubMed] [Google Scholar]
- 11.Fleury D, Barrere B, Bizebard T, Daniels RS, Skehel JJ, Knossow M. 1999. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat Struct Biol 6:530–534. doi: 10.1038/9299. [DOI] [PubMed] [Google Scholar]
- 12.Knossow M, Gaudier M, Douglas A, Barrere B, Bizebard T, Barbey C, Gigant B, Skehel JJ. 2002. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 302:294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- 13.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 14.Garcia NK, Guttman M, Ebner JL, Lee KK. 2015. Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure 23:665–676. doi: 10.1016/j.str.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calder LJ, Rosenthal PB. 2016. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat Struct Mol Biol 23:853–858. doi: 10.1038/nsmb.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran EE, Podolsky KA, Bartesaghi A, Kuybeda O, Grandinetti G, Wohlbold TJ, Tan GS, Nachbagauer R, Palese P, Krammer F, Subramaniam S. 2016. Cryo-electron microscopy structures of chimeric hemagglutinin displayed on a universal influenza vaccine candidate. mBio 7:e00257-. doi: 10.1128/mBio.00257-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris AK, Meyerson JR, Matsuoka Y, Kuybeda O, Moran A, Bliss D, Das SR, Yewdell JW, Sapiro G, Subbarao K, Subramaniam S. 2013. Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc Natl Acad Sci U S A 110:4592–4597. doi: 10.1073/pnas.1214913110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klasse P. 2014. Neutralization of virus infectivity by antibodies: old problems in new perspectives. Adv Biol 2014:157895. doi: 10.1155/2014/157895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otterstrom JJ, Brandenburg B, Koldijk MH, Juraszek J, Tang C, Mashaghi S, Kwaks T, Goudsmit J, Vogels R, Friesen RHE, Van Oijen AM. 2014. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proc Natl Acad Sci U S A 111:E5143–E5148. doi: 10.1073/pnas.1411755111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor H, Armstrong S, Dimmock N. 1987. Quantitative relationships between an influenza virus and neutralizing antibody. Virology 159:288–298. doi: 10.1016/0042-6822(87)90466-1. [DOI] [PubMed] [Google Scholar]
- 21.Laver WG, Valentine RC. 1969. Morphology of the isolated hemagglutinin and neuraminidase subunits of influenza virus. Virology 38:105–119. doi: 10.1016/0042-6822(69)90132-9. [DOI] [PubMed] [Google Scholar]
- 22.McCraw DM, Gallagher JR, Harris AK. 2016. Characterization of influenza vaccine hemagglutinin complexes by cryo-electron microscopy and image analyses reveals structural polymorphisms. Clin Vaccine Immunol 23:483–495. doi: 10.1128/CVI.00085-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc Natl Acad Sci U S A 79:968–972. doi: 10.1073/pnas.79.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. 2012. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J Virol 86:2919–2929. doi: 10.1128/JVI.06698-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Outlaw MC, Dimmock NJ. 1991. Insights into neutralization of animal viruses gained from study of influenza virus. Epidemiol Infect 106:205–220. doi: 10.1017/S0950268800048354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortiz JO, Brandt F, Matias VR, Sennels L, Rappsilber J, Scheres SH, Eibauer M, Hartl FU, Baumeister W. 2010. Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ. J Cell Biol 190:613–621. doi: 10.1083/jcb.201005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liljeroos L, Huiskonen JT, Ora A, Susi P, Butcher SJ. 2011. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc Natl Acad Sci U S A 108:18085–18090. doi: 10.1073/pnas.1105770108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang JT, Schmid MF, Rixon FJ, Chiu W. 2007. Electron cryotomography reveals the portal in the herpesvirus capsid. J Virol 81:2065–2068. doi: 10.1128/JVI.02053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 30.White JM, Wilson IA. 1987. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J Cell Biol 105:2887–2896. doi: 10.1083/jcb.105.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. 2013. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. Elife 2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danieli T, Pelletier SL, Henis YI, White JM. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol 133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee PS, Yoshida R, Ekiert DC, Sakai N, Suzuki Y, Takada A, Wilson IA. 2012. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc Natl Acad Sci U S A 109:17040–17045. doi: 10.1073/pnas.1212371109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong M, Lee PS, Hoffman RM, Zhu X, Krause JC, Laursen NS, Yoon SI, Song L, Tussey L, Crowe JE Jr, Ward AB, Wilson IA. 2013. Antibody recognition of the pandemic H1N1 influenza virus hemagglutinin receptor binding site. J Virol 87:12471–12480. doi: 10.1128/JVI.01388-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brandenburg B, Koudstaal W, Goudsmit J, Klaren V, Tang C, Bujny MV, Korse HJ, Kwaks T, Otterstrom JJ, Juraszek J, van Oijen AM, Vogels R, Friesen RH. 2013. Mechanisms of hemagglutinin targeted influenza virus neutralization. PLoS One 8:e80034. doi: 10.1371/journal.pone.0080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. 2012. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards MJ, Dimmock NJ. 2000. Two influenza A virus-specific Fabs neutralize by inhibiting virus attachment to target cells, while neutralization by their IgGs is complex and occurs simultaneously through fusion inhibition and attachment inhibition. Virology 278:423–435. doi: 10.1006/viro.2000.0631. [DOI] [PubMed] [Google Scholar]
- 38.Al-Majhdi FN. 2007. Structure of the sialic acid binding site in influenza A virus: hemagglutinin. J Biol Sci 7:113–122. doi: 10.3923/jbs.2007.113.122. [DOI] [Google Scholar]
- 39.Lafferty KJ. 1963. The interaction between virus and antibody. III. Examination of virus-antibody complexes with the electron microscope. Virology 21:91–99. doi: 10.1016/0042-6822(63)90307-6. [DOI] [Google Scholar]
- 40.Dimmock NJ, Hardy SA. 2004. Valency of antibody binding to virions and its determination by surface plasmon resonance. Rev Med Virol 14:123–135. doi: 10.1002/rmv.419. [DOI] [PubMed] [Google Scholar]
- 41.Almeida J, Cinader B, Howatson A. 1963. The structure of antigen-antibody complexes. A study by electron microscopy. J Exp Med 118:327–340. doi: 10.1084/jem.118.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossman JS, Leser GP, Lamb RA. 2012. Filamentous influenza virus enters cells via macropinocytosis. J Virol 86:10950–10960. doi: 10.1128/JVI.05992-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallis C, Melnick JL. 1967. Virus aggregation as the cause of the non-neutralizable persistent fraction. J Virol 1:478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madel B. 1978. Neutralization of animal viruses. Adv Virus Res 23:205–268. doi: 10.1016/S0065-3527(08)60101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullarkey CE, Bailey MJ, Golubeva DA, Tan GS, Nachbagauer R, He W, Novakowski KE, Bowdish DM, Miller MS, Palese P. 2016. Broadly neutralizing hemagglutinin stalk-specific antibodies induce potent phagocytosis of immune complexes by neutrophils in an Fc-dependent manner. mBio 7:e01624-16. doi: 10.1128/mBio.01624-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DiLillo DJ, Tan GS, Palese P, Ravetch JV. 2014. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med 20:143–151. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas AA, Vrijsen R, Boeyé A. 1986. Relationship between poliovirus neutralization and aggregation. J Virol 59:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith TJ, Mosser AG, Baker TA. 1995. Structural studies of the mechanisms of antibody-mediated neutralization of human rhinovirus. Virology 6:233–242. [Google Scholar]
- 50.Stieh DJ, King DF, Klein K, Liu P, Shen X, Hwang KK, Ferrari G, Montefiori DC, Haynes B, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Robb ML, Kim JH, Denny TN, Tomaras GD, Shattock RJ. 2014. Aggregate complexes of HIV-1 induced by multimeric antibodies. Retrovirology 11:78. doi: 10.1186/s12977-014-0078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wasilewski S, Calder LJ, Grant T, Rosenthal P. 2012. Distribution of surface glycoproteins on influenza A virus determined by electron cryotomography. Vaccine 30:7368–7373. doi: 10.1016/j.vaccine.2012.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edwards MJ, Dimmock NJ. 2001. A haemagglutinin (HA1)-specific FAb neutralizes influenza A virus by inhibiting fusion activity. J Gen Virol 82:1387–1395. doi: 10.1099/0022-1317-82-6-1387. [DOI] [PubMed] [Google Scholar]
- 55.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, Jensen GJ. 2009. Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol 167:11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kremer JR, Mastronarde DN, McIntosh JR. 1996. Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]