

ABSTRACT
The increased prevalence of drug-resistant, nosocomial Acinetobacter infections, particularly from pathogenic members of the Acinetobacter calcoaceticus-baumannii complex, necessitates the exploration of novel treatments such as phage therapy. In the present study, we characterized phage Petty, a novel podophage that infects multidrug-resistant Acinetobacter nosocomialis and Acinetobacter baumannii. Genome analysis reveals that phage Petty is a 40,431-bp ϕKMV-like phage, with a coding density of 92.2% and a G+C content of 42.3%. Interestingly, the lysis cassette encodes a class I holin and a single-subunit endolysin, but it lacks canonical spanins to disrupt the outer membrane. Analysis of other ϕKMV-like genomes revealed that spaninless lysis cassettes are a feature of phages infecting Acinetobacter within this subfamily of bacteriophages. The observed halo surrounding Petty's large clear plaques indicated the presence of a phage-encoded depolymerase capable of degrading capsular exopolysaccharides (EPS). The product of gene 39, a putative tail fiber, was hypothesized to possess depolymerase activity based on weak homology to previously reported phage tail fibers. The 101.4-kDa protein gene product 39 (gp39) was cloned and expressed, and its activity against Acinetobacter EPS in solution was determined. The enzyme degraded purified EPS from its host strain A. nosocomialis AU0783, reducing its viscosity, and generated reducing ends in solution, indicative of hydrolase activity. Given that the accessibility to cells within a biofilm is enhanced by degradation of EPS, phages with depolymerases may have enhanced diagnostic and therapeutic potential against drug-resistant Acinetobacter strains.
IMPORTANCE Bacteriophage therapy is being revisited as a treatment for difficult-to-treat infections. This is especially true for Acinetobacter infections, which are notorious for being resistant to antimicrobials. Thus, sufficient data need to be generated with regard to phages with therapeutic potential, if they are to be successfully employed clinically. In this report, we describe the isolation and characterization of phage Petty, a novel lytic podophage, and its depolymerase. To our knowledge, it is the first phage reported to be able to infect both A. baumannii and A. nosocomialis. The lytic phage has potential as an alternative therapeutic agent, and the depolymerase could be used for modulating EPS both during infections and in biofilms on medical equipment, as well as for capsular typing. We also highlight the lack of predicted canonical spanins in the phage genome and confirm that, unlike the rounding of lambda lysogens lacking functional spanin genes, A. nosocomialis cells infected with phage Petty lyse by bursting. This suggests that phages like Petty employ a different mechanism to disrupt the outer membrane of Acinetobacter hosts during lysis.
KEYWORDS: phage therapy, Acinetobacter nosocomialis, Acinetobacter baumannii, depolymerase, genome analysis, lysis, spanins
INTRODUCTION
Pathogenic members of the Acinetobacter genus, particularly from the Acinetobacter calcoaeticus-baumannii complex, are considered a significant threat to human health (1, 2). The prevalences of infections caused by these Gram-negative bacteria, specifically A. baumannii, A. pittii, and A. nosocomialis (3), have increased both in hospital settings (4–6) and in the community (7). The high number of multidrug-resistant (MDR) strains reported, and the capability of these opportunistic pathogens to colonize and persist on catheters and other medical equipment (8, 9), hinders effective clinical intervention strategies and outcomes. Although the mechanism by which Acinetobacter establishes infection is not fully characterized, it is known that the presence of a capsule in clinical strains, resistance to desiccation, and ability to form biofilm promote pathogen colonization and survival (10–12).
Clinical strains of the A. calcoaeticus-baumannii complex display capsular exopolysaccharides (EPS), which have been shown to contribute to biofilm formation (13), enhance bacterial colonization, and promote survival in mammalian tissues (14). By covering components on the otherwise-exposed bacterial outer membrane, EPS thwarts recognition of host immune mechanisms and diminishes the action of antimicrobial compounds. Together, these factors have been correlated with the development of systemic infections (15). EPS also plays a role as a phage resistance mechanism: by masking the receptors on the membrane, bacteria limit the access to potential phage binding sites and resist infection (16). To circumvent this barrier, some bacteriophages express a depolymerase that degrades capsular polysaccharides, which enables viral particles to access the bacterial surface (17). It has been asserted (18, 19) that phage depolymerases are either integral components of the phage particles or soluble proteins generated during host cell lysis with either hydrolase or lyase activity.
In clinical settings, phage depolymerases have been used to determine capsule types of different Klebsiella strains with epidemiological and diagnostic purposes (20, 21). A serotyping system for Acinetobacter based on monoclonal antibodies was developed (22, 23) but was not widely used. Since capsular variations and strain adaptation require both a phenotypic and genotypic characterization of clinical isolates (24), depolymerases are being evaluated as tools for capsular typing of A. calcoaeticus-baumannii complex clinical isolates (14, 25, 26).
There is evidence that exposure of A. baumannii to subinhibitory concentrations of antibiotic results in an increased capsule locus gene expression and a concomitant increase in bacterial virulence (27). Associated with the variable susceptibility pattern of different Acinetobacter strains (28, 29), this may complicate treatment and clinical outcome. It has been shown that intraperitoneal administration of a phage-derived endosialidase in Escherichia coli-infected mice successfully prevented bacteremia and death from systemic infection (30, 31). Recently, phages with depolymerase activity isolated against A. baumannii and Pseudomonas were shown to degrade biofilms of MDR strains (32–34) and reduce colonization. Thus, modulation of EPS has therapeutic potential (33, 35), which is why phage depolymerases could also be used as part of the novel strategies to treat MDR isolates of Acinetobacter.
We have isolated a novel bacteriophage against a multidrug-resistant clinical strain of A. nosocomialis, AU0783. The phage produces plaques surrounded by a turbid halo, suggesting the presence of a virion-associated protein with depolymerase activity. In phages such as E. coli K1, Pseudomonas AF, or Erwinia L1, the depolymerase activity is encoded in the tail fiber (36–38). We cloned and characterized Petty's tail fiber gene and show that the tail fiber protein is able to hydrolyze EPS. The use of bacteriophages (phages) to treat and/or control multidrug-resistant infections is being reconsidered an alternative strategy for therapeutic and prophylactic applications, but its implementation requires a thorough characterization of candidate phages (39, 40). In this report, we present phage Petty's host range and genome features. To our knowledge, this is the first report of a phage able to infect multiple species of the A. calcoaeticus-baumannii complex.
RESULTS
Phage isolation and characterization.
Bacteriophage Petty was isolated from sewage collected in the summer of 2009 in College Station, TX. When plated on A. nosocomialis AU0783, the phage produces plaques of ∼2 mm in diameter surrounded by a slowly expanding turbid halo (Fig. 1A). Transmission electron microscopy (TEM) of negatively stained virions showed a 60-nm head and a short, stubby 15-nm tail, characteristic of Podoviridae morphology (Fig. 1B). The phage infection was characterized by a 25-min latent period and a burst size of 240 particles per infective center (Fig. 2A). As shown in Fig. 2B, 90% of the phage particles were adsorbed within 10 min of addition; the adsorption constant was calculated to be 1.8 ×10−9 ml/min. Phage-resistant cells were present at a frequency of ∼10−7 CFU in typical cultures.
FIG 1.
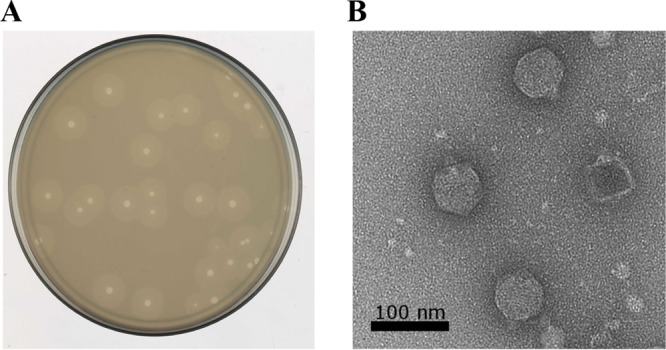
Phage Petty plaque and virion morphology. Phage Petty was isolated from sewage and propagated on A. nosocomialis AU0783. (A) The phage produces plaques with an expanding translucent halo, suggesting exopolysaccharide depolymerase activity. (B) Transmission electron microscopy shows podophage morphology with a capsid diameter of ∼60 nm.
FIG 2.
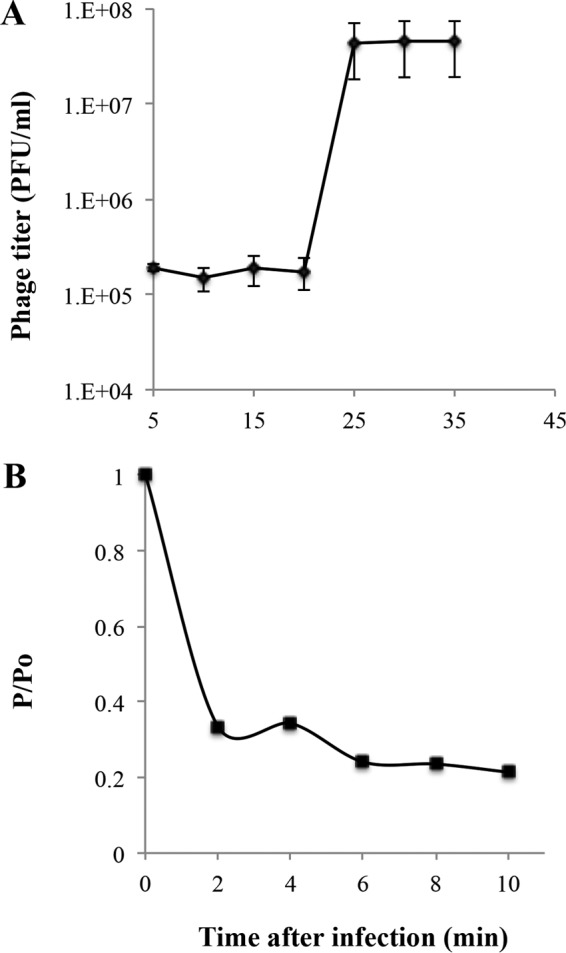
Characterization of Petty adsorption and lysis of its host, A. nosocomialis AU0783. (A) One-step growth curve of phage Petty on to A. nosocomialis AU0783. The cells lyse 22 min after infection, with a burst size of 240 particles per infective center. (B) Adsorption of phage Petty to A. nosocomialis AU0783. Error bars represent SDs (n = 3).
Using a spot-dilution assay, the host range of Petty was tested against a panel of 40 Acinetobacter strains, including 38 A. calcoaeticus-baumannii strains, Acinetobacter genospecies 16, and A. baylyi ADP1; 25 of these strains are clinical multidrug-resistant strains (see Table S1 in the supplemental material). Phage Petty plated and produced plaques on 4 drug-resistant A. baumannii strains, isolated from wound and eye infections, with a reduced efficiency of plating (EOP; >10−2). The ocular strains, characterized previously (28), are resistant to chloramphenicol, cefotaxime, tetracycline, and cefazolin and are able to form biofilms; this ability enhances adherence and colonization of the corneal tissue.
In addition, Petty formed clearing zones on one clinical A. baumannii strain (41) when the undiluted phage stock (∼109 PFU/ml) was spotted on the lawn, presumably the result of lysis from without.
Genome analysis.
The 40,431-bp podophage Petty genome was assembled into a single contig at 88.4-fold coverage. It has a coding density of 92.2% and a G+C content of 42.3%. It encodes 45 predicted proteins encoded on the positive strand (Table S2 in the supplemental material), 20 of which have homologs in non-Acinetobacter phage genomes. Of the 23 proteins with unknown function encoded in the Petty genome, 11 are unique to phage Petty. One protein with hypothetical function has homologs only in bacterial genomes. The morphology, genome arrangement, genomic DNA (gDNA) size, and presence of a T7-like single-subunit RNA polymerase gene (gene 26) establish Petty in the T7-like phage subfamily; moreover, the position of the polymerase gene, adjacent to virion structural genes (Fig. 3A), groups it in the ϕKMV-like genus of that extensive subfamily (https://talk.ictvonline.org/taxonomy/). The length of the direct terminal repeats was determined to be 308 bp by primer walking, shorter than for other ϕKMV-like phages (410 bp for Acinetobacter phage ϕAB1, 341 bp for Acinetobacter phage Abp1, and 414 bp for Pseudomonas phage ϕKMV) but 150 bp longer than the terminal repeats of phage T7 (42–44). In a pairwise alignment of the phage Petty genome (Fig. 3B) with those of bacteriophages ϕAB1 and Acibel007 (which share 56% identity between them), Petty was found to share 47% and 53% identity with each of these phages, respectively (42, 45).
FIG 3.

Genome architecture of phage Petty and other ϕKMV-like Acinetobacter phages. (A) Genome architecture and predicted ORFs of phage Petty are shown. The linear map is based on nucleotide sequences of the phage genome and predicted ORFs. ORFs are color-coded based on their functional role category: yellow for phage DNA replication and recombination, orange for regulation, purple for endonucleases, blue for morphogenesis-related proteins, pink for lysis, and green for DNA packaging. Unattributed genes are colored black. Selected gene numbers are shown. Like for other ϕKMV-like phages, the Petty RNA polymerase gene is adjacent to the morphogenesis genes. Interestingly, the lysis cassette of the phage genomes does not have a predicted canonical spanin. (B) Genome maps and protein identity comparison between selected ϕKMV-like phages infecting Acinetobacter. Phages are aligned relative to gp1, which is highly conserved in this genus. Gene products (gp) sharing significant sequence identity are linked in gray (see Materials and Methods).
The genome organization of phage Petty is similar to those of other ϕKMV-like phages (Fig. 3A). Early genes code for proteins of unknown function, with homology only to other Acinetobacter phages with the exception of gene product 1 (gp1), which is homologous to hypothetical proteins of other ϕKMV-like phages infecting different hosts, suggesting that it has a strongly conserved function within phages of this group.
The genes encoding proteins required for replication/recombination are grouped, including a DnaG-like primase (gp13), helicase (gp15), ligase (gp16), and DNA polymerase (gp17). Unlike Acinetobacter AB1, ϕAB1, and Abp1, which encode multiple putative HNH endonucleases (42, 43), phage Petty encodes a single putative DDE endonuclease (gp18) (in purple in Fig. 3A). Despite differences in their domains, both HNH and DDE endonucleases cleave DNA and facilitate a strand transfer reaction. Homing endonucleases such as the ones found in Acinetobacter phages Abp1, ϕAB1, and Fri1 are either “free-standing,” usually found at intergenic regions—where the impact on host gene structure is reduced (46, 47)—or encoded within introns and inteins (48). Unlike the DDE endonuclease of phage Petty, the endonucleases of Acinetobacter phages Abp1, ϕAB1, and Fri1 are either interrupting the DNA polymerase gene or upstream of the endonuclease (Fig. 3B). The DDE endonuclease of Petty lacks the HNH motif, despite having sequence similarity with other HNH endonucleases in the database. Homing endonucleases, both HNH and DDE, confer mobility by recombination, but unlike HNH endonucleases, DDE endonucleases require metal ions for catalysis, which are coordinated by carboxylate residues.
Like Acinetobacter phages ϕAB1, ϕAB6, Adp1, and Acibel007 (Fig. 3B), the genes upstream of the RNA polymerase (gp26) encode a deoxyribonucleotide monophosphate kinase (gp25), a phosphoesterase (gp24), and a DNA exonuclease (gp22). Type VII endonucleases have been shown to resolve mismatch repair and Holliday junctions by recognizing a variety of DNA substrates, from single-pair mismatches to branched DNAs. In T4, endonuclease VII (gp49) is expressed at both the early and late stages of infection, and it is involved in recombination-mediated replication and in resolving branch points prior to DNA packaging (49). Although closely related to gp49 of Acinetobacter phage Acibel007 (65% identity), the recombination endonuclease VII (gp23) of phage Petty shares significant sequence similarity to otherwise distant ϕKMV-like phages infecting Pseudomonas (68% and 59% identity with gp33 of LKA1 and gp21 of ϕ2, respectively), Pectobacterium (53% identity with gp28 of PP16), and Proteus (62% identity with gp24 of PM16).
As noted above, the single-subunit RNA polymerase (gp26) of Petty is adjacent to the phage replication and structural genes. Petty gp26 is closely related to gp32 in Acibel007 (with a 66% identity) and other Acinetobacter ϕKMV-like phages (63% identity), such as Fri1, Abp1, IME200, and ϕAB1. This is reflected in the phylogenetic relations shown in Fig. 4, where despite being grouped with other ϕKMV phages that infect Acinetobacter, both Petty and Acibel007 are relatively distant from the other members of the clade and closely related to each other.
FIG 4.

Petty is genetically distant from other ϕKMV-like Acinetobacter phages. Phylogenetic tree of the RNA polymerase of ϕKMV-like phages was constructed by aligning the protein sequences with MUSCLE (88) and using the pipeline available in Phylogeny.fr (89) to run the maximum likelihood analysis and plot the tree using TreeDyn. Branch support values are displayed in purple, and accession numbers of each RNA polymerase are given. Phage T7 RNA polymerase was included as a reference.
The nine identified structural genes identified by homology are also clustered and located downstream of the gene encoding the RNA polymerase. SDS-PAGE of purified phage particles coupled to mass spectrometry identified 11 structural proteins (Fig. 5), among which 3 have an unknown function (gp10, gp27, and gp29) and 1 is unique to phage Petty (gp27). Proteins homologous to Petty gp29, namely, gp30 of ϕAB1 and gp36 of phage Abp1, have been detected by mass spectrometry previously (42, 43), whereas Petty gp10, which is homologous to gp13 of Acibel007, was detected only in this study and not in the mass spectrometry analysis of Acibel007 (45).
FIG 5.

Gene products of phage Petty, identified by mass spectrometry. (A) Whole-phage particles were run on SDS-PAGE alongside the purified recombinant Dpo1, and the bands were trypsin digested and analyzed by mass spectrometry system. Molecular masses (MM) of the protein ladder are indicated to the left of the gel. (B) The generated peptide spectra were identified by comparison to all potential peptide spectra deduced from a predetermined database which included the predicted gene products of phage Petty. The total number of peptides identified, as well as the number of unique peptides for each inferred gp, the corresponding function of each gp predicted by BLAST, and the protein sequence coverage is indicated. ID, identifier.
Similar to other proteomic analyses of ϕKMV-like phages, the coverage for most of the sequenced proteins is low (30 to 40%), except for the larger (gp38 and gp37) or more abundant (gp32) proteins. Mass spectrometry confirmed the presence of the predicted head-to-tail connector protein (gp30), the major capsid protein (gp32), the tail tubular proteins (gp34 and gp35), the tail fiber (gp39), and capsid core protein (gp38), which were identified in the genome by homology to other T7-like phages. As seen in Pseudomonas phage ϕKMV, Petty gp32 does not contain any detectable ribosomal slippage sites, which are a feature of the major capsid protein genes of phages T7 and T3 (44).
Among the proteins with no known function present within the structural gene cluster of phage Petty (gp33 and 37), only gp37 was detected by mass spectrometry. The gp31 protein, identified as a scaffolding protein by homology to gp37 of Acinetobacter phage Acibel007 (35% identity), gp31 of Pseudomonas phage LKD16 (28% identity), and the scaffolding protein of Klebsiella phage KPV811 (34% identity), is also encoded in the cluster but not present in the mass spectrometry results, as scaffolding proteins are not present in the mature phage particle in T7-like phages. Proteomic analysis done on Acinetobacter phage Acibel007 identified 3 structural proteins, 2 of which are encoded among Acibel007 early genes (gp03 and gp04), which were not detected in this study. Multiple bands were observed at ∼50 kDa and ∼30 kDa which could not be assigned to identified open reading frames (ORFs). These proteins could be proteolytic products of some other phage structural proteins, which could be confirmed by N-terminal sequencing.
The genes encoding the small and large subunit of the terminase (gp42 and gp43) are involved in DNA recognition and packaging. Like the RNA polymerase, the large subunit of the terminase has higher homology to that of Acibel007 (71% identity) than to other T7-like Acinetobacter phages (∼60% identity).
The lysis cassette lacks spanins.
In the lysis systems of phages infecting Gram-negative hosts, there are two established pathways: the canonical holin-endolysin pathway, involving a holin that forms micrometer-scale holes in the inner membrane (IM) and a cytoplasmic endolysin, and the pinholin-signal anchor release (SAR) endolysin pathway, where the pinholin forms nanometer-scale holes in the membrane and the muralytic enzyme is exported by the host sec apparatus (50). In phage Petty, the lysis cassette encodes a holin (gp40) and an endolysin (gp41). The holin has 3 predicted transmembrane domains (TMDs) with a predicted N-out, C-in topology and is thus a member of the class I holins, which have been shown to form micrometer-scale holes (51–53). The gp41 endolysin has one predicted muralytic enzyme domain and lacks any secretion or membrane localization signal. Taken together, these observations strongly suggest that Petty uses the canonical lysis pathway. The Petty holin has homology to other holins of ϕKMV-like Acinetobacter phages (Fig. 3B), such as Acibel007, Fri1, and ϕAB1, with which it shares ∼47% amino acid identity. Unlike the holin, the endolysin of phage Petty is not closely related to the endolysins of other phages infecting Acinetobacter. Instead, the protein shares between 47 and 49% identity with Pseudomonas KPP10 and PaP1-like myophages. This is consistent with previous observations that lysis cassettes are often mosaic, suggesting that the holin and endolysin do not physically interact and can evolve independently in unrelated bacterial hosts.
In the current model for lysis of Gram-negative hosts, after permeabilization of the IM by the holin and the subsequent degradation of the peptidoglycan by the endolysin, a third step, disruption of the outer membrane (OM), is effected by the phage-encoded spanins (54). There are two types of spanins, the 2-component spanins, comprised of an OM lipoprotein and an integral IM protein, and the unimolecular spanins, in which a single polypeptide has both OM lipoprotein motifs and a C-terminal TMD (50, 55). Inspection of the Petty genome revealed that none of the encoded proteins had an OM lipoprotein “lipobox” motif and thus ruled out the possibility that Petty has either type of established OM disruption system. Bioinformatic analysis confirmed that other ϕKMV-like phages of A. calcoaeticus-baumannii complex strains also lack spanins. However, spanins can be identified in T4-like myophages and N4-like podophages of Acinetobacter, and spanin genes can be found in genomes of other ϕKMV-like phages of other hosts, such as Pseudomonas and Ralstonia. The spanin-defective lysis phenotype has been characterized in coliphage lambda infections where the parental phage effects abrupt, explosive polar lysis, whereas infections with spanin-defective mutants terminate in spherical cell forms, bounded by an intact OM (55). To determine if, with no canonical spanins, Petty infections might exhibit a significant lysis defect, single infected cells were examined using phase-contrast video microscopy (Movie S1). Similar to wild-type (wt) lambda lysis, AU0783 cells infected with Petty lyse explosively without exhibiting a spherical intermediate (Fig. 6), but unlike for lambda, there does not seem to be a preference to lyse from the poles. These results indicate that the outer membrane is actively disrupted during lysis. In an attempt to quantify the explosive nature of AU0783 lysis, we measured the average pixel intensity during a lysis event to determine the speed in which lysis occurred, evidenced by the loss of cell refractivity (Fig. 7). Acinetobacter cells lyse in an average of 5 s (Fig. 7A), a time frame comparable to that of lambda lysis (Fig. 7B), suggesting that Petty and other ϕKMV-like phages of Acinetobacter manage to disrupt the OM via a different pathway than the 2-component or unimolecular spanins used by other ϕKMV-like phages as well as A. calcoaeticus-baumannii complex phages of different subfamilies.
FIG 6.

Lysis of A. nosocomialis cells infected by phage Petty. AU0783 cells were grown to an A550 of ∼0.4 and infected with phage Petty at a multiplicity of infection (MOI) of 2. Samples were taken 2 min before the onset of lysis, to be observed with phase-contrast microscopy in order to document the morphology of phage lysis at 5 frames/s. A time-lapse series of 2 representative lysis events is shown. The zero time point (in seconds) was established at the frame before cell modifications leading to lysis were observed.
FIG 7.
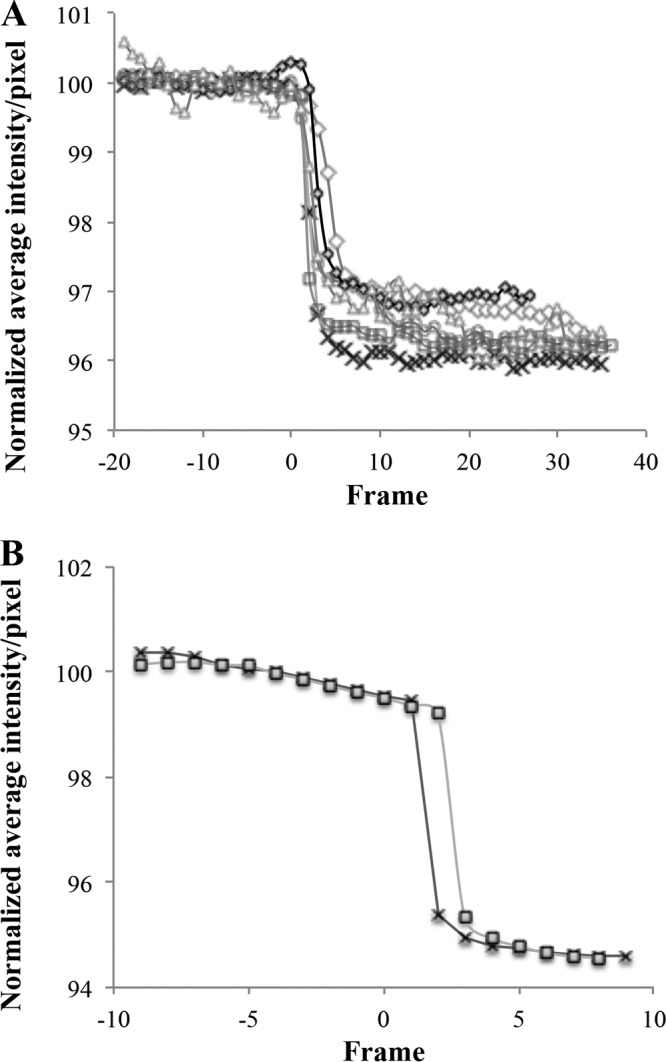
Phage Petty causes explosive lysis of AU0783 cells. After selecting sections of equal size (in pixels), a subset of images of single lysis events (taken at 5 frames/s) were extracted from the video microscopy using Zeiss image analysis software v. 2.3 (Zeiss, NY). The stack of images obtained was used to quantify the average intensity per pixels per frame using ImageJ (90, 91). Relative time was established using time zero as the time in the frame taken before the first lytic lesion was visible. The experiment was performed with both AU0783 cells infected by Petty (A) and E. coli cells infected by lambda (B). Each series represents one independent lysis event. The results were normalized to the average initial intensity of the images before cell lysis (before time zero).
The simplest explanation is that the lysis proteins of Petty somehow bypass the requirement for a spanin to disrupt the OM. The gp40 holin has no exceptional features that distinguish it from other class I holins, with no predicted periplasmic domains capable of interacting across the periplasm. Based on the notion that any protein involved in disrupting the OM would have to have some kind of secretory signal, the Petty genome was examined for proteins predicted to have TMDs or signal sequences. Other than the gp40 holin, three proteins, encoded by genes 4, 27, and 36, were found, with one, two, and one predicted TMD, respectively; no proteins with a secretory signal were found. gp4 is only 24 residues long, and its predicted periplasmic N terminus spans only 4 residues. gp36 is a virion structural protein, probably involved in DNA injection at the start of infection. gp27 has two predicted TMDs flanking a substantial (∼50-residue) periplasmic domain and is thus is a candidate for a protein that could interact with the OM. Interestingly, the C-terminal domain of this protein has homology to a bacterial acyltransferase (39% identity) and an autolysin from Bacillus phage JL. However, there are no gp27 homologs in the database, much less in other ϕKMV-like phages. Recently, some endolysins of phages infecting Gram-negative hosts have been found to contain membrane-penetrating domains consisting of a pair of amphipathic helices at the C terminus of the enzyme (56, 57). Although no such domains are predicted for gp41, and its close homologs in phages of Pseudomonas and Ralstonia function with spanin systems, it is nevertheless possible that there are novel motifs in the Petty endolysin that allow it to interact with and destabilize the OM during lysis.
Gene 39 encodes a putative tail fiber with depolymerase activity.
Phages that infect encapsulated bacteria, such as clinical A. calcoaeticus-baumannii complex isolates, may possess specific mechanisms that allow them to infect cells. In the case of phage Petty, we observed plaques surrounded by expanding translucent halos, indicative of phage-associated capsular depolymerase activity. Phage depolymerase activity is often encoded in the tail fiber or tail spike; hence, gene 39, encoding a putative tail fiber, was hypothesized to possess this activity. Bioinformatic analysis of gp39 shows that the protein shares ∼13% identity with other phage tail fibers, except for the putative tail fiber of Abp1 and ϕAB1 (35% and 34% identity, respectively). The protein has 2 conserved domains: an N-terminal T7-like tail fiber domain and a centrally located metal-dependent hydrolase domain with a pectin lyase fold (Fig. 8A). Secondary-structure prediction (58, 59) shows the N-terminal domain being mostly alpha-helical, while the hydrolase domain is mostly composed of beta-sheets connected by coils (Fig. 8A).
FIG 8.

Dpo1 protein structure and purification. (A) Diagram of the predicted domains of Dpo1. Numbers indicate the position of the amino acid residue in the polypeptide chain, when counted from the amino terminus. The predicted N-terminal virion-binding domain, coiled-coil domain, sugar hydrolase domain, C-terminal chaperone domain, and hexahistidine tag (black) are labeled. (B) Gene 39 was cloned into pET28b in frame to a C-terminal histidine tag [lane 1, empty vector control; lane 2, uninduced BL21(DE3)/pET-Dpo1 after cell disruption]. BL21(DE3)/pET-Dpo1 cells were induced with IPTG, lysed by French press, and centrifuged to clear cell debris (lane 3, supernatant; lane 4, pellet). The putative depolymerase gp39 was purified using immobilized-metal affinity chromatography (IMAC) (loading flowthrough; lanes 5 and 6). The column was washed with 10 bed volumes of buffer (lane 7) and eluted in 400 mM imidazole (lane 8). MM, prestained protein molecular mass marker (Invitrogen). (C) Purified recombinant Dpo1 was run on a Superdex 200 10/300 size exclusion column under native conditions, with elution monitored by absorbance at 280 nm. Arrows at the top indicate elution of molecular mass standards, in kilodaltons. Activity was recovered from fractions eluted at a 9- to 12-ml volume. While the monomer molecular mass is predicted to be 101.4 kDa, the protein is eluted from the column with an apparent molecular mass of ∼600 kDa.
Gene 39 was cloned into pET28b in frame with a C-terminal hexahistidine tag and confirmed by sequencing. The 102.4-kDa protein, renamed Dpo1, was expressed in E. coli BL21(DE3) and Dpo1 was purified by immobilized-metal affinity chromatography (IMAC) (Fig. 8B). The protein concentration was determined spectrophotometrically, and protein yield was calculated to be 39 mg/liter of culture. As can be seen in Fig. 7B, the purified protein has an apparent molecular mass of 100 kDa, which is in close agreement with the predicted molecular mass of 102.4 kDa. The recombinant enzyme was also able to form translucent zones on lawns of AU0783 similar to the halos formed around phage plaques, as well as in the other multidrug-resistant A. baumannii strains that are sensitive to the phage (data not shown). Size exclusion chromatography was performed to predict the oligomeric state of Dpo1 in solution and further purify the protein. Fractions collected were tested for activity and analyzed by SDS-PAGE; the protein was eluted in a single peak at an equivalent molecular mass of ∼600 kDa, suggesting that it may form a stable hexamer or a dimer of trimers in solution (Fig. 8C).
Characterization of Dpo1 activity.
The ability of Dpo1 to depolymerize EPS was determined by measuring the generation of reducing ends as determined by a colorimetric copper-bicinchoninate assay. Purified EPS was incubated with Dpo1; samples were taken every 5 min, and reducing activity was measured. As shown in Fig. 9A, the generation of reducing ends from the EPS substrate suggests that Dpo1 possesses hydrolase activity. Adding 5 mM EDTA to the buffer reduced the activity of the enzyme by almost half (data not shown), which shows that enzyme activity is enhanced by the presence of divalent cations. To assess if the production of reducing ends correlated with the degradation of the substrate, the viscosity of EPS solutions was measured qualitatively using an Ostwald viscometer, which measures solution viscosity as a function of travel time through a narrow channel. As can be observed in Fig. 9B, the viscosity of the EPS solution was reduced to a level comparable to buffer alone when incubated with active enzyme, suggesting that the tail fiber binds and degrades the substrate backbone to reduce overall carbohydrate chain length. Viscosity was retained in the untreated EPS solution and in EPS incubated with inactive enzyme. The effect of Dpo1 on biofilms of Acinetobacter strains was assessed using a colorimetric microtiter plate method (Fig. 10). Although Dpo1 was able to remove biofilm of some of the tested Acinetobacter strains, the removal amounted to only ∼20%. Interestingly, biofilm formed by the phage host AU0783 was not removed significantly, even though Dpo1 is active against this strain's EPS in vitro. These results suggest that simple degradation of capsular polysaccharide may not be sufficient for removal of Acinetobacter cells adhered to solid substrates.
FIG 9.
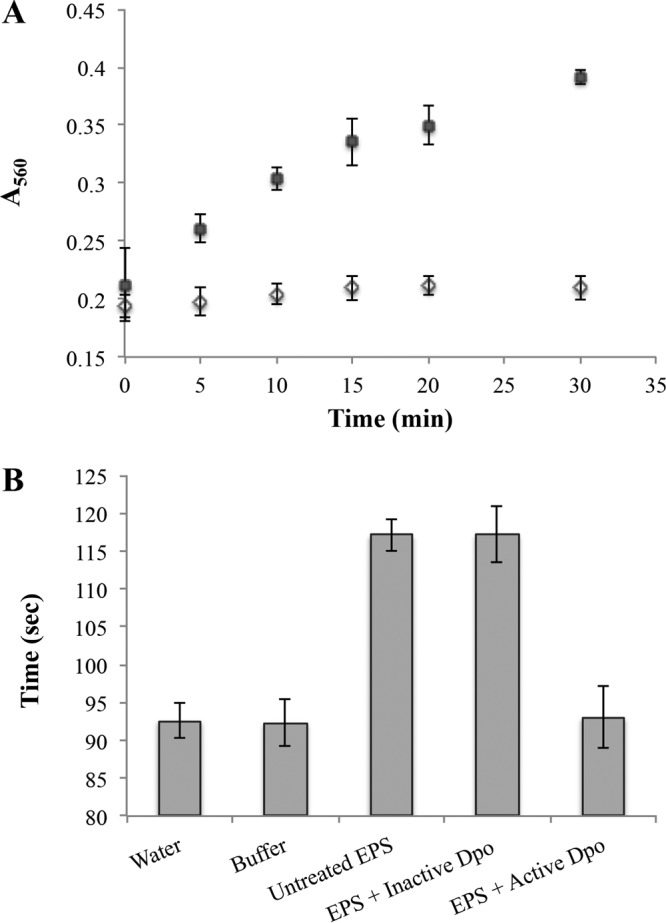
Dpo1 is able to reduce EPS viscosity in vitro and generates reducing ends. (A) Purified EPS (1 mg/ml) was incubated either with active enzyme (0.5 μg/ml) (■) or with previously boiled enzyme (◇) at 37°C. Samples were taken at the times indicated, and the generation of reducing ends was determined by a colorimetric copper-bicinchoninate assay measured at 560 nm. Data shown represent results from 3 independent assays. The generation of reducing ends from the EPS substrate suggests that Dpo1 possesses hydrolase activity. (B) EPS (2.5 mg/ml) was purified from the phage host A. nosocomialis AU0783 and incubated with purified enzyme (17 μg/ml) for 1 h at 37°C. EPS viscosity was measured qualitatively using an Ostwald viscometer, which measures solution viscosity as a function of travel time through a narrow channel. Untreated EPS and EPS incubated with boiled enzyme were used as controls and retained viscosity.
FIG 10.
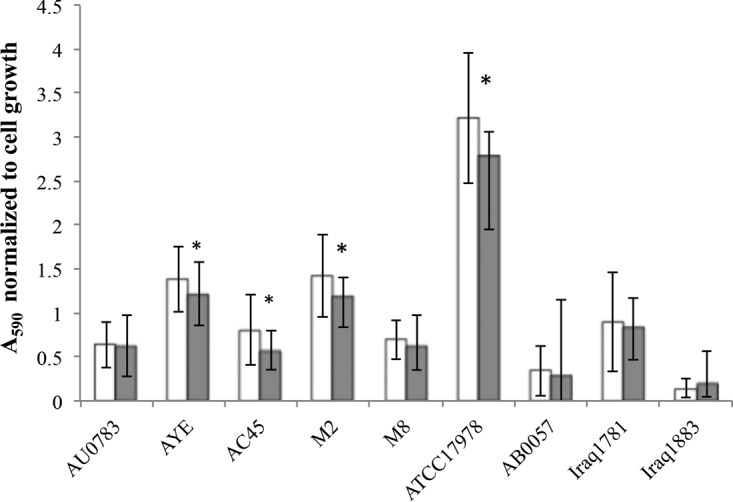
Removal of static Acinetobacter biofilms by Dpo1. Biofilms of different A. calcoaeticus-baumannii strains, either left untreated (white bars) or treated with purified active Dpo1 (gray bars), were quantified. Data shown for each strain (x axis) are normalized to cell growth and represent results from 3 independent assays; error bars indicate SDs. Biofilm was significantly reduced (t test, P < 0.05) in four strains (marked with asterisks).
DISCUSSION
Multidrug-resistant strains of Acinetobacter baumannii have been isolated more frequently in clinical settings, especially from patients who have severe underlying diseases or are hospitalized for extended periods, immunosuppressed, subjected to invasive procedures, or treated with broad-spectrum antibiotics (2). Phage therapy has been contemplated as an alternative to antibiotic treatment in scenarios of infections with multidrug-resistant pathogens (39, 60). However, knowledge about mechanisms of phage infection and phage resistance for phages of A. baumannii remains limited. Phage Petty is a novel podophage able to infect multidrug-resistant strains of A. nosocomialis and A. baumannii. Although its host range is limited, it has potential use in therapeutic phage preparations. Although Petty's genome is smaller than those of known ϕKMV-like Acinetobacter phages (26, 42, 43), it contains 8 genes with hypothetical function that share homology to genes exclusively found in other Acinetobacter phages of this group.
One benefit of the small genome was that it was possible to demonstrate unambiguously that no lipoprotein genes, and thus no spanin genes, were present in Petty. Nevertheless, we were able to show that Petty caused saltatory, explosive lysis and the lysis morphology exhibited no evidence of a defect in OM disruption. The fact that this lack of spanin genes was common to other ϕKMV-like phages of Acinetobacter but not for ϕKMV-like phages in general or for other phages of Acinetobacter suggests that this group of phages has evolved a different mechanism for OM disruption in lysis. No protein predicted to have a periplasmic domain or other secretory signal that would render it capable of interacting with the OM was found to be shared among these phages. The recent identification of membrane-penetrating domains in endolysins of phages of Gram-negative hosts (56, 57) raises the possibility that the ϕKMV-like phages have endolysins that are capable of direct destabilization of the OM during lysis. Although primary-structure analysis of Petty gp41 did not reveal predicted amphipathic helices that are characteristic of the demonstrated membrane-active endolysins, it is interesting that the Petty endolysin is not related to the endolysins of the other Acinetobacter ϕKMV-like phages, despite the fact that the holins of these phages are similar. This suggests that a common progenitor of the ϕKMV-like phages of Acinetobacter lost its spanin function and that descendants diverged in terms of acquiring genes that would complement the lysis defect. It will be interesting to determine whether the Petty endolysin or the endolysins of other ϕKMV-like Acinetobacter phages are OM active.
The capsule, an important virulence factor, protects the bacterium from the host immune response (61, 62). A. baumannii strains have been grouped based on the structural variability of capsular polysaccharides (63, 64). Although it has not yet been fully explored in Acinetobacter, serotyping has been used as a clinical indicator of pathogenesis and possible clinical prognosis, such as Klebsiella pneumoniae (65). Given the central role of capsular EPS in the pathogenesis of Acinetobacter, the potential use of phage-encoded proteins with depolymerization activity for therapeutic and diagnostic applications is worth evaluating. In this study, a novel depolymerase enzyme, encoded by Acinetobacter phage Petty, has been identified and characterized. The depolymerase shares a conserved N-terminal region found in the tail fiber proteins of T7-like phages, which suggests that this domain mediates the interaction with the phage head. We found that unlike previously reported tail fibers with depolymerase activity in E. coli (66, 67), Dpo1 is not processed before being assembled on the phage head, suggesting different structural requirements for folding in these podophages. A pectin lyase fold fragment, found in the metal binding domain by sequence alignment, indicates the involvement of Dpo1 in the cleavage of glycoside bonds in the capsular polysaccharide. Previously, this domain has been reported to be present in other phage tail spike proteins targeting and degrading EPS, such as in K5 lyase of coliphage K5A and Tsp2 of phage ΦSH19 (67, 68). The C-terminal domain of the protein has very low amino acid identity (∼11%) with other tail fibers in the database. It has been suggested that in other tail fibers with depolymerase activity, this region is responsible for recognizing polysaccharides and binding to the host cell receptor, which highlights the potential to use this depolymerase as a tool for capsular characterization of clinical strains of A. calcoaeticus-baumannii. Phage receptor binding proteins have been proposed as potential targets for genetic modification to broaden the host range, thus contributing to practical applications of phage therapy and the detection of bacteria by their specific phages.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The Acinetobacter strains used in this study were kindly provided by John J. LiPuma, University of Michigan Medical School (Ann Arbor, MI), Ashok Kumar, Wayne State University (Detroit, MI), Philip Rather, Emory School of Medicine (Atlanta, GA), UCSD Medical Hospital, and Carlos Gonzales, Texas A&M University (College Station, TX). All Acinetobacter strains were routinely cultured on tryptic soy broth (TSB; 17 g/liter of Bacto tryptone, 3 g/liter of soytone, 2.5 g/liter of d-glucose, 5 g/liter of NaCl, 2.5 g/liter of disodium phosphate) and tryptic soy agar (TSA; TSB plus 1.5% [wt/vol] Bacto agar). Recombinant strains of E. coli XL1-Blue and BL21(DE3) were cultured on LB (10 g/liter of Bacto tryptone [BD], 5 g/liter of yeast extract, 10 g/liter of NaCl) with kanamycin (40 μg/ml). For all plaque assays, a 0.75% TB agar overlay (10 g/liter of tryptone, 5 g/liter of NaCl, and 0.75% Bacto agar) was plated over TSA plates (69). All strains were grown at 37°C.
Phage isolation and preparation of high-titer phage lysates.
Activated sludge was collected from a municipal wastewater treatment plant located in College Station, TX. The sample was centrifuged (8,000 × g for 40 min), and the supernatant was filtered (0.45 μm). The clarified supernatant was enriched for phages against A. nosocomialis AU0783 as previously described (70). Isolated plaques obtained on lawns of AU0783 were recovered, resuspended in SM buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 8 mM MgSO4, 0.01% [wt/vol] gelatin), and subcultured three times to ensure clonality.
High-titer phage stocks were obtained by inoculating TSB 1:100 with an overnight (ON) AU0783 culture, subsequently grown to an A550 of 0.2. Phages were then added to a multiplicity of infection (MOI) of 0.1, and A550 was monitored after infection. When onset of lysis was observed, sodium citrate was added to a final concentration of 10 mM. Lysates were harvested after complete lysis, centrifuged (10,000 × g for 20 min at 4°C), and filtered (0.22 μm). Phage titer was determined by plating serial dilutions of phage using the double-layer method (69).
Phage particles were further concentrated by pelleting the lysates (24 h at 10,000 × g and 4°C). Phage pellets were resuspended in 3 ml of SM buffer and purified by ultracentrifugation in a CsCl step gradient (ρ 1.2, ρ 1.45, ρ 1.5, and ρ 1.65 g/cm3) at 100,000 × g for 4 h at 4°C in an SW28 rotor (Beckman Coulter, USA). The collected phage band was extracted and dialyzed stepwise, first against SM salt buffer (gelatin-free SM buffer plus 1 M NaCl) and then against gelatin-free SM buffer, in a 3.5-kDa dialysis cassette (Thermo Scientific). Concentrated phage samples were stored at 4°C.
TEM.
Phage morphology was examined by transmission electron microscopy (TEM) of negatively stained preparations using the Valentine method (71). Grids were stained with 2% uranyl acetate (Sigma-Aldrich), and images were collected on a JEOL 1200 EX TEM at a 100-kV accelerating voltage at the Microscopy and Imaging Center at Texas A&M University.
Latency period and burst size determination.
A one-step growth experiment (72, 73) was performed in triplicate to determine the latent period and burst size of phage Petty. AU0783 was grown overnight in TSB, and a high-titer phage stock was prepared. Forty microliters of overnight-cultured bacteria was added to 20 ml of fresh medium and grown at 37°C on a shaking incubator to an optical density at 550 nm (OD550) of 0.2 (∼2 × 108 CFU/ml). The bacterial culture was then infected with phage at an MOI of 0.01. The number of unadsorbed phages was determined by plating serial dilutions of a sample taken 4 min after phage infection. Postadsorption, the infected culture was diluted 103 and 105 in prewarmed medium and samples from those dilution tubes were taken every 5 min. All plating was done using 100 μl of ON culture of AU0783 using the double-layer method. The burst size was calculated as the difference in average phage titer after and before the onset of lysis, measured in the plaque assay, divided by the number of infective centers.
Adsorption and host range assays.
Adsorption assays were performed as previously described (74). Briefly, A. nosocomialis AU0783 cells were gown to an OD550 of ∼0.2 and infected with phage at an MOI of 0.01. Samples (100 μl) were collected every 2 min, diluted 10-fold in cold TSB, centrifuged (12,000 × g for 5 min at 4°C), and filtered (0.22 μm). The phage particles remaining in the supernatant were quantified by plaque assay. The adsorbed phage fraction was calculated as the residual titer in supernatant divided by the initial titer.
To evaluate the host range of phage Petty, 10-μl quantities of serial dilutions of high-titer lysates were plated on lawns of clinical Acinetobacter strains using the double-layer method. A strain was considered sensitive to phage Petty if single plaques were observed at higher lysate dilutions. Efficiency-of-plating (EOP) assays were conducted on strains sensitive to phage infection as previously described (69, 75). In brief, Acinetobacter cells were grown overnight in TSB at 37°C, and 100 μl of each of those cultures was used to seed soft-agar overlays over which 10-μl quantities of serial dilutions of phage lysate were plated. Plaques were counted after overnight incubation at 37°C. EOP was calculated as the ratio of plaques appearing on the target bacterial lawn to the number of plaques in the lawn of the host strain. All experiments were replicated three times.
DNA isolation, genome sequencing, and annotation.
A crude lysate (∼109 PFU/ml) was treated with 10 μg/ml of DNase I and RNase A (Sigma-Aldrich) for 1 h at 37°C, and phage DNA was extracted as previously described (76).
Phage DNA was sequenced by 454 pyrosequencing at the Emory GRA Genome Center (Emory University, Atlanta, GA). The trimmed FLX Titanium reads were assembled into a single contig using the Newbler assembler version 2.5.3 (454 Life Sciences). The contig was confirmed to be complete by PCR, using custom primers designed to amplify outwards from both the 3′ and 5′ ends of the initial assembly, and standard Sanger sequencing of the amplicon (77). Genes were predicted using GeneMark S (78) and corrected in Artemis (79). Functional annotation was done using software tools available on the Center for Phage Technology (CPT) portal (https://cpt.tamu.edu/galaxy-pub).
Comparative genomics of the closely related Acinetobacter phages was done by generating a protein database of the coding sequences of each genome. Each phage protein was queried against that database using a BLASTP with an E value cutoff of 10−20 (80). A greedy algorithm was then used to group the coding sequences into clusters, which were aligned with ClustalW (81) and output as locally colinear blocks (LCBs) in XMFA format. Adjacent LCBs within a threshold distance (50 nucleotides) were further consolidated. To visualize these relationships in genomic context, X-Vis, a custom XMFA visualization tool written in JavaScript rendered each genome, its proteins, and associated LCB alignments (https://cpt.tamu.edu/galaxy-pub).
The boundaries of genomic direct terminal repeats were determined by primer walking across the repeats using genomic DNA as the template (82). The phage genome is available in GenBank under accession number KF669656.1.
Proteome analysis of phage structural proteins.
CsCl-purified phage particles were diluted 1:1 in Laemmli loading buffer (83) boiled at 95°C for 5 min and analyzed by SDS-PAGE on a 4 to 20% Tris-glycine gel (Bio-Rad). Approximately 1 × 1011 PFU/ml were loaded per lane. The gel was stained with Coomassie blue (0.1% Coomassie R250, 10% acetic acid, 40% methanol). Prestained SeeBlue Plus2 (Invitrogen) was used as a molecular weight marker. Bands were cut from the gel and used for trypsin digestion and mass spectrometry analysis as previously described (70). Peptides obtained were compared to a database including the predicted gene products from the genome of phage Petty.
Cloning, expression, and purification of the phage depolymerase (Dpo1).
Gene 39 (nucleotides [nt] 33863 to 36619 in the phage genome) was amplified by PCR with primers DpoNco (5′-TAGCTTTGGAGGATTACCATGGCTTATGTAGAAAAGG) and DpoXho (5′-GGGTTTAAAAAGGTTTTACCTCGAGACTAGTTTGCACTCACCTC), which introduced restriction sites for NcoI and XhoI (New England Biolabs [NEB]) on each side of the amplified fragment. After digestion with these restriction enzymes, the PCR product was cloned in a pET28b backbone in frame with a C-terminal hexahistidine tag. The resulting pET-Dpo1 plasmid was transformed into chemically competent E. coli BL21(DE3). Transformant selection was done by plating the cells on LB-kanamycin (LB-Kan; 40 μg/ml).
BL21(DE3)/pET-Dpo1 cells were grown in to an A550 of ∼0.6. The protein was expressed by chilling the culture for 20 min on ice and induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) followed by incubation for 16 h at 16°C. Induced cells were collected by centrifugation at 4,000 × g for 30 min after the A550 was measured. Cells were resuspended at a calculated equivalent A550 of 30. A protease inhibitor cocktail (1×; Sigma-Aldrich), RNase, and DNase (10 μg/ml; Sigma-Aldrich) were added to the resuspension, and cells were disrupted by passage through an Aminco French press cell at 16,000 lb/in2. Cell debris was removed by centrifugation at 10,000 × g for 30 min at 4°C. The protein was purified by immobilized-metal affinity chromatography (IMAC) using a Talon metal affinity resin (Clontech) followed by size exclusion chromatography on a Sephadex S300 column. Fractions from each of the purification steps were analyzed on 10% Tris-lysine SDS-PAGE gels stained with Coomassie blue.
Bacterial EPS purification.
A. nosocomialis AU0783 was grown to confluence on TSB plates supplemented with 0.5% (wt/vol) glucose and incubated at 37°C for ∼120 h. EPS purification was carried out as described previously (84). Briefly, cells were harvested by adding 10 ml of saline solution (0.9% [wt/vol] NaCl) per plate and scraping the cells from the agar surface. Equilibrated phenol was added to the resulting cell suspension to a final concentration of 5% (vol/vol) and agitated for 5 h at room temperature (RT). Cells were cleared by centrifugation at 10,000 × g for 30 min, and the supernatant was precipitated overnight with 5 volumes of 95% ethanol. The EPS-containing precipitate was pelleted by centrifugation at 15,000 × g for 20 min at 4°C and washed twice with 25 ml of 95% ethanol. The pellet was allowed to air dry overnight prior to resuspension in 25 ml of sterile double-distilled water (ddH2O). After treatment with DNase I and RNase A (1 μg/ml) for 30 min, the EPS mixture was dialyzed overnight in a 6- to 8-kDa molecular-mass-cutoff membrane (Spectra-por) against water. The dialyzed EPS material was lyophilized and weighed.
Depolymerase activity assays.
To qualitatively assay the activity of the purified enzyme, a modification of the method described by Adams and Park (85) was employed. Ten-microliter quantities of a serial 10-fold dilution of recombinant Dpo1 were placed on lawns of AU0783 and incubated at 37°C for 18 h. The endpoint was taken as the reciprocal of the highest dilution at which a zone of reduced turbidity was observed in the bacterial lawn.
The ability of the enzyme to degrade exopolysaccharides was also evaluated by measuring the viscosity of EPS solutions following incubation with Dpo1. Untreated EPS was dissolved at a final concentration of 2.5 mg/ml in 1× assay buffer (25 mM Tris, 150 mM NaCl [pH 7.2]) and incubated for 1 h at 37°C either with active enzyme (15 μg/ml) or with inactive Dpo1 (boiled at 95°C for 10 min). After incubation, the reactions were heat inactivated at 95°C for 5 min. As a control, the viscosity of the untreated EPS solution (2.5 mg/ml) and that of the assay buffer were measured before and after incubation and heat inactivation. The viscosity measurements were taken at 20°C (n = 5).
A bicinchoninic acid (BCA) assay was used to determine the generation of reducing ends in an EPS solution incubated in the presence of purified protein (86). The BCA reagent was prepared fresh in a 1:1 ratio of solution A (5 mM BCA, 513 mM Na2CO3, and 288 mM NaHCO3) and solution B (5 mM CuSO4 and 12 mM l-serine) in ddH2O. A reaction master mix was prepared by mixing 100 μl of 10× assay buffer, 100 μl of 5-mg/ml EPS, and 800 μl of ddH2O. The master mix was aliquoted into microcentrifuge tubes (95 μl per tube) which were preincubated at 37°C for 5 min prior to addition of 5 μl of enzyme (200 nM) to each tube. After 5, 10, 20, and 30 min of incubation at 37°C, a pair of tubes was removed and placed at 95°C for 5 min to inactivate the enzyme. After cooling, 1 ml of BCA reagent was added to each tube. The samples were mixed and incubated at 95°C for 15 min, cooled to room temperature, and measured for absorbance at 560 nm in a spectrophotometer to measure reducing ends. As controls, assay buffer and 0.2 mM glucose in assay buffer were used.
Biofilm removal of Acinetobacter strains by Dpo1 was measured using a crystal violet microtiter plate assay (87). Bacteria were grown for 48 h at 37°C in TSB in untreated 96-well plates, and OD550 was measured. Planktonic cells were removed and attached cells were incubated with Dpo1 (17 μg/ml) or buffer alone for 3 h at 37°C. Biofilm removal was assessed by crystal violet staining measured at 590 nm.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge J. J. LiPuma, A. Kumar, and C. F. Gonzales for providing the clinical strains used in this study. We also thank Mikhail Shneider for very productive discussions and the members of the Center for Phage Technology (CPT) for their help and support, especially Eric Rasche for the development of bioinformatic workflows. The CPT is supported by Texas AgriLife Research and Texas A&M University.
This research was supported by NIH research grant R01 GM27099-36 and by NSF grant EF-0949351.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01064-17.
REFERENCES
- 1.Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- 2.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wisplinghoff H, Paulus T, Lugenheim M, Stefanik D, Higgins PG, Edmond MB, Wenzel RP, Seifert H. 2012. Nosocomial bloodstream infections due to Acinetobacter baumannii, Acinetobacter pittii and Acinetobacter nosocomialis in the United States. J Infect 64:282–290. doi: 10.1016/j.jinf.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Boyle DP, Zembower TR. 2015. Epidemiology and management of emerging drug-resistant Gram-negative bacteria: extended-spectrum beta-lactamases and beyond. Urol Clin North Am 42:493–505. doi: 10.1016/j.ucl.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Roca I, Espinal P, Vila-Farres X, Vila J. 2012. The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front Microbiol 3:148. doi: 10.3389/fmicb.2012.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network (NHSN) Team, Participating NHSN Facilities. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 7.Farrugia DN, Elbourne LD, Hassan KA, Eijkelkamp BA, Tetu SG, Brown MH, Shah BS, Peleg AY, Mabbutt BC, Paulsen IT. 2013. The complete genome and phenome of a community-acquired Acinetobacter baumannii. PLoS One 8:e58628. doi: 10.1371/journal.pone.0058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obeidat N, Jawdat F, Al-Bakri AG, Shehabi AA. 2014. Major biologic characteristics of Acinetobacter baumannii isolates from hospital environmental and patients' respiratory tract sources. Am J Infect Control 42:401–404. doi: 10.1016/j.ajic.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Rodríguez-Baño J, Marti S, Soto S, Fernandez-Cuenca F, Cisneros JM, Pachon J, Pascual A, Martinez-Martinez L, McQueary C, Actis LA, Vila J, Spanish Group for the Study of Nosocomial Infections (GEIH). 2008. Biofilm formation in Acinetobacter baumannii: associated features and clinical implications. Clin Microbiol Infect 14:276–278. doi: 10.1111/j.1469-0691.2007.01916.x. [DOI] [PubMed] [Google Scholar]
- 10.Antunes LC, Imperi F, Carattoli A, Visca P. 2011. Deciphering the multifactorial nature of Acinetobacter baumannii pathogenicity. PLoS One 6:e22674. doi: 10.1371/journal.pone.0022674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eijkelkamp BA, Stroeher UH, Hassan KA, Papadimitrious MS, Paulsen IT, Brown MH. 2011. Adherence and motility characteristics of clinical Acinetobacter baumannii isolates. FEMS Microbiol Lett 323:44–51. doi: 10.1111/j.1574-6968.2011.02362.x. [DOI] [PubMed] [Google Scholar]
- 12.Espinal P, Marti S, Vila J. 2012. Effect of biofilm formation on the survival of Acinetobacter baumannii on dry surfaces. J Hosp Infect 80:56–60. doi: 10.1016/j.jhin.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Longo F, Vuotto C, Donelli G. 2014. Biofilm formation in Acinetobacter baumannii. New Microbiol 37:119–127. [PubMed] [Google Scholar]
- 14.Russo TA, Luke NR, Beanan JM, Olson R, Sauberan SL, MacDonald U, Schultz LW, Umland TC, Campagnari AA. 2010. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect Immun 78:3993–4000. doi: 10.1128/IAI.00366-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campos MA, Vargas MA, Regueiro V, Llompart CM, Alberti S, Bengoechea JA. 2004. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect Immun 72:7107–7114. doi: 10.1128/IAI.72.12.7107-7114.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samson JE, Magadan AH, Sabri M, Moineau S. 2013. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol 11:675–687. doi: 10.1038/nrmicro3096. [DOI] [PubMed] [Google Scholar]
- 17.Bayer ME, Thurow H, Bayer MH. 1979. Penetration of the polysaccharide capsule of Escherichia coli (Bi161-42) by bacteriophage K29. Virology 94:95–118. doi: 10.1016/0042-6822(79)90441-0. [DOI] [PubMed] [Google Scholar]
- 18.Pires DP, Oliveira H, Melo LDR, Sillankorva S, Azeredo J. 2016. Bacteriophage-encoded depolymerases—their diversity and biotechnological applications. Appl Microbiol Biotechnol 100:2141–2151. doi: 10.1007/s00253-015-7247-0. [DOI] [PubMed] [Google Scholar]
- 19.Drulis-Kawa Z, Majkowska-Skrobek G, Maciejewska B, Delattre AS, Lavigne R. 2012. Learning from bacteriophages—advantages and limitations of phage and phage-encoded protein applications. Curr Protein Pept Sci 13:699–722. doi: 10.2174/138920312804871193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niemann H, Frank N, Stirm S. 1977. Klebsiella serotype-13 capsular polysaccharide: primary structure and depolymerization by a bacteriophage-borne glycanase. Carbohydr Res 59:165–177. doi: 10.1016/S0008-6215(00)83303-0. [DOI] [PubMed] [Google Scholar]
- 21.Lin TL, Hsieh PF, Huang YT, Lee WC, Tsai YT, Su PA, Pan YJ, Hsu CR, Wu MC, Wang JT. 2014. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: implication in typing and treatment. J Infect Dis 210:1734–1744. doi: 10.1093/infdis/jiu332. [DOI] [PubMed] [Google Scholar]
- 22.Traub WH, Leonhard B, Bauer D. 1996. Clusters of nosocomial cross-infection due to Acinetobacter baumannii and genospecies 3: Comparison of serotyping with macrorestriction analysis of genomic DNA with pulsed-field gel electrophoresis. Zentralbl Bakteriol 284:115–123. [DOI] [PubMed] [Google Scholar]
- 23.Traub WH, Bauer D. 2000. Surveillance of nosocomial cross-infections due to three Acinetobacter genospecies (Acinetobacter baumannii, genospecies 3 and genospecies 13) during a 10-year observation period: serotyping, macrorestriction analysis of genomic DNA and antibiotic susceptibilities. Chemotherapy 46:282–292. doi: 10.1159/000007300. [DOI] [PubMed] [Google Scholar]
- 24.Hu D, Liu B, Dijkshoorn L, Wang L, Reeves PR. 2013. Diversity in the major polysaccharide antigen of Acinetobacter baumannii assessed by DNA sequencing, and development of a molecular serotyping scheme. PLoS One 8:e70329. doi: 10.1371/journal.pone.0070329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes KA, Sutherland IW, Clark J, Jones MV. 1998. Bacteriophage and associated polysaccharide depolymerases—novel tools for study of biofilms. J Appl Microbiol 85:583–590. [DOI] [PubMed] [Google Scholar]
- 26.Lai MJ, Chang KC, Huang SW, Luo CH, Chiou PY, Wu CC, Lin NT. 2016. The tail associated protein of Acinetobacter baumannii phage PhiAB6 is the host specificity determinant possessing exopolysaccharide depolymerase activity. PLoS One 11:e0153361. doi: 10.1371/journal.pone.0153361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geisinger E, Isberg RR. 2015. Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog 11:e1004691. doi: 10.1371/journal.ppat.1004691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talreja D, Muraleedharan C, Gunathilaka G, Zhang Y, Kaye KS, Walia SK, Kumar A. 2014. Virulence properties of multidrug resistant ocular isolates of Acinetobacter baumannii. Curr Eye Res 39:695–704. doi: 10.3109/02713683.2013.873055. [DOI] [PubMed] [Google Scholar]
- 29.Fishbain J, Peleg AY. 2010. Treatment of Acinetobacter infections. Clin Infect Dis 51:79–84. doi: 10.1086/653120. [DOI] [PubMed] [Google Scholar]
- 30.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. 2005. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J Antimicrob Chemother 56:160–165. doi: 10.1093/jac/dki177. [DOI] [PubMed] [Google Scholar]
- 31.Mushtaq N, Redpath MB, Luzio JP, Taylor PW. 2004. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob Agents Chemother 48:1503–1508. doi: 10.1128/AAC.48.5.1503-1508.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thawal ND, Yele AB, Sahu PK, Chopade BA. 2012. Effect of a novel podophage AB7-IBB2 on Acinetobacter baumannii biofilm. Curr Microbiol 65:66–72. doi: 10.1007/s00284-012-0127-2. [DOI] [PubMed] [Google Scholar]
- 33.Glonti T, Chanishvili N, Taylor PW. 2010. Bacteriophage-derived enzyme that depolymerizes the alginic acid capsule associated with cystic fibrosis isolates of Pseudomonas aeruginosa. J Appl Microbiol 108:695–702. doi: 10.1111/j.1365-2672.2009.04469.x. [DOI] [PubMed] [Google Scholar]
- 34.Cornelissen A, Ceyssens PJ, T'Syen J, Van Praet H, Noben JP, Shaburova OV, Krylov VN, Volckaert G, Lavigne R. 2011. The T7-related Pseudomonas putida phage phi15 displays virion-associated biofilm degradation properties. PLoS One 6:e18597. doi: 10.1371/journal.pone.0018597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu CR, Lin TL, Pan YJ, Hsieh PF, Wang JT. 2013. Isolation of a bacteriophage specific for a new capsular type of Klebsiella pneumoniae and characterization of its polysaccharide depolymerase. PLoS One 8:e70092. doi: 10.1371/journal.pone.0070092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stummeyer K, Schwarzer D, Claus H, Vogel U, Gerardy-Schahn R, Muhlenhoff M. 2006. Evolution of bacteriophages infecting encapsulated bacteria: lessons from Escherichia coli K1-specific phages. Mol Microbiol 60:1123–1135. doi: 10.1111/j.1365-2958.2006.05173.x. [DOI] [PubMed] [Google Scholar]
- 37.Cornelissen A, Ceyssens PJ, Krylov VN, Noben JP, Volckaert G, Lavigne R. 2012. Identification of EPS-degrading activity within the tail spikes of the novel Pseudomonas putida phage AF. Virology 434:251–256. doi: 10.1016/j.virol.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 38.Bartell PF, Orr TE. 1969. Distinct slime polysaccharide depolymerases of phage infected Pseudomonas aeruginosa—evidence of close association with the structured phage particle. J Virol 4:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García-Quintanilla M, Pulido MR, Lopez-Rojas R, Pachon J, McConnell MJ. 2013. Emerging therapies for multidrug resistant Acinetobacter baumannii. Trends Microbiol 21:157–163. doi: 10.1016/j.tim.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Gill JJ, Hyman P. 2010. Phage choice, isolation, and preparation for phage therapy. Curr Pharm Biotechnol 11:2–14. doi: 10.2174/138920110790725311. [DOI] [PubMed] [Google Scholar]
- 41.Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones J, Salka S, Bishop-Lilly KA, Young R, Hamilton T. 2017. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob Agents Chemother 61:e00954-17. doi: 10.1128/AAC.00954-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang KC, Lin NT, Hu A, Lin YS, Chen LK, Lai MJ. 2011. Genomic analysis of bacteriophage phiAB1, a phiKMV-like virus infecting multidrug-resistant Acinetobacter baumannii. Genomics 97:249–255. doi: 10.1016/j.ygeno.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Huang G, Le S, Peng Y, Zhao Y, Yin S, Zhang L, Yao X, Tan Y, Li M, Hu F. 2013. Characterization and genome sequencing of phage Abp1, a new phiKMV-like virus infecting multidrug-resistant Acinetobacter baumannii. Curr Microbiol 66:535–543. doi: 10.1007/s00284-013-0308-7. [DOI] [PubMed] [Google Scholar]
- 44.Lavigne R, Burkal'tseva MV, Robben J, Sykilinda NN, Kurochkina LP, Grymonprez B, Jonckx B, Krylov VN, Mesyanzhinov VV, Volckaert G. 2003. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 312:49–59. doi: 10.1016/S0042-6822(03)00123-5. [DOI] [PubMed] [Google Scholar]
- 45.Merabishvili M, Vandenheuvel D, Kropinski AM, Mast J, De Vos D, Verbeken G, Noben JP, Lavigne R, Vaneechoutte M, Pirnay JP. 2014. Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS One 9:e104853. doi: 10.1371/journal.pone.0104853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nolan JM, Petrov V, Bertrand C, Krisch HM, Karam JD. 2006. Genetic diversity among five T4-like bacteriophages. Virol J 3:30. doi: 10.1186/1743-422X-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedrich NC, Torrents E, Gibb EA, Sahlin M, Sjöberg B-M, Edgell DR. 2007. Insertion of a homing endonuclease creates a genes-in-pieces ribonucleotide reductase that retains function. Proc Natl Acad Sci U S A 104:6176–6181. doi: 10.1073/pnas.0609915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gogarten JP, Senejani AG, Zhaxybayeva O, Olendzenski L, Hilario E. 2002. Inteins: structure, function, and evolution. Annu Rev Microbiol 56:263–287. doi: 10.1146/annurev.micro.56.012302.160741. [DOI] [PubMed] [Google Scholar]
- 49.Center MS, Richardson CC. 1970. An endonuclease induced after infection of Escherichia coli with bacteriophage T7. I. Purification and properties of the enzyme. J Biol Chem 245:6285–6291. [PubMed] [Google Scholar]
- 50.Young R. 2013. Phage lysis: do we have the hole story yet? Curr Opin Microbiol 16:790–797. doi: 10.1016/j.mib.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dewey JS, Savva CG, White RL, Vitha S, Holzenburg A, Young R. 2010. Micron-scale holes terminate the phage infection cycle. Proc Natl Acad Sci U S A 107:2219–2223. doi: 10.1073/pnas.0914030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savva CG, Dewey JS, Moussa SH, To KH, Holzenburg A, Young R. 2014. Stable micron-scale holes are a general feature of canonical holins. Mol Microbiol 91:57–65. doi: 10.1111/mmi.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang IN, Smith DL, Young R. 2000. Holins—the protein clocks of bacteriophage infections. Annu Rev Biochem 54:799–825. [DOI] [PubMed] [Google Scholar]
- 54.Young R. 2014. Phage lysis—three steps, three choices, one outcome. J Microbiol 52:243–258. doi: 10.1007/s12275-014-4087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berry J, Rajaure M, Pang T, Young R. 2012. The spanin complex is essential for lambda lysis. J Bacteriol 194:5667–5674. doi: 10.1128/JB.01245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai MJ, Lin NT, Hu A, Soo PC, Chen LK, Chen LH, Chang KC. 2011. Antibacterial activity of Acinetobacter baumannii phage phiAB2 endolysin (LysAB2) against both gram-positive and gram-negative bacteria. Appl Microbiol Biotechnol 90:529–539. doi: 10.1007/s00253-011-3104-y. [DOI] [PubMed] [Google Scholar]
- 57.Lood R, Winer BY, Pelzek AJ, Diez-Martinez R, Thandar M, Euler CW, Schuch R, Fischetti VA. 2015. Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother 59:1983–1991. doi: 10.1128/AAC.04641-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. 2013. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. [DOI] [PubMed] [Google Scholar]
- 60.Projan S. 2004. Phage-inspired antibiotics? Nat Biotechnol 22:167–168. doi: 10.1038/nbt0204-167. [DOI] [PubMed] [Google Scholar]
- 61.Moxon ER, Kroll JS. 1990. The role of bacterial polysaccharide capsules as virulence factors. Curr Top Microbiol Immunol 150:65–85. [DOI] [PubMed] [Google Scholar]
- 62.Scorpio A, Chabot DJ, Day WA, O'Brien DK, Vietri NJ, Itoh Y, Mohamadzadeh M, Friedlander AM. 2007. Poly-gamma-glutamate capsule-degrading enzyme treatment enhances phagocytosis and killing of encapsulated Bacillus anthracis. Antimicrob Agents Chemother 51:215–222. doi: 10.1128/AAC.00706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kenyon JJ, Shneider MM, Senchenkova SN, Shashkov AS, Siniagina MN, Malanin SY, Popova AV, Miroshnikov KA, Hall RM, Knirel YA. 2016. K19 capsular polysaccharide of Acinetobacter baumannii is produced via a Wzy polymerase encoded in a small genomic island rather than the KL19 capsule gene cluster. Microbiology 162:1479–1489. doi: 10.1099/mic.0.000313. [DOI] [PubMed] [Google Scholar]
- 64.Kenyon JJ, Hall RM. 2013. Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii genomes. PLoS One 8:e62160. doi: 10.1371/journal.pone.0062160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen L, Chavda KD, Findlay J, Peirano G, Hopkins K, Pitout JD, Bonomo RA, Woodford N, DeLeo FR, Kreiswirth BN. 2014. Multiplex PCR for identification of two capsular types in epidemic KPC-producing Klebsiella pneumoniae sequence type 258 strains. Antimicrob Agents Chemother 58:4196–4199. doi: 10.1128/AAC.02673-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leggate DR, Bryant JM, Redpath MB, Head D, Taylor PW, Luzio JP. 2002. Expression, mutagenesis and kinetic analysis of recombinant K1E endosialidase to define the site of proteolytic processing and requirements for catalysis. Mol Microbiol 44:749–760. doi: 10.1046/j.1365-2958.2002.02908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson JE, Pourhossein M, Waterhouse A, Hudson T, Goldrick M, Derrick JP, Roberts IS. 2010. The K5 lyase KflA combines a viral tail spike structure with a bacterial polysaccharide lyase mechanism. J Biol Chem 285:23963–23969. doi: 10.1074/jbc.M110.127571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hooton SP, Timms AR, Rowsell J, Wilson R, Connerton IF. 2011. Salmonella Typhimurium-specific bacteriophage ΦSH19 and the origins of species specificity in the Vi01-like phage family. Virol J 8:498. doi: 10.1186/1743-422X-8-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams MH. 1959. Bacteriophages. Interscience Publishers, New York, NY. [Google Scholar]
- 70.Gill JJ, Summer EJ, Russell WK, Cologna SM, Carlile TM, Fuller AC, Kitsopoulos K, Mebane LM, Parkinson BN, Sullivan D, Carmody LA, Gonzalez CF, LiPuma JJ, Young R. 2011. Genomes and characterization of phages Bcep22 and BcepIL02, founders of a novel phage type in Burkholderia cenocepacia. J Bacteriol 193:5300–5313. doi: 10.1128/JB.05287-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Valentine RC, Shapiro BM, Stadtman ER. 1968. Regulation of glutamine synthetase XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7:2143–2152. [DOI] [PubMed] [Google Scholar]
- 72.Choi C, Kuatsjah E, Wu E, Yuan S. 2010. The effect of cell size on the burst size of T4 bacteriophage infections of Escherichia coli B23. J Exp Microbiol Immunol 14:85–91. [Google Scholar]
- 73.Carlson K, Miller ES. 1994. Working with T4, p 421–455. In Karam JD. (ed), Molecular biology of bacteriophage T4. ASM Press, Washington, DC. [Google Scholar]
- 74.Kropinski A, Mazzocco A, Waddell T, Lingohr E, Johnson R. 2009. Enumeration of bacteriophages by double agar overlay plaque assay, p 77–80. In Clokie MJ, Kropinski A (ed), Bacteriophages: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 75.Kutter E. 2009. Phage host range and efficiency of plating, p 141–149. In Clokie MJ, Kropinski A (ed), Bacteriophages: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 76.Summer EJ. 2009. Preparation of a phage DNA fragment library for whole genome shotgun sequencing. Methods Mol Biol 502:27–46. doi: 10.1007/978-1-60327-565-1_4. [DOI] [PubMed] [Google Scholar]
- 77.Farmer NG, Wood TL, Chamakura KR, Kuty Everett GF. 2013. Complete genome of Acinetobacter baumannii N4-like podophage Presley. Genome Announc 1(6):e00852-13. doi: 10.1128/genomeA.00852-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 80.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 81.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 82.Casjens SR, Gilcrease EB. 2009. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol 502:91–111. doi: 10.1007/978-1-60327-565-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 84.Steinmetz I, Rohde M, Brenneke B. 1995. Purification and characterization of an exopolysaccharide of Burkholderia (Pseudomonas) pseudomallei. Infect Immun 63:3959–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adams MH, Park BH. 1956. An enzyme produced by a phage-host cell system. II. The properties of the polysaccharide depolymerase. Virology 2:719–736. [DOI] [PubMed] [Google Scholar]
- 86.Waffenschmidt S, Jaenicke L. 1987. Assay of reducing sugars in the nanomole range with 2,2′-bicinchoninate. Anal Biochem 165:337–340. doi: 10.1016/0003-2697(87)90278-8. [DOI] [PubMed] [Google Scholar]
- 87.Knezevic P, Petrovic O. 2008. A colorimetric microtiter plate method for assessment of phage effect on Pseudomonas aeruginosa biofilm. J Microbiol Methods 74:114–118. doi: 10.1016/j.mimet.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 88.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abramoff MD, Magalhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 91.Rasband WS. 2017. ImageJ. https://imagej.nih.gov/ij/index.html Accessed 1 October 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.