

ABSTRACT
The innate immune response plays a pivotal role during human cytomegalovirus (HCMV) primary infection. Indeed, HCMV infection of primary fibroblasts rapidly triggers strong induction of type I interferons (IFN-I), accompanied by proinflammatory cytokine release. Here, we show that primary human foreskin fibroblasts (HFFs) infected with a mutant HCMV TB40/E strain unable to express UL83-encoded pp65 (v65Stop) produce significantly higher IFN-β levels than HFFs infected with the wild-type TB40/E strain or the pp65 revertant (v65Rev), suggesting that the tegument protein pp65 may dampen IFN-β production. To clarify the mechanisms through which pp65 inhibits IFN-β production, we analyzed the activation of the cGAS/STING/IRF3 axis in HFFs infected with either the wild type, the revertant v65Rev, or the pp65-deficient mutant v65Stop. We found that pp65 selectively binds to cGAS and prevents its interaction with STING, thus inactivating the signaling pathway through the cGAS/STING/IRF3 axis. Consistently, addition of exogenous cGAMP to v65Rev-infected cells triggered the production of IFN-β levels similar to those observed with v65Stop-infected cells, confirming that pp65 inactivation of IFN-β production occurs at the cGAS level. Notably, within the first 24 h of HCMV infection, STING undergoes proteasome degradation independently of the presence or absence of pp65. Collectively, our data provide mechanistic insights into the interplay between HCMV pp65 and cGAS, leading to subsequent immune evasion by this prominent DNA virus.
IMPORTANCE Primary human foreskin fibroblasts (HFFs) produce type I IFN (IFN-I) when infected with HCMV. However, we observed significantly higher IFN-β levels when HFFs were infected with HCMV that was unable to express UL83-encoded pp65 (v65Stop), suggesting that pp65 (pUL83) may constitute a viral evasion factor. This study demonstrates that the HCMV tegument protein pp65 inhibits IFN-β production by binding and inactivating cGAS early during infection. In addition, this inhibitory activity specifically targets cGAS, since it can be bypassed via the addition of exogenous cGAMP, even in the presence of pp65. Notably, STING proteasome-mediated degradation was observed in both the presence and absence of pp65. Collectively, our data underscore the important role of the tegument protein pp65 as a critical molecular hub in HCMV's evasion strategy against the innate immune response.
KEYWORDS: human cytomegalovirus, IFI16, STING, cGAS, innate immunity, interactome, interferons, pp65
INTRODUCTION
Human cytomegalovirus (HCMV), a member of the family Herpesviridae, is a widespread pathogen with a seroprevalence that ranges from 50% to 90% depending on the geographical area and socioeconomic factors. The virus establishes a lifelong infection that is asymptomatic in the immunocompetent host despite the occurrence of periodic reactivation and subsequent virus-shedding episodes. However, reactivation in the immunocompromised host or infection of the immunologically naive fetus in utero can cause significant morbidity and mortality. Indeed, congenital HCMV infection can lead to abortion or dramatic disabilities, such as deafness and mental retardation (1, 2).
Pathogen recognition receptors (PRRs) detect viral components, such as viral nucleic acid, and subsequently lead to the induction of type I interferons (IFN-I), including IFN-α/β, which in turn triggers the expression of numerous IFN-stimulated genes (ISGs) (3). PRRs may be located either on the cell membrane, e.g., Toll-like receptors (TLRs), or in the cytosol and the nucleus; the latter group includes retinoic acid-inducible gene I (RIG-I)-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors, and several proteins involved in the DNA damage response (4–7). Intracellular double-stranded DNA (dsDNA) receptors, such as DNA-dependent activator of interferon-regulatory factors (DAI; also known as ZBP1) (8, 9) and IFN-γ-inducible protein 16 (IFI16), have been shown to recognize HCMV components (10–16). More recent studies have demonstrated that the dsDNA receptor cyclic GMP/AMP (cGAMP) synthase (cGAS) is activated upon HCMV DNA binding and synthesizes the second messenger cGAMP (17–21). Subsequently, cGAMP binds to the endoplasmic reticulum (ER) transmembrane adaptor protein stimulator of IFN genes (STING) and triggers its translocation from the ER to perinuclear punctate structures in order to induce IFN-I induction via TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3) (13, 22, 23).
On the opposing side, viruses have adopted various strategies to evade host innate immune responses and establish persistent infection (14, 24–26). In particular, herpesviruses have evolved strategies that target distinct steps in the signaling of IFN-I production (27–29). Su and Zheng (30) showed that the herpes simplex virus 1 (HSV-1) tegument protein UL41 was involved in counteracting the cGAS/STING-mediated DNA-sensing pathway. HSV-1 ICP0, a viral E3 ubiquitin ligase, has been shown to promote proteasome-dependent degradation of the DNA sensor IFI16 (31, 32). Another HSV-1 immediate-early gene product, ICP27, has been demonstrated to antagonize IFN-I signaling by targeting the TBK1-activated STING signalosome, followed by accumulation of IRF3 in the nucleus (33), or by downregulating STAT-1 phosphorylation and accumulation in the nucleus (34). Finally, VP16, an abundant 65-kDa HSV-1 virion phosphoprotein, has been shown to inhibit IRF3 recruiting of its coactivator CREB-binding protein, thereby blocking its transactivation activity (35). With respect to HCMV, previous studies by Abate at al. (36) demonstrated that during virus infection, pp65 prevented IRF3 activation in the IFN-I response by inhibiting its nuclear accumulation associated with a reduced IRF3 phosphorylation state. Moreover, pp65 has been shown to bind directly to the nuclear DNA sensor IFI16, which impairs its DNA-dependent oligomerization and triggers its nuclear delocalization, followed by inhibition of subsequent immune signals (26, 37). Consistent with these results, another HCMV tegument protein, pUL82/pp71, was recently identified as a negative regulator of the STING-dependent antiviral response, impairing its cellular trafficking and formation of the TBK1/IRF3 complex (29).
Although these studies demonstrate that tegument proteins can downregulate type I interferon production during HCMV infection, they currently offer only limited insight into the mechanisms these viral proteins rely on to counteract the cGAS/STING/IRF3 axis, leading to reduced IFN-I production. Thus, to fill this gap, we sought to compare the IFN-β response in human foreskin fibroblasts (HFFs) upon infection with a pp65 mutant HCMV that is unable to express UL83-encoded pp65 (v65Stop) with that observed with the wild-type TB40/E strain and the pp65 revertant (v65Rev), viruses that express normal levels of pp65.
We show that primary HFFs infected with HCMV v65Stop produce significantly higher IFN-β levels than HFFs infected with HCMV wild type or v65Rev, suggesting that the tegument protein pp65 impairs IFN-β production. Furthermore, when we analyzed the mode of action, we found that pp65 binds to cGAS and inhibits the release of biologically active cGAMP, preventing its interaction with STING and thus interfering with the cGAS/STING signaling pathway. The finding that the response to exogenous cGAMP is not affected by pp65 confirms that cGAS is the key target of pp65. Finally, we demonstrate that STING undergoes rapid degradation early during infection independently of pp65. Altogether, this work defines a previously unrecognized mechanism underlying HCMV evasion, i.e., cGAS inactivation, which assigns a critical role to pp65 in determining the outcome of host defense and viral pathogenesis.
RESULTS
The HCMV tegument protein pp65 inhibits IFN-β induction early during infection.
It has been demonstrated that infection of HFFs with HCMV induces the production of IFN-I through activation of the cGAS/STING/IRF3 signaling pathway (13, 21, 26, 29). On the opposing side, the HCMV tegument protein pp65 has been shown to inhibit IFN-I expression by preventing IRF3 activation or by inhibiting IFI16-mediated DNA sensing for immune evasion (36), suggesting that the cGAS/STING/IRF3 and/or IFI16 signaling pathways become blocked. Here, we sought to determine whether the immunosuppressive function of pp65 could also be extended to other components of these signaling pathways, i.e., cGAS and STING, thereby interfering with the activation of IFN-I production. For this purpose, we compared IFN-β induction in HFFs infected with a pp65 mutant virus unable to express UL83-encoded pp65 (v65Stop), the TB40/E wild-type virus, or the revertant virus (v65Rev) (37, 38). To this end, HFFs were mock infected, infected with the wild type or v65Rev, or infected with v65Stop at a multiplicity of infection (MOI) of 1, and total RNA, harvested at 6 h postinfection (hpi), was analyzed by reverse transcription-quantitative PCR (RT-qPCR). As shown in Fig. 1A, the IFN-β mRNA levels observed with v65Stop were ∼2.7-fold higher than those observed with wild-type- or v65Rev virus-infected cells, suggesting that HCMV pp65 impairs IFN-β production. Interestingly, HFF treatment with the synthetic dsRNA poly(I·C), which mimics RNA virus infection and induces antiviral immune responses by promoting the production of both type I IFNs (39), induced IFN-β levels similar to those observed in cells infected with v65Stop.
FIG 1.
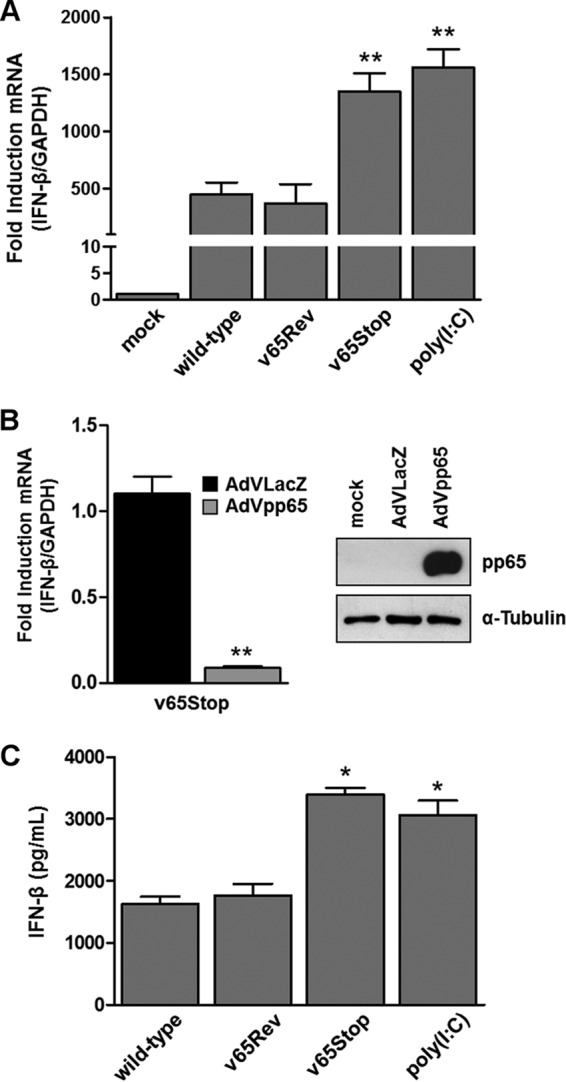
Inhibition of IFN-β response by HCMV pp65. (A) HFFs were infected at an MOI of 1 with wild-type, v65Rev, or v65Stop virus and processed by RT-qPCR. Kinetics analysis results for IFN-β mRNA expression following HCMV versus mock infection were normalized to those for GAPDH expression and are shown as mean fold changes plus SD (**, P < 0.01; one-way ANOVA followed by Bonferroni's posttest for comparison of treated versus untreated cells). (B) (Left) HFFs were transduced with AdVLacZ or AdVpp65 at an MOI of 50. Afterward, the cells were infected with v65Stop (MOI = 1). At 6 hpi, IFN-β mRNA expression was normalized to that of GAPDH and is shown as the mean fold change plus SD (**, P < 0.01; unpaired t test for comparison of AdVpp65- versus AdVLacZ-transduced cells). (Right) The efficiency of pp65 overexpression was analyzed by Western blotting with anti-pp65 monoclonal antibody; α-tubulin was included as a loading control. Experiments were repeated at least three times, and one representative result is shown. (C) HFFs were infected with the wild type, v65Rev, or v65Stop at an MOI of 1 or stimulated with poly(I·C) (4 μg/ml). Supernatants were collected at the indicated times postinfection and assessed by ELISA for IFN-β production. The results are shown as mean fold change plus SD [*, P < 0.05; one-way ANOVA followed by Bonferroni's posttest for comparison of wild-type/v65Rev- versus v65Stop/poly(I·C)-treated cells].
To provide further evidence supporting the physiological relevance of pp65 in the regulation of IFN-β production, HFFs transduced with an adenovirus-derived vector (AdV) constitutively expressing pp65 protein (AdVpp65) or with control vector (AdVLacZ) at an MOI of 50 for 24 h were infected with v65Stop for 6 h (MOI = 1). RT-qPCR analysis revealed that IFN-β mRNA transcription was significantly decreased in AdVpp65-infected cells compared with AdVLacZ-infected cells (∼90% reduction) (Fig. 1B). Taken together, these results support the immunosuppressive role of pp65 in downregulating IFN-β production in HCMV-infected cells and its contribution to innate immune response evasion.
Next, we wanted to verify whether higher levels of IFN-β mRNA in v65Stop-infected HFFs correlated with an increase in the production of biologically active IFN-β protein. For this purpose, supernatants from HFFs infected with wild-type, v65Rev, and v65Stop viruses were harvested at 24 hpi and assessed by enzyme-linked immunosorbent assay (ELISA) for IFN-β production. In parallel, HFFs were stimulated with poly(I·C). As shown in Fig. 1C, consistent with the results obtained with RT-qPCR, when cells were infected with the v65Stop virus or stimulated with poly(I·C), the levels of IFN-β secreted at 24 hpi were significantly higher than those observed with HFFs infected with wild-type or v65Rev virus, confirming that in the presence of HCMV pp65, the transduction pathway leading to IFN-β production is impaired (Fig. 1C).
HCMV pp65 dampens IFN-β production by inactivating the cGAS/STING/IRF3 axis.
HCMV infection can be detected by multiple innate sensing pathways, including TLR2 and TLR9, in diverse cell types (14, 40, 41). In addition to TLRs, the HCMV dsDNA genome directly engages cytosolic or nuclear DNA-sensing pathways, including DAI/ZBP1 (42), AIM2 (43, 44), the cGAS/STING/IRF3 axis, and IFI16 (13, 20, 21, 33, 39, 45–47).
To better understand how pp65 dampens IFN-β production during HCMV infection, we generated gene knockout (KO) variants in HFFs through clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology. Primary cell lines carrying mutations in genes encoding cGAS (cGAS KO), STING (STING KO), and IFI16 (IFI16 KO) were generated based on three different guide RNAs (gRNAs). Verification of genetic KO was carried out by RT-qPCR, Western blotting, and TIDE (tracking of indels by decomposition) analysis (48). As shown in Fig. 2, expression of cGAS, STING, and IFI16 genes was efficiently abrogated at both the mRNA (Fig. 2A) and protein (Fig. 2B) levels. Moreover, a TIDE analysis of the STING and IFI16 knockdown populations showed overall knockdown efficiencies of 87% (Fig. 2C) and 82% (Fig. 2D), respectively. This shows that over 80% of the cells carry an indel (insertion or deletion) in the targeted gene that leads to a frameshift in the reading frame, rendering the gene nonfunctional. Unfortunately, due to a technical limitation of the TIDE analysis, we have been unable to confirm cGAS knockout in CRISPR/Cas9 expressors. This limitation is likely due to the high number of repeats upstream and downstream of the region of the cGAS gene (MB21D1) targeted by the gRNA, which makes it nearly impossible to generate a clean target side-spanning PCR amplicon. Furthermore, this region displays an unusually high GC content, which, coupled with the high repeat number, is likely responsible for the low signal-to-noise ratio and the ensuing early sequencing termination that has hampered our attempts. However, the Western blot and qPCR data clearly show that the majority of the cells are silenced for cGAS (Fig. 2A and B).
FIG 2.

Generation of specific gene knockout cell lines by CRISPR/Cas9-mediated genome editing. Knockout gene variants in HFFs for cGAS (cGAS KO), STING (STING KO), and IFI16 (IFI16 KO), were generated using CRISPR-Cas9 technology. The efficiency of IFI16, cGAS, and STING protein depletion was assayed by RT-qPCR for cGAS, STING, IFI16, and the GADPH housekeeping gene (the data are shown as mean fold changes plus SD; ***, P < 0.001 by two-way ANOVA followed by Bonferroni's posttest for comparison of KO versus WT cells) (A); by Western blot analysis for cGAS, STING, IFI16, and vinculin as a loading control (B); and by TIDE analysis to quantify indel frequencies and composition using PCR amplicons spanning the sgRNA target sites for Sanger sequencing and subsequent analysis with TIDE software (http://tide.nki.nl) (C and D).
We therefore sought to determine whether the KO cell lines exhibited a dependence on these three genes for HCMV-induced IFN-β expression. Consistent with the above-mentioned results (Fig. 1), elevated IFN-β mRNA induction was observed at 6 hpi in normal HFFs infected with v65Stop, whereas infection with wild-type virus or v65Rev virus triggered lower, albeit significant, levels of IFN-β mRNA expression. In contrast, induction of IFN-β mRNA expression was completely abolished in HFFs lacking cGAS or STING independent of the type of virus used for infection. Interestingly, although dramatically decreased, minor residual IFN-β induction was still observed in IFI16 KO cells (Fig. 3A).
FIG 3.

The cGAS/STING axis mediates IFN-β production during HCMV infection. (A) Control (WT), cGAS KO, STING KO, and IFI16 KO HFFs were infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1. Six hours later, IFN-β mRNA expression was processed by RT-qPCR. The values were normalized to GAPDH mRNA and plotted as fold induction over WT HFFs. The RT-qPCR data are shown as mean fold changes plus SD (*, P < 0.05; ***, P < 0.001; two-way ANOVA followed by Bonferroni's posttest for comparison of KO versus WT cells). (B) WT, cGAS KO, STING KO, or IFI16 KO HFFs were infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1. Supernatants from the cells were collected at 24 hpi and analyzed by IFN-β ELISA. The results are shown as the mean fold change plus SD (*, P < 0.05; ***, P < 0.001; two-way ANOVA followed by Bonferroni's posttest for comparison of KO versus WT cells). (C and D) WT, cGAS KO, STING KO, and IFI16 KO HFFs were transfected with poly(I·C). IFN-β mRNA modulation was assessed by RT-qPCR 6 hpt (C) or IFN-β ELISA 24 hpt (D). The results are shown as the mean fold change plus SD [***, P < 0.001; two-way ANOVA followed by Bonferroni's posttest for comparison of poly(I·C)-transfected cells versus untransfected cells].
Taken together, these results confirm that cGAS and STING, and to a lesser extent IFI16, are required for early immune activation by HCMV infection, consistent with the results reported by other investigators (21, 49).
Next, to address whether disruption of the IFI16/cGAS/STING pathway affected the secretion of IFN-β, we performed an ELISA specific for IFN-β using supernatants obtained from HFFs permanently depleted of IFI16, cGAS, or STING, respectively, and then infected with the wild type, v65Rev, or v65Stop for 24 h. A significant decrease in IFN-β production was observed in cells depleted of cGAS and STING and then infected with wild-type, v65Rev, or v65Stop virus (Fig. 3B), thus confirming that these proteins are necessary for the effective production of IFN-β during HCMV infection, independent of the presence of pp65. In agreement with the RT-qPCR results, residual induction of IFN-β release was observed in HCMV-infected IFI16-deficient HFFs compared with HFFs devoid of cGAS and STING. Altogether, these results indicate that cGAS and STING, and to a lesser extent IFI16, are essential to mount abundant IFN-β responses to HCMV.
To exclude potential off-target effects of the cGAS-, STING-, and IFI16-directed CRISPR knockout, we assessed innate immune responses to the synthetic dsRNA poly(I·C), known to trigger RIG-I activation (39). In this case, both IFN-β expression and release were not affected by knocking out the above-mentioned genes, thereby ruling out nonspecific off-target effects (Fig. 3C and D).
pp65 inhibits enzymatic activity of cGAS.
To define the relative roles of cGAS and pp65 interplay in innate sensing, we examined the production of cGAMP—the product of cGAS—in HFFs upon infection with wild-type, v65Rev, or v65Stop virus. cGAMP activity was measured in cell extracts using a modified bioassay based on the original method of Orzalli et al. (50) that analyzes the induction of IFN-β transcripts, as a marker of cGAMP activity, in permeabilized secondary reporter cells (HFFs) at 24 hpi. As shown in Fig. 4A, an increase in cGAMP activity was observed in HCMV-infected HFFs. However, the lack of pp65 in cells infected with v65Stop resulted in cGAMP activity more robust than that in wild-type and v65Rev-infected cells, indicating that interaction of pp65 with cGAS impairs cGAMP production.
FIG 4.

HCMV pp65 inhibits cGAS activity. (A) HFFs were infected with the wild type, v65Rev, or v65Stop at an MOI of 1 for 24 h. Extracts from the infected cells were prepared, DNase and heat treated, and incubated with permeabilized HFFs for 6 h. IFN-β RNA induction was analyzed by RT-qPCR and normalized to that of GAPDH and is shown as the mean fold change plus SD following HCMV versus mock infection (**, P < 0.01; one-way ANOVA followed by Bonferroni's posttest). (B) HFFs were electroporated with pp65 Halo-WT or left untransfected and then infected with v65Stop at an MOI of 1 for 24 h. cGAMP was harvested at 24 hpi and assayed on HFFs. IFN-β mRNA induction was measured in HFFs at 6 hpi by RT-qPCR (***, P < 0.001; one-way ANOVA followed by Bonferroni's posttest for comparison of v65Stop-HaloTag-transfected cells versus v65Stop-untransfected cells). (C) Cells were transduced as described for panel B and 24 h later transfected with poly(dA-dT) (4 μg/ml) for 24 h and assayed on HFFs. IFN-β mRNA induction was measured in HFFs at 6 h by RT-qPCR [***, P < 0.001; one-way ANOVA followed by Bonferroni's posttest for comparison of poly(dA-dT)-HaloTag-transfected cells versus poly(dA-dT)-untransfected cells]. (D) WT, cGAS KO, STING KO, and IFI16 KO HFFs were infected with the wild type, v65Rev, or v65Stop at an MOI of 1. cGAMP was harvested at 24 hpi and assayed on HFFs. IFN-β mRNA induction was measured in HFFs at 6 hpi by RT-qPCR (***, P < 0.001; two-way ANOVA followed by Bonferroni's posttest). (E) HFFs were transfected with synthetic 2′3′-cGAMP or 2′3′-cGAMP control (2 μg/ml) or HFFs were electroporated with pp65 Halo-WT and then transfected with 2′3′-cGAMP. IFN-β mRNA induction was analyzed by RT-qPCR at the time points indicated and normalized to that of GAPDH. The experiment was repeated six times, and no statistically significant differences by unpaired t test analysis were observed between cells transfected with cGAMP alone versus cGAMP plus pp65 Halo-WT.
To understand whether pp65 is able to modulate cGAMP activity by itself during viral replication, HFFs transfected with a HaloTag fusion plasmid expressing the full-length pp65 (pp65 Halo-WT) were infected with HCMV v65Stop. At 6 hpi, the induction of IFN-β transcripts in permeabilized secondary reporter cells (HFFs) was measured (Fig. 4B). As expected, HFFs infected with v65Stop exhibited cGAMP activity significantly higher than that of mock-infected HFFs. Of note, the ability of v65Stop to induce IFN-β was almost completely ablated (∼80%) after the expression of pp65 Halo-WT, further confirming that pp65 is crucial for cGAS activity suppression.
Next, we sought to determine whether the suppression exerted by pp65 was specific for HCMV DNA or whether it could be extended to other cyclic dinucleotides that are sensed by cGAS. Interestingly, a similar pattern of cGAMP activity modulation was observed following transfection of HFFs with the dsDNA synthetic analog poly(dA-dT), indicating that pp65 acts directly on cGAS activity independently of the type of viral DNA, whether viral or synthetic (Fig. 4C).
As the DNA sensor IFI16 has been recently reported to cooperate with cGAS for DNA sensing in fibroblasts (50), human keratinocytes (46), and human macrophages (39) and to modulate STING activation in keratinocytes (46), we next tested whether IFI16 would be able to influence cGAS activity for the production of the second messenger cGAMP. For this purpose, cGAS KO, STING KO, and IFI16 KO HFFs (39) were infected with wild-type, v65Rev, or v65Stop virus, and whole-cell extracts were harvested at 24 hpi and analyzed for the presence of cGAMP activity. As shown in Fig. 3D, depletion of cGAS, STING, and IFI16 resulted in a significant reduction in IFN-β production, confirming the cooperation of cGAS and IFI16 in cGAMP production upon HCMV infection.
Finally, to understand if the inhibitory activity of pp65 is limited to cGAS or could be extended to downstream components of the cGAS/STING/IRF3 axis, we tested whether pp65 affects the activation of STING by exogenous addition of cGAMP (39, 46). For this purpose, HFFs were transfected with synthetic 2′3′-cGAMP or with the linearized 2′3′-cGAMP control, as described by Jønsson et al. (39) and Almine et al. (46), and the IFN-β gene expression response over time was quantified. As shown in Fig. 4E, the delivery of synthetic cGAMP induced the expression of IFN-β mRNA, peaking at 24 h posttransfection (hpt), even in the presence of pp65 Halo-WT, indicating that pp65 inhibitory activity is limited to cGAS and can be bypassed by direct addition of 2′3′-cGAMP. As expected, HFFs transfected with 2′3′-cGAMP control exhibited a severely blunted response.
pp65 selectively interacts with cGAS.
To investigate whether cGAS localization could be regulated during HCMV infection, HFFs were mock infected or infected with wild-type, v65Rev, or v65Stop virus (MOI = 1). The intracellular localization of pp65 and cGAS was assessed by confocal microscopy at 2 hpi. In both mock- and HCMV-infected cells, cGAS was localized in the cytoplasm, as previously reported (17, 27, 45, 51). Interestingly, cGAS-defined puncta also occurred in the nucleus (Fig. 5A), in accordance with the results of Orzalli et al. (50), where they colocalized with pp65. Analysis of three-dimensional (3D) reconstructions created using confocal z stacks to enhance the colocalization analysis confirmed this observation (Fig. 5A, far right). Because pp65 and a subset of cGAS showed colocalization, we hypothesized that they could form a heterocomplex. To learn more about the basis of a pp65-cGAS interaction, we attempted to monitor the pp65-cGAS interaction during HCMV infection in situ using a proximity ligation assay (PLA). PLA allows the detection of adjacent proteins through the use of antibodies that recognize two proteins located within a maximum distance of 40 nm from each other (37). Mock-, wild-type-, v65Rev-, and v65Stop-infected HFFs (MOI = 1) were fixed at 2 hpi and stained with anti-pp65 and anti-cGAS antibodies, and their interaction was analyzed by PLA. As shown in Fig. 5B, cGAS and pp65 were mostly found in close proximity early during HCMV infection with the wild type and v65Rev.
FIG 5.

pp65/cGAS interaction. (A) HFFs were infected with wild-type, v65Rev, or v65Stop virus (MOI = 1) or left uninfected (mock) and subjected to IIF at 2 hpi. pp65 (green) and cGAS (red) were visualized using primary antibodies, followed by secondary-antibody staining in the presence of 10% HCMV-negative human serum. Nuclei were counterstained with TO-PRO-3 (blue). Images were generated by confocal microscopy; the far-right column shows 3D image reconstruction using the confocal z stacks. Digitally reconstructed 3D images were generated for at least 5 fields per condition; representative images are shown. (B) A PLA was performed to detect protein-protein interactions using fluorescence microscopy. The signal was detected as distinct fluorescent dots in the Texas Red channel when cells reacted with the indicated pairs of primary antibodies, followed by PLA to assess the interactions between pp65 and cGAS. (C) Coimmunoprecipitation from virus-infected or mock-infected cell lysates. HFFs were infected with wild-type, v65Rev, or v65Stop virus (MOI = 1) and harvested at 2 hpi. Immunoprecipitations were performed using antibodies against pp65 or without antibody as a negative control (CTRL). Immunoprecipitated proteins were detected by Western blotting analyses using antibodies against pp65, cGAS, and STING. Nonimmunoprecipitated whole-cell extracts (Input) were immunoblotted (IB) using anti-pp65, anti-cGAS, and anti-STING antibodies. (D) (Left) Immunoprecipitation was performed as described for panel C, except that the samples were split in two and half were treated with benzonase (1 U/μl) for 2 h on ice, followed by immunoprecipitation using antibodies against pp65. (Right) The mock cell lysate used in the IP depicted on the left was run on an ethidium bromide-stained (0.8%) agarose gel. −, IP in the absence of benzonase; +, IP in the presence of benzonase. (E) Mapping the region of pp65 required for its interaction with cGAS. Wild-type pp65 (pp65 Halo-WT) and serial deletion mutants of pp65 (pp65 Halo-ΔN and pp65 Halo-ΔC) were used to immunoprecipitate lysates of HFFs transiently expressing pp65 HaloTag. Interaction was detected by Western blotting using antibodies against cGAS.
Given the novelty of these results, pp65-cGAS interaction was also confirmed by the immunoprecipitation (IP) of pp65 from lysates of infected cells at 2 hpi with an anti-pp65 monoclonal antibody (MAb) or with an unrelated MAb of the same isotype as a negative control (CTRL). The immunoprecipitates were then analyzed by immunoblotting with antibodies directed against cGAS or pp65. The presence of pp65 and cGAS in all the protein extracts was monitored by the analysis of input control samples (nonimmunoprecipitated whole-cell extracts) (Fig. 5C). As shown in Fig. 5C (left and middle), a band corresponding to cGAS was detectable in precipitates of wild-type- and v65Rev-infected cell lysates when pp65 was precipitated with an antibody against virus pp65. The specificity of this interaction was verified by the observation that no signal was detected in immunoprecipitates obtained from v65Stop-infected cell lysates (Fig. 4C, right) or in samples with CTRL antibody (Fig. 5C). Moreover, in coimmunoprecipitation experiments with STING, no interaction with pp65 could be observed (Fig. 5C), suggesting that pp65 interacts with cGAS but not with STING during HCMV infection.
It has been demonstrated by our group and others that pp65 binds to HCMV DNA (26, 37). DNA-binding proteins can associate during immunoprecipitation due to their adjacent binding on DNA rather than due to protein-protein interactions. To determine whether nucleic acid is required for the pp65/cGAS association, pp65 was immunoprecipitated from HCMV-infected cell lysates using anti-pp65 MAb in the presence or absence of the DNA-degrading enzyme benzonase and then probed by Western blotting using an anti-cGAS antibody. cGAS was detected in immunoprecipitates from infected lysates in both the absence and presence of benzonase, whereas no migrating bands corresponding to cGAS protein were detected in the absence of primary antibodies (Fig. 5D, left). The activity of benzonase on nucleic acid was confirmed when the cell lysates shown in Fig. 4D were examined on an ethidium bromide-stained agarose gel (Fig. 5D, right).
pp65 displays a bipartite structure, with a conserved N-terminal domain (∼386 residues), a divergent linker region, and a conserved C terminus (C-terminal domain [CTD]; ∼90 residues) (26). Thus, to learn more about the basis of the pp65/cGAS interaction, we employed HaloTag technology to generate HaloTag fusion plasmids expressing the full-length pp65 (pp65 Halo-WT), the N-terminal domain (pp65 Halo-ΔC; deletion of residues 371 to 561), or the C terminus (pp65 Halo-ΔN; deletion of residues 1 to 414). HFFs were transfected with the above-described constructs, and 72 h later, IP was carried out using a MAb recognizing HaloTag or control antibodies. Immunoprecipitated proteins were examined by Western blotting using antibodies recognizing cGAS. As depicted in Fig. 4E, constructs harboring the N-terminal residues 1 to 414 could efficiently bind cGAS (Fig. 5E, left and right lanes), while lack of this domain (middle lane) abolished cGAS binding. We can therefore conclude that the N-terminal domain is necessary and sufficient for pp65 interaction with cGAS.
pp65 does not interfere with STING proteasome degradation during HCMV infection.
Recent work by Fu et al. (29) has shown that the HCMV tegument protein pUL82/pp71 binds to and inhibits STING-mediated signaling to evade innate antiviral immunity. To investigate whether pp65 could also interfere with STING—the downstream component of cGAS in the signaling pathway leading to IFN-β production—we examined the levels of STING expression at different times postinfection in mock-infected HFFs or upon v65Rev or v65Stop infection (MOI = 1). As shown in Fig. 6A, cells infected with wild-type, v65Rev, and v65Stop viruses displayed comparable levels of cGAS up to 24 hpi that were similar to those in mock-infected HFFs. In contrast, infection with both v65Rev and v65Stop induced degradation of STING within 24 hpi, suggesting that STING expression is independent of the presence of pp65. Moreover, the finding that STING is activated in the first hours posttransfection and then disappears within 24 h in cells transfected with the synthetic dsDNA poly(dA-dT) (Fig. 6B) suggests that DNA sensing, independent of pp65, is responsible for STING disappearance (29, 52–55).
FIG 6.

STING undergoes proteasome degradation. (A) HFFs were infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1. Lysates were prepared at the indicated time points and subjected to Western blot analysis for pp65, cGAS, STING, and α-tubulin. (B) Western blot analysis for STING in cells transfected with poly(dA-dT) (4 μg/ml) at the indicated time points. Lysates were also stained for α-tubulin as a loading control. (C) HFFs infected with wild-type, v65Rev, and v65Stop viruses (MOI = 1) were treated with MG132 or DMSO. Cells were harvested at 6 and 24 hpi and processed for Western blot analyses with antibodies against STING. Lysates were also stained for pp65 and with α-tubulin as a loading control. (D) Coimmunoprecipitation from virus-infected or mock-infected cell lysates. HFFs were infected with wild-type, v65Rev, or v65Stop virus (MOI = 1) and harvested at 2 hpi. At the indicated time points, total cell protein extracts were immunoprecipitated for ubiquitin and stained with anti-STING antibodies. Immunoprecipitation of ubiquitin-conjugated proteins was performed using a UbiQapture-Q kit (Enzo Life Science). (E) Cells were infected as described for panel D. Immunoprecipitations were performed using ubiquitin-K48-specific antibodies. The immunoprecipitated proteins were detected by Western blot analyses using antibodies against STING. Nonimmunoprecipitated whole-cell extracts (Input) were immunoblotted using anti-STING and with α-tubulin antibody as a loading control.
We hypothesized that disappearance of STING before 24 hpi could be caused by rapid proteasomal degradation. To test this hypothesis, the effect of proteasomal inhibition on STING protein levels was assessed early during HCMV infection. HFFs were pretreated with the proteasome inhibitor MG132 for 30 min and then infected with either the wild type, v65Rev, or v65Stop for 6 or 24 h. As shown in Fig. 6C, STING levels were rescued by MG132, in contrast to cells treated with the solvent dimethyl sulfoxide (DMSO), indicating that STING undergoes proteasomal degradation early during HCMV infection.
To test whether ubiquitination is responsible for proteasomal degradation of STING, as previously reported (56), HFFs were infected with wild-type, v65Rev, or v65Stop virus (MOI = 1), and ubiquitinated proteins were immunoprecipitated from the cell lysates by anti-ubiquitin antibodies, followed by detection of STING with specific anti-STING antibodies. As shown in Fig. 6D, top, STING was immunoprecipitated by anti-ubiquitin antibodies from HCMV-infected cell lysates, suggesting that HCMV infection induces ubiquitination and thus degradation of STING. K48-linked ubiquitination is normally associated with proteasome-mediated protein degradation (56). By using a specific antibody for immunoprecipitation and detection of STING in precipitates of HCMV-infected cells, we confirmed the ubiquitination of STING on K48-linked residues that is apparently induced by HCMV infection (Fig. 6E).
DISCUSSION
Viral DNA is recognized by various PRRs, including DAI/ZBP1 (42), AIM2 (43, 44), IFI16, and cGAS (13, 20, 21, 33, 39, 45–47). cGAS HCMV DNA sensing during the early phase of infection leads to the stimulation of the STING/TBK1/IRF3 signaling pathway, followed by IFN production. The IFN response antagonizes virus infection via the repression of viral replication, elimination of virus-infected cells, and activation of adaptive immune responses (16, 20, 21, 51, 57, 58). On the opposing side are diverse immune evasion strategies that viruses, including herpesviruses, have evolved in order to inhibit the activation of PRRs, such as IFI16 and cGAS, and their downstream signaling cascades (59–63).
Although data are available on the activation of IFN production by HCMV, the mechanisms HCMV relies on to counteract the cGAS signaling pathway are only partially defined. In order to fill this gap, here, we took advantage of three HCMVs: the wild-type TB40/E and the revertant v65Rev, both displaying intact pp65 expression capacity, and v65Stop, unable to express pp65 (38). Our data demonstrate, for the first time, that the HCMV tegument protein pp65 directly binds and inhibits cGAS enzymatic activity, leading to downregulation of IFN-β production (Fig. 7). The interaction of pp65 with cGAS required the presence of the N-terminal domain of pp65, whereas it appeared to be independent of viral DNA, since the digestion of viral DNA by benzonase did not prevent protein interaction, as shown by immunoprecipitation experiments. Consistent with this, addition of exogenous cGAMP to v65Rev-infected cells triggered the production of IFN levels similar to those observed with v65Stop-infected cells, confirming that pp65 inactivation of IFN-β production occurs at the cGAS level. Thus, the functional relevance of the inhibitory activity of viral pp65 against cGAS can be inferred by the observation that infection with a mutant virus that is unable to express pp65 results in a significant increase in cGAS activity accompanied by a significant increase in IFN production.
FIG 7.
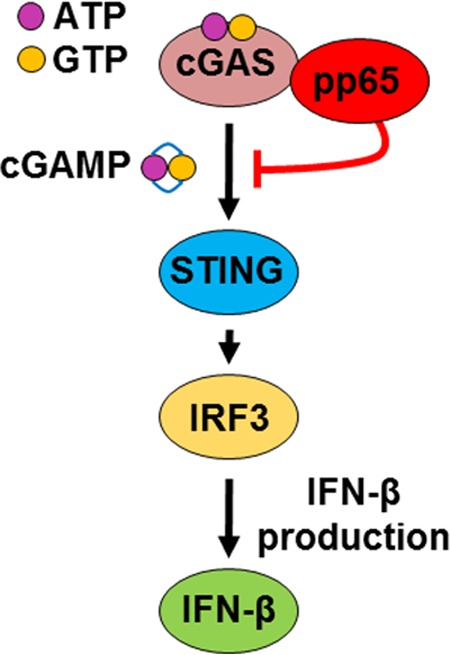
Model depicting the proposed functional role of pp65 modulation of IFN-β activity during HCMV infection.
Recent studies have demonstrated that IFI16 and cGAS cooperate in the activation of STING during the response to exogenous DNA sensing in human keratinocytes (46). In addition, it has been demonstrated that IFI16 is required for early DNA sensing in human macrophages by promoting cGAMP production (39). However, recent findings by Stetson's group (49) demonstrated that IFI16 is not essential for the IFN response to human cytomegalovirus infection. Collectively, these results indicate that the role of IFI16 in the IFN pathway is still a matter of debate. When we infected IFI16-deficient HFFs in an attempt to verify whether IFI16 also participates in the modulation of the IFN-β response to HCMV, residual IFN-β production was observed compared to HFFs depleted of STING and cGAS, suggesting that in human fibroblasts IFI16 is less involved in the signaling transduction pathway leading to IFN production to HCMV. Partially consistent with our results, Paijo et al. (21) demonstrated that HCMV-treated cGAS- or STING-deficient THP-1 monocytes exhibit significantly impaired IFN-β production. In contrast, IFI16-deficient THP-1 monocytes infected with HCMV mounted a robust IFN-β response that was moderately enhanced compared with that of wild-type THP-1 cells. All these discrepancies might be explained by the different methods used to ablate IFI16 (i.e., CRISPR/Cas9, small interfering RNA [siRNA], and short hairpin RNA [shRNA]-mediated IFI16 knockdown); by the different viruses and synthetic DNA employed to stimulate the IFN-I response; and, finally, by the different target cell types employed (i.e., HFFs, keratinocytes, monocytes, and plasmacytoid dendritic cells).
Having demonstrated that HCMV pp65 inhibits cGAS activation, we next investigated whether pp65 could also inactivate STING, the downstream partner of cGAS. Recently, Fu et al. (29) demonstrated that another HCMV tegument protein, namely, pUL82/pp71, binds to and inhibits STING-mediated signaling, leading to viral innate immune evasion. Our results showed that pp65 does not bind to STING and that regulation of its activity is independent of pp65, as its degradation is observed within 24 hpi in both the presence and absence of the pp65 protein. This deduction was generated from the following observations: (i) STING degradation is observed upon infection with the wild type, v65Rev, or v65Stop; (ii) immunoprecipitation experiments showed that pp65 does not bind to STING; (iii) STING degradation is also observed in the presence of unrelated dsDNA, such as synthetic poly(dA-dT); and (iv) STING degradation is not modulated by pp65. In addition, in the present study, we report that STING undergoes ubiquitination followed by degradation in HFFs infected with the wild type, v65Rev, or v65Stop within 6 h of infection. The following two findings led us to this conclusion: first, pretreatment with MG132, a proteasome inhibitor, prevented STING degradation; second, STING was ubiquitinated at Lys48, a marker of degradation by ubiquitination. Our results are consistent with those of previous studies that delineated the following model of STING activation and downstream signaling (Fig. 7). Intracellular dsDNA induces autophagy and the trafficking of STING/TBK1 through the Golgi network to endosomal compartments that harbor members of the IRF and NF-κB family. These transcription factors become activated, and numerous immune-related genes are induced, such as those encoding type I IFN and a variety of cytokines and chemokines. STING is then degraded, and the signaling process is halted. These events ensure the transient production of host defense genes required for direct antimicrobial effects, as well as stimulating the adaptive immune response. Eventual suppression of STING function also ensures that the chronic production of cytokines is prevented, thus avoiding the consequences of inflammatory disease.
In conclusion, our data identify a previously unknown role of pp65 in downregulating the cGAS/STING/IRF3 axis and thus IFN-β production (a proposed model is shown in Fig. 7). The finding that HCMV, by means of its tegument proteins, i.e., pp65 and pUL82/pp71, interferes with the activities of all the components of this signaling pathway (i.e., cGAS, STING, and IRF3) in order to evade the IFN response underlines the relevance of the IFN system in blocking virus replication. Therefore, further delineation of the mechanisms through which HCMV inhibits cGAS/STING/IRF3 signaling not only will help our comprehension of this pathway, but may also facilitate the development of therapeutics aimed at ameliorating the diseases in which the pathway is altered.
MATERIALS AND METHODS
Cells and viruses.
Primary HFFs (ATCC strain SCRC-1041) and human embryonic kidney 293 (HEK 293) cells (Microbix Biosystems Inc.) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS) (Sigma-Aldrich) as previously described (39, 64). The HCMVs used in this study were all bacterial artificial chromosome (BAC) clones. The clones of the endotheliotropic HCMV strain TB40/E (the wild type), the revertant virus (v65Rev), and a mutant virus unable to express UL83-encoded pp65 (v65Stop) were previously generated (38). The viruses were propagated on HFFs and titrated by standard plaque assay (37, 65). HCMV infections were all performed at an MOI of 1, where more than 50% of the cells should be infected at 24 hpi (20, 38).
Antibodies and reagents.
The following primary antibodies were used: rabbit polyclonal anti-cGAS (Sigma-Aldrich), anti-ubiquitin-K48-specific (Millipore), anti-IFI16 (Santo Landolfo), and mouse MAbs and anti-STING (R&D), anti-pp65 (Virusys), anti-HaloTag (Promega), anti-vinculin (Sigma-Aldrich), and anti-α-tubulin (Active-Motive) antibodies. The following conjugated secondary antibodies were used: Alexa Fluor 488 anti-mouse or Alexa Fluor 568 anti-rabbit antibodies (Life Technologies) and horseradish peroxidase-labeled anti-mouse and anti-rabbit antibodies (GE Healthcare). The proteasome inhibitor MG132 (Calbiochem) was used at a concentration of 30 μM (66). Poly(dA-dT) and poly(I·C) (4 μg/ml; InvivoGen) (39, 67) were transfected into the cells using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
Recombinant adenoviral vectors.
AdV expressing pp65 was generated by means of a replacement strategy using recombineering methods (68). Briefly, the UL83 open reading frame (ORF) was amplified using a specific set of primers (forward, AACCGTCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTGGATCCATGGAGTCGCGCGGTCGCCG, and reverse, TATAGAGTATACAATAGTGACGTGGGATCCCTACGTAGAATCAAGACCTAGGAGCGGGTTAGGGATTGGCTTACCAGCGCTACCTCGATGCTTTTTGGGC). In order to accomplish homologous recombination, approximately 200 ng of DNA derived from HCMV-infected cells was electroporated into SW102 bacteria harboring pAdZ5-CV5 vectors. The cells were then plated on minimal medium agar plates containing 5% sucrose and chloramphenicol and incubated at 32°C for 1 day. The colonies that appeared were inoculated into LB broth containing ampicillin and chloramphenicol and LB broth containing chloramphenicol only. In the colonies grown in chloramphenicol only, the UL83 ORF replaced the ampicillin resistance sequence at multiple cloning sites. Colonies were checked by PCR and sequencing. AdVpp65 was cotransfected into HEK 293 cells. To obtain the recombinant adenoviruses, AdZ vectors were transfected into HEK 293 packaging cells. The transfected cells were maintained at 37°C with 5% CO2 until an extensive cytopathic effect was obtained. Viruses were then purified from the infected cultures by freeze-thaw-vortex cycles and assessed for pp65 expression by Western blotting. For cell transduction, HFFs were washed once with phosphate-buffered saline (PBS) and incubated with AdVpp65 at an MOI of 50 in DMEM. After 2 h at 37°C, the virus was washed off, and fresh medium was applied. For all the experiments, a recombinant adenovirus expressing the Escherichia coli β-galactosidase gene (AdVLacZ) was used as a control (65).
Plasmid construction.
The HCMV UL83 sequences were amplified using specific sets of primers (pp65 Halo-WT forward, CGGAATTCATGGAGTCGCGCGGTCG, and reverse, AACTCGAGACCTCGATGCTTTTTGGGCGT; pp65 Halo-ΔC forward, CGGAATTCATGGAGTCGCGCGGTCGCC, and reverse, GCTCTAGAGGTGGTTACGAGTTCTTCGT; pp65 Halo-ΔN forward, GCGAATTCATGCAGTATCGCATCCAGGGCAAG, and reverse, GCTCTAGAACCTCGATGCTTTTTGGGCGT). The 5′ and 3′ primers were engineered to contain EcoRI and XbaI restriction sites. The PCR fragments were subsequently digested and directionally cloned into the corresponding sites of the pHTC HaloTag CMV-neo vector (Promega). The following HaloTag fusion plasmids were constructed: plasmids expressing full-length pp65 (pp65 Halo-WT), the N domain (pp65 Halo-ΔC; deletion of residues 371 to 561), or the C domain (pp65 Halo-ΔN; deletion of residues 1 to 414). The plasmids were then tested by Western blotting, and the nucleotide sequences were confirmed by sequencing. HFFs were transiently transfected using a MicroPorator (Digital Bio) according to the manufacturer's instructions (1,200 V; 30-ms pulse width; one impulse).
RNA isolation and semiquantitative RT-qPCR.
Total RNA was extracted using a NucleoSpin RNA kit (Macherey-Nagel), and 1 μg was retrotranscribed using a Revert-Aid H-Minus First Strand cDNA synthesis kit (Fermentas) according to the manufacturer's protocol. Comparison of mRNA expression between samples (i.e., infected versus untreated) was performed by SYBR green-based RT-qPCR using an Mx3000P apparatus (Stratagene) with the following primers: IFN-β forward, AAACTCATGAGCAGTCTGCA, and IFN-β reverse, AGGAGATCTTCAGTTTCGGAGG and, for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene, GAPDH forward, AGTGGGTGTCGCTGTTGAAGT, and GADPH reverse, AACGTGTCAGTGGTGGACCTG.
Transduction of HFFs with lentiviral CRISPR/Cas9.
The CRISPR/Cas9 system was employed to generate specific gene knockouts in primary human fibroblasts. Specifically, we used a lentiviral CRISPR/Cas9 vector 54 that carries a codon-optimized nuclear-localized Cas9 gene N-terminally fused to the puromycin resistance gene via a T2A ribosome-skipping sequence. Additionally, the vector contains a human U6 promoter driving expression of a gRNA consisting of a gene-specific CRISPR RNA (crRNA) fused to the trans-activating crRNA (tracrRNA) and a terminator sequence.
The gene-specific crRNA sequences cloned were as follows: for IFI16 knockout, 5′-GTACCAACGCTTGAAGACC-3′; for cGAS knockout, 5′-GACTCGGTGGGATCCATCG-3′; and for STING knockout, 5′-GAGCACACTCTCCGGTACC-3′.
Vesicular stomatitis virus G (VSVg)-pseudotyped lentiviral vector-based clustered regularly interspaced short palindromic repeats (lenti-CRISPR) virions were produced by transfecting HEK293T cells with the following plasmids: CRISPR/Cas9 vector, pMD.2G, pRSV-REV, and pMDlg/p-RRE. Viral supernatants were harvested after 72 h and used to transduce fibroblasts by infection in the presence of 4 mg/ml Polybrene. Transduced cells were selected with increasing dosages of puromycin (0.5 μg/ml, 1 μg/ml, and 2 μg/ml) over the course of 14 days postransduction. After selection, successful knockout was confirmed using qPCR and immunoblotting. Additionally, indel frequencies were quantified using TIDE (48); genomic DNA was extracted, and PCR amplicons spanning the single guide RNA (sgRNA) target site were generated. The purified PCR products were then Sanger sequenced, and indel frequencies were quantified using the TIDE software (http://tide.nki.nl). A reference sequence (WT cells) was used as a control.
Immunofluorescence microscopy.
Indirect immunofluorescence (IIF) analysis was performed as previously described (12) using the appropriate dilution of primary antibodies for 1 h at room temperature, followed by 1 h with secondary antibodies in the dark at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) or TO-PRO-3. Finally, coverslips were mounted with Vectashield mounting medium (Vector). Samples were observed using a fluorescence microscope (Olympus IX70) equipped with cellSens Standard microscopy imaging software, or a confocal microscope (Leica TCS SP2). ImageJ software was used for image processing.
PLA.
Proximity ligation assay (DuoLink; Sigma-Aldrich) was performed with a DuoLink PLA kit to detect protein-protein interactions, using fluorescence microscopy according to the manufacturer's protocol. Briefly, HFFs were infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1 for 2 h, fixed for 15 min at room temperature, permeabilized with 0.2% Triton X-100, and blocked with 10% HCMV-negative human serum for 30 min at room temperature. The cells were then incubated with primary antibodies diluted in Tris-buffered saline (TBS)-0.05% Tween for 1 h, washed, and then incubated for another hour at 37°C with species-specific PLA probes under hybridization conditions and in the presence of 2 additional oligonucleotides to facilitate hybridization only in close proximity (∼40 nm). A ligase was then added to join the two hybridized oligonucleotides, thus forming a closed circle. Using the ligated circle as the template, rolling-circle amplification was initiated by adding an amplification solution, generating a concatemeric product extending from the oligonucleotide arm of the PLA probe. Lastly, a detection solution consisting of fluorophore-labeled oligonucleotides was added, and the labeled oligonucleotides were hybridized to the concatemeric products. The signal was detected as distinct fluorescent dots in the Texas Red channel and analyzed by fluorescence microscopy (Olympus IX70). Negative controls consisted of mock-infected cells that were otherwise treated in the same way as the infected cells.
Western blot analysis.
Whole-cell protein extracts were prepared and subjected to Western blot analysis as previously described (12, 69). Briefly, equal amounts of cell extracts were fractionated by electrophoresis on sodium dodecyl sulfate-polyacrylamide gels and transferred to Immobilon-P membranes (Bio-Rad). After blocking with 5% nonfat dry milk in TBS-0.05% Tween, the membranes were incubated overnight at 4°C with the appropriate primary antibodies. The membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. Proteins were detected using an enhanced chemiluminescence detection kit (SuperSignal West Pico chemiluminescent substrate; Thermo Scientific).
Immunoprecipitation assay.
Uninfected or HCMV-infected (MOI = 1) cells were washed with PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.1% SDS, 0.5% deoxycholate, protease inhibitors). Two hundred micrograms of proteins was incubated with 2 μg of specific antibody, or without antibody as a negative control, for 1 h at room temperature with rotation, followed by overnight incubation at 4°C with protein G-Sepharose (Sigma-Aldrich). Immune complexes were collected by centrifugation and washed with RIPA buffer. The Sepharose beads were pelleted and washed three times with RIPA buffer, resuspended in reducing sample buffer (50 mM Tris, pH 6.8, 10% glycerol, 2% SDS, 1% 2-mercaptoethanol), boiled for 5 min, and resolved on an SDS-PAGE gel to assess protein binding by Western blotting. Where indicated, ∼1 U/μl benzonase (Sigma-Aldrich) was added for 2 h on ice as described by Wu et al. (27). Immunoprecipitation of ubiquitin-conjugated proteins was performed using a UbiQapture-Q kit (Enzo Life Science). A total of 25 μg of lysates from cultured cells was used per assay. Samples were added to the tubes containing 20 μl UbiQapture-Q matrix and incubated for 3 h at 4°C in a horizontal rotor mixer. The matrix was then carefully washed, and the ubiquitin-protein conjugates were eluted by addition of 50 μl sample buffer and heating at 95°C for 10 min. The eluted fraction was clarified from the matrix and analyzed by immunoblotting.
ELISA.
The IFN-β secreted in culture supernatants was analyzed using single-analyte human ELISA kits for IFN-β (Human IFN Beta ELISA kit; PBL Assay Science) according to the manufacturer's instructions. All absorbance readings were measured at 450 nm using a Victor X4 multilabel plate reader (PerkinElmer).
cGAMP activity assay.
cGAMP activity assay was performed as previously described (50, 51). Briefly, HFFs were infected with the wild type, v65Rev, or v65Stop or transfected with poly(dA-dT) (4 μg/ml; InvivoGen) or pp65 Halo-WT using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. At the indicated times, cells were washed with PBS and lysed in hypotonic buffer (10 mM Tris, pH 7.4, 10 mM KCl, 1.5 mM MgCl2). Cell extracts were incubated with ∼1 U/μl benzonase (Sigma-Aldrich) for 30 min at 37°C. The cell extracts were then heated at 95°C for 5 min and centrifuged for 5 min at maximum speed (16,100 × g) in an Eppendorf microcentrifuge. HFFs were used as reporter cells to measure cGAMP production. The HFFs were permeabilized as previously described (70) with modifications. Briefly, medium was aspirated from the HFFs, and digitonin permeabilization solution (50 mM HEPES, pH 7.0, 100 mM KCl, 85 mM sucrose, 3 mM MgCl2, 0.2% bovine serum albumin [BSA], 1 mM ATP, 0.1 mM dithiothreitol [DTT], and 10 μg/ml digitonin) was added to the treated cell extracts. Reporter HFFs were incubated with the extracts for 30 min at 37°C, and the extracts were then replaced with supplemented medium. RNA was harvested 6 h after the initial addition of extracts, and RT-qPCR for IFN-β, as a marker of cGAMP activity, and the GADPH gene, as a housekeeping gene, was performed as described above.
Response to exogenous cGAMP.
Synthetic 2′3′-cGAMP (2 μg/ml; InvivoGen) or 2′3′-cGAMP control (2 μg/ml; InvivoGen), also known as 2′5′-GpAp, a linear dinucleotide analog after hydrolysis of 2′3′-cGAMP by phosphodiesterases, were transfected into HFFs using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. At the indicated time points, RNA was harvested, and the IFN-β gene expression response was quantified over time by RT-qPCR.
Statistical analysis.
All statistical tests were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA, USA). The data are presented as means and standard deviations (SD). For comparisons consisting of two groups, means were compared using two-tailed Student t tests; for comparisons consisting of three groups, means were compared using one-way or two-way analysis of variance (ANOVA) with Bonferroni's posttest. Differences were considered statistically significant for P values of <0.05.
ACKNOWLEDGMENTS
This study was supported by the Italian Ministry of Education, University and Research-MIUR (PRIN 2015 to M.D.A., 2015W729WH; PRIN 2015 to V.D., 2015RMNSTA); by research funding from the University of Turin 2017 to M.D.A., S.L., and V.D.; by Regione Piemonte (Italy) (PAR-FCS 2007/2013) to S.L.; by Compagnia di San Paolo (CSP 2014) to C.B.; by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG 2016) to M.G.; and by the Danish Council for Independent Research (4183-00275B) to M.R.J. and C.K.
REFERENCES
- 1.Britt WJ. 2017. Congenital human cytomegalovirus infection and the enigma of maternal immunity. J Virol 91:e02392-. doi: 10.1128/JVI.02392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 3.McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. 2015. Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luecke S, Paludan SR. 2017. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine 98:4–14. doi: 10.1016/j.cyto.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Dempsey A, Bowie AG. 2015. Innate immune recognition of DNA: a recent history. Virology 479-480:146–152. doi: 10.1016/j.virol.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komatsu T, Nagata K, Wodrich H. 2016. The role of nuclear antiviral factors against invading DNA viruses: the immediate fate of incoming viral genomes. Viruses 8:E290. doi: 10.3390/v8100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zevini A, Olagnier D, Hiscott J. 2017. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol 38:194–205. doi: 10.1016/j.it.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 9.Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li T, Diner BA, Chen J, Cristea IM. 2012. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horan KA, Hansen K, Jakobsen MR, Holm CK, Søby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR. 2013. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol 190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dell'Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Lo Cigno I, Biolatti M, Vallino M, Marschall M, Gariglio M, Landolfo S. 2014. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol 88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diner BA, Lum KK, Toettcher JE, Cristea IM. 2016. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 7:e01553-. doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orzalli MH, Knipe DM. 2014. Cellular sensing of viral DNA and viral evasion mechanisms. Annu Rev Microbiol 68:477–492. doi: 10.1146/annurev-micro-091313-103409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knipe DM. 2015. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 479-480:153–159. doi: 10.1016/j.virol.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diner BA, Lum KK, Cristea IM. 2015. The emerging role of nuclear viral DNA sensors. J Biol Chem 290:26412–26421. doi: 10.1074/jbc.R115.652289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. 2013. Cyclic [G(2′,5′)pA(3′,5′)p] Is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhat N, Fitzgerald KA. 2014. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol 44:634–640. doi: 10.1002/eji.201344127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lio C-WJ, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, Sharma S. 2016. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J Virol 90:7789–7797. doi: 10.1128/JVI.01040-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paijo J, Döring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, Kalinke U. 2016. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog 12:e1005546. doi: 10.1371/journal.ppat.1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen MH, Paludan SR. 2017. Viral evasion of DNA-stimulated innate immune responses. Cell Mol Immunol 14:4–13. doi: 10.1038/cmi.2016.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Z, Damania B. 2016. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19:150–158. doi: 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T, Chen J, Cristea IM. 2013. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Stürzl M, Whitby D, Li H, Zhu F. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crow MS, Lum KK, Sheng X, Song B, Cristea IM. 2016. Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit Rev Biochem Mol Biol 51:452–481. doi: 10.1080/10409238.2016.1226250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Y-Z, Su S, Gao Y-Q, Wang P-P, Huang Z-F, Hu M-M, Luo W-W, Li S, Luo M-H, Wang Y-Y, Shu H-B. 2017. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21:231–243. doi: 10.1016/j.chom.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Su C, Zheng C. 2017. HerpeAEs cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein UL41. J Virol 91:e02414-. doi: 10.1128/JVI.02414-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008-17. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orzalli MH, Broekema NM, Knipe DM. 2016. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J Virol 90:8351–8359. doi: 10.1128/JVI.00939-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson KE, Song B, Knipe DM. 2008. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 374:487–494. doi: 10.1016/j.virol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-κB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87:9788–9801. doi: 10.1128/JVI.01440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 78:10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biolatti M, Dell'Oste V, Pautasso S, von Einem J, Marschall M, Plachter B, Gariglio M, De Andrea M, Landolfo S. 2016. Regulatory interaction between the cellular restriction factor IFI16 and viral pp65 (pUL83) AD IFI16 protein stability. J Virol 90:8238–8250. doi: 10.1128/JVI.00923-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chevillotte M, Landwehr S, Linta L, Frascaroli G, Lüske A, Buser C, Mertens T, von Einem J. 2009. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J Virol 83:2480–2490. doi: 10.1128/JVI.01818-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, Sørensen LK, Sun C, Jensen SB, Holm CK, Lebbink RJ, Johannsen M, Nyegaard M, Mikkelsen JG, Kirchhoff F, Paludan SR, Jakobsen MR. 2017. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun 8:14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathinam VAK, Fitzgerald KA. 2011. Innate immune sensing of DNA viruses. Virology 411:153–162. doi: 10.1016/j.virol.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Früh K. 2010. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol 84:585–598. doi: 10.1128/JVI.01748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y, Ma D, Huang H, Lu Y, Liao Y, Liu L, Liu X, Fang F. 2017. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol J 14:34. doi: 10.1186/s12985-016-0673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Liu L, Ma D, Liao Y, Lu Y, Huang H, Qin W, Liu X, Fang F. 2017. Human cytomegalovirus triggers the assembly of AIM2 inflammasome in THP-1-derived macrophages. J Med Virol 89:2188–2195. doi: 10.1002/jmv.24846. [DOI] [PubMed] [Google Scholar]
- 45.Xia P, Wang S, Gao P, Gao G, Fan Z. 2016. DNA sensor cGAS-mediated immune recognition. Protein Cell 7:777–791. doi: 10.1007/s13238-016-0320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almine JF, O'Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA. 2014. Interferon γ-inducible protein (IFI) 16 transcriptionally regulates type I interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289:23568–23581. doi: 10.1074/jbc.M114.554147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinkman EK, Chen T, Amendola M, van Steensel B. 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. 2016. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45:255–266. doi: 10.1016/j.immuni.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773-81. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao D, Wu J, Wu Y-T, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Li J, Chen J, Li Y, Wang W, Du X, Song W, Zhang W, Lin L, Yuan Z. 2015. Hepatitis B virus polymerase disrupts K63-AF STING to block innate cytosolic DNA-sensing pathways. J Virol 89:2287–2300. doi: 10.1128/JVI.02760-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, Lin G, Lian Z, Jiang Z, Sun B. 2015. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog 11:e1005012. doi: 10.1371/journal.ppat.1005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castillo Ramirez JA, Urcuqui-Inchima S. 2015. Dengue virus control of type I IFN responses: a history of manipulation and control. J Interferon Cytokine Res 35:421–430. doi: 10.1089/jir.2014.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalamvoki M, Roizman B. 2014. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci U S A 111:E611-17. doi: 10.1073/pnas.1323414111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis ME, Gack MU. 2015. Ubiquitination in the antiviral immune response. Virology 479-480:52–65. doi: 10.1016/j.virol.2015.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ablasser A. 2016. ReGLUation of cGAS. Nat Immunol 17:347–349. doi: 10.1038/ni.3397. [DOI] [PubMed] [Google Scholar]
- 58.Paludan SR. 2015. Activation and regulation of DNA-driven immune responses. Microbiol Mol Biol Rev 79:225–241. doi: 10.1128/MMBR.00061-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diner BA, Cristea IM. 2015. Blowing off steam: virus inhibition of cGAS DNA sensing. Cell Host Microbe 18:270–272. doi: 10.1016/j.chom.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan YK, Gack MU. 2016. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su C, Zhan G, Zheng C. 2016. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J 13:38. doi: 10.1186/s12985-016-0495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dell'Oste V, Gatti D, Giorgio AG, Gariglio M, Landolfo S, De Andrea M. 2015. The interferon-inducible DNA-sensor protein IFI16: a key player in the antiviral response. New Microbiol 38:5–20. [PubMed] [Google Scholar]
- 63.Landolfo S, De Andrea M, Dell'Oste V, Gugliesi F. 2016. Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World J Virol 5:87–96. doi: 10.5501/wjv.v5.i3.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pignoloni B, Fionda C, Dell'Oste V, Luganini A, Cippitelli M, Zingoni A, Landolfo S, Gribaudo G, Santoni A, Cerboni C. 2016. Distinct roles for human cytomegalovirus immediate early proteins IE1 and IE2 in the transcriptional regulation of MICA and PVR/CD155 expression. J Immunol 197:4066–4078. doi: 10.4049/jimmunol.1502527. [DOI] [PubMed] [Google Scholar]
- 65.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Evers DL, McCarville JF, Dantonel J-C, Huong S-M, Huang E-S. 2006. Evidence that the human cytomegalovirus IE2-86 protein binds mdm2 and facilitates mdm2 degradation. J Virol 80:3833–3843. doi: 10.1128/JVI.80.8.3833-3843.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abe T, Barber GN. 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88:5328–5341. doi: 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. 1996. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol 70:4805–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gugliesi F, Mondini M, Ravera R, Robotti A, de Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2005. Up-regulation of the interferon-inducible IFI16 gene by oxidative stress triggers p53 transcriptional activity in endothelial cells. J Leukoc Biol 77:820–829. doi: 10.1189/jlb.0904507. [DOI] [PubMed] [Google Scholar]
- 70.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]