

ABSTRACT
Interferon-induced transmembrane proteins (IFITMs) are restriction factors that inhibit the infectious entry of many enveloped RNA viruses. However, we demonstrated previously that human IFITM2 and IFITM3 are essential host factors facilitating the entry of human coronavirus (HCoV) OC43. In a continuing effort to decipher the molecular mechanism underlying IFITM differential modulation of HCoV entry, we investigated the roles of structural motifs important for IFITM protein posttranslational modifications, intracellular trafficking, and oligomerization in modulating the entry of five HCoVs. We found that three distinct mutations in IFITM1 or IFITM3 converted the host restriction factors to enhance entry driven by the spike proteins of severe acute respiratory syndrome coronavirus (SARS-CoV) and/or Middle East respiratory syndrome coronavirus (MERS-CoV). First, replacement of IFITM3 tyrosine 20 with either alanine or aspartic acid to mimic unphosphorylated or phosphorylated IFITM3 reduced its activity to inhibit the entry of HCoV-NL63 and -229E but enhanced the entry of SARS-CoV and MERS-CoV. Second, replacement of IFITM3 tyrosine 99 with either alanine or aspartic acid reduced its activity to inhibit the entry of HCoV-NL63 and SARS-CoV but promoted the entry of MERS-CoV. Third, deletion of the carboxyl-terminal 12 amino acid residues from IFITM1 enhanced the entry of MERS-CoV and HCoV-OC43. These findings suggest that these residues and structural motifs of IFITM proteins are key determinants for modulating the entry of HCoVs, most likely through interaction with viral and/or host cellular components at the site of viral entry to modulate the fusion of viral envelope and cellular membranes.
IMPORTANCE The differential effects of IFITM proteins on the entry of HCoVs that utilize divergent entry pathways and membrane fusion mechanisms even when using the same receptor make the HCoVs a valuable system for comparative investigation of the molecular mechanisms underlying IFITM restriction or promotion of virus entry into host cells. Identification of three distinct mutations that converted IFITM1 or IFITM3 from inhibitors to enhancers of MERS-CoV or SARS-CoV spike protein-mediated entry revealed key structural motifs or residues determining the biological activities of IFITM proteins. These findings have thus paved the way for further identification of viral and host factors that interact with those structural motifs of IFITM proteins to differentially modulate the infectious entry of HCoVs.
KEYWORDS: IFITM, viral entry, coronavirus
INTRODUCTION
The interferon (IFN)-mediated innate immune response is the first line of defense against virus infections in vertebrates (1, 2). IFNs execute antiviral activity by binding to their cognate receptors on the cell surface to activate a signaling cascade leading to induction of hundreds of IFN-stimulated genes (ISGs) (3, 4). Among the ISGs, those encoding IFN-induced transmembrane (IFITM) proteins, including IFITM1, IFITM2, and IFITM3, are widely expressed and can be induced by all three types of IFNs in many cell types. The IFITMs localize at the cell plasma membrane and endocytic vesicles and restrict the entry of enveloped RNA viruses from nine viral families (5), including some medically important human pathogens, such as influenza A virus (IAV), dengue virus (DENV), West Nile virus, Zika virus, chikungunya virus, Ebola virus (EBOV), Rift Valley fever virus, human immunodeficiency virus (HIV), and hepatitis C virus (3, 6–15).
Coronaviruses (CoVs) are a large family of enveloped, positive-strand RNA viruses with a broad host range and primarily cause respiratory or enteric diseases, but some of them cause hepatitis, neurological disorders, or cardiomyopathy (16, 17). Human coronaviruses (HCoVs) 229E, OC43, NL63, and HKU1 circulate globally and cause mild upper respiratory tract infections (18) but are occasionally associated with more severe lower respiratory tract diseases in elderly and immunocompromised patients (19). On the other hand, the recently emerging HCoVs, such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), cause severe diseases among infected individuals (20, 21). Thus far, several groups have reported that IFITMs inhibited entry of HCoV-229E, HCoV-NL63, SARS-CoV, and MERS-CoV into their host cells with varying efficiency (8, 22, 23).
Concerning the molecular mechanism underlying IFITM restriction of virus entry, the currently favored hypothesis postulates that the existence of IFITM proteins in the endocytic membranes alters either the membrane curvature or fluidity to make the endosomal membrane rigid and less able to fuse with viral envelopes (24, 25–28). However, several recent findings challenge this hypothesis. First, human IFITM2 and IFITM3 efficiently enhance the infectious entry of HCoV-OC43 via a post-receptor binding/endocytosis mechanism (29). Second, mutation of the SVKS motif in the CD225 domain required for IFITM1 to inhibit HIV-1 entry and for IFITM3 to restrict IAV and dengue virus infection (24, 30) enhances Lloviu virus (LLOV) glycoprotein-mediated entry (see Fig. 11) (31). Third, human IFITM proteins are required for the formation of the human cytomegalovirus virion assembly complex (VAC) and infectious virion secretion (32). The VAC is a perinuclear membrane structure in which the vesicles with endosomal markers occupy the central area and the vesicles with Golgi markers are wrapped around it to form a circle (33). It is possible that IFITMs modulate endosomal trafficking/fusion during VAC formation. All these findings strongly suggest that the IFITM-induced membrane curvature and/or fluidity alterations may not always make the endocytic membranes too “rigid” to fuse but, at least under certain conditions, may facilitate membrane fusion.
FIG 11.

Illustration of IFITM protein structural domains and motifs important for subcellular trafficking, oligomerization, and posttranslational modification. An alignment of human IFITM1 (hIFITM1), IFITM2, and IFITM3 protein sequences using Vector NTI 8.0 software is shown. Five structural domains, i.e., the N-terminal domain (NTD), intramembrane domain (IMD), intracellular loop (CIL), transmembrane domain (TMD), and C-terminal domain (CTD), are indicated. The IMD and CIL domain comprise the canonical CD225 domain. The 20YXXΦ23 motif required for IFITM3 endocytosis and the 122KRXX125 motif that serves as a sorting signal for IFITM1 are underlined in purple. The putative phosphorylation, palmitoylation, and ubiquitination sites are indicated. Two phenylalanine residues in the IMD that promote IFITM oligomerization are indicated by red triangles.
In order to further understand the molecular mechanism underlying the differential modulation of HCoV entry by IFITM proteins, we set out to identify structural motifs important for IFITM protein posttranslational modifications, intracellular trafficking, and oligomerization in modulating the entry of five HCoVs. We found that although both SARS-CoV and NL63 use angiotensin-converting enzyme 2 (ACE2) as their entry receptor, IFITMs differentially modulate the entry of the two viruses. We also found that three distinct mutations in IFITM1 or IFITM3 converted the host restriction factors to enhancers of SARS-CoV and/or MERS-CoV entry. These findings imply that restriction or promotion of virus entry by an IFITM may rely on its fine-tuned interaction with viral and host cellular factors via the structural motifs at the site of viral envelope and cellular membrane fusion. Moreover, posttranslation modification of those structural motifs by host cellular factors may alter IFITM interaction with the components of viral entry machinery and consequently change its potency and/or the nature of modulating the entry of a virus.
RESULTS
Host cellular factors other than viral receptors have a strong impact on antiviral activity of IFITM proteins.
Viral envelope proteins and cellular receptors are the major players in virus entry into their host cells. It is conceivable that IFITM interaction with the viral envelope and/or cellular receptors may play an important role in restriction of virus entry. ACE2, as the common receptor for both SARS-CoV and NL63, provides a unique opportunity to investigate the role of viral receptors in IFITM modulation of HCoV entry (34). Accordingly, we examined the effects of three human IFITM proteins on the entry of four HCoVs, with Lassa fever virus (LASV) and IAV as negative and positive controls, in HEK293 cells and the Huh7.5 hepatoma cell line, which express detectable and undetectable basal levels of IFITMs, respectively (29) (Fig. 1). As anticipated, expression of any of the three IFITMs in either HEK293 or Huh7.5 cells did not inhibit infection by lentiviral particles pseudotyped with the envelope proteins of Lassa fever virus (LASVpp) (Fig. 1B and C). However, all three IFITMs significantly inhibited the infection by lentiviral particles pseudotyped with IAV hemagglutinin 1 (H1) and neuraminidase 1 (N1) (IAVpp), Spike protein (S) of HCoV-229E (229Epp), HCoV-NL63 (NL63pp), SARS-CoV (SARSpp), and MERS-CoV (MERSpp) in both HEK293 (Fig. 1B) and Huh7.5 (Fig. 1C) cells. Comparing the extent of IFITM inhibition in the two cell lines (Fig. 1B and C), IFITM2 and IFITM3 inhibition of IAVpp infection was 4- and 20-fold more potent, respectively, in HEK293 cells than in Huh7.5 cells. On the other hand, IFITM2 and IFITM3 more efficiently inhibited entry of all four HCoVpp, particularly MERSpp and 229Epp, in Huh7.5 cells. The steady-state levels of IFITM1 were lower than those of IFITM2 and IFITM3 in both HEK293 and Huh7.5 cells, which may, at least in part, explain its lower ability to inhibit infection by all the tested pseudoviruses except 229Epp. Interestingly, IFITM1 more potently inhibited 229Epp infection than IFITM2/3 in HEK293 cells but was less effective in Huh7.5 cells. While the viral envelope proteins are obviously the primary determinants of the potency of IFITMs to restrict virus entry, the more potent inhibition by all three IFITMs of infection by NL63pp than by SARSpp suggests that host cellular factors other than viral receptors have a strong impact on antiviral activity of IFITMs.
FIG 1.

IFITM proteins inhibit infection by pseudoviruses in FLIP-IN T Rex 293 and Huh7.5 cells. (A) The expression of FLAG-tagged IFITMs in FLIP-IN T Rex 293 cells cultured in the presence of 1 μg/ml of Tet for 24 h and in Huh7.5-derived stable cell lines was detected by a Western blot assay using a monoclonal antibody against FLAG tag. β-Actin served as a loading control. (B) FLIP-IN T Rex 293 cells expressing CAT or the indicated IFITM protein were cultured in the presence or absence of Tet for 24 h and then infected with 229Epp, NL63pp, SARSpp, MERSpp, IAVpp, or LASVpp. Luciferase activities were determined at 48 h postinfection (hpi). Relative infection efficiency is the ratio of luciferase activity in cells cultured in the presence of Tet over that in cells cultured in the absence of Tet. The error bars indicate standard deviations (n = 6). IFITMs did not significantly affect infection by LASVpp but significantly (P < 0.001) inhibited infection by all the other tested pseudotyped viruses. (C) Huh7.5 cells expressing N-terminally FLAG-tagged human IFITM proteins or transduced with an empty vector (pQCXIP) were infected with the indicated pseudoviruses. Luciferase activities were determined at 48 hpi. Relative infection is the ratio of the luciferase activity of cells expressing the indicated IFITM protein over that of cells transduced with the empty vector. The error bars indicate standard deviations (n = 6). All three IFITM proteins significantly (P < 0.001) inhibited infection by all the pseudoviruses except LASVpp.
Replacement of Y20 in IFITM3 enhances SARS-CoV and MERS-CoV entry.
We next aimed at identifying IFITM structural motifs that control the modulation of HCoV entry. IFITM proteins contain a conserved CD225 domain flanked by sequence-divergent N- and C-terminal variable regions (5). The N-terminal 21 amino acid residues unique to IFITM2 or -3 have been demonstrated to be important for IFITM3 to inhibit IAV infection in cultured cells and in vivo in humans (35–38). It has been shown recently that the N-terminal region of IFITM3 contains a 20YEML23 tetrapeptide that is consistent with the canonical YXXΦ endocytic sorting signal (where X can be any amino acid and Φ denotes Val, Leu, or Ile) (39–41). Furthermore, Y20 can be phosphorylated by the tyrosine kinase Fyn, which regulates IFITM3 trafficking and metabolism (42). We therefore investigated how the phosphorylation of IFITM3 at Y20 may regulate its function of modulating HCoV entry. The results showed that, compared to wild-type IFITM3, replacement of Y20 with alanine (A) and aspartic acid (D) or glutamic acid (E) to mimic the nonphosphorylated (Y20A) or phosphorylated (Y20E or Y20D) IFITM3, respectively, did not alter the steady-state levels of expression (Fig. 2A and F) and activity to enhance the entry of HCoV-OC43 in both HEK293 and Huh7.5 cells (Fig. 2B and G). However, the mutant IFITM3 proteins showed significantly reduced activity to inhibit NL63pp and 229Epp infection (Fig. 2C, D, H, and I). On the other hand, mutant IFITM3 proteins enhanced infection by SARSpp and MERSpp in both cell lines (Fig. 2C, E, H, and J). Consistent with previous reports (39, 40), wild-type IFITM3 accumulated in the perinuclear region and primarily colocalized with Rab9, a later endosome marker (43) (Fig. 3A). In contrast, IFITM3 proteins bearing a Y20A or Y20D mutation primarily accumulated in the regions close to the plasma membrane (Fig. 3B and C). These results indicate that Y20 is critical for endocytic sorting, which is regulated by tyrosine phosphorylation.
FIG 2.

Y20 of IFITM3 plays a critical role in modulating the entry of 229E, MERS-CoV, SARS-CoV, and NL63 in both 293 and Huh7.5 cells. (A) FLIP-IN T Rex cells expressing CAT or the indicated wild-type or mutant IFITM proteins were cultured in the presence of 1 μg/ml of Tet for 24 h. Expression of FLAG-tagged IFITM mutants was detected by Western blotting assay using anti-FLAG monoclonal antibody. β-Actin served as a loading control. (B to E) The above-mentioned FLP-IN T Rex-derived cell lines were cultured in the presence or absence of Tet for 24 h and then infected with HCoV-OC43pp (B), SARSpp or NL63pp (C), 229Epp (D), or MERSpp and LASVpp (E). Luciferase activities were determined at 48 hpi. Relative infection efficiency is the ratio of the luciferase activity in cells cultured in the presence of Tet over that in cells cultured in the absence of Tet. The error bars indicate standard deviations (n = 6). (F) Huh7.5 cells were stably transduced with an empty retroviral vector (pQCXIP) or vectors expressing wild-type or mutant IFITM3 proteins. Expression of FLAG-tagged IFITM proteins was detected by Western blotting assay using an anti-FLAG monoclonal antibody. β-Actin served as a loading control. (G to K) The above-mentioned Huh7.5-derived cell lines were infected with OC43pp (G), SARSpp and NL63pp (H), 229Epp (I), MERSpp (J), or LASVpp (K). Luciferase activities were determined at 48 hpi. Relative infection is the ratio of the luciferase activity of cells expressing the indicated IFITM protein over that of cells transduced with the empty vector. The error bars indicate standard deviations (n = 6).
FIG 3.

Y20 mutation alters IFITM3 subcellular localization. FLP-IN T Rex cells expressing the indicated wild-type and mutant IFITM3 proteins were cultured in the presence of Tet for 24 h to induce IFITM expression. The localization of FLAG-tagged IFITM3 (A), IFITM3/Y20A (B), and IFITM3/Y20D (C) was detected by immunofluorescent staining with an anti-FLAG monoclonal antibody (red). EEA1, Rab5, or Rab9 was visualized by immunofluorescent staining with the respective antibodies (green). Cell nuclei were stained with DAPI (blue).
In order to investigate whether the enhanced infection by SARSpp and MERSpp by the mutant IFITM3 proteins is due to the induction of membrane fusion on the plasma membrane, we examined the effect of endosomal pH on HCoVpp infection of Huh7.5 cells expressing wild-type or Y20A IFITM3 (Fig. 4). As shown in Fig. 4A, among the five tested HCoVpp, MERSpp and NL63pp infections were less sensitive to NH4Cl treatment that elevated endosomal pH, suggesting that membrane fusion for the two viruses may occur in early endosomal compartments. Interestingly, IFITM3 Y20A-enhanced SARSpp and MERSpp infections were efficiently inhibited by NH4Cl treatment in a concentration-dependent manner (Fig. 4B and C). The results thus suggest that although Y20 mutant IFITM3 proteins primarily accumulate in the plasma membrane regions, enhanced infection by SARSpp and MERSpp still occurs in low-pH endosomal compartments.
FIG 4.

IFITM3/Y20A-enhanced SARSpp and MERSpp infection is low pH dependent. (A) Huh7.5 cells were infected with OC43pp, MERSpp, SARSpp, NL63pp, or 229Epp in the absence (mock) or presence of the indicated concentrations of NH4Cl. Luciferase activities were determined at 48 hpi. Relative infection is the ratio of the luciferase activity in cells treated with NH4Cl over that in the mock-treated cells. The error bars indicate standard deviations (n = 6). (B and C) Huh7.5 cells stably expressing the indicated wild-type or Y20A mutant IFITM3 proteins or transduced with the empty vector were infected with MERSpp (B) or SARSpp (C) in the absence (mock) or presence of the indicated concentrations of NH4Cl. Luciferase activities were determined at 48 hpi. Relative infection is the ratio of the luciferase activity in cells treated with NH4Cl over that in the mock-treated cells. The error bars indicate standard deviations (n = 6).
Replacement of Y99 in IFITM3 enhances MERS-CoV entry.
In addition to Y20, Y99 had been shown by mass spectrometry analysis to be phosphorylated in cells and to play a role in restricting the infectious entry of IAV, but not dengue virus (24). Therefore, we performed phosphomimetic analysis on this amino acid residue. As shown in Fig. 5A, the Y99A or Y99D mutant IFITM3 was expressed to a level similar to that of wild-type IFITM3. Compared with wild-type IFITM3, Y99A or Y99D mutants had slightly increased activity to enhance OC43pp infection but significantly reduced activity to inhibit infection by SARSpp, NL63pp, IAVpp, and vesicular stomatitis virus (VSVpp) (Fig. 5B to E). Intriguingly, both Y99A and Y99D IFITM3 enhanced MERSpp infection by approximately 10-fold (Fig. 5F). The results imply that Y99 plays a critical role in IFITM3 modulation of the entry of different HCoVs.
FIG 5.

Y99 of IFITM3 plays a critical role in modulating the entry of HCoVs. (A) FLIP-IN T Rex 293 cells expressing CAT, IFITM3, or the indicated mutant IFITM3 proteins were cultured in the presence of 1 μg/ml of Tet for 24 h. Expression of FLAG-tagged IFITM proteins was detected by Western blotting assay using anti-FLAG monoclonal antibody. β-Actin served as a loading control. (B to F) FLP-IN T Rex 293-derived cells were cultured in the presence of Tet for 24 h to induce IFITM mutant expression. The cells were then infected with HCoV-OC43pp (B), SARSpp or NL63pp (C), 229Epp (D), VSVpp or IAVpp (E), or MERSpp (F). Luciferase activities were determined at 48 hpi. Relative infection efficiency represents the luciferase activity of cells cultured with Tet normalized to that of cells cultured in the absence of Tet. The error bars indicate standard deviations (n = 6).
Oligomerization of IFITM3 is essential for its suppression of the entry of HCoVs, except for NL63.
In addition to phosphorylation, the function of IFITM proteins is regulated by cysteine palmitoylation (44, 45), ubiquitination (46, 47), and homo- and hetero-oligomerization (24, 29). To investigate the roles of these posttranslational modifications in IFITM3 inhibition of HCoV entry, HEK293 cell lines inducibly expressing wild-type or mutant IFITM3 proteins bearing mutations that preclude cysteine palmitoylation or ubiquitination were established. Specifically, the conserved cysteine residues C71 and C72 or one additional cysteine, C105, which are critical for IFITM3 palmitoylation, were replaced with alanine to yield two mutants, IFITM3/2CA and IFITM3/3CA, respectively. As shown in Fig. 6A and B, the mutations had minimal impacts on protein expression but completely abolished activity to restrict infection by all five pseudoviruses sensitive to IFITM3. However, IFITM3/4KA, which were generated by replacement of K24, K83, K88, and K104 with alanines, accumulated at a significantly reduced level in cells and failed to inhibit infection by all the pseudoviruses examined. Moreover, IFITM3 containing F75A and F78A mutations (IFITM3/2FA), which disrupt its oligomerization (29), completely lost the ability to inhibit infection by SARSpp, 229Epp, MERSpp, and IAVpp but could still significantly inhibit infection by NL63pp, despite reduced activity (Fig. 6B). The results thus suggest that, unlike other viruses (24, 29), suppression of NL63 spike protein-triggered membrane fusion does not absolutely require the oligomerization of IFITM3.
FIG 6.
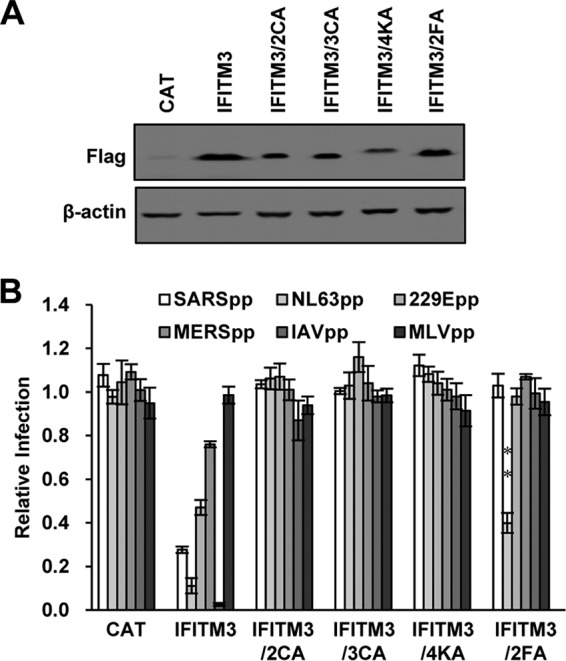
Palmitoylation, ubiquitination, and oligomerization are important for IFITM3 restriction of HCoV entry. (A) FLIP-IN T Rex 293 cells expressing CAT or the indicated wild-type and mutant IFITM3 proteins were cultured in the presence of Tet for 24 h. Expression of the IFITM proteins was detected by Western blotting assay using anti-FLAG monoclonal antibody. β-Actin served as a loading control. (B) The above-mentioned FLP-IN T Rex-derived cells were cultured in the presence or absence of Tet for 24 h and then infected with SARSpp, NL63pp, 229Epp, MERSpp, IAVpp, or MLVpp. Luciferase activities were determined at 48 hpi. Relative infection efficiency is the ratio of the luciferase activity in cells cultured in the presence of Tet over that in cells cultured in the absence of Tet. The error bars indicate standard deviations (n = 6).
The C-terminal domain of IFITM1 differentially regulates the entry of HCoVs.
We showed previously that sequential truncation of the C-terminal 18 amino acid residues from IFITM1 did not apparently affect its activity to inhibit infection by IAVpp but converted the antiviral protein into an increasingly potent enhancer of OC43pp infection (29). In our efforts to further dissect the role of the C-terminal domain (CTD) in IFITM1 modulation of HCoV entry, we found that deletion of the C-terminal 3, 6, or 9 amino acids did not apparently affect the activity of IFITM1 to inhibit infection by SARSpp, but further deletion of the C-terminal 12, 15, or 18 amino acids significantly compromised or abolished the ability of IFITM1 to inhibit infection by SARSpp (Fig. 7B). In contrast, deletion of the C-terminal 3, 6, and 9 amino acids enhanced the activity of IFITM1 to inhibit infection by NL63pp by 5-, 10-, and 3-fold, respectively. However, further truncation of the C-terminal 12, 15, or 18 amino acids abolished the enhanced inhibitory effect on NL63pp infection (Fig. 7B). Interestingly, sequential truncation of the CTD gradually attenuated and ultimately abolished the activity of IFITM1 to inhibit 229Epp infection (Fig. 7C). On the other hand, sequential truncation of the C-terminal 12 amino acids gradually increased its activity to enhance MERSpp infection, but further deletion of the C-terminal 15 or 18 amino acids reduced its activity to enhance MERSpp infection (Fig. 7D).
FIG 7.

Role of the IFITM1 C-terminal motifs in modulating the entry of HCoVs. (A) FLIP-IN T Rex cells expressing CAT or wild-type or mutant IFITM1 proteins were cultured in the presence of Tet for 24 h. The expression of FLAG-tagged IFITM1 proteins was detected by Western blotting assay using an anti-FLAG monoclonal antibody. β-Actin served as a loading control. (B to D) The above-mentioned cell lines were left untreated or treated with 1 μg/ml of Tet for 24 h to induce IFITM expression. The cells were then infected with SARSpp, NL63pp, or MLVpp (B); 229Epp (C); or MERSpp (D). Luciferase activities were determined at 48 hpi. Relative infection efficiency is the ratio of the luciferase activity in cells cultured in the presence of tetracycline over that in cells cultured in the absence of tetracycline. The error bars indicate standard deviations (n = 6). **, P < 0.001 compared to wild-type IFITM1.
To rule out the potential interference of endogenous IFITM proteins with mutant IFITM1 in HEK 293 cells (29), we further confirmed the observation in Huh7.5 cells that expressed undetectable endogenous IFITM proteins. Four Huh7.5 cell lines were established by transduction with an empty retroviral vector or a retroviral vector expressing wild-type IFITM1, IFITM1/TC6, or IFITM1/TC18 protein. Consistent with the observations made in HEK293 cells, deletion of the C-terminal 18, but not 6, amino acids significantly compromised IFITM1's ability to suppress SARSpp infection, and deletion of the C-terminal 6, but not 18, amino acids significantly enhanced activity to inhibit NL63pp infection (Fig. 8B). In agreement with the results obtained with NL63pp infection, we also observed that deletion of the C-terminal 6, but not 18, amino acids significantly increased the ability of IFITM1 to inhibit infection by HCoV-NL63, as judged by significant reduction in the percentages of infected cells and intracellular viral RNA (Fig. 8C and D).
FIG 8.

Role of the IFITM1 C-terminal motifs in modulating infection by HCoV-NL63 in Huh7.5 cells. (A) Huh7.5 cells were stably transduced with retroviral vectors expressing wild-type IFITM1, IFITM1/TC6, IFITM1/TC18, or an empty vector (pQCXIP). Expression of FLAG-tagged IFITM1 proteins was detected by Western blotting assay using anti-FLAG monoclonal antibody. β-Actin served as a loading control. (B) The above-mentioned cell lines were infected with SARSpp, NL63pp, or MLVpp. Luciferase activities were determined at 48 hpi. Relative infection is the ratio of the luciferase activity of cells expressing the indicated IFITM protein over that of cells transduced with the empty vector. The error bars indicate standard deviations (n = 6). (C) The above-mentioned cell lines were infected with HCoV-NL63 at a multiplicity of infection (MOI) of 5. Infected cells were visualized by immunofluorescent staining using NL63 NP monoclonal antibody (green). Cell nuclei were stained with DAPI (blue). The percentages of cells infected by NL63 are expressed as averages ± standard deviations. (D) The amounts of intracellular HCoV-NL63 RNA at 24 hpi were quantified by a qRT-PCR assay and expressed as the ratio of the viral RNA in IFITM-expressing cells over that in the cells transduced with an empty vector. The error bars indicate standard deviations (n = 4).
Taken together, the results suggest that two partially overlapping functional motifs exist in the CTD of IFITM1. While its C-terminal 9 to 12 amino acid residues contain a motif that downregulates antiviral activity against HCoV-NL63 and suppresses activity enhancing MERS-CoV infection, the motif located in the N-terminal 9 amino acid residues of the CTD is important for IFITM1 to suppress SARS-CoV and HCoV-229E entry.
To investigate the molecular mechanisms underlying the differential modulation of HCoV entry by the CTD, we examined the subcellular localization of wild-type and mutant IFITM1 proteins. As shown in Fig. 9, similar to wild-type IFITM1, IFITM1/TC6 and IFITM1/TC18 primarily accumulate in regions close to or at the plasma membrane. However, modestly increased intracellular localization of both IFITM1/TC6 and IFITM1/TC18 is evident. Specifically, IFITM1/TC6 tends to more frequently colocalize with Rab5 and Rab9, whereas IFITM1/TC18 is more frequently colocalized with EEA1, an early endosomal marker (48), and Rab9, a later endosomal marker. Moreover, as shown in Fig. 10, compared to wild-type IFITM1, IFITM1/TC6, but not IFITM1/C18, demonstrated reduced mono- and diubiquitination. As anticipated, replacement of four lysine residues in IFITM3 (IFITM3/4KA) completely abolished ubiquitination. The results thus indicate that the CTD of IFITM1 contains a structural motif(s) that regulates its subcellular trafficking and ubiquitination, which may consequentially affect its activity to modulate the entry of HCoVs.
FIG 9.

Subcellular localization of wild-type and C-terminally truncated IFITM1 proteins. FLP-IN T Rex 293 cells expressing the indicated wild-type and mutant IFITM1 proteins were treated with 1 μg/ml of tetracycline for 24 h to induce IFITM expression. The localization of FLAG-tagged IFITM1 (A), IFITM1/TC6 (B), and IFITM1/TC18 (C) was detected by immunofluorescent staining with an anti-FLAG monoclonal antibody (red). EEA1, Rab5, and Rab9 were visualized by immunofluorescent staining with the respective antibodies (green). Cell nuclei were stained with DAPI (blue).
FIG 10.

The C-terminal motif of IFITM1 is required for its ubiquitination. 293T cells were transfected with a plasmid expressing the indicated FLAG-tagged wild-type (WT) and mutant IFITM1 or IFITM3 proteins. Immunoprecipitation was performed by using an anti-FLAG monoclonal antibody. The precipitated proteins were resolved by SDS-PAGE and blotted onto a membrane. IFITM proteins and their ubiquitinated species were visualized by probing with an anti-FLAG rabbit polyclonal antibody (A) or anti-ubiquitin rabbit polyclonal antibody (B). The monoubiquitinated and diubiquitinated forms of IFITM are indicated by single and double asterisks, respectively. IFITM proteins without ubiquitination with a molecular mass smaller than 15 kDa served as loading controls.
DISCUSSION
In spite of the relatively broad spectrum of antiviral activities, IFITM proteins do not restrict infection by MLV, Sendai virus, and several members of the family Arenaviridae, as well as all the DNA viruses tested thus far (6, 8, 49, 50). The molecular determinants that control the viral specificity and potency of IFITMs remain to be fully understood. Because viral envelope proteins and cellular receptors are the two major players in viral membrane fusion, it is plausible to consider that modulating the interaction between viral envelope proteins and cellular receptors might be the key mechanism of IFITM restriction of virus entry. Indeed, it was reported recently that the IFITM sensitivity of HIV-1 strains is determined by the coreceptor usage of viral envelope glycoproteins (51, 52). However, the difference in the potency and the requirement for IFITM oligomerization in inhibition of SARSpp and NL63pp infection (Fig. 1 and 6) and, particularly, the results showing that IFITM3/Y20 phosphomimetic mutations enhanced only infection by SARSpp, but not by NL63pp (Fig. 2), strongly suggest that IFITM interaction with viral and host cellular factors, other than viral receptors, such as ACE2, plays a critical role in IFITM modulation of virus entry. Furthermore, our findings that IFITM2 and IFITM3 promoted HCoV-OC43 infection and that three distinct mutations converted IFITM1 and IFITM3 from inhibitors to enhancers of SARS-CoV and/or MERS-CoV spike protein-mediated entry challenge the “rigid-membrane” hypothesis and suggest that IFITM proteins may also promote membrane fusion, under select conditions, to facilitate virus entry.
Based on these new findings, we hypothesize that, depending on the fine-tuned interaction with the entry machinery of a given virus, which consists of viral envelope components, as well as viral receptors and other host entry factors at the site of membrane fusion, IFITM proteins can either promote or arrest the fusion between viral envelope and endosomal membranes (29). It is possible that the three structural motifs identified here mediate interactions with key host factors to determine either to arrest or to enhance membrane fusion. Along these lines, recent studies revealed that zinc metallopeptidase STE24 forms complexes with IFITM proteins and is required for IFITMs to inhibit the entry of many different viruses (53). In addition, the sensitivity of IAVs to IFITM3 appears to depend on the pH value at which the viral hemagglutinin (HA) undergoes a conformational transition and mediates membrane fusion (54). More interestingly, IFITM expression promotes the uptake of avian sarcoma leukosis virus (ASLV) and the acidification of endosomal compartments, resulting in accelerated membrane fusion when driven by the glycosylphosphatidylinositol-anchored, but not by the transmembrane, isoform of the ASLV receptor (55). These recent findings clearly highlight the fact that multiple viral and host cellular components regulate IFITM activity in the fusion of viral envelope and endosomal membranes.
Our new hypothesis predicts that in order to modulate virus entry, IFITM proteins ought to be at the site of viral membrane fusion (26–28). Indeed, previous studies demonstrated in a variety of virus infection systems that localization in the subcellular compartment where a virus enters the cytoplasm is important for the IFITM protein to inhibit its infectious entry (5). For instance, disruption of the canonical endocytic signal in IFITM3 by Y20A/E/D mutations resulted in its plasma membrane accumulation (Fig. 11). As a consequence, the mutant IFITM3 demonstrated reduced antiviral activity against IAV, which enters cells by fusion with the lysosomal membrane (39–41), but enhanced the activity to restrict infection by parainfluenza virus 3, a virus that enters cells by fusion with the plasma membrane (56). However, the subcellular localization of IFITM proteins is not always strictly correlated with antiviral activity. For example, while IFITM3 Y99A mutation does not apparently alter subcellular localization, the mutation significantly compromises the antiviral activity of IFITM3 against IAV, but not dengue virus (24). In this study, we further demonstrated that although Y20A mutant IFITM3 predominantly accumulated in the plasma membrane region, its enhancement of infection by SARSpp, MERSpp, and OC43pp was still low pH dependent (Fig. 4) (29), suggesting that the enhanced entry of the viruses still occurs in low-pH intracellular endosomal compartments. The apparent contradiction between the subcellular localization of the mutant IFITM3 proteins and the site of membrane fusion implies that a small fraction of the mutant IFITM3 might still traffic to the sites where the viral spike protein-induced membrane fusion occurs. In addition, phosphomimetic analyses suggest that Y20 or Y99 tyrosine phosphorylation regulates the metabolism, trafficking, and biological function of IFITM3.
In addition to tyrosine phosphorylation, IFITM3 can also be posttranslationally modified at more than 8 different amino acid residues with at least three different types of modifications: palmitoylation, ubiquitination, and methylation (42, 44–47, 57). Our mutagenesis studies showed that both palmitoylation and ubiquitination are absolutely required for IFITM3 to modulate the entry of all the tested HCoVs (Fig. 6). Moreover, oligomerization of IFITM proteins has been demonstrated to be essential for their restriction of IAV and dengue virus infection, as well as enhancement of HCoV-OC43 infection (24, 29). Interestingly, in hetero-oligomerization between IFITM1 and IFITM3, which inhibit and enhance HCoV-OC43 infection, respectively, the two proteins antagonize each other's functions (29). In this study, we further revealed that IFITM3 bearing F75A and F78A mutations, which disrupt its oligomerization, completely lost its ability to inhibit infection by SARSpp, 229Epp, and MERSpp but still partially inhibited NL63pp infection (Fig. 6 and 11). While the results reinforce the notion that oligomerization is important for IFITMs to modulate the entry of many viruses, NL63 appears to be an exception.
It was reported previously that the CTD of IFITM1, illustrated in Fig. 11, plays an important role in modulating the entry of HCoV-OC43 (29), HIV-1 (58), Jaagsiekte sheep retrovirus, and 10A1 amphotropic murine leukemia virus (59). In this study, we showed that the CTD of IFITM1 plays distinct roles in modulating the entry of different HCoVs. It appears that the CTD contains two overlapping functional motifs. While the C-terminal 9 and 12 amino acid residues negatively regulate IFITM restriction of HCoV-NL63 entry and enhancement of MERS-COV infection, the motif located in the N-terminal 9 amino acid residues of the CTD is important for IFITM1 to suppress SARS-CoV and HCoV-229E entry. In a search for the underlying mechanism, a study identified a dibasic 122KRXX125 motif at the C terminus of IFITM1 that regulates IFITM1 intracellular trafficking with reduced localization in LAMP1-positive lysosomes but increased levels in CD63-positive multivesicular bodies (59). IFITM1 binds to cellular adaptor protein complex 3 (AP-3), an association that is lost when the dibasic motif is altered (59). However, we found that partial or complete deletion of the CTD does not dramatically alter its subcellular distribution (Fig. 9). Instead, deletion of 6, but not 18, amino acid residues from the C terminus reduced IFITM1 ubiquitination (Fig. 10). The results collectively indicate that the CTD is a key regulator of IFITM function. However, its effects on the entry of different viruses imply versatile functions of the CTD. Further investigation into the structure, posttranslational modification, and membrane topology of the CTD, as well as identification of the cellular and/or viral proteins interacting with the CTD, will shed light on the mechanism by which the CTD regulates the function of IFITM1.
In summary, we demonstrated in this study that, in addition to viral envelope proteins and cellular receptors, IFITM protein oligomerization, posttranslational modification, and intracellular trafficking, which can be regulated by host cellular pathobiological cues, play critical roles in determining the extent and nature of IFITM modulation of the entry of HCoVs. More importantly, identification of the three structural motifs that reverse the functions of IFITM1 and IFITM3 on virus entry paves the way for uncovering viral and host factors that interact with those structural motifs to differentially modulate the infectious entry of HCoVs and other viruses (53).
MATERIALS AND METHODS
Cell lines, viruses, and antibodies.
LLC-MK2 cells were cultured in minimal essential medium (MEM), which was prepared by mixing Hanks MEM (Invitrogen; catalog no. 11575-032) and Earle's MEM (Invitrogen; catalog no. 11095-080) in a 2:1 ratio and supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen). Huh7.5 cells were cultured with Dulbecco's modified Eagle's medium (DMEM) (Corning) supplemented with 10% FBS, 1× nonessential amino acids (Invitrogen), and 2 mM l-glutamine (Invitrogen). The human colon cancer cell line HCT-8 was grown in RPMI 1640 medium (ATCC; catalog no. 30-2001) supplemented with 10% FBS. GP2-293 and Lenti-X 293T cell lines were purchased from Clontech and cultured in DMEM supplemented with 10% FBS and 1 mM sodium pyruvate (Invitrogen). FLP-IN T Rex 293 cells were purchased from Invitrogen and maintained in DMEM supplemented with 10% FBS, 10 μg/ml blasticidin (Invitrogen), and 100 μg/ml zeocin (Invivogen) (60). HCoV-NL63 was purchased from ATCC through BEI Resource (catalog no. NR470) and amplified in LLC-MK2 cells. Virus titers were determined by plaque assay (29). Monoclonal antibody against FLAG tag (anti-FLAG M2), rabbit anti-FLAG polyclonal antibody, and β-actin antibody were purchased from Sigma (catalog no. F1804, F7425, and A2228, respectively). A mouse monoclonal antibody against HCoV-NL63 nucleocapsid (N) protein was purchased from Ingenansa, Spain (catalog no. M.30.HCo.I2D4).
Plasmids.
pcDNA5/FRT-derived plasmids expressing chloramphenicol acetyltransferase (CAT) and N-terminally FLAG-tagged human IFITM1, IFITM2, and IFITM3, as well as C-terminally truncated IFITM1 mutants, were reported previously (7, 29, 60, 61). Plasmids expressing N-terminally FLAG-tagged mutant IFITM3 proteins with point mutations were constructed by overlap extension PCR (60). All the resulting plasmids were sequenced to verify the desired mutation(s). N-terminally FLAG-tagged human IFITM1, IFITM2, and IFITM3 and their mutants were also cloned into the pQCXIP vector (Clontech) between the NotI and BamHI sites (29). Plasmids expressing HCoV-OC43 S and HE proteins, VSV G protein, H1N1 IAV (A/WSN/33) HA and neuraminidase (NA), Ebola virus (EBOV) GP protein, LASV GP protein, murine leukemia virus (MLV) envelope protein, and HCoV-NL63 and SARS-CoV spike proteins were described previously (62, 63). The MERS-CoV spike gene (GenBank accession number AFS88936) was synthesized by GeneScript, cloned into the pCAGGS vector, and confirmed by DNA sequence analyses. Plasmid pNL4-3.Luc.R−E− was obtained through the NIH AIDS Research and Reference Reagent Program (64, 65). ACE2, aminopeptidase N (APN), and dipeptidyl peptidase 4 (DPP4) cDNA clones were obtained from Origene and cloned into a pcDNA3 vector (Invitrogen) to yield plasmids pcDNA3/ACE2, pcDNA3/APN, and pcDNA3/DDP4, respectively (66).
Package of pseudotyped retroviral particles.
The various viral envelope protein-pseudotyped lentiviruses bearing luciferase reporter genes, as well as VSV G protein-pseudotyped retroviruses expressing wild-type and mutant IFITM proteins, were packaged as reported previously (66, 67). Each pseudotype was titrated by infection of cells with a serial dilution of pseudotype preparations. The modulation by IFITM of the transduction of a given pseudotype was determined with a titrated amount of pseudotypes that yielded a luciferase signal between 10,000 and 1,000,000 light units per well of 96-well plates. For a given pseudotype, the input of pseudoviral particles was consistent across all the experiments.
Establishment of cell lines stably expressing wild-type and mutant IFITM proteins.
Huh7.5 cells in each well of 6-well plates were incubated with 2 ml of Opti-MEM medium containing pseudotyped retroviruses and centrifuged at 20°C for 30 min at 4,000 × g. Forty-eight hours postransduction, the cells were cultured with medium containing 2 μg/ml of puromycin for 2 weeks. The antibiotic-resistant cells were pooled and expanded into cell lines stably expressing human wild-type or mutant IFITM proteins (66). FLP-IN T Rex 293-derived cell lines expressing mutant IFITM proteins in a tetracycline (Tet)-inducible manner were established as previously described (7, 60).
Western blot assay.
Cell monolayers were washed once with phosphate-buffered saline (PBS) and lysed with 1× Laemmli buffer. An aliquot of cell lysate was separated on a NuPAGE Novex 4 to 12% Bis-Tris gel (Invitrogen) and electrophoretically transferred onto a nitrocellulose membrane (Invitrogen). The membranes were blocked with PBS containing 5% nonfat dry milk and probed with the desired antibody. The bound antibodies were visualized with IRDye secondary antibodies and by imaging with the Li-Cor Odyssey system (Li-Cor Biotechnology).
Real-time RT-PCR.
Total cellular RNA was extracted using TRIzol reagent (Invitrogen) and reverse transcribed using SuperScript III (Invitrogen). Quantitative PCR (qPCR) was performed as previously described on a LightCycler 480II (Roche) with a modified forward primer (5′-AAA CCT CGT TGG AAG CGT GTTC-3′) and reverse primer (5′-CTG TGG AAA ACC TTT GGC ATC-3′) under the following conditions: denaturing at 95°C for 10 min and 45 cycles of amplification (15 s at 95°C and 1 min at 60°C). The PCR amplified a fragment of the HCoV-NL63 N gene (GenBank Gene ID 2943504).
Luciferase assay.
T Rex 293-derived IFITM-expressing cell lines were seeded into 96-well plates with black walls and clear bottoms and transfected with plasmids encoding ACE2, APN, or DPP4 to express viral receptors. For Huh7.5-derived IFITM-expressing cell lines, cells were seeded into black-wall 96-well plates. The cells were infected at 24 h posttransfection by seeding with the desired pseudotyped lentiviral particles for 2 h and then replenished with fresh medium. Two days postinfection, the medium was removed and the cells were lysed with 20 μl/well of cell lysis buffer (Promega) for 15 min, followed by adding 50 μl/well of luciferase substrate (Promega). The firefly luciferase activities were measured by luminometry in a TopCounter (PerkinElmer) (66).
Immunofluorescence.
To visualize HCoV-NL63-infected cells, the infected cultures were fixed with 4% paraformaldehyde for 20 min. After permeabilization with 0.1% Triton X-100, the cells were stained with a monoclonal antibody recognizing HCoV-NL63 N protein (Ingenansa, Spain; catalog no. M.30.HCo.I2D4). Bound antibodies were visualized by using Alexa Fluor 488-labeled goat anti-mouse IgG (Abcam; catalog no. ab150113). Cell nuclei were counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI).
For determination of mutant IFITM1 or IFITM3 subcellular localization, T Rex 293-derived cell lines inducibly expressing mutant IFITM proteins were fixed and permeabilized as described above. The cells were then stained with anti-FLAG monoclonal antibody, together with a rabbit-derived polyclonal antibody against EEA1 (Cell Signaling; catalog no. 2411), Rab5 (Cell Signaling; catalog no. 2143), or Rab9 (Cell Signaling; catalog no. D52G8). The bound antibodies were visualized using Alexa Fluor 594-labeled goat anti-mouse IgG (red) and Alex Fluor 488-labeled goat anti-rabbit IgG (green). Cell nuclei were counterstained with DAPI. Images were sequentially acquired on an FV1000 confocal microscope (Olympus) with a PlanApoN 60×/1.42 numerical aperture objective (Olympus). The pinhole size was adjusted to 1 Airy unit. The optimal diffraction-limited spatial resolution was obtained using a pixel size of 82 nm/pixel. DAPI was excited at 405 nm, and its fluorescence emission was collected between 430 nm and 470 nm. Alexa Fluor 488 was excited at 488 nm, and its fluorescence emission was collected between 505 and 525 nm. Alexa Fluor 594 was excited at 543 nm, and its fluorescence emission was collected between 560 and 660 nm. Negative controls were performed to make sure that there were no significant spectral bleedthrough artifacts between channels.
TCID50.
Confluent LLC-MK2 cells cultured in 96-well plates were infected with 200 μl of Opti-MEM containing serial 10-fold dilutions of viral stock for 2 h at 33°C. The cells were then cultured at 33°C with MEM containing 2.5% FBS for 6 days. The cells from each well with cytopathic effect (CPE) were visualized and counted under a microscope. The 50% tissue culture infective dose (TCID50) of virus stock was measured and converted to PFU per milliliter (68).
IP.
To detect ubiquitination of IFITM protein and its mutants, immunoprecipitation (IP)-Western blotting was performed as reported previously (29, 44). Briefly, 293T cells were transfected with plasmids expressing FLAG-tagged IFITM1, IFITM3, and their mutants. The cells were lysed at 48 h posttransfection with lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and protease inhibitors (Roche) and immunoprecipitated with a monoclonal antibody against FLAG epitope, followed by incubation with protein A/G-agarose beads (Pierce) and washing with Tris-buffered saline (TBS). The immunocomplexes were resolved in a NuPAGE Novex 4 to 12% Bis-Tris gel in morpholineethanesulfonic acid (MES) buffer (Invitrogen) and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with rabbit polyclonal antibody against FLAG epitope (Sigma; catalog no. F7425) or anti-ubiquitin rabbit polyclonal antibody (Proteintech; catalog no. 10201-2-AP). The bound antibodies were visualized with IRDye secondary antibodies and imaged with a Li-Cor Odyssey system (Li-Cor Biotechnology).
ACKNOWLEDGMENTS
This work was supported by grants from the U.S. National Institutes of Health (AI113267), the National Natural Science Foundation of China (81571976), and the Commonwealth of Pennsylvania through the Hepatitis B Foundation.
REFERENCES
- 1.Cullen BR, Cherry S, ten Oever BR. 2013. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 14:374–378. doi: 10.1016/j.chom.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Chang J, Block TM, Guo JT. 2012. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antiviral Res 96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey CC, Zhong G, Huang IC, Farzan M. 2014. IFITM-family proteins: the cell's first line of antiviral defense. Annu Rev Virol 1:261–283. doi: 10.1146/annurev-virology-031413-085537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perreira JM, Chin CR, Feeley EM, Brass AL. 2013. IFITMs restrict the replication of multiple pathogenic viruses. J Mol Biol 425:4937–4955. doi: 10.1016/j.jmb.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang D, Weidner JM, Qing M, Pan XB, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi PY, Block TM, Guo JT. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J Virol 84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mudhasani R, Tran JP, Retterer C, Radoshitzky SR, Kota KP, Altamura LA, Smith JM, Packard BZ, Kuhn JH, Costantino J, Garrison AR, Schmaljohn CS, Huang IC, Farzan M, Bavari S. 2013. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J Virol 87:8451–8464. doi: 10.1128/JVI.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anafu AA, Bowen CH, Chin CR, Brass AL, Holm GH. 2013. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J Biol Chem 288:17261–17271. doi: 10.1074/jbc.M112.438515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M Jr. 2013. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep 13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL. 2016. The IFITMs inhibit Zika virus replication. Cell Rep 15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 14.Gorman MJ, Poddar S, Farzan M, Diamond MS. 2016. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J Virol 90:8212–8225. doi: 10.1128/JVI.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poddar S, Hyde JL, Gorman MJ, Farzan M, Diamond MS. 2016. The interferon-stimulated gene IFITM3 restricts infection and pathogenesis of arthritogenic and encephalitic alphaviruses. J Virol 90:8780–8794. doi: 10.1128/JVI.00655-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss SR, Navas-Martin S. 2005. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 18.Gaunt ER, Hardie A, Claas EC, Simmonds P, Templeton KE. 2010. Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J Clin Microbiol 48:2940–2947. doi: 10.1128/JCM.00636-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dijkman R, Jebbink MF, Gaunt E, Rossen JW, Templeton KE, Kuijpers TW, van der Hoek L. 2012. The dominance of human coronavirus OC43 and NL63 infections in infants. J Clin Virol 53:135–139. doi: 10.1016/j.jcv.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilgenfeld R, Peiris M. 2013. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res 100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham RL, Donaldson EF, Baric RS. 2013. A decade after SARS: strategies for controlling emerging coronaviruses. Nat Rev Microbiol 11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wrensch F, Winkler M, Pohlmann S. 2014. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: evidence for cholesterol-independent mechanisms. Viruses 6:3683–3698. doi: 10.3390/v6093683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pohlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87:6150–6160. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.John SP, Chin CR, Perreira J, Feeley EM, Aker A, Savidis G, Smith SE, Elia AE, Everitt AR, Vora M, Pertel T, Elledge SJ, Kellam P, Brass AL. 2013. The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and dengue virus replication. J Virol 87:7837–7852. doi: 10.1128/JVI.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU. 2013. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13:452–464. doi: 10.1016/j.chom.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li K, Markosyan RM, Zheng YM, Golfetto O, Bungart B, Li M, Ding S, He Y, Liang C, Lee JC, Gratton E, Cohen FS, Liu SL. 2013. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog 9:e1003124. doi: 10.1371/journal.ppat.1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. 2014. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog 10:e1004048. doi: 10.1371/journal.ppat.1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin TY, Chin CR, Everitt AR, Clare S, Perreira JM, Savidis G, Aker AM, John SP, Sarlah D, Carreira EM, Elledge SJ, Kellam P, Brass AL. 2013. Amphotericin B increases influenza A virus infection by preventing IFITM3-mediated restriction. Cell Rep 5:895–908. doi: 10.1016/j.celrep.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT. 2014. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A 111:6756–6761. doi: 10.1073/pnas.1320856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J Virol 85:2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wrensch F, Karsten CB, Gnirss K, Hoffmann M, Lu K, Takada A, Winkler M, Simmons G, Pohlmann S. 2015. Interferon-induced transmembrane protein-mediated inhibition of host cell entry of ebolaviruses. J Infect Dis 212(Suppl 2):S210–S218. doi: 10.1093/infdis/jiv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie M, Xuan B, Shan J, Pan D, Sun Y, Shan Z, Zhang J, Yu D, Li B, Qian Z. 2015. Human cytomegalovirus exploits interferon-induced transmembrane proteins to facilitate morphogenesis of the virion assembly compartment. J Virol 89:3049–3061. doi: 10.1128/JVI.03416-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, GenISIS Investigators, MOSAIC Investigators, Smyth RL, Openshaw PJ, Dougan G, Brass AL, Kellam P. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey CC, Huang IC, Kam C, Farzan M. 2012. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog 8:e1002909. doi: 10.1371/journal.ppat.1002909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang YH, Zhao Y, Li N, Peng YC, Giannoulatou E, Jin RH, Yan HP, Wu H, Liu JH, Liu N, Wang DY, Shu YL, Ho LP, Kellam P, McMichael A, Dong T. 2013. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat Commun 4:1418. doi: 10.1038/ncomms2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, Ren Y, Wang J, Dong Y, Bao M, Li L, Zhou M, Yuan S, Sun J, Zhu Z, Chen L, Li Q, Zhang Z, Zhang X, Lu S, Doherty PC, Kedzierska K, Xu J. 2014. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci U S A 111:769–774. doi: 10.1073/pnas.1321748111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia R, Pan Q, Ding S, Rong L, Liu SL, Geng Y, Qiao W, Liang C. 2012. The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J Virol 86:13697–13707. doi: 10.1128/JVI.01828-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia R, Xu F, Qian J, Yao Y, Miao C, Zheng YM, Liu SL, Guo F, Geng Y, Qiao W, Liang C. 2014. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell Microbiol 16:1080–1093. doi: 10.1111/cmi.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Compton AA, Roy N, Porrot F, Billet A, Casartelli N, Yount JS, Liang C, Schwartz O. 2016. Natural mutations in IFITM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep 17:1657–1671. doi: 10.15252/embr.201642771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chesarino NM, McMichael TM, Hach JC, Yount JS. 2014. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J Biol Chem 289:11986–11992. doi: 10.1074/jbc.M114.557694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganley IG, Carroll K, Bittova L, Pfeffer S. 2004. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol Biol Cell 15:5420–5430. doi: 10.1091/mbc.E04-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yount JS, Karssemeijer RA, Hang HC. 2012. S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J Biol Chem 287:19631–19641. doi: 10.1074/jbc.M112.362095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yount JS, Moltedo B, Yang YY, Charron G, Moran TM, Lopez CB, Hang HC. 2010. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol 6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chesarino NM, McMichael TM, Yount JS. 2015. E3 ubiquitin ligase NEDD4 promotes influenza virus infection by decreasing levels of the antiviral protein IFITM3. PLoS Pathog 11:e1005095. doi: 10.1371/journal.ppat.1005095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesarino NM, McMichael TM, Yount JS. 2014. Regulation of the trafficking and antiviral activity of IFITM3 by post-translational modifications. Future Microbiol 9:1151–1163. doi: 10.2217/fmb.14.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawe DC, Sitouah N, Hayes S, Chawla A, Virbasius JV, Tuft R, Fogarty K, Lifshitz L, Lambright D, Corvera S. 2003. Essential role of Ca2+/calmodulin in early endosome antigen-1 localization. Mol Biol Cell 14:2935–2945. doi: 10.1091/mbc.E02-09-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hach JC, McMichael T, Chesarino NM, Yount JS. 2013. Palmitoylation on conserved and nonconserved cysteines of murine IFITM1 regulates its stability and anti-influenza A virus activity. J Virol 87:9923–9927. doi: 10.1128/JVI.00621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, Pyeon D. 2014. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One 9:e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster TL, Wilson H, Iyer SS, Coss K, Doores K, Smith S, Kellam P, Finzi A, Borrow P, Hahn BH, Neil SJ. 2016. Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe 20:429–442. doi: 10.1016/j.chom.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu WL, Grotefend CR, Tsai MT, Wang YL, Radic V, Eoh H, Huang IC. 2017. Delta20 IFITM2 differentially restricts X4 and R5 HIV-1. Proc Natl Acad Sci U S A 114:7112–7117. doi: 10.1073/pnas.1619640114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu B, Wang L, Li S, Dorf ME. 2017. ZMPSTE24 defends against influenza and other pathogenic viruses. J Exp Med 214:919–929. doi: 10.1084/jem.20161270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlach T, Hensen L, Matrosovich T, Bergmann J, Winkler M, Peteranderl C, Klenk HD, Weber F, Herold S, Pohlmann S, Matrosovich M. 2017. pH optimum of hemagglutinin-mediated membrane fusion determines sensitivity of influenza A viruses to the interferon-induced antiviral state and IFITMs. J Virol 91:e00246-17. doi: 10.1128/JVI.00246-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desai TM, Marin M, Mason C, Melikyan GB. 2017. pH regulation in early endosomes and interferon-inducible transmembrane proteins control avian retrovirus fusion. J Biol Chem 292:7817–7827. doi: 10.1074/jbc.M117.783878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rabbani MA, Ribaudo M, Guo JT, Barik S. 2016. Identification of IFN-stimulated gene (ISG) proteins that inhibit human parainfluenza virus type 3. J Virol 90:11145–11156. doi: 10.1128/JVI.01551-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yount JS, Zhang MM, Hang HC. 2013. Emerging roles for protein S-palmitoylation in immunity from chemical proteomics. Curr Opin Chem Biol 17:27–33. doi: 10.1016/j.cbpa.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jia R, Ding S, Pan Q, Liu SL, Qiao W, Liang C. 2015. The C-terminal sequence of IFITM1 regulates its anti-HIV-1 activity. PLoS One 10:e0118794. doi: 10.1371/journal.pone.0118794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li K, Jia R, Li M, Zheng YM, Miao C, Yao Y, Ji HL, Geng Y, Qiao W, Albritton LM, Liang C, Liu SL. 2015. A sorting signal suppresses IFITM1 restriction of viral entry. J Biol Chem 290:4248–4259. doi: 10.1074/jbc.M114.630780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang D, Guo H, Xu C, Chang J, Gu B, Wang L, Block TM, Guo JT. 2008. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol 82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang J, Warren TK, Zhao X, Gill T, Guo F, Wang L, Comunale MA, Du Y, Alonzi DS, Yu W, Ye H, Liu F, Guo JT, Mehta A, Cuconati A, Butters TD, Bavari S, Xu X, Block TM. 2013. Small molecule inhibitors of ER alpha-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res 98:432–440. doi: 10.1016/j.antiviral.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin HX, Feng Y, Tu X, Zhao X, Hsieh CH, Griffin L, Junop M, Zhang C. 2011. Characterization of the spike protein of human coronavirus NL63 in receptor binding and pseudotype virus entry. Virus Res 160:283–293. doi: 10.1016/j.virusres.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 69:6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 66.Zhao X, Guo F, Comunale MA, Mehta A, Sehgal M, Jain P, Cuconati A, Lin H, Block TM, Chang J, Guo JT. 2015. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob Agents Chemother 59:206–216. doi: 10.1128/AAC.03999-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang J, Warren TK, Zhao X, Gill T, Guo F, Wang L, Comunale MA, Du Y, Alonzi DS, Yu W, Ye H, Liu F, Guo JT, Mehta A, Cuconati A, Butters TD, Bavari S, Xu X, Block TM. 2013. Small molecule inhibitors of ER alpha-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res 98:432–440. doi: 10.1016/j.antiviral.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. 2014. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol 88:13221–13230. doi: 10.1128/JVI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]