

ABSTRACT
Herpesviruses replicate and package their genomes into capsids in replication compartments within the nuclear interior. Capsids then move to the inner nuclear membrane for envelopment and release into the cytoplasm in a process called nuclear egress. We previously found that nuclear F-actin is induced upon infection with the betaherpesvirus human cytomegalovirus (HCMV) and is important for nuclear egress and capsid localization away from replication compartment-like inclusions toward the nuclear rim. Despite these and related findings, it has not been shown that any specific motor protein is involved in herpesvirus nuclear egress. In this study, we have investigated whether the host motor protein, myosin Va, could be fulfilling this role. Using immunofluorescence microscopy and coimmunoprecipitation, we observed associations between a nuclear population of myosin Va and the viral major capsid protein, with both concentrating at the periphery of replication compartments. Immunoelectron microscopy showed that nearly 40% of assembled nuclear capsids associate with myosin Va. We also found that myosin Va and major capsid protein colocalize with nuclear F-actin. Importantly, antagonism of myosin Va with RNA interference or a dominant negative mutant revealed that myosin Va is important for the efficient production of infectious virus, capsid accumulation in the cytoplasm, and capsid localization away from replication compartment-like inclusions toward the nuclear rim. Our results lead us to suggest a working model whereby human cytomegalovirus capsids associate with myosin Va for movement from replication compartments to the nuclear periphery during nuclear egress.
IMPORTANCE Little is known regarding how newly assembled and packaged herpesvirus capsids move from the nuclear interior to the periphery during nuclear egress. While it has been proposed that an actomyosin-based mechanism facilitates intranuclear movement of alphaherpesvirus capsids, a functional role for any specific myosin in nuclear egress has not been reported. Furthermore, the notion that an actomyosin-based mechanism facilitates intranuclear capsid movement is controversial. Here we show that human cytomegalovirus capsids associate with nuclear myosin Va and F-actin and that antagonism of myosin Va impairs capsid localization toward the nuclear rim and nuclear egress. Together with our previous results showing that nuclear F-actin is induced upon HCMV infection and is also important for these processes, our results lend support to the hypothesis that nascent human cytomegalovirus capsids migrate to the nuclear periphery via actomyosin-based movement. These results shed light on a poorly understood viral process and the cellular machinery involved.
KEYWORDS: actin, cytomegalovirus, intranuclear movement, myosin, nuclear egress, replication compartment
INTRODUCTION
Important steps of the herpesvirus replication cycle take place in the host cell nucleus. Among these is nuclear egress, an unusual process by which nascent capsids translocate from the nucleus to the cytoplasm. Nuclear egress includes movement of capsids from the nuclear interior to the rim, disruption of the nuclear lamina, primary capsid envelopment at the inner nuclear membrane (INM), and, finally, deenvelopment at the outer nuclear membrane (1–3). For human cytomegalovirus (HCMV), processes at the nuclear rim are orchestrated by a virus-encoded two-subunit nuclear egress complex comprising INM-anchored UL50 and its nucleoplasmic binding partner, UL53, both of which are required for the production of infectious virus and nuclear egress (4–6). While progress has been made toward understanding steps of nuclear egress that take place at the INM, much less is known about earlier steps of nuclear egress (1).
During HCMV infection, viral DNA synthesis occurs in the interior of the nucleus at the periphery of subnuclear structures called replication compartments (RCs) (7–10). The presence of capsid and DNA packaging proteins in RCs suggests that capsid assembly and packaging are spatially coordinated with DNA synthesis in the nuclear interior (11–14). How HCMV capsids then migrate from RCs to the INM for primary capsid envelopment is poorly understood.
Several studies have suggested functions for nuclear myosins in intranuclear movements of herpesvirus capsids. Specifically, herpes simplex virus 1 (HSV-1) capsids were found to undergo intranuclear motility in a manner that was antagonized by temperature reduction or by inhibitors of ATP, myosin, or actin filaments (F-actin) (15). Another group showed that HSV-1 and pseudorabies virus (PRV) capsids associate with a nuclear population of myosin V and with nuclear F-actin (16). It was also reported that at least some HCMV capsids move rapidly from the nuclear interior to the periphery and eventually into the cytoplasm (17). The mean velocity reported (0.3 to 0.4 μm/s) is similar to what was reported for HSV-1 intranuclear capsid movement (0.5 μm/s) (15) and is consistent with myosin V-based movement (18). More recently, we found that nuclear F-actin is induced during HCMV infection and is important for nuclear egress, specifically at the step of capsid localization from RC-like inclusions toward the nuclear rim (19). Despite these findings, it has not been shown that any specific myosin is important for herpesvirus nuclear egress. Furthermore, the notion that an actomyosin-based mechanism facilitates herpesvirus intranuclear capsid motility is controversial (20–22). It has been reported that alphaherpesvirus (PRV and HSV-1) infection remodels nuclear architecture so that capsids can diffuse efficiently to the nuclear periphery (21). Resolving the potential role of the nuclear actomyosin system for herpesvirus capsid motility is important not only for virology but also for cell biology, as little is known regarding functions of nuclear actin and its associated myosin motor proteins.
In this study, we investigated a role for myosin Va in HCMV nuclear egress. Immunofluorescence and immunoelectron microscopy analyses revealed that myosin Va concentrates in RCs, where it colocalizes with capsids. We also found that myosin Va and major capsid protein colocalize with nuclear F-actin at the RC periphery and between RCs and the nuclear rim. Importantly, RNA interference (RNAi) and dominant negative (DN) approaches indicated that myosin Va is required for the efficient production of infectious virus, nuclear egress, and capsid localization from RC-like inclusions toward the nuclear periphery. These results lead us to suggest a working model in which HCMV capsids associate with myosin Va for capsid movement to the nuclear periphery.
RESULTS
Myosin Va localizes to RCs, where it associates with capsid protein.
We previously found that nuclear F-actin is induced upon HCMV infection and is important for capsid localization toward the nuclear periphery and for nuclear egress (19). However, we did not investigate the involvement of host motor proteins in that study. Myosin Va possesses structural characteristics that make it well suited for processive movement of diverse cargos (23), has been reported to associate with capsids in the nuclei of alphaherpesvirus-infected cells (16), and is thus an attractive candidate for facilitating capsid movement. Moreover, in a mass spectrometry experiment, we detected possible associations of myosin Va and major capsid protein (MCP) with the HCMV nuclear egress complex subunit, UL53 (unpublished data).
We began by investigating the subcellular distribution of myosin Va using cellular fractionation followed by Western blotting. At 72 h postinfection (hpi), mock-infected or HCMV-infected cells were separated into cytoplasmic and nuclear fractions which were probed for myosin Va, and for tubulin and lamin B as markers for cytoplasm and nucleus, respectively, using Western blot analysis. Myosin Va was found in both the cytoplasmic and nuclear fractions in both mock-infected and infected cells (Fig. 1). We also observed an increase in myosin Va levels in the cytoplasm of infected cells, consistent with reports that HCMV upregulates host gene expression (24). Using a nuclear localization signal (NLS) prediction program (http://nls-mapper.iab.keio.ac.jp), we found that human myosin Va contains a predicted monopartite NLS (RELKKLKIE) starting at residue 903 with a score indicative of proteins present in both the cytoplasm and nucleus, consistent with these results.
FIG 1.
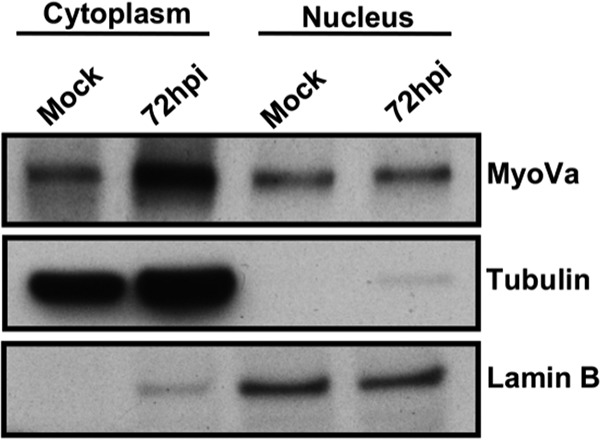
HFFs were either mock infected or infected with WT HCMV (MOI of 1). At 72 hpi, nuclear and cytoplasmic fractions were prepared and then analyzed by Western blotting to assess myosin Va (MyoVa) expression levels, with tubulin and lamin B serving as fractionation controls for the cytoplasm and nucleus, respectively.
To further explore the distribution of myosin Va in infected cells, we used immunofluorescence assays (IFA) and confocal microscopy. We mock infected or infected human foreskin fibroblasts (HFFs) with HCMV encoding a FLAG-tagged version of UL44 (44-F), the viral DNA polymerase subunit that has previously been shown to concentrate at the periphery of RCs (8). We then stained these cells with anti-FLAG, anti-MCP, and anti-myosin Va antibodies and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) to mark the nucleus. In mock-infected cells, myosin Va was fairly evenly distributed across the cell (Fig. 2A). However, upon HCMV infection a population of myosin Va redistributed to RCs marked by UL44. Interestingly, at 48 hpi myosin Va already colocalized with UL44 in RCs in cells lacking visible MCP expression (Fig. 2B). In cells displaying MCP expression at this time point, MCP was distributed throughout the nucleoplasm but exhibited little colocalization with myosin Va and UL44 in RCs. By 72 hpi, however, MCP could be found at the periphery of RCs, where it colocalized extensively with myosin Va and UL44 (Fig. 2C).
FIG 2.

(A to C) HFFs were mock infected or infected with HCMV 44-F (MOI of 1) and fixed at 48 or 72 hpi. Coverslips were then stained with DAPI (blue), anti-FLAG (blue), anti-MCP (green), and anti-MyoVa (red) antibodies and imaged with spinning-disk confocal microscopy. In panels B and C, DAPI is excluded from the merged images. In panel B, arrows indicate MyoVa in RCs prior to detectable MCP expression. In panel C, white color indicates colocalization between UL44, MCP, and MyoVa. All images are single optical sections. Scale bars are 10 mm. (D) HFFs were infected with HCMV (MOI of 1). At 72 hpi, nuclear lysates were prepared and immunoprecipitated with resin conjugated to either an anti-Myosin Va or an IgG isotype control antibody. Lysates and eluates were then analyzed by Western blotting using antibodies against the proteins indicated at the right.
Finally, we utilized coimmunoprecipitation (co-IP) to investigate whether myosin Va associates with MCP in nuclear lysates. HFFs were infected with HCMV (multiplicity of infection [MOI] = 1) and nuclear lysates were prepared at 72 hpi, followed by immunoprecipitation with resin conjugated to either an anti-myosin Va antibody or an IgG isotype control antibody. We detected a band corresponding to MCP in myosin Va but not in IgG immunoprecipitates. As an additional negative control, we probed for the host nuclear protein PCNA, which we did not detect in myosin Va or IgG immunoprecipitates, as expected (Fig. 2D). Collectively, these results suggest that a population of nuclear myosin Va concentrates in RCs, where it associates with capsid protein.
Immuno-EM reveals that myosin Va associates with assembled capsids.
Our findings that myosin Va colocalizes and associates with MCP suggested an association between myosin Va and assembled capsids. To investigate this possibility, we utilized immunoelectron microscopy (immuno-EM). HFFs were infected with HCMV (MOI = 1), and cells were fixed at 72 hpi. The samples were further processed by primary staining with either an anti-myosin Va primary antibody or, as a control, a primary antibody against the HCMV gene regulatory proteins IE 1 and 2, followed by secondary staining with 10-nm protein A-gold (Fig. 3A and B). Consistent with our IFA results, we found that most myosin Va in the nucleus was present in the nuclear interior, where it associated with electron-dense inclusions reminiscent of RCs. Other groups have considered these inclusions to be RCs (19, 25, 26); we term these structures RC-like inclusions (19). Interestingly, myosin Va appeared to associate with capsids in RC-like inclusions (Fig. 3A). Conversely, while IE 1/2 was present in the RC-like structures, it rarely associated with capsids (Fig. 3B). To quantify these results, we calculated the percentage of nuclear capsids associated with at least 1 gold particle in samples stained with primary antibodies against myosin Va or IE 1/2 (Fig. 3C). We found that 39% of capsids associated with myosin Va (n = 95), whereas only 2% of capsids associated with IE 1/2 (n = 127), which was highly significant (P < 0.0001). Thus, a substantial fraction of nuclear capsids associate with myosin Va.
FIG 3.

(A and B) HFFs were infected with HCMV (MOI of 1) and fixed for immuno-EM at 72 hpi. The cells were further processed by primary staining with anti-MyoVa or anti-IE 1/2 (negative control) antibodies, followed by secondary staining with 10-nm protein A-gold. Imaging was conducted using a transmission electron microscope. In the rightmost image in panel A, the white arrowheads indicate capsids (without DNA) associated with MyoVa and the black arrowhead indicates a capsid (without DNA) that is not associated with MyoVa. In the rightmost image in panel B, the black arrowheads indicate capsids that are not associated with IE 1/2 (leftmost capsids contain DNA; rightmost capsid does not contain DNA). Scale bars are 100 nm. (C) The percentage of capsids associated with at least one gold particle was calculated for each condition (MyoVa, n = 95; IE 1/2, n = 127). The P value was calculated using Fisher's exact test. ****, P < 0.0001.
Myosin Va and capsid protein colocalize with nuclear F-actin.
We wondered whether myosin Va and capsid protein would colocalize with nuclear actin filaments. We therefore mock infected or infected HFFs stably expressing LifeAct-GFP-NLS, an actin binding peptide that we have previously used to visualize nuclear F-actin in infected cells (19), and stained for myosin Va and MCP and with DAPI. In mock-infected cells, we observed diffuse LifeAct-GFP-NLS signal in the nucleus, with no F-actin apparent (Fig. 4A, left). Conversely, at 72 hpi we observed thick nuclear F-actin structures that localized along the periphery of RCs and extended between RCs and the nuclear rim (Fig. 4A, right), as we did previously (19). We also observed puncta of colocalization of myosin Va and MCP with nuclear F-actin along the periphery of RCs and close to the nuclear rim (Fig. 4A, right), which we verified by measuring the fluorescence intensity of each channel across the indicated line (Fig. 4B). Thus, MCP and myosin Va can be found together on nuclear actin filaments, consistent with the possibility that HCMV capsids utilize myosin Va to traffic on nuclear F-actin.
FIG 4.

(A) HFFs stably expressing LifeAct-GFP-NLS (green) were either mock infected (left) or infected with WT HCMV (MOI = 1) (right). At 72 hpi, cells were fixed, stained with anti-MCP (shown as blue [imaged in far red]) and anti-MyoVa (red) antibodies and DAPI (blue), and imaged with spinning-disk confocal microscopy. White color indicates colocalization between MCP, actin, and MyoVa. Scale bar is 10 mm. (B) To measure colocalization, the fluorescence intensity of each channel was plotted across the white line shown in the merged image. Images are single optical sections.
Depletion of myosin Va antagonizes the production of infectious virus and nuclear egress.
We then set out to investigate whether myosin Va has a functional role during HCMV infection, particularly for nuclear egress. HFFs were transfected with a pool of myosin Va short interfering RNA (siRNA) or nontargeting (NT) siRNA, followed by infection with wild-type (WT) HCMV (MOI = 1). At 72 or 96 hpi, media were harvested to evaluate the production of infectious virus, and cell monolayers were fixed for EM analysis to examine nuclear egress. At 72 hpi, we found that myosin Va was depleted ∼5-fold by myosin Va siRNA compared to NT siRNA (Fig. 5A). Notably, titration of media revealed a 3.6-fold reduction in the production of infectious virus at 72 hpi and a 3.1-fold reduction at 96 hpi in cells transfected with myosin Va siRNA compared to NT siRNA, which were significant (72 hpi, P < 0.0001; 96 hpi, P = 0.045), suggestive of a role for myosin Va at some stage of the HCMV replication cycle (Fig. 5B).
FIG 5.

HFFs were transfected with a pool of MyoVa siRNA or NT siRNA (control), followed by infection with WT HCMV (MOI = 1). (A) Whole-cell lysates were harvested at 72 hpi and analyzed by Western blotting. Undiluted (neat) NT siRNA lysates were diluted as shown to quantify MyoVa knockdown. The numbers under the top portion represent band intensities relative to the neat lysates. (B) At 72 or 96 hpi, media were removed for titration to measure the production of infectious virus. The P values were calculated using an unpaired t test (3 independent experiments). ****, P < 0.0001; *, P = 0.045. (C) Cell monolayers were fixed and processed for EM. Capsids were counted in the nucleus and cytoplasm of whole-cell sections of 10 (72 hpi) or 7 (96 hpi) cells for each condition, with each point representing the number in a compartment in an individual cell section. Bars and the numbers alongside them indicate mean numbers of capsids. P values were calculated using the Mann-Whitney test. ns, not significant. P values were as follows: Nucleus, 72 hpi, P = 0.31; 96 hpi, P > 0.99; Cytoplasm, *, P = 0.04 (72 hpi) and P = 0.01 (96 hpi). (D) Cytoplasmic capsids as a percentage of total capsids (cytoplasmic and nuclear) for each cell. The P values were calculated using an unpaired t test. *, P = 0.02 (72 hpi) and P = 0.04 (96 hpi). (E) Lysates derived from NT or MyoVa siRNA-transfected cells were analyzed by Western blotting to assess late viral gene expression using antibodies against proteins indicated on the right.
To measure the effect of myosin Va knockdown on nuclear egress, capsids in the cytoplasm and nucleus were counted in representative whole-cell EM sections (Fig. 5C). As observed before in HCMV-infected cells (27–30), there were markedly fewer capsids in the cytoplasm than in the nucleus in the NT siRNA-treated samples. This contrasts with findings for HSV-1-infected cells, in which there are similar numbers of capsids in the two compartments (31, 32). Regardless, we observed a 2.6-fold (72 hpi) and a 2.8-fold (96 hpi) decrease in the mean number of capsids in the cytoplasm in cells treated with myosin Va siRNA relative to those treated with NT siRNA, which were significant (72 hpi, P = 0.04; 96 hpi, P = 0.01), but only a 20% decrease (72 hpi) and a 0.01% increase (96 hpi) in the mean number of capsids in the nucleus, which were not significant (72 hpi, P = 0.31; 96 hpi, P > 0.99). We also quantified cytoplasmic capsids as a percentage of total (cytoplasmic and nuclear) capsids for each cell and condition and observed a 2.5-fold (72 hpi) and a 2-fold (96 hpi) decrease in the percentage of cytoplasmic capsids in myosin Va versus NT siRNA-treated cells, which were significant (72 hpi, P = 0.02; 96 hpi, P = 0.04) (Fig. 5D). Finally, as defects in viral production and nuclear egress with myosin Va knockdown could conceivably be caused by a decrease in late viral gene expression, we analyzed lysates from the above-described experiment using Western blotting, which indicated that myosin Va knockdown did not discernibly affect expression of the late proteins MCP, UL53, and pp28 (Fig. 5E). These results suggest that myosin Va knockdown does not affect earlier events in the replication cycle, including entry, capsid movement to the nucleus, immediate early and early gene expression, and DNA replication.
To help assess whether off-target effects from the myosin Va siRNA pool contributed to defects in the production of infectious virus and nuclear egress, HFFs were transfected with NT siRNA, the pool of myosin Va siRNA, or each of the 4 individual siRNAs that comprise the pool and processed as described above. We found that siRNA 4 knocked down myosin Va as well as or more than the pool, while siRNAs 1, 2, and 3 had lesser effects on knockdown (Fig. 6A). We then compared the effects of siRNA 1 and siRNA 4 on viral production and nuclear egress. In this experiment, we found that relative to siRNA 1, siRNA 4 caused a 3.7-fold reduction in the production of infectious virus (Fig. 6B) and a 2.6-fold decrease in the mean number of cytoplasmic capsids, which was significant (P = 0.04), but only a 14% decrease in the mean number of nuclear capsids, which was not significant (P = 0.31) (Fig. 6C). We also measured cytoplasmic capsids as a percentage of total capsids for each cell and observed a 2-fold decrease in the percentage of cytoplasmic capsids in cells transfected with siRNA 4 versus siRNA 1, which was statistically significant (P = 0.02) (Fig. 6D). Collectively, the results in Fig. 5 and 6 provide evidence that myosin Va is important for the efficient production of infectious virus and nuclear egress.
FIG 6.

(A) HFFs were transfected with NT siRNA, the pool of myosin Va siRNA, or each of the siRNAs that comprise the pool and then infected with WT HCMV (MOI = 1). (A) Whole-cell lysates were harvested at 72 hpi and analyzed by Western blotting to assess myosin Va knockdown. Also at 72 hpi, media were removed for titration to measure virus production (one experiment) (B), and cell monolayers were fixed and processed for EM. Capsids were counted in the nucleus and cytoplasm of whole-cell sections in 10 cells for each condition (C). P values were calculated using the Mann-Whitney test. ns, not significant (P = 0.31). *, P = 0.04. (D) Cytoplasmic capsids as a percentage of total capsids for each cell. The P value was calculated using an unpaired t test. *, P = 0.04.
Expression of a myosin Va DN mutant causes defects in the production of infectious virus and nuclear egress.
We next evaluated whether expression of a myosin Va dominant negative (DN) mutant would similarly affect the production of infectious virus and nuclear egress. For these experiments, we utilized the “long-tail” (LT) myosin Va DN mutant that encodes the spliced isoform of the myosin Va tail domain predominantly found in the brain, plus the cargo-binding globular tail domain (33–35). However, this DN mutant lacks the amino acid sequences that we predict functions as an NLS. As we wished to examine the role of nuclear myosin Va during HCMV infection, we fused an NLS from simian virus 40 (SV40) to the N terminus of a green fluorescent protein (GFP)-tagged version of the LT DN mutant, creating LT-GFP-NLS. LT-GFP-NLS or GFP-NLS (control) was cloned into a doxycycline (Dox)-inducible lentiviral vector and introduced into HFFs by transduction. Western blot analysis and IFA showed that LT-GFP-NLS and GFP-NLS were expressed at roughly similar levels in HFFs and localized mainly to the nucleus following Dox induction (Fig. 7A and B). LT-GFP-NLS- or GFP-NLS-transduced HFFs were then infected with HCMV (MOI = 1), and Dox was added to the medium at 24 hpi to induce expression. Medium was removed at 72 hpi and titrated to measure the production of infectious virus, which showed that relative to control GFP-NLS expression, LT-GFP-NLS expression caused a 3.2-fold defect in the production of infectious virus, which was significant (P = 0.007) (Fig. 7C). Furthermore, we observed a 2.9-fold defect in the accumulation of cytoplasmic capsids, which was significant (P = 0.003), and only a 12% decrease in the accumulation of nuclear capsids, which was not significant (P = 0.183) (Fig. 7D). We also measured cytoplasmic capsids as a percentage of total capsids for each cell and observed a 2-fold decrease in the percentage of cytoplasmic capsids in LT-GFP-NLS- versus GFP-NLS-expressing cells, which was significant (P = 0.02) (Fig. 7E). Thus, in agreement with our RNAi results, inhibition of myosin Va by a DN mutant causes defects in the production of infectious virus and nuclear egress.
FIG 7.

Lentiviruses expressing LT-GFP-NLS or GFP-NLS (control) were generated and used to transduce HFFs. Doxycycline (Dox) was added to media to induce DN expression. Cells were harvested to assess DN mutant expression levels with Western blot analysis using antibodies against GFP (top) or actin as a loading control (bottom) (A) or fixed, stained with DAPI (blue), and imaged with spinning-disk confocal microscopy to assess subcellular localization of LT-GFP-NLS or GFP-NLS relative to DAPI (B). (C) LT-GFP-NLS- and GFP-NLS-transfected HFFs were infected with WT HCMV (MOI = 1) and Dox was added to media at 24 hpi to induce DN mutant expression. At 72 hpi, media were removed and titrated to measure the production of infectious virus. The P value was calculated using an unpaired t test (4 independent experiments). **, P = 0.007. (D) Cell monolayers were fixed and processed for EM to assess nuclear egress. Capsids were counted in the nucleus and cytoplasm of whole-cell sections in 12 cells for each condition. P values were calculated using the Mann-Whitney test. ns, not significant (P = 0.183); **, P = 0.003. (E) Cytoplasmic capsids as a percentage of total capsids (cytoplasmic and nuclear) for each cell. The P value was calculated using an unpaired t test. *, P = 0.02.
Myosin Va is important for capsid localization away from RC-like inclusions.
We previously found that inhibition of nuclear F-actin impaired capsid localization from RC-like inclusions toward the nuclear rim (19). As we observed in immuno-EM that myosin Va associates with assembled capsids in these inclusions, we wondered whether antagonism of myosin Va would similarly cause capsids to accumulate in RC-like inclusions away from the nuclear rim. We therefore counted and calculated the percentages of capsids inside and outside these structures in cell sections from the previous experiments in which cells were transfected with individual siRNAs, or in cells expressing the nucleus-localized myosin Va DN mutant. We found that 28% of capsids were located outside RC-like inclusions in cells expressing GFP-NLS, compared to 11% in LT-GFP-NLS-expressing cells (2.5-fold decrease), which was highly significant (P < 0.0001; Fig. 8A and B). Similarly, 16% of capsids were present outside RC-like inclusions in cells transfected with siRNA 1, compared to 8% in cells transfected with siRNA 4 (2-fold decrease), which was also highly significant (P = 0.003) (Fig. 8C). We did not detect disruption of RC-like inclusions in LT-GFP-NLS-expressing cells (Fig. 8A) or in cells transfected with siRNA 4. Thus, antagonism of myosin Va reduces capsid localization away from the RC-like inclusions toward the nuclear periphery.
FIG 8.

(A) LT-GFP-NLS- or GFP-NLS-expressing HFFs were infected with WT HCMV (MOI = 1). At 24 hpi, doxycycline was added to media to induce DN expression, and at 72 hpi cell monolayers were fixed for EM analysis. Capsids associated or not associated with electron-dense RC-like inclusions were counted in representative nuclear sections of 16 cells for each condition. White arrows indicate capsids located within RC-like inclusions; black arrows indicate capsids located outside RC-like inclusions. Nuc., nuclear compartment; Cyt., cytoplasmic compartment. (B) The percentage of capsids not associated with RC-like inclusions was calculated in each nucleus and plotted. (C) HFFs were transfected with siRNA 1 or 4 and then infected with WT HCMV (MOI = 1). At 72 hpi, cell monolayers were fixed for EM analysis. Capsids were counted in the nuclei of 10 cells for each condition and plotted. All P values were calculated using the Mann-Whitney test. ****, P < 0.0001; **, P = 0.003.
DISCUSSION
How nascent herpesvirus capsids move from the nuclear interior to the periphery during nuclear egress is poorly understood and controversial. We previously reported that nuclear F-actin is induced upon HCMV infection and is important for nuclear egress, specifically at the step of capsid localization from RC-like inclusions toward the nuclear periphery (19). These results suggested a possible role for a myosin motor protein in this process, which we have investigated in this study. Upon infection, nuclear myosin Va concentrated at the periphery of RCs, where we observed colocalization with MCP using IFA and an association between myosin Va and MCP in nuclear lysates with co-IP. Immuno-EM showed that a substantial fraction of assembled nuclear capsids associate with myosin Va. We also found that myosin Va and MCP colocalize with nuclear F-actin at the RC periphery and between RCs and the nuclear rim. Crucially, RNAi and DN approaches indicated that myosin Va is important for the efficient production of infectious virus, nuclear egress, and capsid localization away from RC-like inclusions toward the nuclear periphery. We discuss these results and suggest a working model in which HCMV capsids associate with myosin Va in RCs for actomyosin-based movement to the nuclear periphery.
Association of myosin Va with capsids.
Cellular fractionation, co-IP, confocal IFA, and immuno-EM analyses revealed substantial amounts of myosin Va in the nucleus during HCMV infection, where it associated with MCP (co-IP and IFA) and with assembled capsids (immuno-EM). Notably, IFA and immuno-EM revealed that these associations largely occurred in RCs and RC-like inclusions, respectively. With IFA, we found that myosin Va had already localized to RCs in cells that had not yet visibly expressed MCP, raising the possibility that myosin Va is poised to facilitate capsid localization toward the nuclear periphery at an early stage of infection. Interestingly, once expressed, MCP initially localized throughout the nucleoplasm, but it was later recruited to the periphery of RCs, where it colocalized with myosin Va. Our lab previously found that viral DNA synthesis occurs at the periphery of RCs (7, 8); thus, it is tempting to speculate that viral DNA synthesis, capsid assembly, and the initiation of myosin Va-dependent capsid movement to the nuclear periphery are coupled at the RC periphery. Similar to our results, it was recently shown that nuclear myosin Va localizes to adenovirus replication centers; however, whether myosin Va associates with capsids or is important for infection was not reported (36).
Importance of myosin Va for nuclear egress.
We observed defects in capsid accumulation in the cytoplasm and capsid localization away from RC-like inclusions with RNAi or DN antagonism of myosin Va. To our knowledge, this is the first report of a role for a particular myosin in herpesvirus nuclear egress. These defects were similar to the overall defects in the production of infectious virus; thus, it is reasonable to suggest that myosin Va is principally involved in nuclear egress, particularly at the step of capsid localization to the nuclear periphery. Furthermore, as RNAi depleted myosin Va by a magnitude similar to the fold decrease in nuclear egress, and the myosin Va DN mutant might have incompletely antagonized its target, it is also formally possible that myosin Va is required for nuclear egress. Alternatively, it may enhance nuclear egress efficiency. Regardless, the simplest explanation of our results together with our previous finding that nuclear F-actin is induced upon HCMV infection and is important for nuclear egress (19), and others' findings that HCMV capsids rapidly move toward the nuclear rim (17), that PRV capsids colocalize with nuclear myosin V (16), and that a small-molecule inhibitor of myosins inhibits directed intranuclear movements of HSV-1 capsids (15), is that myosin Va promotes capsid movement to the nuclear periphery.
It is also possible that some capsids reach the nuclear periphery without the aid of myosin Va, for example, by diffusion (particularly late in HCMV infection when RCs fill most of the nucleus so that capsids might only need to move short distances), as suggested for alphaherpesviruses by Bosse et al. (20–22). These authors reported that PRV and HSV-1 capsids do not use directed movement in the nucleus but instead take advantage of remodeled nuclear architecture to diffuse freely (21). These findings appear to contrast with our results, which suggest that myosin Va is important for HCMV nuclear egress by promoting capsid localization near the nuclear periphery. We speculate that these discrepancies may be due either to alphaherpesviruses and HCMV differing in their capacities to utilize actomyosin-based capsid transport or to myosin Va being important for remodeling of nuclear architecture, which could enable capsid diffusion from RCs to the nuclear periphery.
Some studies addressing the involvement of nuclear myosins in herpesvirus infection have also suggested roles for nuclear F-actin (15, 16, 37). Yet whether herpesvirus infection induces nuclear F-actin is also controversial (19, 20, 22). We previously found that HCMV, but not HSV-1 infection, induces nuclear F-actin in HFFs and is important for nuclear egress (19). In the present study, we found that myosin Va and MCP colocalize with nuclear F-actin at the RC periphery and near the nuclear rim. This finding, coupled with our findings that antagonism of nuclear F-actin (19) or myosin Va (this study) both reduced capsid localization toward to the nuclear rim, suggests that HCMV capsids utilize the actomyosin system for directed capsid transport. Additional live-cell imaging studies are needed to rigorously test whether HCMV capsids undergo directed intranuclear movement and, if so, whether the actomyosin system is important for such movement.
In the HSV-1 system, expression of myosin Va LT DN mutants caused a decrease in secreted infectivity but an increase in total (intracellular) infectivity and also impaired glycoprotein expression at the plasma membrane, leading to the conclusion that myosin Va principally functions in the cytoplasm during HSV-1 infection (38). However, the LT DN mutants utilized in that previous study encode a region of myosin Va lacking the predicted NLS, and they thus may have been largely excluded from the nucleus. Using a nucleus-localized LT DN mutant in HCMV-infected cells, we found similar defects in the production of infectious virus, capsid accumulation in the cytoplasm, and capsid localization outside RC-like inclusions, highlighting the importance of myosin Va in nuclear egress. We cannot discount the possibility, however, that cytoplasmic myosin Va functions to some extent during HCMV infection, as the nuclear egress defects upon RNAi antagonism did not completely account for the detects in viral production.
Model.
We propose the following working model that explains our results: nascent HCMV capsids associate with myosin Va at the periphery of RCs. Since most nuclear capsids are present in RC-like inclusions under steady-state conditions, we speculate that movement of capsids to the nuclear periphery is a rate-limiting step. Based on our previous findings that nuclear F-actin is induced upon HCMV infection and is important for capsid localization toward the nuclear periphery and nuclear egress (19), and our current findings that myosin Va colocalizes with nuclear F-actin and capsid protein in RCs and between RCs and the nuclear rim, the model posits that HCMV capsids move to the nuclear periphery via myosin Va and nuclear F-actin-based directed movement.
As myosin Va localization to RCs and nuclear F-actin induction (19) occur prior to nuclear egress, it is possible that the nuclear actomyosin system participates in steps such as RC movement and/or maturation, as proposed for HSV-1 (37). It is also possible that myosin Va and nuclear F-actin help remodel chromatin to allow capsids to move efficiently to the nuclear periphery by diffusion, as proposed for alphaherpesviruses (21). This mechanism and directed movements are not mutually exclusive. However, a diffusion-only model would not explain our finding that assembled HCMV capsids associate with myosin Va or why we observed colocalization of myosin Va with capsid protein and nuclear F-actin.
Further studies of how HCMV capsids move from RCs toward the nuclear rim should distinguish among these possibilities and shed light on this step of herpesvirus nuclear egress and on the still poorly understood host nucleoskeleton.
MATERIALS AND METHODS
Cells and viruses.
Human foreskin fibroblasts (HFFs) (ATCC; CRL-1684) and human embryonic kidney (293T) cells (ATCC; CRL-11268) were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). Generation of HFFs stably expressing LifeAct-GFP-NLS was described previously (19). The HCMV laboratory strain AD169 was used in all experiments. Recombinant viruses encoding FLAG-tagged versions of UL53 (53-F) (4) or UL44 (44-F) (39) were described previously. Viruses were propagated and titrated as described previously (40).
Cellular fractionation.
A total of 2 × 105 HFFs/well were seeded in a 12-well plate and either mock-infected or infected with WT HCMV (MOI = 1). At 72 hpi, cells were fractionated using an NE-PER extraction kit (Thermo). Nuclear and cytoplasmic fractions were analyzed by Western blotting using the protocol described below.
Immunofluorescence analysis.
A total of 1 × 105 HFFs or LifeAct-GFP-NLS-expressing HFFs/well were seeded on glass coverslips in a 24-well plate, followed by either mock infection or infection with WT, 53-F, or 44-F HCMV (MOI = 1). At the desired time points, cells were fixed at room temperature (RT) in 3.7% formaldehyde–Dulbecco's phosphate-buffered saline (DPBS). Cells were then permeabilized at RT in 0.1% Triton X-100–DPBS, washed 3 three times with DPBS, and blocked overnight in a mixture of 1% bovine serum albumin (BSA; Sigma) and 5% human serum (Sigma) in DPBS. The following antibodies and dilutions were used for primary staining: rabbit anti-myosin Va (Abcam; ab11094), 1:50; mouse anti-FLAG M2 (Sigma; F1804), 1:500; and mouse anti-MCP (generous gift from William Britt, University of Alabama, Birmingham), 1:250. Antibodies were diluted in a mixture of 1% BSA of 5% human serum in DPBS and added to coverslips for 1 h at RT with rocking. Primary antibodies were removed and coverslips washed 3 times with DPBS for 5 min with rocking at RT. The staining procedure was repeated with the appropriate fluorescently labeled Alexa Fluor secondary antibodies (Invitrogen), and DAPI was applied in the last 10 min of the secondary antibody incubation. After the final washes, coverslips were mounted on glass slides using ProLong antifade (Invitrogen). Imaging was done at the Nikon Imaging Center (NIC) at Harvard Medical School using a Nikon Ti spinning-disk confocal laser microscope. Postacquisition image analysis was conducted using Metamorph and ImageJ software packages.
Immunoprecipitation.
For myosin Va IP, 2.5 × 106 HFFs were infected with HCMV 53-F (MOI = 1) and nuclear lysates were prepared using a nuclear complex co-IP kit (Active Motif). Nuclear lysates were precleared with 50 μl of EZ-View Red protein A affinity gel (Sigma) combined with 10 μg of IgG isotype control antibody (Cell Signaling; 3900) by rotation overnight at 4°C prior to IP. Precleared lysate was then split into 2 equal volumes, and each was mixed with 25 μl of protein A affinity gel combined with either 1 μg of the isotype control antibody or rabbit anti-myosin Va antibody (Cell Signaling; 3402). Reaction mixtures were rotated overnight at 4°C, and resin was centrifuged and washed 4 times in 750 μl of ice-cold EBC buffer (50 mM Tris [pH 8.0], 300 mM NaCl, 0.5% NP-40 containing one complete EDTA-free protease inhibitor tablet [Roche] per 50 ml) by rotation for 20 min between washes. After the final spin, the resin pellet was mixed with 40 μl of EBC buffer, and protein was eluted from resin by incubation with 50 μl of 2× Laemmli buffer at 95°C for 5 min.
Western blotting.
For IP Western blotting, lysates and eluates in Laemmli buffer were separated on a 4 to 20% SDS-polyacrylamide gel (Bio-Rad). Proteins were then transferred onto a polyvinylidene difluoride (PVDF) membrane, blocked with 5% milk in DPBS-T (DPBS with 0.5% Tween 20), and probed with primary antibodies (see below for sources and dilutions) overnight (o/n) at 4°C with rocking. Membranes were washed 3 times with DPBS-T for 10 min at RT with rocking. Membranes were then incubated with TrueBlot secondary antibodies conjugated to horseradish peroxidase (HRP; Rockland) at 1:1,000 for 1 h at RT with rocking, followed by washing. Finally, chemiluminescence solution (Pierce) was added to membranes, and signal was detected with film. Where indicated, band intensities were measured using ImageJ software.
For Western blotting following cellular fractionation, nuclear or cytoplasmic lysates were mixed with an equal volume of 2× Laemmli buffer, heated at 95°C for 5 min, and further processed as described above.
For all other Western blotting, cells were harvested by washing with DPBS followed by the addition of 2× Laemmli buffer with protease inhibitors (Thermo Scientific) directly to the monolayer. Lysates were scraped off the plate, heated at 95°C for 5 min, and further processed as described above.
Primary antibody dilutions were as follows: rabbit anti-myosin Va (Cell Signaling; 3402), 1:1,000; mouse anti-β-actin (Sigma; A5441), 1:5,000; rabbit anti-PCNA (Abcam; ab18197), 1:700; mouse anti-MCP (generous gift from William Britt, University of Alabama, Birmingham), 1:200; goat anti-lamin B (Santa Cruz C-20), 1:200; and mouse antitubulin (GeneTex; GT114), 1:5,000.
Immunoelectron microscopy.
A total of 2 × 105 HFFs/well were seeded in a 12-well plate and infected with either WT or 53-F HCMV (MOI = 1). At 72 hpi, cells were washed with DPBS, trypsinized, and harvested. The cells were pelleted and fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4) at RT. After 2 h, the fixative was replaced with DPBS. Prior to freezing in liquid nitrogen, the cell pellets were cryoprotected in 2.3 M sucrose in DPBS for 15 min. Frozen samples were sectioned at −120°C, and the sections were transferred to Formvar carbon-coated copper grids. The gold labeling was carried out at RT. All antibodies and protein A gold were diluted in 1% BSA in DPBS. After blocking in 1% BSA for 10 min at RT, the grids were incubated in primary antibody (anti-myosin Va, 1:10; anti-IE1/2, 1:10; anti-FLAG M2, 1:50) for 30 min. The grids were then washed in 4 drops of DPBS, transferred to 10-nm protein A gold for 20 min, and washed in 4 drops and 6 drops of double-distilled water. Contrasting/embedding of the labeled grids was carried out in 0.3% uranyl acetate in 2% methyl cellulose for 5 min. The grids were examined on a JEOL 1200EX electron microscope, and images were recorded with an AMT 2k charge-coupled-device (CCD) camera.
RNA interference.
A total of 1 × 105 HFFs/well were seeded in a 12-well plate, followed by transfection with ON-TARGET-plus SMARTpool human myosin Va siRNA or nontargeting (NT) siRNA (Dharmacon) the following day. The 4 siRNAs that comprised the pool were transfected individually. For transfection, 2.5 μl of 100 μM siRNA and 2.5 μl Dharmafect transfection reagent (Dharmacon) (per well) were individually incubated in 97.5 μl of Opti-MEM medium (Invitrogen) for 5 min at RT. The solutions were then mixed and incubated for 20 min at RT before 800 μl of prewarmed Opti-MEM was added. Culture medium was removed from cells and replaced with transfection mixture. After 6 h, the transfection mixture was replaced with DMEM containing 5% FBS. The above-described procedure was repeated on 2 more consecutive days to ensure that most cells received siRNA (∼70 to 90% of cells based on reference 7 and this study). Seventy-two hours after the initial transfection, cells were infected with WT HCMV (MOI = 1) in duplicate. At 72 hpi, medium was removed and pooled for virus titration and cells were either lysed for Western blot analysis (to assess myosin Va knockdown) or fixed for EM analysis (to measure nuclear egress and subnuclear capsid distribution) as described in the text.
TEM.
Transmission electron microscopy (TEM) was utilized to assess nuclear egress and subnuclear capsid distribution by counting capsids in the nuclei and cytoplasm in representative whole-cell sections under the desired conditions using a TecnaiG2 Spirit BioTWIN electron microscope equipped with an AMT 2k CCD camera.
Processing for image acquisition and data analysis were performed essentially as described previously (41). Capsids were counted in a blinded manner such that it was not known which cell sections received which treatment. Statistical significance was assessed using Mann-Whitney or unpaired t tests (GraphPad Prism software).
Nucleus-localized myosin Va dominant negative mutant.
To generate LT-GFP-NLS and GFP-NLS (control) lentiviral vectors, a plasmid encoding the brain long tail (BR-LT) myosin Va DN mutant N-terminally fused to GFP (generous gift from John Hammer III, National Institutes of Health [NIH]/National Heart, Lung, and Blood Institute [NHLBI]) was used as a template for PCR amplification. An NLS was fused to the N terminus of LT-GFP or just GFP using the following primers containing the SV40 NLS sequence and Kpn1 restriction sites: for LT-GFP-NLS and GFP-NLS, forward primer 5′-TAAGCAGGTACCATGCCGCCTAAGAAAAAGCGGAAGGTGGGGGTGAGCAAGGGCGAGGAGCTGTTCACCGGG-3′; for LT-GFP-NLS, reverse primer 5′-TGCTTAGGTACCCTGCAGCCCGGGGGATCCACTAGTTCTAGAGCGGCCGCAATCGACCAA-3′; and for GFP-NLS, reverse primer 5′-TGCTTAGGTACCGAAGCTTGAGCTCGAGATCTGAGTCCGGACTTGTACAGCTCGTCCATGCCGAGAGTGATC-3′. The PCR products were then digested with Kpn1 (New England BioLabs) and ligated into the pENTR1a entry vector (Addgene plasmid 17398; generous gift from Eric Campeau). The entry vector was recombined with pLIX_402 inducible lentiviral destination vector (Addgene plasmid 41394; generous gift from Daniel Root) using Gateway cloning (Thermo). To generate lentiviruses, 5 × 105 293T cells/well were plated in a 6-well plate (3 wells/condition) and transfected with LT-GFP-NLS or GFP-NLS lentiviral vector along with psPAX2 (Addgene plasmid 12260) and pMDG.2 (Addgene plasmid 12259) packaging plasmids (generous gifts from Didier Trono) using Roche XTREME gene HD transfection reagent (according to the manufacturer's lentivirus production protocol). Forty-eight hours after transfection, supernatants containing lentiviruses (6 ml/condition) were harvested and centrifuged at 1,000 × g for 5 min to remove cellular debris. Two milliliters of lentiviral supernatant was mixed with Polybrene (Sigma; final concentration of 8 μg/ml) and then used to transduce 3 × 105 HFFs/well in a 6-well plate. Twenty-four hours later, the lentiviral mixture was removed and replaced with new mixture. This was repeated the following day to achieve 3 total transductions. Transduced HFFs were then selected with puromycin (Sigma; final concentration of 1 μg/ml) and expanded in tissue culture. To induce DN expression, doxycycline (Sigma; final concentration of 1 μg/ml) was added directly to media at 24 hpi. Subsequent measurements of viral production, nuclear egress, and subnuclear capsid distribution were conducted as described above.
ACKNOWLEDGMENTS
We thank John Hammer III for generously providing the myosin Va DN plasmid and Bill Britt for the kind gift of the anti-MCP antibody. We are also grateful for the assistance of staff and the availability of equipment at various core facilities at Harvard Medical School, including the Nikon Imaging Center for acquisition and analysis of spinning-disk confocal micrographs and the Electron Microscopy Facility (particularly Elizabeth Benecchi and Margaret Coughlin) for the processing, acquisition, and analysis of EM data. Finally, we thank Blair Strang, Jeremy Kamil, David Knipe, Samara Reck-Peterson, James Hogle, Thomas Schwarz, Frederick Wang, Sandra Weller, Lee Gehrke, and all members of the Coen laboratory for helpful discussions.
This work was supported by National Institutes of Health grants R01 AI026077 to D.M.C. and F31 AI20651 to A.R.W.
REFERENCES
- 1.Lye MF, Wilkie AR, Filman DJ, Hogle JM, Coen DM. 2017. Getting to and through the inner nuclear membrane during herpesvirus nuclear egress. Curr Opin Cell Biol 46:9–16. doi: 10.1016/j.ceb.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bigalke JM, Heldwein EE. 2016. Nuclear exodus: herpesviruses lead the way. Annu Rev Virol 3:387–409. doi: 10.1146/annurev-virology-110615-042215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Müller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 4.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2014. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strang BL, Boulant S, Kirchhausen T, Coen DM. 2012. Host cell nucleolin is required to maintain the architecture of human cytomegalovirus replication compartments. mBio 3:e00301-11. doi: 10.1128/mBio.00301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strang BL, Boulant S, Chang L, Knipe DM, Kirchhausen T, Coen DM. 2012. Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J Virol 86:2089–2095. doi: 10.1128/JVI.06720-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bender BJ, Coen DM, Strang BL. 2014. Dynamic and nucleolin-dependent localization of human cytomegalovirus UL84 to the periphery of viral replication compartments and nucleoli. J Virol 88:11738–11747. doi: 10.1128/JVI.01889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penfold ME, Mocarski ES. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46–61. doi: 10.1006/viro.1997.8848. [DOI] [PubMed] [Google Scholar]
- 11.Borst EM, Wagner K, Binz A, Sodeik B, Messerle M. 2008. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J Virol 82:2065–2078. doi: 10.1128/JVI.01967-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giesen K, Radsak K, Bogner E. 2000. Targeting of the gene product encoded by ORF UL56 of human cytomegalovirus into viral replication centers. FEBS Lett 471:215–218. doi: 10.1016/S0014-5793(00)01407-1. [DOI] [PubMed] [Google Scholar]
- 13.Giesen K, Radsak K, Bogner E. 2000. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J Gen Virol 81:2231–2244. doi: 10.1099/0022-1317-81-9-2231. [DOI] [PubMed] [Google Scholar]
- 14.Dittmer A, Drach JC, Townsend LB, Fischer A, Bogner E. 2005. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-d-ribonucleosides. J Virol 79:14660–14667. doi: 10.1128/JVI.79.23.14660-14667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forest T, Barnard S, Baines JD. 2005. Active intranuclear movement of herpesvirus capsids. Nat Cell Biol 7:429–431. doi: 10.1038/ncb1243. [DOI] [PubMed] [Google Scholar]
- 16.Feierbach B, Piccinotti S, Bisher M, Denk W, Enquist LW. 2006. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog 2:e85. doi: 10.1371/journal.ppat.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. 2010. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J Biol Chem 285:13979–13989. doi: 10.1074/jbc.M109.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierobon P, Achouri S, Courty S, Dunn AR, Spudich JA, Dahan M, Cappello G. 2009. Velocity, processivity, and individual steps of single myosin V molecules in live cells. Biophys J 96:4268–4275. doi: 10.1016/j.bpj.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkie AR, Lawler JL, Coen DM. 2016. A role for nuclear F-actin induction in human cytomegalovirus nuclear egress. mBio 7:e01254-16. doi: 10.1128/mBio.01254-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, Ruzsics Z, Koszinowski UH, Enquist LW. 2014. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 5:e01909-14. doi: 10.1128/mBio.01909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosse JB, Hogue IB, Feric M, Thiberge SY, Sodeik B, Brangwynne CP, Enquist LW. 2015. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc Natl Acad Sci U S A 112:E5725–E5733. doi: 10.1073/pnas.1513876112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosse JB, Enquist LW. 2016. The diffusive way out: herpesviruses remodel the host nucleus, enabling capsids to access the inner nuclear membrane. Nucleus 7:13–19. doi: 10.1080/19491034.2016.1149665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammer JA, Sellers JR. 2012. Walking to work: roles for class V myosins as cargo transporters. Nat Rev Mol Cell Biol 13:13–26. [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Cong J-P, Mamtora G, Gingeras T, Shenk T. 1998. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci U S A 95:14470–14475. doi: 10.1073/pnas.95.24.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pogoda M, Bosse JB, Wagner FM, Schauflinger M, Walther P, Koszinowski UH, Ruzsics Z. 2012. Characterization of conserved region 2-deficient mutants of the cytomegalovirus egress protein pM53. J Virol 86:12512–12524. doi: 10.1128/JVI.00471-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tandon R, Mocarski ES, Conway JF. 2015. The A, B, Cs of herpesvirus capsids. Viruses 7:899–914. doi: 10.3390/v7030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma M, Coen DM. 2014. Comparison of effects of inhibitors of viral and cellular protein kinases on human cytomegalovirus disruption of nuclear lamina and nuclear egress. J Virol 88:10982–10985. doi: 10.1128/JVI.01391-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM. 2015. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J Virol 89:523–534. doi: 10.1128/JVI.02426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strang BL, Bender BJ, Sharma M, Pesola JM, Sanders RL, Spector DH, Coen DM. 2012. A mutation deleting sequences encoding the amino terminus of human cytomegalovirus UL84 impairs interaction with UL44 and capsid localization. J Virol 86:11066–11077. doi: 10.1128/JVI.01379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krosky PM, Baek M-C, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol 77:905–914. doi: 10.1128/JVI.77.2.905-914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leach NR, Roller RJ. 2010. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 406:127–137. doi: 10.1016/j.virol.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seperack PK, Mercer JA, Strobel MC, Copeland NG, Jenkins NA. 1995. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO J 14:2326–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, Wu XS, Hammer JA, Xu T, Lippincott-Schwartz J. 2012. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol 198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Wang F, Rao K, Sellers JR, Hammer JA. 2002. Rab27a is an essential component of melanosome receptor for myosin Va. Mol Biol Cell 13:1735–1749. doi: 10.1091/mbc.01-12-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuchsova B, Serebryannyy LA, de Lanerolle P. 2015. Nuclear actin and myosins in adenovirus infection. Exp Cell Res 338:170–182. doi: 10.1016/j.yexcr.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang L, Godinez WJ, Kim I-H, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc Natl Acad Sci U S A 108:E136–E144. doi: 10.1073/pnas.1103411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts KL, Baines JD. 2010. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J Virol 84:9889–9896. doi: 10.1128/JVI.00732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strang BL, Boulant S, Coen DM. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for efficient viral replication. J Virol 84:1771–1784. doi: 10.1128/JVI.01510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. 2015. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci U S A 112:9010–9015. doi: 10.1073/pnas.1511140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reim NI, Kamil JP, Wang D, Lin A, Sharma M, Ericsson M, Pesola JM, Golan DE, Coen DM. 2013. Inactivation of retinoblastoma protein does not overcome the requirement for human cytomegalovirus UL97 in lamina disruption and nuclear egress. J Virol 87:5019–5027. doi: 10.1128/JVI.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]