

ABSTRACT
Combinations of drugs that affect distinct mechanisms of HIV latency aim to induce robust latency reversal leading to cytopathicity and elimination of the persistent HIV reservoir. Thus far, attempts have focused on combinations of protein kinase C (PKC) agonists and pan-histone deacetylase inhibitors (HDIs) despite the knowledge that HIV gene expression is regulated by class 1 histone deacetylases. We hypothesized that class 1-selective HDIs would promote more robust HIV latency reversal in combination with a PKC agonist than pan-HDIs because they preserve the activity of proviral factors regulated by non-class 1 histone deacetylases. Here, we show that class 1-selective agents used alone or with the PKC agonist bryostatin-1 induced more HIV protein expression per infected cell. In addition, the combination of entinostat and bryostatin-1 induced viral outgrowth, whereas bryostatin-1 combinations with pan-HDIs did not. When class 1-selective HDIs were used in combination with pan-HDIs, the amount of viral protein expression and virus outgrowth resembled that of pan-HDIs alone, suggesting that pan-HDIs inhibit robust gene expression induced by class 1-selective HDIs. Consistent with this, pan-HDI-containing combinations reduced the activity of NF-κB and Hsp90, two cellular factors necessary for potent HIV protein expression, but did not significantly reduce overall cell viability. An assessment of viral clearance from in vitro cultures indicated that maximal protein expression induced by class 1-selective HDI treatment was crucial for reservoir clearance. These findings elucidate the limitations of current approaches and provide a path toward more effective strategies to eliminate the HIV reservoir.
IMPORTANCE Despite effective antiretroviral therapy, HIV evades eradication in a latent form that is not affected by currently available drug regimens. Pharmacologic latency reversal that leads to death of cellular reservoirs has been proposed as a strategy for reservoir elimination. Because histone deacetylases (HDACs) promote HIV latency, HDAC inhibitors have been a focus of HIV cure research. However, many of these inhibitors broadly affect multiple classes of HDACs, including those that promote HIV gene expression (class 1 HDACs). Here, we demonstrate that targeted treatment with class 1-selective HDAC inhibitors induced more potent HIV latency reversal than broadly acting agents. Additionally, we provide evidence that broadly acting HDIs are limited by inhibitory effects on non-class 1 HDACs that support the activity of proviral factors. Thus, our work demonstrates that the use of targeted approaches to induce maximum latency reversal affords the greatest likelihood of reservoir elimination.
KEYWORDS: HIV, Hsp90, latency reversal, shock and kill, histone deacetylase inhibitors
INTRODUCTION
Latent HIV within long-lived cells constitutes a reservoir of persistent virus that cannot be eliminated by antiretroviral therapy (ART) and is a major barrier to a cure (1, 2). Pharmacologic reversal of HIV latency has been proposed as a means to induce viral gene expression, rendering infected cells susceptible to viral cytopathic effect and immunological clearance (3). Thus far, work toward this goal has led to the identification of mechanistically distinct drugs that affect several regulatory factors of HIV gene expression, including histone deacetylase inhibitors (HDIs) and protein kinase C (PKC) agonists, such as bryostatin-1 (4–6, 24). Despite promising in vitro results, attempts to reduce the viral reservoir in vivo using these latency reversal agents (LRAs) have failed at clinically achievable levels.
High-level viral gene expression is needed for viral proteins to accumulate to toxic levels and kill the infected cells (7). Both HDIs and PKC agonists have the capacity to activate viral gene expression in patient-derived resting CD4pos (rCD4pos) T cells as measured by cell-associated HIV RNA, but only PKC agonists used at supratherapeutic levels have the capacity to induce viral gene expression sufficient for viral outgrowth (8). In attempts to lower the concentrations of individual agents to clinically achievable levels while still maximizing viral gene expression, efforts have focused on combinations of HDIs and PKC agonists, which demonstrate the synergistic induction of cell-associated HIV mRNA expression in cell lines and primary cell models of HIV latency. However, the HDI combinations tested thus far have failed to maximally enhance gene expression as assessed by viral outgrowth assays using patient rCD4pos T cells tested ex vivo (9).
There are three main classes of histone deacetylases (HDACs), and efforts to reverse HIV latency have focused largely on HDIs that act on a broad range of HDAC classes (pan-HDIs). Because broadly acting agents inhibit a wider range of HDACs, they are more likely to have deleterious effects that could limit maximal latency reversal. A more narrowly focused regimen that targets the subset of HDACs (class 1) involved in HIV latency (10) may allow more efficient activation of HIV gene expression. Indeed, previous work has demonstrated that class 1-selective HDIs can reverse HIV latency in cell line models and are superior to pan-HDIs when used in combination with PKC agonists in in vitro primary cell HIV latency model systems (11–13). These studies, however, are significantly limited by the use of latency models that do not fully recapitulate the effects of LRA treatment in rCD4pos T cells from HIV-infected people and by focusing on the frequency of reactivated proviruses rather than the potency of the induced viral gene expression, viral outgrowth, and clearance of infected cells (8). Moreover, the efficacy of selective agents in hematopoietic stem and progenitor cells (HSPCs), which may constitute a small but important long-lived reservoir in vivo (14, 16), has not been demonstrated.
Therefore, using an in vitro model system of HIV latency in HSPCs and rCD4pos T cells from optimally treated HIV-infected donors, we compared the efficacy of pan-HDIs to that of class 1-selective HDIs on viral reactivation and outgrowth from latently infected cells. We show that class 1-selective HDIs are unique among HDIs tested thus far in that they enhance the ability of PKC agonists to induce viral outgrowth from latently infected HSPCs and from rCD4pos T cells from HIV-infected people. Moreover, our results provide the first evidence that class 1-selective HDIs are superior at inducing viral outgrowth due to reduced off-target inhibition of cellular factors necessary for optimal HIV gene expression.
RESULTS
A robust model of HIV latency in HSPCs allows measurement of reactivation frequency and viral protein production per reactivated cell.
To compare the levels of ability of HDIs to induce HIV latency reversal, we used a latency model system in which purified umbilical cord blood HSPCs from healthy donors were infected with a replication-defective, NL4-3-based HIV molecular clone that expresses green fluorescent protein fused to a short Env peptide (E-GFP; NL4-3-ΔGPE-GFP) (Fig. 1a and b). Three days postinfection, actively infected (E-GFPpos) HSPCs were removed using fluorescence-activated cell sorting (Fig. 1c), and the remaining cells, which included latently infected cells, were immediately treated with each LRA regimen for 24 h. An integration inhibitor (raltegravir) was also added to block new integration events, ensuring that we scored only reversal of postintegration latency. Because latency occurs relatively frequently in the HSPC model system, the relative amount of viral protein production in cells bearing a reactivated provirus (E-GFPpos) could be accurately measured by flow cytometry via the E-GFP mean fluorescence intensity (MFI).
FIG 1.

Model system of HIV latency in HSPCs. (a) Schematic illustration of HIV activation assay in HSPCs. (b) Schematic diagram of NL4-3-based HIV molecular clone used for activation studies. Filled black and green rectangles indicate genes that have been deleted from or added to the wild-type molecular clone, respectively. (c) Flow cytometric analysis of mock-infected and sorted, infected HSPCs used for activation studies. Numbers above gates indicate the frequency of live E-GFPpos cells. Live cells were gated based on FSC and SSC parameters. (d) Representative flow cytometric analysis of infected HSPCs treated for 24 h with the indicated LRAs. Live cells were gated based on forward scatter (FSC), side scatter (SSC), and 7-AAD. Numbers above gates indicate the frequency of CD34pos, E-GFPpos cells. Numbers in parentheses indicate the E-GFP MFI of cells within the gate. Gates are based on mock-infected cells and staining with an isotype control antibody. (e to g) Summary graphs showing the frequency of E-GFPpos cells (e), E-GFP MFI (f), and the frequency of CD34pos HSPCs (g) following 24 h treatment with medium and 0.1% (vol/vol) DMSO. (h) Flow-cytometric analysis of HSPCs and PBMCs for cell surface CD34 and lineage markers (markers of cells committed to the T, B, NK, myeloid, and erythroid lineages [anti-CD2, CD3, CD14, CD16, CD19, CD56 and CD235a]). (i) Immunoblot analysis of whole-cell lysates from HSPCs treated for 9 h under the indicated conditions, demonstrating HDI selectivity.
Consistent with our previous work (17), when latently infected HSPCs were treated with tumor necrosis factor alpha (TNF-α), we observed potent latency reversal compared to 0.1% dimethyl sulfoxide (DMSO) solvent control (Fig. 1d). The capacity to quantitatively assess the effects of LRAs on viral protein expression in an unmodified primary cell model of latency is a unique feature of our system and may allow the identification of combinations of drugs that will maximally reverse latency and have the greatest capacity to promote eradication.
Based on our previous work (18), reactivation occurring in the solvent control is due to spontaneous differentiation of HSPCs and does not reflect latency reversal activity by the DMSO solvent. Compared to untreated cells, 0.1% DMSO did not significantly affect the frequency of E-GFPpos cells (Fig. 1e) or the E-GFP MFI (Fig. 1f). In addition, the concentration of DMSO used in our assay was not sufficient to induce acetylation of histone or tubulin (Fig. 1i).
HSPCs gradually lose some expression of the CD34 cell surface marker under culture conditions that support HSPC proliferation. Moreover, differentiation as assessed by CD34 expression is associated with reactivation from latency in the HSPC model system (18). However, the solvent DMSO does not affect differentiation of HSPCs as assessed by cell surface CD34 expression (Fig. 1g) and all cells remain lineage negative (Fig. 1h).
Class 1-selective HDIs induce more viral Env (E-GFP) production per infected cell than pan-HDIs.
To assess the efficacy of HDIs on latency reversal in HSPCs, we performed a titration over a range of drug concentrations and assessed the frequency of GFPpos cells (Fig. 2a) and the amount of GFP expressed per cell (E-GFP MFI) (Fig. 2b). Optimal concentrations were those that maximized latency reversal while minimizing toxicity and were consistent with results from other studies (6, 11, 19). Work from other groups has shown that selectivity of class 1-selective HDIs is maintained at concentrations well above those tested here (20–22). Nevertheless, to confirm selectivity of HDIs at the concentrations chosen for our assays, we assessed their effects on histone H4 and tubulin acetylation. Histone H4 is deacetylated by class 1 HDACs; therefore, both pan-HDIs and class 1-selective HDIs should promote histone H4 acetylation. In contrast, tubulin is deacetylated by the class 2 HDAC HDAC6 and should be affected by pan-HDIs but not class 1-selective HDIs. Indeed, we observed that at the concentrations chosen for our study, the pan-HDI vorinostat, but not the class 1-selective HDI entinostat, inhibited deacetylation of tubulin, whereas both drugs inhibited deacetylation of histone H4 (Fig. 1i).
FIG 2.

HDI combinations reveal an inhibitory effect of pan-HDIs that limits HIV protein production in HSPCs. Summary graphs of frequency of E-GFPpos, CD34pos HSPCs (a) and E-GFP MFI (b) in CD34pos cells bearing activated HIV following 24-h treatment over the range of HDI concentrations indicated (means ± standard errors of the means [SEM], n = 3). (c) Summary graph of the frequency of E-GFPpos, CD34pos cells following treatment with the indicated LRAs at the concentrations described in Materials and Methods and used in all subsequent assays. The frequency of spontaneous reactivation observed under DMSO solvent conditions was subtracted from each experiment to reflect the actual frequency of reactivated provirus. Each symbol represents data from an independent experiment (mean ± SEM, n ≥ 4). (d) Summary graph of E-GFP MFI in E-GFPpos, CD34pos cells (mean ± SEM, n ≥ 4). (e) Summary graph of frequency of viable HSPCs following 24-h treatment with HDIs alone and in combinations at the concentrations used for HIV latency reversal studies. Cell viability was calculated using the percentage of cells within the live-cell gating strategy based on FSC, SSC, and 7-AAD. Data are presented as percentages of the effect of TNF-α. P values were calculated by two-tailed, unpaired Student's t test. *, P < 0.05; ****, P < 0.0001. Solid bars indicate statistically significant differences between groups subjected to different conditions.
A summary of results for HSPCs from at least four independent donors indicate that, on average, every HDI tested induced E-GFP expression in a smaller fraction of latently infected cells than the positive control, TNF-α (Fig. 2c). Among HDIs, pan-HDIs (vorinostat, panobinostat, and romidepsin) induced E-GFP expression in a significantly greater fraction of latently infected cells than the class 1-selective HDIs (entinostat, tacedinaline, and mocetinostat) at the concentrations tested (Fig. 2c).
In contrast, all class 1-selective HDIs tested induced significantly more viral protein expression per cell than pan-HDIs based on E-GFP MFI (1.5-fold) (Fig. 2d). Moreover, this difference in efficacy was observed across a range of concentrations; increasing the concentration of pan-HDIs above that used in Fig. 2d did not overcome this limitation (Fig. 2b). In addition, these differences were not due to toxicity as cell viability was similar at HDI concentrations chosen for our assays (Fig. 2e).
We also assayed cell surface major histocompatibility complex class I (MHC-I) expression in cells bearing reactivated virus, which has been shown to be inversely proportional to Nef expression levels (23). We observed that treatment of latently infected HSPCs with entinostat induced an 8-fold Nef-dependent downmodulation of MHC-I in cells bearing reactivated virus compared to E-GFPneg cells (Fig. 3a). Compared to vorinostat, we observed significantly more downmodulation of cell surface MHC-I on cells treated with entinostat (1.3-fold) (Fig. 3b), which corresponds to the degree of induction of viral protein (E-GFP) production (Fig. 2d). Together these data demonstrate that the class 1-selective HDI entinostat induces greater expression of both early and late HIV genes than pan-HDIs.
FIG 3.

Entinostat induces more Nef-dependent downmodulation of MHC-I than pan-HDIs. (a) Representative flow cytometric analysis of MHC-I downmodulation on latently infected HSPCs treated with the indicated LRAs. The magnitude of MHC-I downmodulation in HSPCs with induced viral gene expression was calculated by dividing the MHC-I median fluorescence intensity (MedFI) of the E-GFPneg population by that of the E-GFPpos population for each sample (E-GFPnegMHC-I MedFI/E-GFPposMHC-I MedFI). (b) Summary graph of fold MHC-I downmodulation. Data are presented relative to DMSO (means ± SEM, n ≥ 4). P values were calculated by two-tailed, unpaired Student's t test. *, P < 0.05; ***, P < 0.001. Asterisks above each condition indicate statistically significant differences compared to DMSO. Solid bar indicates statistically significant differences between groups subjected to different conditions.
Pan-HDIs suppress HIV protein production induced by class 1-selective HDIs.
The ideal LRA would maximally reverse latency in all infected cells to promote cytopathicity and clearance. Thus, we next asked whether combinations of pan- and class 1-selective HDIs would maximize both the frequency of cells expressing E-GFP and the amount of E-GFP expressed per infected cell. Indeed, we observed that the addition of pan-HDIs to class 1-selective HDIs maintained the higher frequencies of E-GFPpos cells observed with pan-HDIs alone (Fig. 2c). However, these combinations failed to achieve the high E-GFP MFIs observed with class 1-selective HDI treatment alone (Fig. 2d). The inhibitory effect of pan-HDIs could not be attributed to additive toxicity, as there was no significant difference in the frequency of viable HSPCs treated with HDIs alone or in combination (Fig. 2e). Based on these results, we hypothesized that the broad inhibitory effects of pan-HDIs had undesirable negative effects on HIV gene expression that did not occur with class 1-selective HDIs.
Class 1-selective HDIs plus a PKC agonist maximally reactivate viral gene expression, and pan-HDIs are inhibitory when added to these combinations.
Reversal of HIV latency has also been observed by activating protein kinase C (PKC) and NF-κB using bryostatin-1. Recent studies have shown that bryostatin-1 acts synergistically with pan-HDIs to increase HIV cell-associated RNA transcription, but not viral protein production or viral outgrowth in latently infected primary CD4pos T lymphocytes (9, 15, 25). To determine whether the higher HIV protein expression observed with class 1-selective HDIs could overcome the limitations observed with pan-HDIs, we next investigated the effect of bryostatin-1 combinations. To accomplish this, we used an HIV molecular clone that expresses full-length Gag and the human placental alkaline phosphatase (PLAP) gene within the HIV env open reading frame (HXB2-ΔE-PLAP) (Fig. 4a and b). HSPCs latently infected with HXB2-ΔE-PLAP were isolated using magnetic bead sorting to deplete actively infected cells (PLAPpos) (Fig. 4c). We then treated PLAPneg cells and scored them for HIV latency reversal (Fig. 4d).
FIG 4.

Class 1-selective HDIs plus bryostatin-1 maximally induce latency reversal and viral protein production, whereas pan-HDIs are inhibitory when added to these combinations. (a) Schematic illustration of modified HIV latency model system in HSPCs. (b) Schematic diagram of the HXB2-based HIV molecular clone. Filled black and green rectangles indicate genes that have been deleted from or added to the wild-type molecular clone, respectively. Filled gray rectangles indicate genes that are dysfunctional in the wild-type HXB2 molecular clone. (c) Flow-cytometric analysis of mock-infected and sorted, infected HSPCs. Numbers above gates indicate the frequency of live PLAPpos cells. Live cells were gated based on FSC and SSC parameters. (d) Representative flow-cytometric analysis of infected HSPCs treated for 24 h with the indicated LRAs. Live cells were gated based on FSC and SSC. Numbers above gates indicate the frequencies of PLAPpos, Gagpos cells. Numbers in brackets indicate PLAP MFI of cells within the double-positive gate. Gates are based on mock-infected cells and staining with an isotype control antibody. Summary graphs of frequency of PLAPpos, Gagpos cells (e) and PLAP MFI of PLAPpos, Gagpos cells (f) and relative cell viability following treatment with the indicated LRAs (g). The frequency of spontaneous reactivation observed under DMSO solvent conditions was subtracted from each experiment to reflect the actual frequency of reactivated provirus. Data are presented as a percentage of the effect of TNF-α (mean ± SEM, n ≥ 3). P values were calculated by two-tailed, unpaired Student's t test. *, P < 0.05; ***, P < 0.001.
Using this system, we again observed that all HDIs in combination with bryostatin-1 had similar frequencies of PLAPpos cells (Fig. 4e). In addition, we confirmed that class 1-selective agents induced the highest levels of viral protein per infected cell when used alone or in combination with bryostatin-1 (Fig. 4f).
We next asked whether the inhibitory effect that we observed with pan-HDIs on class 1-selective HDI-mediated E-GFP expression from Fig. 2 was also observed when added to combinations of bryostatin-1 and class 1-selective HDIs in this modified latency model system. Indeed, when any of the pan-HDIs tested were added in combination with bryostatin-1 plus a class 1-selective HDI, the PLAP MFI was significantly reduced in almost every case (Fig. 4f), an effect that could not be attributed to additive toxicity (Fig. 4g).
Entinostat plus bryostatin-1 maximally induces viral outgrowth, and pan-HDIs are inhibitory when added to this combination.
Complete HIV latency reversal is considered to be the induction of virus outgrowth from latently infected cells, which is needed for maximal cytopathicity. Therefore, we tested the ability of LRA combinations to induce virus outgrowth from latently infected HSPCs. To accomplish this, we performed experiments as illustrated in Fig. 4 and assayed culture supernatants for HIV virion-associated RNA by quantitative reverse transcription-PCR (RT-qPCR) with primers specific for genomic HIV mRNA. Alone, HDIs induced only modest viral outgrowth, whereas bryostatin-1 induced it more than any HDI (P < 0.01) (Fig. 5). When combined with bryostatin-1, entinostat significantly increased the potency of viral outgrowth compared to either LRA alone (Fig. 5). When pan-HDIs were added to combinations of bryostatin-1 and entinostat, we observed significant suppression of viral outgrowth, concordant with the suppressive effects on viral protein production described earlier (compare Fig. 5 and 4f). Importantly, latently infected HSPCs treated with triple-LRA combinations demonstrated neither dramatic differences in the frequencies of PLAPpos cells induced nor significant cellular toxicity following treatment compared to relevant double combinations (Fig. 4e and f). Therefore, entinostat plus bryostatin-1 induced the greatest viral protein production and outgrowth, which was suppressed by cotreatment with pan-HDIs.
FIG 5.
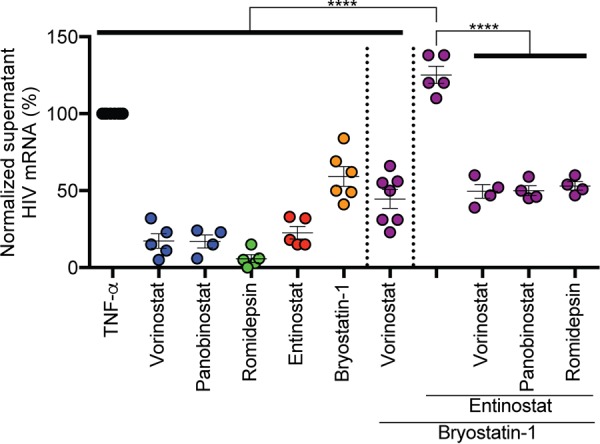
Entinostat plus bryostatin-1 induces maximum viral outgrowth from latently infected cells, whereas pan-HDIs are inhibitory when added in combination. Summary graph of genomic HIV mRNA copies/ml measured in supernatants from cultures of latently infected HSPCs treated as described for Fig. 4. Viral outgrowth observed with spontaneous reactivation under DMSO solvent conditions was subtracted from each experiment. Data are presented as a percentage of the effect of TNF-α (means ± SEM, n ≥ 4). P values were calculated by two-tailed, unpaired Student's t test. ****, P < 0.0001.
Entinostat plus bryostatin-1 induces maximal viral outgrowth and gene expression from latently infected resting CD4pos T cells from optimally treated HIV-infected individuals.
While previous work has demonstrated that pan-HDIs enhance the ability of bryostatin-1 to stimulate cell-associated RNA from patient-derived rCD4pos T cells ex vivo, pan-HDIs did not significantly increase bryostatin-1-induced virus outgrowth (9). To determine whether class 1-selective HDIs could achieve this important goal, we treated rCD4pos T lymphocytes obtained from the donors whose characteristics are listed in Table 1 with bryostatin-1 alone or in combination with entinostat and assayed culture supernatants at 24 and 48 h for virion-associated genomic HIV mRNA. We also measured cell-associated HIV mRNA from cell pellets harvested at 48 h. Remarkably, in three of four donors, we detected 5- to 11-fold-greater virus outgrowth with the combination of bryostatin-1 plus entinostat than with bryostatin-1 alone (Fig. 6a). Concomitant with this increased viral outgrowth detected in the culture supernatant, we also detected more cell-associated HIV mRNA under bryostatin-1 plus entinostat treatment than under treatment with bryostatin-1 alone (Fig. 6b). Interestingly, in the one donor from whom we did not detect bryostatin-1-induced virus outgrowth (ID 419000), we nevertheless observed induction of cell-associated HIV mRNA with bryostatin-1 treatment as well as enhanced expression with the combination of bryostatin-1 plus entinostat (Fig. 6b).
TABLE 1.
HIV-infected donor characteristicsa
| Donor ID | Age | Sex | Race | Yr of diagnosis | ART regimen | Time on ART (mo) | Duration of viral suppression (mo) |
|---|---|---|---|---|---|---|---|
| 412406 | 45 | M | W | 2007 | 3TC, AZT, EFV | 62 | 60 |
| 419000 | 53 | M | W | 1986 | FTC, TDF, ATV/r | 192 | 28 |
| 445000 | 59 | M | B | 2009 | FTC, TDF, EFV | 32 | 13 |
| 457413402 | 50 | M | B | 2011 | FTC, TDF, EFV | 59 | 55 |
M, male; F, female; W, white; B, black; 3TC, lamivudine; AZT, zidovudine; ATV/r, atazanavir boosted with ritonavir; EFV, efavirenz; FTC, emtricitabine; TDF, tenofovir.
FIG 6.

Entinostat plus bryostatin-1 induces maximum viral outgrowth and gene expression in latently infected rCD4pos T cells from HIV-infected individuals. (a) Graphical analysis of HIV released by patient rCD4pos T cells following treatment with indicated LRAs (means ± SD for experimental duplicates). The limit of quantification is indicated by a dotted gray horizontal line. (b) Induced cell-associated HIV mRNA in rCD4pos T cells treated for 48 h with indicated LRAs (means ± SD for experimental duplicates).
In the two donors from whom sufficient cells were available (ID 412406 and ID 445000), we observed a 2- to 5-fold reduction in viral outgrowth with the addition of vorinostat to entinostat plus bryostatin-1 (Fig. 6a). This was correlated with similar reductions of cell-associated RNA expression (Fig. 6b). Collectively, these data suggest that even in ex vivo models, class 1-selective HDIs enhance bryostatin-1-mediated viral outgrowth, whereas pan-HDIs have an inhibitory effect that limits maximum viral gene expression and virion outgrowth.
Entinostat plus bryostatin-1 leads to the greatest reduction of LRA-induced actively infected cells.
The ultimate test of the effectiveness of an LRA is the ability to reduce the HIV reservoir. To compare the ability of LRAs to reduce the inducible latent viral reservoir in vitro, we modified our HSPC viral outgrowth assay by washing and culturing LRA-treated cells an additional 3 days following treatment and quantified the frequency of PLAPpos cells that remained in culture 4 days after treatment (Fig. 7a). We found that the combination of entinostat plus bryostatin-1 was the only combination that led to a significant reduction of infected (PLAPpos) cells based on flow cytometry (Fig. 7b and c). Interestingly, we observed an inverse correlation when we compared the frequency of residual PLAPpos cells that remained in culture 4 days after the initial LRA regimen treatment to the potency of latency reversal (Fig. 7d), suggesting that more potent latency reversal is more likely to induce clearance of affected cellular reservoirs. Finally, to compare the amount of residual inducible latent HIV 4 days after initial LRA treatment, we treated every condition with TNF-α and quantified viral outgrowth after 24 h (Fig. 7a). Remarkably, the only condition that led to a significant reduction in the relative amount of induced viral outgrowth between the first and second treatments was bryostatin-1 plus entinostat (Fig. 7e). Thus, the capacity of LRAs to maximally induce virus release correlates with the likelihood of affected cellular reservoirs being eliminated from culture.
FIG 7.

Potency of latency reversal is correlated with survival of affected cells bearing reactivated HIV. (a) Schematic of in vitro assay to test the survival of latently infected HSPCs following treatment with LRAs. (b) Representative flow-cytometric analysis of HSPCs 1 and 4 days after treatment with LRAs. (c) Summary graph showing the relative frequency of residual PLAPpos cells in culture 4 days post-LRA treatment. (d) Summary graph showing inverse correlation between frequency of residual infected cells on 4 days after LRA treatment and potency of induced viral outgrowth by the indicated LRAs. Data were fit to a least-squares nonlinear regression. (e) Box and whisker plot (5th to 95th percentiles) of virus released into culture supernatant by HSPCs treated with TNF-α 4 days after initial LRA treatment. To facilitate comparisons across multiple experiments, data were normalized to TNF-α (n = 5). P values were calculated by two-tailed, unpaired Student's t test. *, P < 0.05; ***, P < 0.001.
Vorinostat inhibits NF-κB and Hsp90.
HDIs regulate the acetylation of many proteins, including nonhistone substrates that can negatively affect HIV reactivation and replication (26). For example, NF-κB, which is required for HIV transcription, is known to be regulated by acetylation. Thus, differential effects of pan- and class 1-selective HDIs on NF-κB could, in part, explain the inhibitory effects of pan-HDIs (27, 28). Interestingly, we found that vorinostat significantly inhibited bryostatin-1-induced NF-κB/p65 activation in PBMCs whereas entinostat did not (Fig. 8a). This modest inhibition of bryostatin-1-mediated NF-κB/p65 activation by vorinostat is concordant with the greater suppression of viral outgrowth observed for this combination than for bryostatin-1 treatment alone (Fig. 5 and 6).
FIG 8.
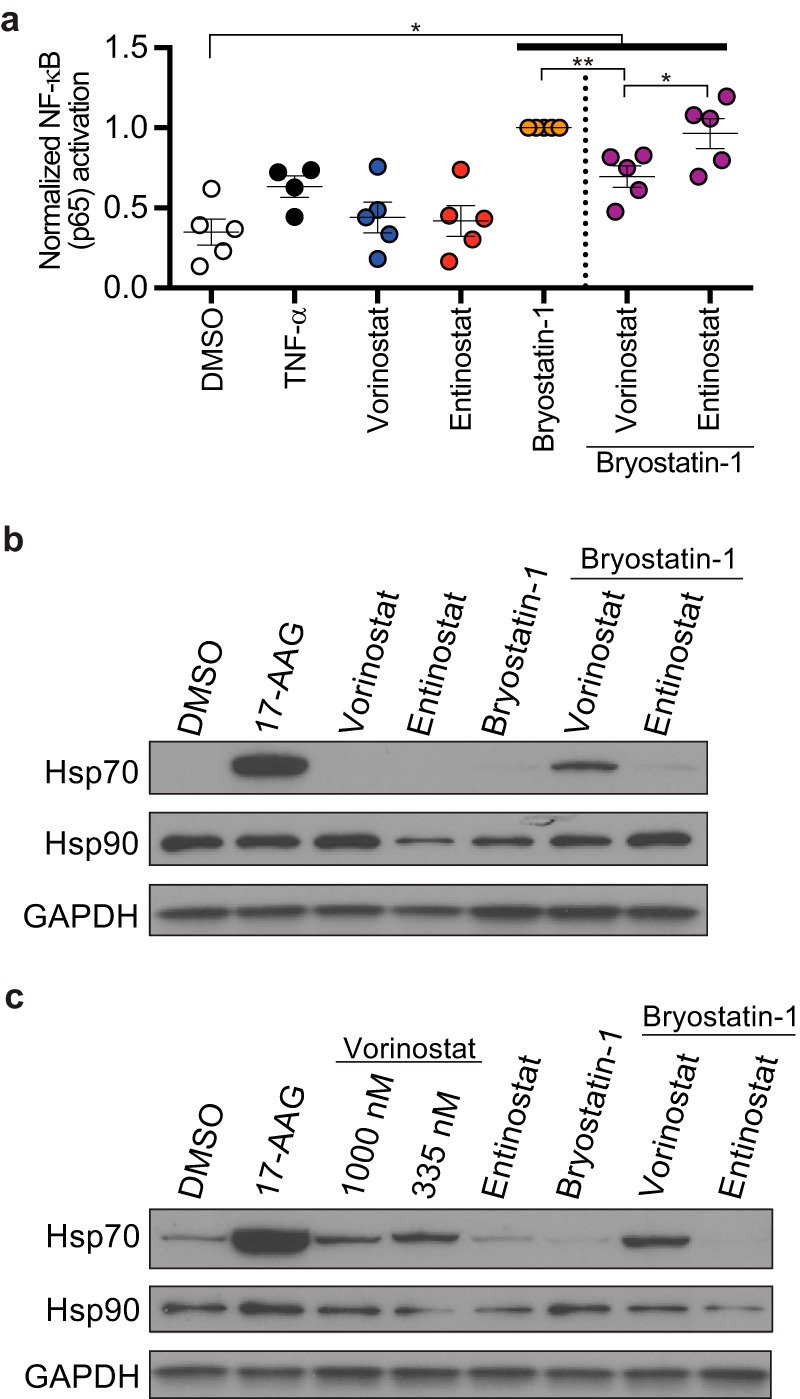
Vorinostat inhibits cellular factors important for robust HIV gene expression. (a) Summary graph showing abundance of activated NF-κB/p65 in PBMCs treated for 24 h with indicated LRAs. Data were normalized to the effect of bryostatin-1 (means ± SEM, n ≥ 4). Immunoblot analysis of whole-cell lysates from HSPCs (b) and PBMCs (c) treated for 24 h with indicated LRAs. P values were calculated by two-tailed, unpaired Student's t test. *, P < 0.05; **, P < 0.01.
Heat shock protein 90 (Hsp90) is a molecular chaperone that is required for the normal functioning of a number of client proteins, and recent studies have demonstrated that Hsp90 activity is required for reversal of HIV latency (29–33). Additional work has also shown that Hsp90 deacetylation by a class 2 HDAC, HDAC6, activates Hsp90 activity (34, 35) and that vorinostat, which inhibits HDAC6, causes acetylation of Hsp90 and inhibits its function (36, 37). In contrast, the class 1-selective HDI entinostat does not affect the acetylation of HDAC6 client proteins under the conditions of our assay (Fig. 1i). Consistent with this, an acetylome analysis that compared protein acetylation by entinostat versus vorinostat directly demonstrated that a subset of lysine residues on Hsp90 are acetylated by vorinostat but not by entinostat treatment (38). To confirm that vorinostat inhibits Hsp90 activity in HIV-infected HSPCs and PBMCs, we assessed Hsp70 levels, which are induced upon inhibition of Hsp90 activity in a heat shock factor-1-dependent pathway (39–41). As a positive control, we treated HSPCs and PBMCs with a specific Hsp90 inhibitor (17-AAG), which induced a dramatic increase in Hsp70, compared to the solvent control (Fig. 8b and c). In addition, we observed that vorinostat, but not entinostat, induced Hsp70 expression in both HSPCs and PBMCs when used in combination with bryostatin-1 (Fig. 8b and c). Treatment with vorinostat alone also induced Hsp70 expression in PBMCs, which was not observed with entinostat treatment (Fig. 8c). Together these results provide evidence that vorinostat but not entinostat inhibits cellular factors necessary for robust viral gene expression and outgrowth.
DISCUSSION
Multiple failed attempts to reduce the viral reservoir in vivo using pan-HDI-based LRA regimens emphasize a need for a change in strategy. Shan and colleagues demonstrated that latency reversal regimens that induced greater viral gene expression led to the more rapid death of infected cells by viral cytopathicity (7). Here, we describe effects of pan-HDIs that limit potent HIV latency reversal and provide evidence for a superior and effective strategy using class 1-selective HDIs.
Notably, we observed potent inhibition of bryostatin-1 plus entinostat-induced viral protein production and outgrowth by all three pan-HDIs tested. Importantly, these effects of pan-HDIs were not due to the combined toxicity of the three drugs. In fact, mocetinostat plus bryostatin-1 treatment was the most toxic of the LRA combinations tested and addition of pan-HDIs modestly increased cell viability relative to the two-LRA combination. Interestingly, inhibition by pan-HDIs affected protein expression and viral outgrowth but not the frequency of reactivated cells. As clearance in our study correlated with the amount of protein expressed per cell, our results highlight potential limitations of previously published work that compared only the frequencies of proviruses affected by HDI treatment. While it will ultimately be important to reverse latency in all latently infected cells, it is critical that reversal be sufficient to promote reservoir elimination. Thus, assays measuring the potency of viral latency reversal within each cell are important to inform successful LRA regimen development.
In addition, we show that pan-HDIs inhibited at least two cellular factors that are necessary for robust viral gene expression and outgrowth (NF-κB and Hsp90). To our knowledge, this is the first study to identify these limitations of pan-HDIs on potent latency reversal and clearance of infected cells. Together with previously described negative effects of pan-HDIs on cytotoxic T lymphocytes and NK cells (42–44), these results suggest that LRA regimens using pan-HDIs are not likely to lead to robust reservoir elimination in vivo.
While we describe a significant limitation of pan-HDIs in reservoir elimination strategies, we also provide evidence for an alternative strategy employing class 1-selective HDIs in combination with bryostatin-1 that maximally induce viral protein production, viral outgrowth, and infected cell death. The demonstration that HDI combinations can promote viral clearance without added cytotoxic T lymphocytes (CTLs) is important because we also, for the first time to our knowledge, demonstrate Nef-dependent downmodulation of cell surface MHC-I on cells bearing reactivated provirus. If we consider CTL-mediated killing of reservoirs as the major mechanism of reservoir elimination, a paradoxical barrier becomes evident, i.e., that the most potent latency reversal that promotes robust antigen production and MHC-I loading will simultaneously induce the greatest Nef-dependent MHC-I downmodulation and limit antigen presentation and CTL killing. Robust viral gene expression is necessary for efficient CTL-mediated killing of reservoirs, a degree of induced viral gene expression that is not achieved by vorinostat treatment alone (7). Therefore, until a specific and effective method to inhibit Nef-dependent MHC-I downmodulation is developed, it will be important to focus on latency reversal strategies that induce robust cytotoxic viral protein production and CTL-independent killing of reservoir cells through cytopathicity, as we show is possible in this study.
With the increasing understanding of the diversity of persistent HIV reservoirs, it is important that any proposed latency reversal regimen affects a diverse pool of reservoir cell subtypes, despite their physiologic differences. Our data demonstrate that the combination of a class 1-selective HDI and a PKC agonist is optimal in both rCD4pos T lymphocytes and HSPCs, which along with other cell types may constitute a small but important long-lived reservoir in vivo (14, 16). This result is highly encouraging, especially since HIV latency biology in HSPCs is known to be distinct from that in rCD4pos T cells (17).
Some assumptions and limitations of this study warrant further discussion. First, the selectivity of the HDIs in this study used at the indicated concentrations is an important assumption, but one based on many studies that investigated these HDIs for their ability to inhibit HDAC isoforms at various concentrations. Indeed, entinostat does not significantly inhibit class 2 HDACs even when used at 3- to 10-fold-higher concentrations than used here (20, 21). Nevertheless, we confirmed this selectivity in HSPCs at the concentrations used in our assays by demonstrating that vorinostat, but not entinostat, induced tubulin acetylation, which is mediated by the class 2 HDAC HDAC6 (Fig. 1i).
In addition, our HIV latency model system in HSPCs demonstrates a background of spontaneous latency reversal in the absence of LRAs. We recently determined that this spontaneous latency reversal is associated with HSPC proliferation and differentiation; culture conditions that induce quiescence and prevent differentiation decrease viral gene expression (18). Moreover, undifferentiated cells expressing higher levels of the HSPC markers CD34 and CD133 are more likely to remain latent than more lineage-restricted HSPCs with lower expression of HSPCs markers (16, 18).
Finally, the clearance assay whose results are shown in Fig. 7 also has some limitations as an alternative interpretation is that previously activated (PLAPpos) cells treated with entinostat plus bryostatin-1 preferentially revert to PLAPneg cells that are refractory to a second round of latency reversal stimulus. However, we believe that this explanation is unlikely, as it would require that entinostat plus bryostatin-1 behave in ways that have not previously been described to selectively suppress HIV gene transcription. Attempts to further investigate this possibility by quantifying proviral DNA were confounded by the presence of unintegrated HIV DNA present in all samples, including in the raltegravir-treated control. Nevertheless, the simplest and most logical conclusion based on the potency of latency reversal induced is that the combination of entinostat plus bryostatin-1 killed reactivated HIV-infected cells, clearing them from the culture more rapidly than the other treatments because higher levels of toxic viral proteins were achieved.
In conclusion, while these studies identify a path toward the development of an effective latency reversal regimen, further studies are required to identify less toxic alternatives to bryostatin-1, such as recently identified bryologs that still need to be assessed in clinical studies, as well as other novel, more targeted approaches to latency reversal (45, 46). Ultimately, the identification of a more effective and more targeted latency reversal regimen is an important step toward the goal of eliminating the viral reservoir in HIV-infected people.
MATERIALS AND METHODS
Ethics statement.
HIV-infected individuals were recruited through the University of Michigan HIV AIDS Treatment Program and the Henry Ford Health System. Written informed consent was obtained according to a protocol approved by the University of Michigan Institutional Review Board (U-M IRB number HUM00004959) and the Henry Ford Institutional Review Board (HFH IRB number 7403). Donors were >18 years old, with normal white blood cell counts, and plasma viral loads were <48 copies/ml for at least 6 months on antiretroviral therapy. Samples consisting of 100 ml of peripheral blood and 20 ml of bone marrow were obtained from each donor. All collected samples were coded. Whole umbilical cord blood from uninfected donors was obtained from the New York Blood Center. All collected samples were anonymized.
HSPC isolation and culture.
HSPCs were isolated and cultured as previously described (17). Briefly, mononuclear cells were isolated from whole umbilical cord blood by Ficoll-Paque (GE Healthcare) density gradient centrifugation and were frozen in bovine serum albumin (BSA; 7.5% in phosphate-buffered saline [PBS]; Gibco) and DMSO (10%; Sigma-Aldrich) or used fresh. Mononuclear cells were adherence depleted for 1 to 2 h at 37°C in Stemspan II medium (Stemcell Technologies). CD133pos cells were isolated by magnetic separation using CD133 magnetic beads according to the manufacturer's protocol (Miltenyi Biotec), with the modification that 1.5 times as many magnetic beads were used to increase the purity of sorted cells (≥90% CD133pos). CD133pos cells were cultured at 37°C with 5% CO2 in Stemspan II medium supplemented with a CC110 cytokine cocktail (100 ng/ml stem cell factor, 100 ng/ml thrombopoietin, and 100 ng/ml Flt3 ligand; Stemcell Technologies) and 100 ng/ml insulin-like growth factor binding protein 2 (R&D Systems) (STIF medium).
rCD4pos T lymphocyte isolation and culture.
Peripheral blood mononuclear cells (PBMCs) of HIV-infected donors with <48 HIV copies/ml were purified by density gradient centrifugation from whole blood samples as described above and frozen in fetal bovine serum (FBS) with 10% DMSO. Frozen PBMCs were thawed and washed twice by dropwise addition of RPMI medium (Gibco) plus DNase I (Invitrogen). Thawed PBMCs were enriched for CD4pos T lymphocytes by negative selection using a CD4pos T cell isolation kit (Miltenyi Biotec). CD4pos T lymphocytes were further enriched for resting CD4pos (rCD4pos) T lymphocytes by magnetic bead depletion of cells that expressed CD69, CD25, or HLA-DR (CD69 MicroBead kit II, CD25 MicroBeads, and HLA-DR MicroBeads; Miltenyi Biotec). The purity of rCD4pos T lymphocytes was assessed by flow cytometry and was greater than 90%. Cells were immediately used in viral outgrowth assays in which rCD4pos T lymphocytes were cultured in T cell culture medium (RPMI supplemented with 10% FBS, 20 mM HEPES [Invitrogen], 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine [PSG; Gibco], and 5.6 μg/ml plasmocin).
Virus constructs and infection of HSPCs.
NL4-3-ΔGPEN-E-GFP (ΔGPEN) was generated by disrupting Nef expression from NL4-3-ΔGPE-E-GFP (ΔGPE) (17) by Xho1 digestion followed by Klenow polymerase fill-in and religation. Generation of HXB2-ΔE-PLAP has been previously described (47). Infectious supernatants were prepared by transfection of proviral plasmids into 293T cells (ATCC) using polyethylenimine. Proviral plasmids were cotransfected with plasmid encoding the vesicular stomatitis virus glycoprotein (VSV-G). For plasmids that lacked HIV structural genes (ΔGPE and ΔGPEN), a helper plasmid (pCMV-HIV) was also cotransfected to provide necessary structural proteins in trans and permit infectious particle formation. Forty-eight to 72 h after transfection, culture supernatants were harvested and filtered through a 0.45-μm syringe filter (GE Healthcare). 293T cells were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% FBS (Gibco), PSG, and 5.6 μg/ml plasmocin (Invivogen). HSPCs were infected by spin inoculation at 1,049 × g for 2 h at room temperature. Mock-infected controls were treated with 293T cell culture medium. Following spin infection, viral supernatant was removed, and cells were cultured in STIF medium for 3 days.
Sorting infected HSPCs.
HSPCs infected with ΔGPE or ΔGPEN viruses were sorted by fluorescence-activated cell sorting (FACS) using a FACSAria III (BD Biosciences) flow cytometer to remove E-GFPpos (actively infected) cells and enrich for E-GFPneg (latently and uninfected) cells. Gating was determined based on the mock-infected control. HSPCs infected with HXB2-ΔE-PLAP were sorted using magnetic beads to isolate PLAPneg cells. Briefly, infected HSPC cultures were incubated on ice in magnetic-activated cell sorting (MACS) buffer (PBS, 0.5% BSA, and 2 mM EDTA) for 20 min with FcR blocking reagent (Miltenyi Biotec) and biotinylated antibody raised against PLAP (Serotec). Cells were washed with MACS buffer and then incubated at 4°C for 15 min in MACS buffer with streptavidin-conjugated magnetic beads (Miltenyi Biotec). Cells were washed and placed on a magnetic column, and cells that did not bind to the column (PLAPneg) were collected. Column-bound cells were also collected by elution with 5 ml of MACS buffer and used to check sorting efficiencies.
Treatment of sorted E-GFPneg HSPCs.
Sorted E-GFPneg cells (50,000 to 80,000 cells) were incubated for 24 h at 37°C with 5% CO2 in 200 μl of Stemspan II medium plus 8 μM raltegravir (Selleck Chemicals) with the following drugs and controls: 0.1% (vol/vol) DMSO (Sigma-Aldrich), 3 ng/ml TNF-α (R&D Chemicals), 1 μM vorinostat (Cayman Chemical or Selleck Chemicals), 25 nM panobinostat (Selleck Chemicals), 25 nM romidepsin (Selleck Chemicals), 10 μM entinostat (Cayman Chemical or Selleck Chemicals), 10 μM tacedinaline (Selleck Chemicals), 10 μM mocetinostat (Selleck Chemicals). Unless otherwise indicated, concentrations of drugs remained the same under all experimental conditions, including for those testing drug combinations.
Treatment of sorted PLAPneg HSPCs.
Sorted PLAPneg cells (250,000 to 750,000 cells) were incubated for 24 h at 37°C with 5% CO2 in 1 ml of Stemspan II medium plus 8 μM raltegravir and with the following drugs and controls: 0.1% (vol/vol) DMSO, 3 ng/ml TNF-α, 1 μM vorinostat, 25 nM panobinostat, 25 nM romidepsin, 10 μM entinostat, 10 μM tacedinaline, 10 μM mocetinostat, 5 nM bryostatin-1 (Sigma-Aldrich). Unless otherwise indicated, concentrations of drugs remained the same under all experimental conditions, including for those testing drug combinations.
For experiments presented in Fig. 7, sorted PLAPneg cells (800,000 to 1,000,000 cells) were incubated at 37°C with 5% CO2 in 1 ml of Stemspan II medium plus 8 μM raltegravir and with the indicated drugs at the concentrations described above. After 24 h, supernatants were harvested as described below, 10% of cells were collected and stained for flow cytometric analysis, and the rest of the cells were washed once in PBS before being plated in STIF medium (1 ml) and cultured without stimulus for 3 days. After 3 days in culture, 10% of cells were again collected from each sample and stained for flow-cytometric analysis. The rest of the cells were replated in Stemspan 2 medium with raltegravir and TNF-α at the concentrations described above. After 24 h, supernatants were harvested and cells were collected and stained for flow analysis.
Treatment of rCD4pos T lymphocytes.
Five million rCD4pos T lymphocytes were incubated for 24 and 48 h in 1 ml of T cell culture medium with the indicated drugs at the following concentrations: 50 ng/ml PMA (Sigma-Aldrich), 1 μM ionomycin (Sigma-Aldrich), 5 nM bryostatin-1, 10 μM entinostat, 1 μM vorinostat.
Measurement of MHC-I downmodulation on treated E-GFPneg HSPCs.
E-GFPneg HSPCs from ΔGPE- or ΔGPEN-infected cultures were isolated by FACS, treated with the indicated drugs for 24 h, and stained for flow analysis. Fold downmodulation of cell surface MHC-I on infected cells was determined by dividing the median fluorescence intensity (MedFI) of the surface MHC-I stain for E-GFPneg cells by that of the E-GFPpos cells for each condition.
Measurement of supernatant HIV mRNA from treated PLAPneg HSPC cultures.
Culture supernatants were harvested at 24 h after addition of drugs, and cell debris was removed by centrifugation (700 × g; 5 min; 4°C). Supernatant RNA was extracted from clarified supernatant with TRIzol LS reagent according to the manufacturer's protocol (Invitrogen). cDNA synthesis was performed using qScript cDNA Supermix (Quanta Biosciences). Real-time qPCR was performed using TaqMan Gene Expression Mastermix (Applied Biosystems) on an Applied Biosystems 7300 thermocycler, under the following cycling conditions: 95°C for 10 min and then 45 cycles of 95°C for 15 s followed by 60°C for 60 s. Primers and probes have been previously described (8). The molecular standard curve was generated as previously described (9).
Measurement of supernatant HIV mRNA from treated rCD4pos T lymphocyte cultures.
Culture supernatants were harvested at 24 and 48 h after addition of drugs, and cell debris was removed by centrifugation. Supernatant RNA was extracted from clarified supernatant with TRIzol LS reagent according to the manufacturer's protocol. cDNA was synthesized, and real-time qPCR was performed as described above with the modification that TaqMan Fast Advanced Mastermix (Applied Biosystems) was used for real-time qPCR.
Measurement of cell-associated HIV mRNA from treated rCD4pos T lymphocytes.
Cell-associated HIV RNA transcripts were measured as previously described (9). Briefly, cell-associated RNA was extracted from treated rCD4pos T lymphocytes cells with TRIzol reagent according to the manufacturer's protocol. Contaminating genomic DNA was removed by DNase I digest (Invitrogen). cDNA synthesis was performed using qScript cDNA Supermix. Real-time qPCR was performed using TaqMan Fast Advanced Mastermix on an Applied Biosystems 7300 thermocycler. Levels of RNA polymerase II (PolR2A) were measured for each sample as a control. Molecular standard curves for HIV mRNA were generated as described above.
NF-κB/p65 binding enzyme-linked immunosorbent assay (ELISA).
Whole-cell lysates from treated PBMCs were obtained using a commercial kit according to the manufacturer's protocol (Active Motif). Total protein amounts of lysates were quantified by the Bradford assay (Thermo Fisher), and an equal total protein amount was loaded for each sample in subsequent assays. The activated form of the NF-κB monomer p65 was detected using a commercial kit according to the manufacturer's protocol (Active Motif).
Hsp70 immunoblotting.
One to 3 million HSPCs or PBMCs were treated with the indicated drugs for 24 h and lysed with Laemmli lysis buffer. Proteins from whole-cell lysates were separated by gel electrophoresis, transferred to a membrane, and detected using antibodies against Hsp70 (Enzo Life Science), Hsp90 (Abcam), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Sigma-Aldrich).
Acetylated histone H4 and tubulin immunoblotting.
HSPCs (500,000) were treated with the indicated drugs for 9 h and lysed with Laemmli lysis buffer. Proteins from whole-cell lysates were separated by gel electrophoresis, transferred to a membrane, and detected using antibodies against acetylated alpha-tubulin (Sigma-Aldrich), acetylated histone-H4(Lys12) (Millipore), and GAPDH (Sigma-Aldrich).
Flow cytometry antibodies and staining.
Antibodies raised against the following antigens were used for flow cytometry: CD133 (phycoerythrin [PE] conjugated; Miltenyi Biotec), CD34 (fluorescein isothiocyanate [FITC] conjugated, allophycocyanin [APC] conjugated, and PE-Cy7 conjugated; all from Miltenyi Biotec), hematopoietic lineage markers (eBiosciences), MHC-I (clones Bw4 and Bw6, PE-Cy7 conjugated; Miltenyi Biotec), PLAP (eFluor660 conjugated; eBioscience), and HIV Gag (clone KC57, FITC or PE conjugated; Beckman Coulter). Flow data were collected with a BD FACSCanto cytometer or a BD FACScan cytometer with Cytek 6-color upgrade. For staining of surface proteins, cells were suspended in FACS buffer (PBS with 2% FBS, 1% human serum, 2 mM HEPES, and 0.025% sodium azide) and antibodies, incubated on ice for 10 min (CD34 and CD133) or 30 min (MHC-I and PLAP), washed, and then fixed in 2% paraformaldehyde in PBS. Intracellular HIV Gag was stained by permeabilizing fixed cells with 0.1% Triton X-100 in PBS for 5 min at room temperature and then incubating in FACS buffer with antibody against HIV Gag for 30 min at room temperature and washing before analyzing.
Statistics.
Statistical analyses were performed using Microsoft Excel software. The statistical tests used to calculate P values are indicated in the corresponding figure legends.
ACKNOWLEDGMENTS
We are grateful to Mary Reyes and Lisa Mac for recruitment of donors and help with regulatory documentation and to Heather Fox and Henry Ford Hospital physicians for the bone marrow aspirations, and we especially thank the donors themselves. We thank the University of Michigan Flow Cytometry core as well as the Michigan Clinical Research Unit (CTSA grant 2UL1TR000433-06). pCMV-HIV was a gift of S.-J.-K. Yee (City of Hope National Medical Center). The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: catalog number 12666 pVQA from Robert Siliciano (48). We thank Kirsten Peterson and Megan McLeod for creating the NL4-3-ΔGPEN-E-GFP viral construct.
REFERENCES
- 1.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 2.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 3.Deeks SG. 2012. HIV: shock and kill. Nature 487:439–440. doi: 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- 4.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. 2009. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses 25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen TA, Søgaard OS, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, Dinarello C, Østergaard L, Tolstrup M. 2013. Comparison of HDAC inhibitors in clinical development. Hum Vaccin Immunother 9:993–1001. doi: 10.4161/hv.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, Hesselgesser J, Irrinki A, Murry JP, Stepan G, Stray KM, Tsai A, Yu H, Spindler J, Kearney M, Spina CA, McMahon D, Lalezari J, Sloan D, Mellors J, Geleziunas R, Cihlar T. 2014. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog 10:e1004071. doi: 10.1371/journal.ppat.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang H-C, Zhang H, Margolick JB, Blankson JN, Siliciano RF. 2012. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. 2014. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med 20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laird GM, Bullen CK, Rosenbloom DIS, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. 2015. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest 125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. 2009. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J Virol 83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savarino A, Mai A, Norelli S, El Daker S, Valente S, Rotili D, Altucci L, Palamara AT, Garaci E. 2009. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 6:52. doi: 10.1186/1742-4690-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wightman F, Ellenberg P, Churchill M, Lewin SR. 2012. HDAC inhibitors in HIV. Immunol Cell Biol 90:47–54. doi: 10.1038/icb.2011.95. [DOI] [PubMed] [Google Scholar]
- 13.Albert BJ, Niu A, Ramani R, Marshall GR, Wender PA, Williams RM, Ratner L, Barnes AB, Kyei GB. 2017. Combinations of isoform-targeted histone deacetylase inhibitors and bryostatin analogues display remarkable potency to activate latent HIV without global T-cell activation. Sci Rep 7:7456. doi: 10.1038/s41598-017-07814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J, Bixby D, Savona MR, Collins KL. 2010. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 16:446–451. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez-Bonet M, Clemente MI, Serramía MJ, Muñoz E, Moreno S, Muñoz-Fernández MÁ. 2015. Synergistic activation of latent HIV-1 expression by novel histone deacetylase inhibitors and bryostatin-1. Sci Rep 5:16445. doi: 10.1038/srep16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sebastian NT, Zaikos TD, Terry V, Taschuk F, McNamara LA, Onafuwa-Nuga A, Yucha R, Signer RAJ, Riddell J IV, Bixby D, Markowitz N, Morrison SJ, Collins KL. 2017. CD4 is expressed on a heterogeneous subset of hematopoietic progenitors, which persistently harbor CXCR4 and CCR5-tropic HIV proviral genomes in vivo. PLoS Pathog 13:e1006509. doi: 10.1371/journal.ppat.1006509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNamara LA, Ganesh JA, Collins KL. 2012. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-κB activation. J Virol 86:9337–9350. doi: 10.1128/JVI.00895-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Painter MM, Zaikos TD, Collins KL. 2017. Quiescence promotes latent HIV infection and resistance to reactivation from latency with histone deacetylase inhibitors. J Virol 91:e01080-. doi: 10.1128/JVI.01080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan L, Xing S, Yang H-C, Zhang H, Margolick JB, Siliciano RF. 2014. Unique characteristics of histone deacetylase inhibitors in reactivation of latent HIV-1 in Bcl-2-transduced primary resting CD4+ T cells. J Antimicrob Chemother 69:28–33. doi: 10.1093/jac/dkt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. 2008. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 21.Lauffer BEL, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE, Steiner P. 2013. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J Biol Chem 288:26926–26943. doi: 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, Sanders K. 2007. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer 121:1138–1148. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Schrager JA, Lange GD, Marsh JW. 2001. HIV Nef-mediated cellular phenotypes are differentially expressed as a function of intracellular NEF concentrations. J Biol Chem 276:32763–32770. doi: 10.1074/jbc.M101025200. [DOI] [PubMed] [Google Scholar]
- 24.Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. 2010. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One 5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pérez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Muñoz-Fernandez MA, Moreno S, Castor TP, Calzado MA, Muñoz E. 2010. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr HIV Res 8:418–429. doi: 10.2174/157016210793499312. [DOI] [PubMed] [Google Scholar]
- 26.Yang XJ, Seto E. 2008. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Mu Y, Greene WC. 2002. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J 21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buerki C, Rothgiesser KM, Valovka T, Owen HR, Rehrauer H, Fey M, Lane WS, Hottiger MO. 2008. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res 36:1665–1680. doi: 10.1093/nar/gkn003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson I, Low JS, Weston S, Weinberger M, Zhyvoloup A, Labokha AA, Corazza G, Kitson RA, Moody CJ, Marcello A, Fassati A. 2014. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc Natl Acad Sci U S A 111(15):E1528–E1537. doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi P, Maidji E, Stoddart CA. 2016. Inhibition of heat shock protein 90 prevents HIV rebound. J Biol Chem 291:10332–10346. doi: 10.1074/jbc.M116.717538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Keeffe B, Fong Y, Chen D, Zhou S, Zhou Q. 2000. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem 275:279–287. doi: 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- 32.Pan X-Y, Zhao W, Wang C-Y, Lin J, Zeng X-Y, Ren R-X, Wang K, Xun T-R, Shai Y, Liu S-W. 2016. Heat shock protein 90 facilitates latent HIV reactivation through maintaining the function of positive transcriptional elongation factor b (p-TEFb) under proteasome inhibition. J Biol Chem 291(50):26177–26187. doi: 10.1074/jbc.M116.743906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roesch F, Meziane O, Kula A, Nisole S, Porrot F, Anderson I, Mammano F, Fassati A, Marcello A, Benkirane M, Schwartz O. 2012. Hyperthermia stimulates HIV-1 replication. PLoS Pathog 8:35. doi: 10.1371/journal.ppat.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 35.Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. 2007. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mühlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M, Fletcher JA, Bauer S. 2009. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res 69:6941–6950. doi: 10.1158/0008-5472.CAN-08-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Marchenko ND, Moll UM. 2011. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 18:1904–1913. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 39.Guo W, Reigan P, Siegel D, Zirrolli J, Gustafson D, Ross D. 2005. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res 65:10006–10015. doi: 10.1158/0008-5472.CAN-05-2029. [DOI] [PubMed] [Google Scholar]
- 40.Isambert N, Delord J-P, Soria J-C, Hollebecque A, Gomez-Roca C, Purcea D, Rouits E, Belli R, Fumoleau P. 2015. Debio0932, a second-generation oral heat shock protein (HSP) inhibitor, in patients with advanced cancer-results of a first-in-man dose-escalation study with a fixed-dose extension phase. Ann Oncol 26:1005–1011. doi: 10.1093/annonc/mdv031. [DOI] [PubMed] [Google Scholar]
- 41.Yong K, Cavet J, Johnson P, Morgan G, Williams C, Nakashima D, Akinaga S, Oakervee H, Cavenagh J. 2016. Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br J Cancer 114:7–13. doi: 10.1038/bjc.2015.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones RB, O'Connor R, Mueller S, Foley M, Szeto GL, Karel D, Lichterfeld M, Kovacs C, Ostrowski MA, Trocha A, Irvine DJ, Walker BD. 2014. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog 10:e1004287. doi: 10.1371/journal.ppat.1004287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pace M, Williams J, Kurioka A, Gerry AB, Jakobsen B, Klenerman P, Nwokolo N, Fox J, Fidler S, Frater J, CHERUB Investigators, Pace M, Frater J, Wightman F, Ellenberg P, Churchill M, Lewin S, Archin N, Liberty A, Kashuba A, Choudhary S, Kuruc J, Crooks A, Elliott J, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron M, Rasmussen T, Tolstrup M, Brinkmann C, Olesen R, Erikstrup C, Solomon A, Shan L, Deng K, Shroff N, Durand C, Rabi S, Yang H, Jones R, O'Connor R, Mueller S, Foley M, Szeto G, Karel D, Kaufman D, Schoon R, Leibson P, Schaefer M, Wonderlich E, Roeth J, Leonard J, Collins K, Magner W, Kazim A, Stewart C, Romano M, Catalano G, Grande C, Pellicciotta I, Cortez-Gonzalez X, Sasik R, Reiter Y, Hardiman G, Langlade-Demoyen P, Falkenberg K, Johnstone R, Varela-Rohena A, Molloy P, Dunn S, Li Y, Suhoski M, Carroll R, Alter G, Martin M, Teigen N, Carr W, Suscovich T, Schneidewind A, Williams J, Southern P, Lissina A, Christian H, Sewell A, Phillips R, Bullen C, Laird G, Durand C, Siliciano J, Siliciano R, Cooper M, Fehniger T, Caligiuri M, Khan A, Gregorie C, Tomasi T, Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L, Nolting A, Dugast A, Rihn S, Luteijn R, Carrington M, Kane K, Fogli M, Mavilio D, Brunetta E, Varchetta S, Ata K, Roby G, Ward J, Bonaparte M, Sacks J, Guterman J, Fogli M, Mavilio D, Jost S, Altfeld M, Ogbomo H, Michaelis M, Kreuter J, Doerr HJC Jr, Shi P, Yin T, Zhou F, Cui P, Gou S, Wang C, Zhu S, Denman C, Cobanoglu Z, Kiany S, Lau C, Gottschalk S, Wei D, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, Shapiro G, Frank R, Dandamudi U, Hengelage T, Zhao L, Gazi L, Laird G, Bullen C, Rosenbloom D, Martin A, Hill A, Durand C, Pace M, Graf E, Agosto L, Mexas A, Male F, Brady T, Zhang Z, Guan X, Li Y, Zhu M, Yang X, Zou X, Suarez-Alvarez B, Rodriguez R, Calvanese V, Blanco-Gelaz M, Suhr S, Ortega F, Bokhoven M, Stephen S, Knight S, Gevers E, Robinson I, Takeuchi Y. 2016. Histone deacetylase inhibitors enhance CD4 T cell susceptibility to NK cell killing but reduce NK cell function. PLoS Pathog 12:e1005782. doi: 10.1371/journal.ppat.1005782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrido C, Spivak AM, Soriano-Sarabia N, Checkley MA, Barker E, Karn J, Planelles V, Margolis DM. 2016. HIV latency-reversing agents have diverse effects on natural killer cell function. Front Immunol 7:356. doi: 10.3389/fimmu.2016.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosque A, Nilson KA, Macedo AB, Spivak AM, Archin NM, Van Wagoner RM, Martins LJ, Novis CL, Szaniawski MA, Ireland CM, Margolis DM, Price DH, Planelles V. 2017. Benzotriazoles reactivate latent HIV-1 through inactivation of STAT5 SUMOylation. Cell Rep 18:1324–1334. doi: 10.1016/j.celrep.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun T-W, Zack JA, Wender PA. 2013. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci U S A 110:11698–11703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen BK, Gandhi RT, Baltimore D. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol 70:6044–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shan L, Rabi SA, Laird GM, Eisele EE, Zhang H, Margolick JB, Siliciano RF. 2013. A novel PCR assay for quantification of HIV-1 RNA. J Virol 87:6521–6525. doi: 10.1128/JVI.00006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]