

Abstract
Transglutaminase 2 (TG2) is a ubiquitously expressed, intracellular as well as extracellular protein with multiple modes of post-translational regulation, including an allosteric disulfide bond between Cys-370–Cys-371 that renders the enzyme inactive in the extracellular matrix. Although recent studies have established that extracellular TG2 is switched “on” by the redox cofactor protein thioredoxin-1 (TRX), it is unclear how TG2 is switched “off.” Here, we demonstrate that TG2 oxidation by small-molecule biological oxidants, including glutathione, cystine, and hydrogen peroxide, is unlikely to be the inactivation mechanism. Instead, endoplasmic reticulum (ER)–resident protein 57 (ERp57), a protein in the ER that promotes folding of nascent proteins and is also present in the extracellular environment, has the cellular and biochemical characteristics for inactivating TG2. We found that ERp57 colocalizes with extracellular TG2 in cultured human umbilical vein endothelial cells (HUVECs). ERp57 oxidized TG2 with a rate constant that was 400–2000-fold higher than those of the aforementioned small molecule oxidants. Moreover, its specificity for TG2 was also markedly higher than those of other secreted redox proteins, including protein disulfide isomerase (PDI), ERp72, TRX, and quiescin sulfhydryl oxidase 1 (QSOX1). Lastly, siRNA-mediated ERp57 knockdown in HUVECs increased TG2-catalyzed transamidation in the extracellular environment. We conclude that, to the best of our knowledge, the disulfide bond switch in human TG2 represents the first example of a post-translational redox regulatory mechanism that is reversibly and allosterically modulated by two distinct proteins (ERp57 and TRX).
Keywords: transglutaminase, thioredoxin, oxidation-reduction (redox), post-translational modification (PTM), disulfide, ERp57, transglutaminase 2, protein disulfide isomerase family A member 3, PDIA3, redox switch, celiac disease
Introduction
Transglutaminase 2 (TG2)2 is a prominent member of the mammalian transglutaminase family of enzymes responsible for catalyzing the formation of cross-links between glutamine and lysine residues of peptide and protein substrates. TG2 is found in the cytosol as well as the extracellular environment of cells. It lacks a hydrophobic leader sequence and is therefore secreted through a poorly understood non-classical pathway (1–3). At the post-translational level, TG2 is regulated by multiple allosteric mechanisms. Upon binding GTP or GDP, the protein adopts a catalytically inactive (“closed”) conformation (4). In the presence of Ca2+ and absence of guanine nucleotides, TG2 undergoes a large conformational change into its “open,” catalytically active form (5). A vicinal disulfide bond can form between Cys-370 and Cys-371 with complete loss of activity while retaining the open conformation. Formation of this sterically constrained disulfide bond is catalyzed by an internal sulfhydryl group, Cys-230 of human TG2 (6). The redox protein cofactor thioredoxin-1 (TRX) activates extracellular TG2 by reducing the Cys-370–Cys-371 disulfide bond with high specificity in vitro and in vivo (7, 8). Thus, redox regulation of TG2 explains why this extracellular enzyme is catalytically inactive in the small intestine and other organs despite the presence of Ca2+ and paucity of GTP (9). Although the function of TG2 leaves much to be understood, abnormal activity has been implicated in the pathogenesis of celiac disease (10).
Cysteine is the second-most conserved amino acid residue in proteins after tryptophan (11), and it is widely recognized to play important roles in stabilizing tertiary and quaternary structures through disulfide bonds (12). More recently, it has also been observed that some disulfide bonds operate as allosteric redox switches to control protein activity, although the physiological reversibility of this regulatory mechanism is unclear (13, 14). We therefore sought to address this question in the context of redox-dependent regulation of extracellular TG2 activity. Specifically, because TG2 is exported via a non-classical pathway, we hypothesized that TG2 is unlikely to encounter thiol oxidoreductases in the endoplasmic reticulum (ER) during export. Hence, TG2 must be released into the extracellular matrix (ECM) in its active, reduced form and undergo oxidation only subsequently. Consistent with this hypothesis, increased TG2 expression correlates with elevated TG2 export and enzymatic activity (15–17).
Because spontaneous thiol-disulfide oxidation is kinetically slow even under thermodynamically favorable conditions, a promoter will likely be required for TG2 oxidation in the ECM. Both small molecule and protein oxidants could play such a role. Cystine is the most abundant small molecule oxidant in plasma (18) and is therefore a plausible candidate, although glutathione disulfide (GSSG) and hydrogen peroxide are also known to play such roles. Some ER-resident redox protein cofactors, including protein disulfide isomerase (PDI) and its homologs ERp5, ERp57, and ERp72, are also secreted to some extent (19, 20). Although their extracellular roles are not well-understood, their extracellular redox functions have been implicated in thrombosis (20), viral infection (21), and priming of neutrophils (22). Last but not least, the extracellular flavoenzyme, quiescin-sulfhydryl oxidase 1 (QSOX1), also catalyzes disulfide bond formation in ECM proteins with concomitant reduction of molecular oxygen (23, 24). We therefore undertook a systematic evaluation of these candidate mechanisms for physiological oxidation of extracellular TG2.
Results
Oxidation of TG2 by small molecule biological oxidants is kinetically slow
The redox potentials of the two major redox couples, GSH/GSSG and Cys/CySS, have been reported to be −139 and −80 mV in plasma, respectively (14). In the presence of glutathione redox buffer at −140 mV, the oxidation of TG2 is slow in vitro, with complete inactivation occurring over 4–6 h (6). We therefore sought to test whether a small molecule oxidant could promote TG2 oxidation under physiologically relevant conditions. Specifically, reduced TG2 was treated with cystine, GSSG, or H2O2 in a microtiter assay in which TG2 was immobilized on a fibronectin-coated surface (Fig. 1). Cystine and H2O2 oxidized TG2 at similar rates (Fig. 2 and Table 1), whereas glutathione reacted much slower. None of the bimolecular rate constants appear to be capable of inactivating TG2 under physiologically relevant oxidant conditions (25). Thus, the oxidation of TG2 is likely to be a protein-mediated process.
Figure 1.
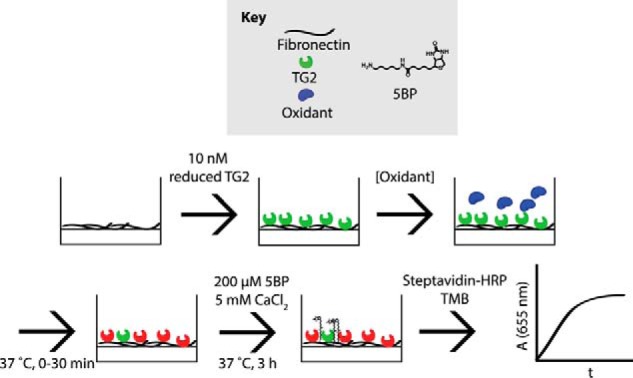
Oxidation of immobilized TG2 activity assay. Reduced, recombinant TG2 (green) was immobilized onto fibronectin-coated plates and incubated with oxidants (blue) for up to 30 min at 37 °C, producing inactivated TG2 (red). Oxidants were then washed away, and 200 μm 5BP and 5 mm CaCl2 were added to analyze TG2 activity. 5BP is a biotinylated primary amine that is a good substrate of TG2 and is therefore cross-linked to Gln residues on extracellular matrix proteins, such as fibronectin, in the presence of catalytically active extracellular TG2 (26). Streptavidin-HRP binds to cross-linked 5BP and turns over TMB, producing a blue color that can be spectrophotometrically monitored at 655 nm. The steady-state slope was extracted and was proportional to TG2 activity.
Figure 2.
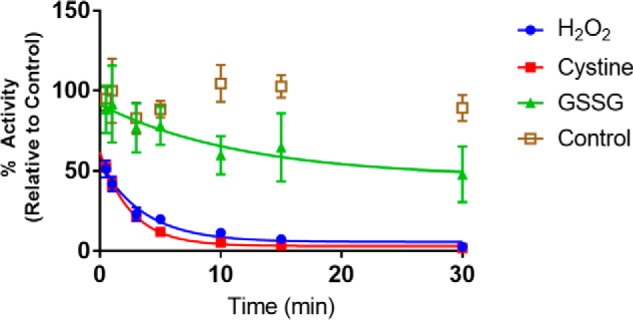
Pseudo-first order kinetic profile of TG2 oxidation by common biological small molecule oxidants at 37 °C (pH 7.5). All experiments were conducted with 10 nm reduced TG2 immobilized on fibronectin-coated plates (Fig. 1). Each oxidant was added at a concentration of 1 mm to the assay mixture. Experiments were performed in quadruplicate with three technical replicates of each time point, and data were fit to a decaying exponential in GraphPad Prism 7.0 to obtain an apparent rate constant, kapp. The bimolecular rate constant was derived by dividing the kapp by the concentration of oxidant (1 mm). Data are presented as average ± S.D.
Table 1.
Rate constants for TG2 oxidation by common biological small molecule oxidants at 37 °C, pH 7.5
| Oxidant | Bimolecular rate constant |
|---|---|
| mm−1 min−1 | |
| H2O2 | 0.27 ± 0.05 |
| Cystine | 0.38 ± 0.03 |
| GSSG | 0.1 ± 0.1 |
Colocalization of CXXC thiol isomerases with extracellular TG2 in HUVEC cultures
Earlier studies have demonstrated that extracellular TG2 is catalytically inactive under basal conditions in most tissue culture models and that the protein redox cofactor TRX activates this enzyme pool in vitro and in vivo with high specificity (kcat/Km = 1.6 μm−1 min−1) via its canonical CXXC motif (6, 7, 26). PDI and its homologs belong to the TRX superfamily of proteins and harbor one or more CXXC domains. In contrast to TRX however, their standard redox potentials render them more well-suited for disulfide bond formation as opposed to disulfide bond reduction (Table 2) (27). Although PDI and its homologs are predominantly localized to the ER, studies have shown that they can exist in the extracellular environment as well (19, 20). Because TG2 is exported via a non-canonical mechanism, we hypothesized that its oxidative agent would be a secreted protein.
Table 2.
Redox potential of TG2 and select CXXC proteins
Unless otherwise stated, the redox potential reflects an average value in cases where multiple CXXC domains are present.
Human umbilical vein endothelial cells (HUVECs) have previously been studied in the context of extracellular TG2 activity (28, 29). These cells are also known to constitutively secrete PDI (30), ERp5 (31), and ERp57 and ERp72 (32, 33), making them a suitable cellular model to study TG2 oxidation. As a preliminary test of this model, we added recombinant human TRX to HUVEC cultures to verify that its extracellular TG2 could be activated. As expected, TRX was able to activate TG2 at a concentration as low as 100 nm (p < 0.01) (26); enzyme activity was abrogated in the presence of the TG2 inhibitor, ERW1041E (Fig. 3) (34). As with most other cultured cell models, basal activity of extracellular TG2 was negligible.
Figure 3.
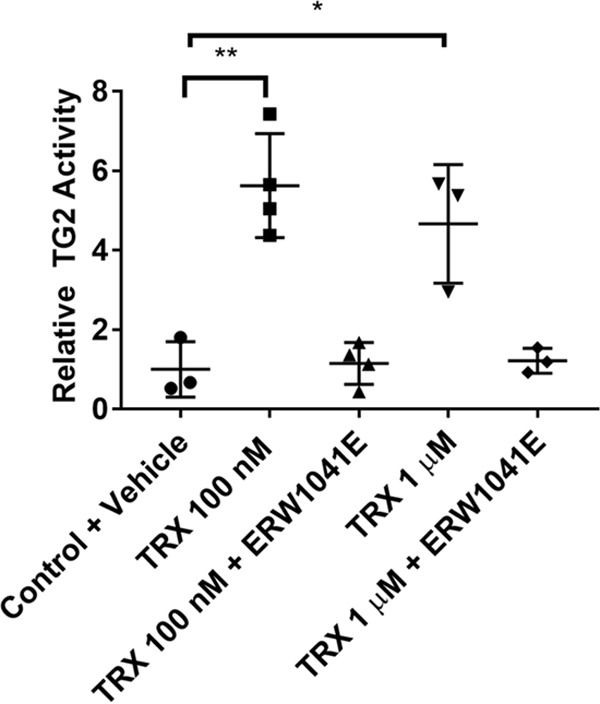
Activation of extracellular TG2 by TRX in HUVECs. Cells were pre-incubated with recombinant, reduced TRX with or without 25 μm ERW1041E for 1 h. 5BP was then added, and TG2 activity was assayed for 3 h. Cells were the washed, fixed without permeabilization, and probed with streptavidin-HRP. TMB was added, and the reaction was followed continuously at 655 nm for 30 min. The data were normalized against the Control + Vehicle condition. Statistical comparisons were performed using Student's t test. A dose of 100 nm (**, p < 0.01) and 1 μm (*, p < 0.05) TRX resulted in increased TG2 activity that was reversed with TG2 inhibitor (ERW1041E). Data are presented as average ± S.D. of at least three replicates per condition.
To analyze the extent to which extracellular TG2 in HUVEC cultures colocalized with various CXXC oxidoreductases, we first validated the specificity of various commercially available antibodies (Fig. S1). Appropriately specific antibodies were then used on fixed but not permeabilized HUVEC cultures. As controls, TG2 was observed to colocalize with fibronectin, presumably due to its affinity for the 42-kDa gelatin-binding domain of the latter protein (35), but not VE-cadherin (Fig. 4). Microscopic analysis revealed that PDI, ERp57, and ERp72 colocalized with extracellular TG2 to varying degrees. In contrast, ERp5 showed no association with TG2 and was therefore eliminated from further consideration as an oxidant of this extracellular protein.
Figure 4.
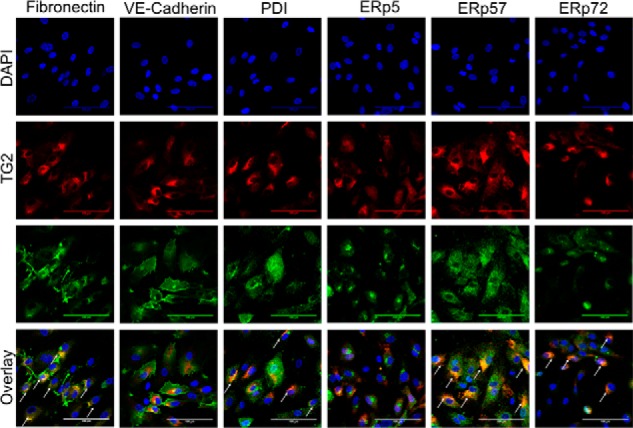
Colocalization of CXXC proteins with extracellular TG2. HUVEC cultures were fixed without permeabilization and stained with antibodies against TG2 (red) and individual CXXC proteins (green). Fibronectin, a known protein partner of extracellular TG2, and VE cadherin, an extracellular protein with no known affinity for TG2, were evaluated as controls. White arrows in the Overlay images denote areas of colocalization between TG2 and the partner protein. Images were taken in triplicate; a representative image is presented. Scale bar, 100 μm.
ERp57 specifically oxidizes TG2 in vitro
The above findings prompted us to biochemically characterize the recognition of TG2 by PDI, ERp57, and ERp72, using TRX as a control. Recombinant forms of each of the latter three human proteins were expressed in Escherichia coli and purified to substantial homogeneity (Fig. 5). Redox activity was verified by their ability to reduce insulin, a standard activity assay for CXXC family members (36). Additionally, the human flavoprotein QSOX1b, which is the soluble isoform of QSOX1, was also expressed and purified (37). QSOX1 is an extracellular enzyme with a single CGHC motif that can oxidize protein substrates using molecular oxygen as an electron acceptor. It has been implicated in cell adhesion and ECM assembly (24). QSOX1b lacks the C-terminal trans-membrane fragment of QSOX1 (37). The recombinant holoprotein was bound to FAD and was able to oxidize TRX. Although TRX is not known to catalyze disulfide bond formation, its inclusion as a control was motivated by the fact that it exhibited high specificity for TG2 and has also been reported to function as a protein disulfide isomerase (38, 39).
Figure 5.
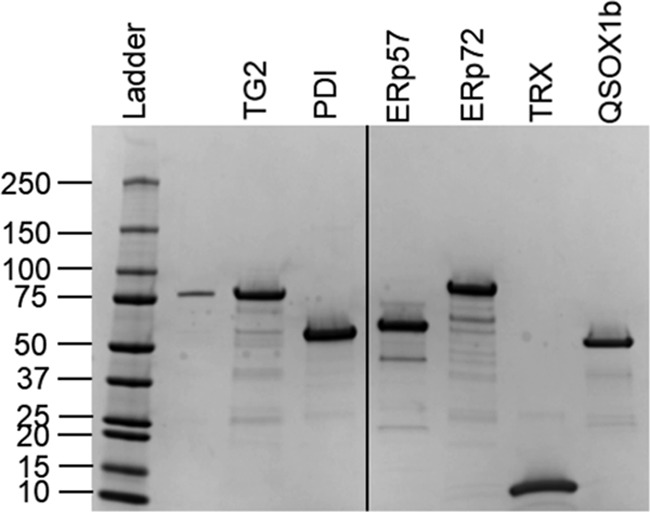
Purified recombinant proteins used in this study. Ladder represents protein molecular mass in kDa.
Analogous to our kinetic analysis of TG2 oxidation by H2O2, cystine, and GSSG, the kinetics of TG2 oxidation by the above CXXC proteins was investigated by varying the concentration of CXXC protein in the presence of reduced, recombinant fibronectin-immobilized TG2. In both dose-dependent assays (Fig. 6A and Table 3) and time-dependent assays (Fig. 6B), ERp57 exhibited the highest degree of specificity toward TG2. The IC50 of ERp57-promoted TG2 inactivation was 60 ± 10 nm, ∼30-fold more specific than ERp72. PDI and QSOX1 showed negligible ability to inactivate TG2 at concentrations up to 1 μm. The bimolecular rate constant for oxidation of TG2 by ERp57 was determined to be 162 mm−1 min−1, a 400-fold rate enhancement over the oxidation of TG2 by cystine. This parameter is also in good agreement with the rate enhancement of PDI-catalyzed oxidation of RNase A. Antibodies against TG2 confirmed that TG2 remained bound to fibronectin plates, implying that the observed loss of activity could not be attributed to protein washout (Fig. 6C).
Figure 6.

ERp57 specifically oxidizes TG2 in vitro. A, dose dependence of TG2 oxidation by CXXC proteins. 10 nm reduced TG2 was immobilized onto fibronectin and incubated with each oxidized CXXC protein for 30 min at 37 °C. After a series of washes with PBS, 200 μm 5BP and 5 mm CaCl2 were added to probe for TG2 activity (see Fig. 1). Experiments were performed in quadruplicate with three technical replicates per concentration. B, time dependence of TG2 oxidation by CXXC proteins. 10 nm reduced TG2 was immobilized onto fibronectin and oxidized with 1 μm PDI, ERp57, or ERp72 for up to 30 min at 37 °C. After a series of PBS washes, 200 μm 5BP and 5 mm CaCl2 was added to probe for TG2 activity. Experiments were performed in triplicate with three technical replicates per time point, and the first-order kinetic profile was fit to obtain an apparent rate constant, kapp. The bimolecular rate constant was derived by dividing the kapp by the initial oxidant concentration (1 μm). C, TG2 remains bound to fibronectin even after oxidation by PDI, ERp57, or ERp72. Immobilized TG2 was incubated with each oxidant for up to 30 min at 37 °C and probed with a rabbit anti-TG2 antibody. The amount of bound TG2 was determined by incorporation of an anti-rabbit HRP-conjugated secondary antibody and TMB liquid substrate system. TMB turnover was monitored continuously for 30 min, and the steady-state slope from 0 to 5 min is presented. D, either CXXC active site of ERp57 can oxidatively inactivate TG2. ERp57 mutants were generated to inactivate the redox-active Cys residues in domain a (SSCC), domain a′ (CCSS), or both domains (SSSS). 10 nm reduced TG2 was immobilized onto fibronectin, and incubated with oxidized proteins for 30 min at 37 °C. After a series of PBS washes, 200 μm 5BP, and 5 mm CaCl2 was added to probe for TG2 activity. Experiments were performed in duplicate with three technical replicates per condition. Data are presented as average ± S.D.
Table 3.
IC50 and bimolecular rate constants of TG2 oxidation by different CXXC proteins
The IC50 value was determined from Fig. 6A, and the bimolecular rate constant was determined from Fig. 6B. ND means not determined.
| Oxidant | IC50 | Bimolecular rate constant |
|---|---|---|
| μm | mm−1 min−1 | |
| PDI | >10 | ND |
| ERp57 | 0.06 ± 0.01 | 162 ± 50 |
| ERp72 | 1.6 ± 0.4 | ND |
| TRX | 5.0 ± 1.9 | ND |
| QSOX1b | >1 | ND |
Single redox-site mutants (SSCC for domain a; CCSS for domain a′) were engineered and expressed to assess whether one of the two redox-active CXXC domains of ERp57 was primarily responsible for oxidation of TG2. (As shown in Table 2, the a′ domain is a stronger oxidant than the a domain.) Both CXXC domains were capable of oxidatively inactivating TG2 to similar degrees, whereas the redox-inactive mutant (ERp57-SSSS) had no appreciable effect on TG2 activity (Fig. 6D).
Mass spectrometric analysis was conducted to verify formation of the allosteric disulfide bond in TG2 (Cys-370–Cys-371). Reduced TG2 and oxidized ERp57 were coincubated in stoichiometric quantities for 0 or 30 min and quenched with iodoacetamide (IAM) to alkylate all residual free cysteine residues. Following separation by non-reducing SDS-PAGE, the disulfide bonds were reduced with DTT and alkylated with iodoacetic acid (IAA). Thus, reduced cysteine residues were mass shifted by 57 Da, whereas oxidized cysteine residues were mass shifted by 58 Da. The trypsin-digested, alkylated Cys-containing peptides were identified (Table S1 and Figs. S2–S5), and their change in redox state was compared between the 0- and 30-min time points following ERp57 treatment. Formation of the Cys-370–Cys-371 disulfide bond was observed, which correlated with reduction of both CXXC domains of ERp57 (Fig. 7).
Figure 7.

ERp57 promotes formation of the Cys-370–Cys-371 disulfide bond of TG2. Reduced (active) TG2 and oxidized ERp57 were coincubated in equimolar amounts for 0 or 30 min at 37 °C. Proteins were quenched with IAM to freeze thiol-disulfide exchange and to modify reduced Cys residues (+57 Da). Oxidized Cys residues were identified following reduction with DTT, and labeling with IAA (+58 Da). Following tryptic digest, peptides were subjected to liquid chromatography-mass spectrometry. Modified peptides were validated by their isotopic distribution and y fragment ions (Figs. S2–S5). The extracted ion chromatogram of each modified peptide (Table S1) is presented at 0 min (top) and 30 min (bottom).
siRNA knockdown of ERp57 gene expression increases extracellular TG2 activity
The biological importance of ERp57 is underscored by the observation that homozygous knock-out mice are embryonic lethal (40). Thus, to study the effects of ERp57 expression on TG2 activity, we employed siRNA to silence ERp57 gene expression (41). HUVECs were transiently transfected with siRNA duplexes for 48 h, leading to ∼65% knockdown in total protein expression, as determined by immunoblotting of whole-cell lysates (Fig. 8, A and B). Global knockdown of ERp57 also resulted in decreased extracellular ERp57 staining (Fig. 8, C and D; p < 0.01) by approximately the same magnitude. In HUVEC cultures treated with ERp57-targeted siRNA, extracellular TG2 activity was increased by nearly 4-fold (Fig. 8, E and F; p < 0.05), presumably due to a decrease in oxidizing equivalents supplied by ERp57. As expected, addition of exogenous oxidized ERp57 reversed the observed increase in TG2 activity (Fig. 8, E and F; p < 0.01).
Figure 8.

siRNA knockdown of ERp57 expression leads to increased extracellular TG2 activity. A, HUVECs were transfected with ERp57-specific siRNA for 48 h and lysed with RIPA buffer. Whole-cell lysates were blotted for total ERp57 expression. B, densitometric analysis of the Western blots shown in A (n = 3). β-Actin was used as the loading control and normalization factor. C, siRNA knockdown of ERp57 leads to a decrease in extracellular ERp57 expression. Images were taken in triplicate with three biological replicates (n = 9). Scale bar, 100 μm. D, quantitative analysis of extracellular ERp57 expression normalized to cell count. Extracellular ERp57 expression was significantly decreased (**, p < 0.01) compared with control siRNA-treated cells. E and F, extracellular TG2 activity is elevated in ERp57 siRNA-knockdown cells compared with control siRNA-treated cells (*, p < 0.05). TG2 activity is restored to basal levels upon addition of recombinant oxidized ERp57 (**, p < 0.01). 200 μm 5BP was added to cells for 3 h to visualize TG2 activity. Exogenous oxidized ERp57 was added 30 min prior to 5BP. During confocal microscopy analysis, at least three images were taken per replicate with three biological replicates per condition. A representative set of images are presented. All images can be found in Figs. S6–S8. 5BP incorporation was normalized to TG2 staining. Scale bar, 100 μm. All quantitative data are presented as average ± S.D., and statistical analyses were performed using Student's t test.
Discussion
Most post-translational modifications (PTMs) in biology, such as protein phosphorylation and acetylation, are reversible events, thereby enabling cells to turn protein function on or off as needed. Whereas disulfide bonds are among the oldest known PTMs, their regulatory significance has not been recognized until relatively recently. For example, HMGB1 (high-mobility group box 1 protein) is also a non-classically secreted protein (42), whose inflammatory activity is allosterically induced by oxidation of a thiol-disulfide redox switch (43). Its inactivation trigger (if any) has not been identified. To our knowledge, this study is the first example of a regulatory disulfide bond that appears to be reversibly switched on or off by recognition events involving distinct proteins.
TG2 is an intracellular as well as extracellular protein whose extracellular activity is allosterically regulated by a disulfide bond (Cys-370–Cys-371). The oxidized protein is catalytically inactive; it is activated upon reduction by TRX via a thiol-disulfide exchange mechanism. Because basal TG2 activity is ordinarily very low in the extracellular matrices of most organs (including the small intestine), robust oxidation mechanism(s) must exist to inactivate the enzyme in conjunction with TG2 export. Identification of this mechanism has been the principal goal of this study.
The cellular and biochemical data presented here argue that ERp57 (also known as PDIA3), a member of the PDI family of redox proteins, promotes oxidative inactivation of reduced TG2 (Fig. 9). The principal function of PDI and its homologs is proper folding of a wide range of nascent proteins in the ER via disulfide bond formation. Early studies by Gilbert (44) and Lyles (45) investigated oxidative refolding of RNase A and revealed modest rate enhancement compared with uncatalyzed oxidation by GSSG (∼400-fold). Our findings with TG2 show comparable activity of ERp57 toward this protein. ERp57 was at least 30-fold more effective than the next-best protein oxidant, ERp72. Its CXXC motifs are required for this activity. The strong colocalization of ERp57 and extracellular TG2 in HUVEC cultures further supports this conclusion. Last but not least, down-regulation of ERp57 expression by siRNA led to a concomitant increase in extracellular TG2 activity but was reverted back upon addition of exogenous ERp57.
Figure 9.
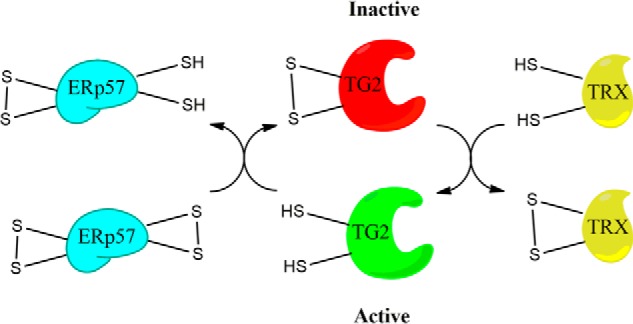
Model for reversible redox regulation of TG2. TG2 is oxidized by ERp57, leading to its catalytic inactivation. The enzyme is reactivated by TRX. Both reactions involve thiol-disulfide exchange events between TG2 and the protein cofactor.
Our finding may have relevance for understanding disease pathogenesis. In celiac disease, extracellular TG2 activity in the small intestinal mucosa is widely thought to play a causative role in the onset and maintenance of inflammatory T cell activity (10). More recent evidence suggests that celiac disease pathogenesis may be associated with aberrantly high intestinal TG2 activity (46). If so, then a clearer understanding of the physiological mechanisms that turn TG2 on and off in the intestine could not only enhance our understanding of celiac disease pathogenesis but also shine light on fundamentally new therapeutic approaches for this widespread but overlooked disorder. Separately, it has been shown that secretion of ERp57 is important in ECM build-up in animal models of renal fibrosis (41). Given that elevated TG2 activity is required for nephropathy in animal models of renal scarring (47), it is possible that the observed increase in ERp57 export evolved as a mechanism to down-regulate aberrantly activated TG2 in the ECM of renal fibrosis patients. If so, a better understanding of this protein–protein interaction could favorably impact the health of a large number of end-stage renal disease patients.
Experimental procedures
Chemicals and other reagents
Unless otherwise noted, chemicals were from Sigma. Cell culture media, trypsin-EDTA, sterile PBS, 7000 MWCO Zeba columns, HiTrap-Q anion-exchange columns, and BSA were from Thermo Fisher Scientific. Ni-NTA resin was from Qiagen. Fetal bovine serum (BenchMark) was from Gemini Bio Products. 5-Biotinoamidopentylamine hydrochloride (5BP) was synthesized and purified as before (48). SDS-polyacrylamide gradient gels were from Bio-Rad. Isopropyl β-d-1-thiogalactopyranoside (IPTG) and antibiotics were from Gold Biotechnology.
All Alexa-Fluor-conjugated secondary antibodies were from Thermo Fisher Scientific. Mouse anti-ERp57 (MaP.ERp57), mouse anti-ERp5 (G-5), mouse anti-ERp72 (B-4), mouse anti-VE cadherin (F-8), and goat anti-fibronectin (C-20) antibodies were from Santa Cruz Biotechnology. Rabbit anti-TG2 was custom-made by Pacific Immunology (7). Mouse anti-PDI (RL90) was from Thermo Fisher Scientific. Streptavidin-HRP, rabbit anti-β-actin (poly6221), and HRP-conjugated secondary antibodies were from Biolegend.
Cell culture
Seed HUVEC cultures were from Lonza (CC-2519). Cells from passages 5 to 10 were used in all experiments. Cells were maintained with EGM-2 growth media from Lonza. At 70% confluence, cells were passaged using trypsin-EDTA. Feeding occurred every 2–3 days.
Expression and purification of human TG2, TRX, PDI, ERp57, ERp72, and QSOX1b
TG2 and TRX were expressed and purified from E. coli Rosetta 2, as described previously (6, 7, 49). Protein concentrations were estimated using the predicted extinction coefficient (ϵ280 = 105.6 mm−1 cm−1 for TG2; ϵ280 = 7.57 mm−1 cm−1 for TRX).
E. coli Rosetta 2 cells expressing human PDI were inoculated and cultured in LB media supplemented with 50 μg/ml kanamycin and 33 μg/ml chloramphenicol at 37 °C until an A600 of 0.6 was attained. Protein expression was induced with 200 μm IPTG and maintained at 18 °C for 16–18 h. Cells were lysed with 50 mm sodium phosphate, 500 mm NaCl, 20 mm imidazole (pH 7.6) and sonicated. The resulting supernatant was incubated with Ni-NTA resin (2 ml resin/liter of culture) for 45 min at 4 °C. The resin was washed with 10 column volumes of lysis buffer, and eluted with 3 volumes of 50 mm sodium phosphate, 300 mm NaCl, 300 mm imidazole (pH 7.6). Eluted protein was buffer-exchanged into 20 mm Tris, 1 mm EDTA (pH 7.6) using an Amicon ultracentrifugal filter (EMD Millipore) and loaded onto a HiTrap-Q anion-exchange column. Anion-exchange chromatography buffers were supplemented with 1 mm DTT. The protein eluted between 300 and 400 mm NaCl. Protein stocks were flash-frozen and stored at −80 °C with 20% glycerol and 1 mm DTT. Protein concentrations were measured using the extinction coefficient (ϵ280 = 45.7 mm−1 cm−1).
hERp57 was expressed in E. coli Rosetta 2 and purified using Ni2+-affinity and anion-exchange chromatography with 1 mm DTT, similar to PDI. The protein eluted between 200 and 300 mm NaCl. Protein stocks were flash-frozen and stored at −80 °C with 20% glycerol and 1 mm DTT. Protein concentrations were determined using the predicted extinction coefficient (ϵ280 = 44.8 mm−1 cm−1).
hERp72 was also expressed in E. coli Rosetta 2, and purified using Ni2+-affinity and anion-exchange chromatography with 1 mm DTT. The protein eluted between 300 and 400 mm NaCl. Protein stocks were flash-frozen and stored at −80 °C with 20% glycerol and 1 mm DTT. Protein concentrations were determined using the extinction coefficient (ϵ280 = 67.7 mm−1 cm−1).
hQSOX1b, the soluble form of QSOX1, was cloned off cDNA (GE Dharmacon, Clone ID: 4447666) using primers 5′-AAACATATGCGGTCGGCGCTCTATTCGC-3′ (forward primer) and 5′-TTTTCTCGAGTCATCAAATAAGCTCAGGTCCCTCAGCC-3′, where the signal sequence was omitted. The amplimer was restriction digested with NdeI and XhoI restriction enzymes and ligated into pET28a to yield NW02. The plasmid was transformed into Rosetta-gami DE3 strain of E. coli by electroporation. Cells were cultured in 50 μg/ml ampicillin, 33 μg/ml chloramphenicol, 15 μg/ml kanamycin, 12.5 μg/ml tetracycline, and 5 μm riboflavin at 37 °C until A600 reached 0.6. Protein expression was induced with 250 μm IPTG, and cells were maintained at 16 °C for 16–18 h. Cell pellets were lysed in 50 mm sodium phosphate, 50 μm FAD (pH 7.5) buffer with sonication. The supernatant was incubated with Ni-NTA resin (2 ml resin/liter of culture) for 1 h at 4 °C. Resin was washed with 5 column volumes of wash buffer I (50 mm sodium phosphate, 1 m NaCl, 50 mm imidazole (pH 7.5)) and 5 column volumes of wash buffer II (50 mm sodium phosphate, 50 mm imidazole (pH 7.5)). Bound protein was eluted with 3 column volumes of 50 mm sodium phosphate, 200 mm imidazole (pH 7.5) and further purified by anion-exchange chromatography using a HiTrap-Q anion-exchange column. The protein eluted between 150 and 200 mm NaCl. Protein aliquots were prepared in 20% glycerol, flash-frozen in liquid nitrogen, and stored at −80 °C. Enzyme concentrations were determined using the extinction coefficient for bound FAD (ϵ280 = 12.5 mm−1 cm−1) as described previously (37).
A quadruple Cys → Ser mutant of ERp57 was prepared via tandem site-directed mutagenesis of the CXXC motifs in the a and a′ catalytic domains. Single a and a′ domain knockouts were prepared in the same manner. Expression and purification of these mutant proteins were very similar to their wildtype counterpart.
Oxidation of PDI, ERp57, ERp72, and TRX
Proteins were purified in reduced monomeric forms, as described above. Afterward, 100 μm PDI, ERp57, or ERp72 was buffer-exchanged into 50 mm Tris, 1 mm EDTA (pH 7.5) containing 20 mm GSSG using a 7000 MWCO Zeba column, and incubated overnight at 4 °C. Oxidized proteins were purified through another round of anion-exchange chromatography in buffers devoid of DTT. Proteins were stored in 20% glycerol aliquots at −80 °C.
TRX was oxidized by H2O2 in a protocol similar to Hashemy and Holmgren (50). Briefly, 40 μm TRX was oxidized with 20 m excess of H2O2 in 50 mm Tris, 1 mm EDTA (pH 7.5) for 15 min at 37 °C, and buffer-exchanged into buffer without H2O2 using a 7000 MWCO Zeba column. Oxidation was verified by determination of protein thiol content using 5,5′-dithiobis(nitrobenzoic acid) or maleimide PEG-5K (Sigma).
Transamidation activity of immobilized TG2
10 μg/ml human plasma fibronectin (fc010, EMD Millipore) was diluted in PBS and coated onto an ELISA 96-well plate overnight at 4 °C. Unbound fibronectin was washed with PBS three times, and unoccupied sites were blocked with 2% BSA in PBS (w/v) for 1 h at room temperature. The plate was washed briefly again with PBS three times. Fully reduced TG2 was added to each well at 10 nm in 50 mm Tris, 1 mm EDTA (pH 7.5) for 30 min at 37 °C. Excess protein was washed off with PBS three times. Oxidants were then added for up to 30 min at 37 °C, and the wells were washed three times with PBS. 200 μm 5BP was then added with 5 mm CaCl2 in Tris-EDTA buffer, and the plate was incubated at 37 °C for 3 h. 5BP is biotinylated primary amine that is a good substrate of TG2 and can therefore be cross-linked to Gln residues on extracellular matrix proteins by catalytically active extracellular TG2 (26). Following three washes with PBS, wells were fixed with 4% paraformaldehyde (PFA) solution for 10 min at room temperature, washed twice with PBS, and blocked overnight at 4 °C with 2% BSA in PBS (w/v). Streptavidin-HRP (1:2500, Biolegend) was diluted in blocking buffer and added to each well for 1 h at room temperature. Afterward, the plate was washed four times with PBS + 0.1% Tween 20 (v/v), and TMB was added. The reaction was monitored continuously using a Synergy HT plate reader (Biotek) at 655 nm for 30 min. Steady-state slopes were taken between 5 and 15 min. Slopes were background subtracted against conditions where no TG2 was included and normalized against a control condition where TG2 was not treated with oxidants.
In situ TG2 activity
Assaying extracellular TG2 activity in tissue culture has been described previously (26). Briefly, HUVECs were seeded into 48-well plates at a density of 20,000 cells/well at 70% confluence. Cells were fed every other day until they reached near-confluence. TRX was reduced for 1 h at room temperature using 10–20 m excess DTT, and buffer-exchanged into PBS using a Zeba 7000 MWCO desalting column. A 3-h incubation with 200 μm 5BP was used to probe biological activity of TG2. 25 μm ERW1041E, an irreversible, active site-directed TG2 inhibitor (34), was used as a negative control. After the 3-h incubation, cells were gently washed three times with sterile HBSS and fixed with 2% PFA for 5 min. After an additional two washes with PBS, the plate was blocked overnight at 4 °C with 5% BSA in PBS (w/v). Streptavidin-HRP (1:1000, Biolegend) was diluted in blocking buffer and added to the wells for a 1-h incubation at room temperature (protected from light). Four sets of washes with PBS + Tween 20 (0.1% v/v) were performed to remove excess and unbound streptavidin-HRP. TMB substrate was added, and the reaction was continuously monitored for 30 min. The steady-state slopes from 5 to 15 min were calculated and background subtracted against conditions where no 5BP was added and normalized against a control condition to yield relative TG2 activity.
Identification of disulfide bonds by mass spectrometry
Reduced wildtype TG2 was buffer-exchanged into 20 mm Tris buffer (pH 7.5) containing 1 mm EDTA and coincubated in equimolar ratios (2 μm each) with oxidized ERp57 for 0 or 30 min at 37 °C. The proteins were quenched with 15 mm IAM in Laemmli buffer. After a 30-min incubation at room temperature in the dark, samples were separated by non-reducing SDS-PAGE. Gel bands were visualized by silver stain, and bands corresponding to ERp57 and TG2 were excised using a clean razor blade. Gel slices were destained, washed with 25 mm NH4HCO3 in 50% acetonitrile, and dehydrated with 100% acetonitrile three times. 25 mm DTT in 50 mm NH4HCO3 was added to the gel fragments and incubated at 55 °C for 30 min. Following another round of dehydration with acetonitrile, the slices were washed with 25 mm NH4HCO3 in 50% acetonitrile three times, and submerged in 50 mm NH4HCO3 containing 15 mm IAA for 1 h at room temperature in the dark. After another three washes, gel slices were subjected to overnight trypsin (Promega) digestion at 37 °C. Peptides were lyophilized and reconstituted with 0.1% formic acid and analyzed by LC-MS/MS using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific).
Peptides harboring cysteine residues were identified by protein database searches against Mascot (51) and manually verified by analysis of the y fragment ions and predicted isotopic distribution using Xcalibur 2.2. Raw mass spectra were analyzed using MaxQuant version 1.6.0.16 (52) against a FASTA file containing the human proteome (UniProt) using a 1% false discovery rate for further validation of observed peptides.
siRNA knockdown of ERp57 gene expression
RNA interference was used to silence ERp57 gene expression. siRNA duplexes were obtained from Santa Cruz Biotechnology (catalog no. sc-35341) in a mixture of three 19–25-nucleotide-long duplexes. Control siRNA, which does not target any gene, was also obtained from Santa Cruz Biotechnology (catalog no. sc-37007). Transient gene silencing was achieved by transfecting HUVECs using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer's recommendations. Briefly, HUVECs were grown to 70% confluence in a 24-well glass bottom plate (catalog no. P24–1.5H-N, CellVis). On the morning of transfection, media were replaced with EGM-2 without antibiotics. RNAi duplexes were formed in Opti-MEM I for 20 min at room temperature using 1 μl of RNAiMAX reagent per well. Cells were incubated with siRNA for 48 h and assayed for protein expression by Western blotting or immunofluorescence.
Immunofluorescence microscopy
For colocalization studies, HUVECs were seeded at a density of 100,000 cells/ml in a 24-well glass-bottom plate (catalog no. P24-1.5H-N, CellVis) and grown to near confluence. Cells were washed briefly with HBSS and fixed with 2% PFA solution for 5 min at room temperature. Following two brief washes with PBS, wells were blocked overnight at 4 °C with 5% BSA diluted in PBS (w/v). Primary antibody was incubated overnight at 4 °C with rabbit anti-TG2 (custom, 1:2000), goat anti-fibronectin (Santa Cruz Biotechnology, C-20, 1:100), mouse anti-ERp57 (Santa Cruz Biotechnology, MaP.ERp57, 1:100), mouse anti-VE cadherin (Santa Cruz Biotechnology, F-8, 1:100), mouse anti-PDI (Thermo Fisher Scientific, RL-90, 1:100), mouse anti-ERp5 (Santa Cruz Biotechnology, G-5, 1:100), and/or mouse anti-ERp72 (Santa Cruz Biotechnology, B-4, 1:100). After three washes with PBS, Alexa-Fluor-conjugated secondary antibodies against mouse, rabbit, or goat (Thermo Fisher Scientific, 1:1000) were incubated for 1 h at room temperature. After four washes with PBS + 0.1% Tween 20 (v/v) and incubation with 300 nm 4,6-diamidino-2-phenylindole (5 min at room temperature on the penultimate wash), wells were kept in PBS, and stored at 4 °C until imaging on a Zeiss LSM780 confocal microscope with ZEN Black acquisition software. All images were taken using a ×40/1.3NA Zeiss EC Plan-Neofluar oil objective or ×20/0.8NA Zeiss Plan-Apochromat objective. Images were post-processed and analyzed using FIJI.
Western blot analysis
HUVEC lysates were obtained by lysing cells with RIPA buffer supplemented with protease inhibitors. Samples were then diluted in Laemmli buffer with or without β-mercaptoethanol. Reduced samples were heated at 95 °C for 15 min and cooled prior to loading onto a 4–20% mini-PROTEAN TGX pre-cast gel (Bio-Rad). Proteins were then transferred onto a nitrocellulose membrane using the Trans-Blot Turbo transfer system (Bio-Rad) and immediately blocked with 5% BSA/PBS + 0.1% Tween 20 overnight at 4 °C. Primary antibodies were incubated with their targets overnight at 4 °C at the manufacturer's recommended dilutions. Following three washes with PBS + Tween 20 (0.1%), membranes were incubated with HRP-conjugated secondary antibodies (1:2000, Biolegend) for 1 h at room temperature. After four washes with PBS + 0.1% Tween 20, blots were developed using the ECL2 substrate (Thermo Fisher Scientific), quenched with MilliQ water, and analyzed on a Typhoon 9500 scanner. Images were analyzed using ImageJ.
Statistical analyses
Student's t test were used, as described in the figure legends. All statistical analyses were performed using GraphPad Prism 7.0.
Author contributions
M. C. Y. and C. K. conceptualization; M. C. Y. data curation; M. C. Y. software; M. C. Y. formal analysis; M. C. Y. validation; M. C. Y., A. V. M., and J. A. O. investigation; M. C. Y. visualization; M. C. Y., A. V. M., and J. A. O. methodology; M. C. Y. writing-original draft; M. C. Y. and C. K. writing-review and editing; C. K. supervision; C. K. funding acquisition; C. K. project administration.
Supplementary Material
Acknowledgments
We thank Brad Palanski, Anwesha Goswami, and Chih-Hisang Weng for their assistance with various experiments. We also acknowledge Brad Palanski and Nicholas Plugis for helpful discussions and Josh Elias for access to an Orbitrap mass spectrometer.
This work was supported by National Institutes of Health Grant R01 DK063158 (to C. K.). C. K. is an advisor to and shareholder of Sitari Pharmaceuticals. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S8 and Table S1.
- TG2
- transglutaminase 2
- TRX
- thioredoxin-1
- HUVEC
- human umbilical vein endothelial cell
- PDI
- protein disulfide isomerase
- QSOX1
- quiescin sulfhydryl oxidase 1
- ER
- endoplasmic reticulum
- ECM
- extracellular matrix
- 5BP
- 5-biotinamidopentylamine
- IAM
- iodoacetamide
- IAA
- iodoacetic acid
- PFA
- paraformaldehyde
- PTM
- post-translational modifications
- HBSS
- Hanks' buffered saline solution
- Ni-NTA
- nickel-nitrilotriacetic acid
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- TMB
- 3,3′,5,5′-tetramethylbenzidine.
References
- 1. Scarpellini A., Germack R., Lortat-Jacob H., Muramatsu T., Billett E., Johnson T., and Verderio E. A. (2009) Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 284, 18411–18423 10.1074/jbc.M109.012948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zemskov E. A., Mikhailenko I., Hsia R.-C., Zaritskaya L., and Belkin A. M. (2011) Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS ONE 6, e19414 10.1371/journal.pone.0019414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adamczyk M., Griffiths R., Dewitt S., Kna V., and Aeschlimann D. (2015) P2X7 receptor activation regulates rapid unconventional export of transglutaminase-2. J. Cell Sci. 7, 4615–4628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu S., Cerione R. A., and Clardy J. (2002) Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. U.S.A. 99, 2743–2747 10.1073/pnas.042454899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinkas D. M., Strop P., Brunger A. T., and Khosla C. (2007) Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 5, e327 10.1371/journal.pbio.0050327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stamnaes J., Pinkas D. M., Fleckenstein B., Khosla C., and Sollid L. M. (2010) Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 285, 25402–25409 10.1074/jbc.M109.097162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin X., Stamnaes J., Klöck C., DiRaimondo T. R., Sollid L. M., and Khosla C. (2011) Activation of extracellular transglutaminase 2 by thioredoxin. J. Biol. Chem. 286, 37866–37873 10.1074/jbc.M111.287490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plugis N. M., Palanski B. A., Weng C. H., Albertelli M., and Khosla C. (2017) Thioredoxin-1 selectively activates transglutaminase 2 in the extracellular matrix of the small intestine: implications for celiac disease. J. Biol. Chem. 292, 2000–2008 10.1074/jbc.M116.767988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siegel M., Strnad P., Watts R. E., Choi K., Jabri B., Omary M. B., and Khosla C. (2008) Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE 3, e1861 10.1371/journal.pone.0001861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klöck C., Diraimondo T. R., and Khosla C. (2012) Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 34, 513–522 10.1007/s00281-012-0305-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong J. W., Ho S. Y., and Hogg P. J. (2011) Disulfide bond acquisition through eukaryotic protein evolution. Mol. Biol. Evol. 28, 327–334 10.1093/molbev/msq194 [DOI] [PubMed] [Google Scholar]
- 12. Thornton J. M. (1981) Disulphide bridges in globular proteins. J. Mol. Biol. 151, 261–287 10.1016/0022-2836(81)90515-5 [DOI] [PubMed] [Google Scholar]
- 13. Cook K. M., and Hogg P. J. (2013) Post-translational control of protein function by disulfide bond cleavage. Antioxid. Redox Signal. 18, 1987–2015 10.1089/ars.2012.4807 [DOI] [PubMed] [Google Scholar]
- 14. Yi M. C., and Khosla C. (2016) Thiol-disulfide exchange reactions in the mammalian extracellular environment. Annu. Rev. Chem. Biomol. Eng. 7, 197–222 10.1146/annurev-chembioeng-080615-033553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Telci D., Collighan R. J., Basaga H., and Griffin M. (2009) Increased TG2 expression can result in induction of transforming growth factor β1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J. Biol. Chem. 284, 29547–29558 10.1074/jbc.M109.041806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fisher M., Jones R. A., Huang L., Haylor J. L., El Nahas M., Griffin M., and Johnson T. S. (2009) Modulation of tissue transglutaminase in tubular epithelial cells alters extracellular matrix levels: a potential mechanism of tissue scarring. Matrix Biol. 28, 20–31 10.1016/j.matbio.2008.10.003 [DOI] [PubMed] [Google Scholar]
- 17. Antonyak M. A., Li B., Regan A. D., Feng Q., Dusaban S. S., and Cerione R. A. (2009) Tissue transglutaminase is an essential participant in the epidermal growth factor-stimulated signaling pathway leading to cancer cell migration and invasion. J. Biol. Chem. 284, 17914–17925 10.1074/jbc.M109.013037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones D. P., Go Y.-M., Anderson C. L., Ziegler T. R., Kinkade J. M. Jr., and Kirlin W. G. (2004) Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 18, 1246–1248 [DOI] [PubMed] [Google Scholar]
- 19. Turano C., Coppari S., Altieri F., and Ferraro A. (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J. Cell. Physiol. 193, 154–163 10.1002/jcp.10172 [DOI] [PubMed] [Google Scholar]
- 20. Schulman S., Bendapudi P., Sharda A., Chen V., Bellido-Martin L., Jasuja R., Furie B. C., Flaumenhaft R., and Furie B. (2016) Extracellular thiol isomerases and their role in thrombus formation. Antioxid. Redox Signal. 24, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gallina A., Hanley T. M., Mandel R., Trahey M., Broder C. C., Viglianti G. A., and Ryser H. J. (2002) Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem. 277, 50579–50588 10.1074/jbc.M204547200 [DOI] [PubMed] [Google Scholar]
- 22. Weisbart R. H. (1992) An antibody that binds a neutrophil membrane protein, ERp72, primes human neutrophils for enhanced oxidative metabolism in response to formyl-methionyl-leucyl-phenylalanine. Implications for ERp72 in the signal transduction pathway for neutrophil priming. J. Immunol. 148, 3958–3963 [PubMed] [Google Scholar]
- 23. Israel B. A., Jiang L., Gannon S. A., and Thorpe C. (2014) Disulfide bond generation in mammalian blood serum: detection and purification of quiescin-sulfhydryl oxidase. Free Radic. Biol. Med. 69, 129–135 10.1016/j.freeradbiomed.2014.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ilani T., Alon A., Grossman I., Horowitz B., Kartvelishvily E., Cohen S. R., and Fass D. (2013) A secreted disulfide catalyst controls extracellular matrix composition and function. Science 341, 74–76 10.1126/science.1238279 [DOI] [PubMed] [Google Scholar]
- 25. Nagy P. (2013) Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 18, 1623–1641 10.1089/ars.2012.4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DiRaimondo T. R., Plugis N. M., Jin X., and Khosla C. (2013) Selective inhibition of extracellular thioredoxin by asymmetric disulfides. J. Med. Chem. 56, 1301–1310 10.1021/jm301775s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lundström J., Krause G., and Holmgren A. (1992) A Pro to His mutation in active site of thioredoxin increases its disulfide-isomerase activity 10-fold: new refolding systems for reduced or randomly oxidized ribonuclease. J. Biol. Chem. 267, 9047–9052 [PubMed] [Google Scholar]
- 28. Wang Z., Perez M., Caja S., Melino G., Johnson T. S., Lindfors K., and Griffin M. (2013) A novel extracellular role for tissue transglutaminase in matrix-bound VEGF-mediated angiogenesis. Cell Death Dis. 4, e808 10.1038/cddis.2013.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Antonella Nadalutti C., Korponay-Szabo I. R., Kaukinen K., Wang Z., Griffin M., Mäki M., and Lindfors K. (2013) Thioredoxin is involved in endothelial cell extracellular transglutaminase 2 activation mediated by celiac disease patient IgA. PLoS ONE 8, e77277 10.1371/journal.pone.0077277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Araujo T. L., Zeidler J. D., Oliveira P. V., Dias M. H., Armelin H. A., and Laurindo F. R. (2017) Protein-disulfide isomerase externalization in endothelial cells follows classical and unconventional routes. Free Radic. Biol. Med. 103, 199–208 [DOI] [PubMed] [Google Scholar]
- 31. Passam F. H., Lin L., Gopal S., Stopa J. D., Bellido-Martin L., Huang M., Furie B. C., and Furie B. (2015) Both platelet- and endothelial cell-derived ERp5 support thrombus formation in a laser-induced mouse model of thrombosis. Blood. 125, 2276–2285 10.1182/blood-2013-12-547208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jasuja R., Furie B., and Furie B. C. (2007) Endothelial cell thiol isomerases: potential role in thrombus formation. Blood 110, 3709 [Google Scholar]
- 33. Holbrook L. M., Watkins N. A., Simmonds A. D., Jones C. I., Ouwehand W. H., and Gibbins J. M. (2010) Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br. J. Haematol. 148, 627–637 10.1111/j.1365-2141.2009.07994.x [DOI] [PubMed] [Google Scholar]
- 34. Watts R. E., Siegel M., and Khosla C. (2006) Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J. Med. Chem. 49, 7493–7501 10.1021/jm060839a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hang J., Zemskov E. A., Lorand L., and Belkin A. M. (2005) Identification of a novel recognition sequence for fibronectin within the NH2-terminal β-sandwich domain of tissue transglutaminase. J. Biol. Chem. 280, 23675–23683 10.1074/jbc.M503323200 [DOI] [PubMed] [Google Scholar]
- 36. Lundström J., and Holmgren A. (1990) Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. J. Biol. Chem. 265, 9114–9120 [PubMed] [Google Scholar]
- 37. Heckler E. J., Alon A., Fass D., and Thorpe C. (2008) Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry 47, 4955–4963 10.1021/bi702522q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mastroberardino P. G., Farrace M. G., Viti I., Pavone F., Fimia G. M., Melino G., Rodolfo C., and Piacentini M. (2006) “Tissue” transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim. Biophys. Acta 1757, 1357–1365 10.1016/j.bbabio.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 39. Malorni W., Farrace M. G., Matarrese P., Tinari A., Ciarlo L., Mousavi-Shafaei P., D'Eletto M., Di Giacomo G., Melino G., Palmieri L., Rodolfo C., and Piacentini M. (2009) The adenine nucleotide translocator 1 acts as a type 2 transglutaminase substrate: implications for mitochondrial-dependent apoptosis. Cell Death Differ. 16, 1480–1492 10.1038/cdd.2009.100 [DOI] [PubMed] [Google Scholar]
- 40. Coe H., Jung J., Groenendyk J., Prins D., and Michalak M. (2010) ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 285, 6725–6738 10.1074/jbc.M109.054015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dihazi H., Dihazi G. H., Bibi A., Eltoweissy M., Mueller C. A., Asif A. R., Rubel D., Vasko R., and Mueller G. A. (2013) Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J. Cell Sci. 126, 3649–3663 10.1242/jcs.125088 [DOI] [PubMed] [Google Scholar]
- 42. Gardella S., Andrei C., Ferrera D., Lotti L. V., Torrisi M. R., Bianchi M. E., and Rubartelli A. (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3, 995–1001 10.1093/embo-reports/kvf198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Venereau E., Casalgrandi M., Schiraldi M., Antoine D. J., Cattaneo A., De Marchis F., Liu J., Antonelli A., Preti A., Raeli L., Shams S. S., Yang H., Varani L., Andersson U., Tracey K. J., et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Gen. Physiol. 140, 1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gilbert H. F. (1989) Catalysis of thiol/disulfide exchange: single-turnover reduction of protein disulfide-isomerase by glutathione and catalysis of peptide disulfide reduction. Biochemistry 28, 7298–7305 10.1021/bi00444a023 [DOI] [PubMed] [Google Scholar]
- 45. Lyles M. M., and Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease a by protein-disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry. 30, 613–619 10.1021/bi00217a004 [DOI] [PubMed] [Google Scholar]
- 46. Bouziat R., Hinterleitner R., Brown J. J., Stencel-Baerenwald J. E., Ikizler M., Mayassi T., Meisel M., Kim S. M., Discepolo V., Pruijssers A. J., Ernest J. D., Iskarpatyoti J. A., Costes L. M., Lawrence I., Palanski B. A., et al. (2017) Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science. 356, 44–50 10.1126/science.aah5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang L., Haylor J. L., Hau Z., Jones R. A., Vickers M. E., Wagner B., Griffin M., Saint R. E., Coutts I. G., El Nahas A. M., and Johnson T. S. (2009) Transglutaminase inhibition ameliorates experimental diabetic nephropathy. Kidney Int. 76, 383–394 10.1038/ki.2009.230 [DOI] [PubMed] [Google Scholar]
- 48. DiRaimondo T. R., Klöck C., Warburton R., Herrera Z., Penumatsa K., Toksoz D., Hill N., Khosla C., and Fanburg B. (2014) Elevated transglutaminase 2 activity is associated with hypoxia-induced experimental pulmonary hypertension in mice. ACS Chem. Biol. 9, 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yi M. C., Palanski B. A., Quintero S. A., Plugis N. M., and Khosla C. (2015) An unprecedented dual antagonist and agonist of human transglutaminase 2. Bioorg. Med. Chem. Lett. 25, 4922–4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hashemy S. I., and Holmgren A. (2008) Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J. Biol. Chem. 283, 21890–21898 10.1074/jbc.M801047200 [DOI] [PubMed] [Google Scholar]
- 51. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data proteomics and 2-DE. Electrophoresis 20, 3551–3567 10.1002/(SICI)1522-2683(19991201)20:18%3C3551::AID-ELPS3551%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]
- 52. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry–based shotgun proteomics. Nat. Protoc. 11, 2301–2319 10.1038/nprot.2016.136 [DOI] [PubMed] [Google Scholar]
- 53. Watson W. H., Pohl J., Montfort W. R., Stuchlik O., Reed M. S., Powis G., and Jones D. P. (2003) Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 278, 33408–33415 10.1074/jbc.M211107200 [DOI] [PubMed] [Google Scholar]
- 54. Chambers J. E., Tavender T. J., Oka O. B., Warwood S., Knight D., and Bulleid N. J. (2010) The reduction potential of the active site disulfides of human protein-disulfide isomerase limits oxidation of the enzyme by Ero1α. J. Biol. Chem. 285, 29200–29207 10.1074/jbc.M110.156596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lundström J., and Holmgren A. (1993) Determination of the reduction—oxidation potential of the thioredoxin-like domains of protein disulfide-isomerase from the equilibrium with glutathione and thioredoxin. Biochemistry 32, 6649–6655 10.1021/bi00077a018 [DOI] [PubMed] [Google Scholar]
- 56. Araki K., Iemura S., Kamiya Y., Ron D., Kato K., Natsume T., and Nagata K. (2013) Ero1-α and pdis constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 202, 861–874 10.1083/jcb.201303027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frickel E. M., Frei P., Bouvier M., Stafford W. F., Helenius A., Glockshuber R., and Ellgaard L. (2004) ERp57 is a multifunctional thiol-disulfide oxidoreductase. J. Biol. Chem. 279, 18277–18287 10.1074/jbc.M314089200 [DOI] [PubMed] [Google Scholar]
- 58. Israel B. A., Kodali V. K., and Thorpe C. (2014) Going through the barrier: Coupled disulfide exchange reactions promote efficient catalysis in quiescin sulfhydryl oxidase. J. Biol. Chem. 289, 5274–5284 10.1074/jbc.M113.536219 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.