

Abstract
Most proteins that reside in the bacterial outer membrane (OM) have a distinctive “β-barrel” architecture, but the assembly of these proteins is poorly understood. The spontaneous assembly of OM proteins (OMPs) into pure lipid vesicles has been studied extensively but often requires non-physiological conditions and time scales and is strongly influenced by properties of the lipid bilayer, including surface charge, thickness, and fluidity. Furthermore, the membrane insertion of OMPs in vivo is catalyzed by a heterooligomer called the β-barrel assembly machinery (Bam) complex. To determine the role of lipids in the assembly of OMPs under more physiological conditions, we exploited an assay in which the Bam complex mediates their insertion into membrane vesicles. After reconstituting the Bam complex into vesicles that contain a variety of different synthetic lipids, we found that two model OMPs, EspP and OmpA, folded efficiently regardless of the lipid composition. Most notably, both proteins folded into membranes composed of a gel-phase lipid that mimics the rigid bacterial OM. Interestingly, we found that EspP, OmpA, and another model protein (OmpG) folded at significantly different rates and that an α-helix embedded inside the EspP β-barrel accelerates folding. Our results show that the Bam complex largely overcomes effects that lipids exert on OMP assembly and suggest that specific interactions between the Bam complex and an OMP influence its rate of folding.
Keywords: Gram-negative bacteria, membrane protein, molecular chaperone, outer membrane, protein folding, protein-lipid interaction, Bam complex, beta-barrel proteins
Introduction
Gram-negative bacteria, a class that includes many pathogenic and emerging antibiotic-resistant organisms, are bound by a double cell membrane. Most of the proteins that reside in the outer membrane (OM),2 which serves as a robust barrier, have a distinctive “β-barrel” architecture. A β-barrel is essentially a β-sheet rolled into a closed cylinder that has a hydrophobic exterior and a hydrophilic interior and is held together by hydrogen bonds between the first and last β-strands. The β-barrel scaffold is found exclusively in the OM of bacteria and organelles of bacterial origin. OM proteins (OMPs) mediate many different functions vital for bacterial survival and range considerably in size from 8 to 26 β-strands (1, 2). Some OMPs form oligomers (3), whereas others contain a segment that is embedded inside the β-barrel or a separately folded domain that is displayed on either the periplasmic or extracellular side of the OM (1).
OMPs must first traverse the inner membrane and the periplasmic space, which is devoid of ATP (4), before they attain their final structure in the OM. After OMPs are transported across the inner membrane in an unfolded conformation through the Sec translocon, a variety of periplasmic chaperones, including SurA, Skp, and DegP, prevent their aggregation in the periplasm (5–7). Integration into the OM is then catalyzed by the heterooligomeric β-barrel assembly machinery (Bam) complex (8, 9). In Escherichia coli, the Bam complex is composed of BamA, a β-barrel protein that contains five periplasmic polypeptide transport-associated (POTRA) domains and four lipoproteins (BamB–E) that bind to the POTRA domains (8–10). BamA and BamD are essential for cell viability and have homologs in all Gram-negative bacteria (8, 11, 12).
Although the structure of the Bam complex was recently solved by both X-ray crystallography and cryo-EM (13–16), the mechanism by which it catalyzes the integration of β-barrel proteins into the OM remains poorly understood. The complex has the shape of a top hat in which the BamA β-barrel forms the crown and the POTRA domains together with the lipoproteins form a brim (14, 15). Based on the structure, it has been proposed that the rotation of the ring-like structure formed by the periplasmic elements of the Bam complex leads to the opening of an unstable junction between the first and last β-strands of the BamA β-barrel (“lateral gate”) and thereby promotes the insertion of OMPs into the lipid bilayer in a stepwise fashion (14–16). The lipoprotein ring would create a protective environment that shields OMPs from repulsive interactions with periplasmic lipid headgroups. Consistent with this model, engineered disulfide bonds that prevent the opening of the putative lateral gate are lethal in E. coli (17). Disulfide locking of the BamA β-barrel, however, does not effectively block OMP assembly in vitro (16, 18, 19). Molecular dynamics simulations (20, 21) and other studies (22) that suggest that the BamA β-barrel causes thinning of the adjacent membrane have led to an alternative model in which the Bam complex catalyzes OMP assembly by creating a local deformation of the lipid bilayer. To further complicate matters, the finding that the absence of non-essential Bam complex subunits or reductions in the level of BamA or BamD differentially affects the assembly of individual OMPs (23, 24) raises the possibility that there are multiple parallel folding pathways that rely on the Bam complex components and periplasmic chaperones in different ways.
Independent of work on the Bam complex, insights into the biophysical and biochemical constraints on OMP assembly have emerged from studies in which the spontaneous folding of urea-denatured β-barrel proteins into pure lipid bilayers, such as large unilamellar vesicles (LUVs), has been analyzed (25–27). These experiments, however, often require non-physiological conditions, including high pH (26, 27), which may influence the net charge of the protein, and timescales of hours to days (26, 28). By comparison, OMPs typically assemble within 1 min in vivo (29–31). Nevertheless, the results suggest that the ability of OMPs to fold is encoded within their amino acid sequence. The results also indicate that properties of lipid bilayers, including fluidity (32, 33), thickness (33), and headgroup charge or type (34), strongly affect the folding of many different β-barrel proteins. Although individual OMPs differ in their propensity to fold in different lipid environments (26, 27), thin, fluid-phase bilayers with charge-neutral lipid headgroups (33) generally facilitate OMP folding, whereas naturally occurring zwitterionic or negatively charged lipid headgroups inhibit or sometimes even preclude folding altogether (26).
Given that the E. coli OM has an abundance of negatively charged lipids in its inner leaflet and a relatively rigid gel-like structure (35), it seems likely that the Bam complex either directly or indirectly attenuates the strong inhibitory properties of native bilayers (and possibly the net charge of OMPs) that have been observed in spontaneous folding experiments. Indeed, by promoting the transfer of client proteins from the pore of the BamA β-barrel into the lipid bilayer through a lateral gate, the Bam complex might prevent interactions with lipid headgroups. Alternatively, by distorting the lipid bilayer, the Bam complex might promote the integration of OMPs based on their intrinsic thermodynamic properties. Consistent with the possibility that the Bam complex directly overcomes inhibitory effects exerted by lipids, BamA alone accelerates the folding of at least some OMPs into liposomes containing small amounts of native headgroups (34, 36) as well as relatively thick bilayers that often inhibit spontaneous assembly (19). More significantly, in the presence of SurA, the Bam complex has been shown to catalyze the assembly of OMPs into proteoliposomes that contain native E. coli lipids near neutral pH and within roughly physiological timescales (37–40). A recent study, however, strongly suggested that the Bam complex is not uniformly distributed, but rather located predominantly in “islands” in the middle of the cell, where it promotes OMP biogenesis (41, 42). Because there is evidence for the existence of membrane domains that have distinct properties in bacteria (43, 44), this observation raises the possibility that the Bam complex also plays a more indirect role in the assembly of OMPs by targeting them to local membrane environments that have favorable lipid compositions.
In this study, we wished to determine the degree to which the lipid environment influences Bam complex–mediated assembly of OMPs. To this end, we first showed that the Bam complex can be efficiently reconstituted into proteoliposomes that contain a variety of synthetic lipids and lipid mixtures. The lipids were chosen to test the effect of headgroup charge, membrane thickness, and membrane fluidity on OMP folding. We then examined the assembly of three different model β-barrel proteins into each type of proteoliposome. Our results show that unlike the spontaneous assembly of OMPs into pure lipid vesicles, the assembly of OMPs catalyzed by the Bam complex is only very slightly affected by the lipid context. Interestingly, we found that the kinetics of assembly of the three model OMPs differed considerably. Taken together, our results indicate that specific interactions between individual OMPs and assembly factors (i.e. molecular chaperones and the Bam complex), as opposed to properties of the lipid bilayer, drive β-barrel assembly.
Results
Effective reconstitution of the Bam complex into synthetic lipid vesicles
Several studies have shown that the purified Bam complex and the molecular chaperone SurA are both necessary and sufficient to catalyze the assembly of at least a subset of denatured OMPs into proteoliposomes within minutes under near physiological conditions (37–40). Initially, the BamAB and BamCDE subcomplexes were produced separately and assembled into a holocomplex before reconstitution into lipid vesicles (38). A subsequent study showed that production of all of the subunits simultaneously generated a significantly more active Bam complex (40). In all of the published studies, however, only the assembly of OMPs into Bam complex-containing proteoliposomes that were generated using commercially available E. coli polar lipid extracts (which contain a variable mix of phospholipids) was examined.
To examine the influence of lipids on Bam complex–mediated OMP folding, we first needed to develop a method to generate proteoliposomes that are comparable despite containing different lipids. In the first step of our protocol, we expressed the Bam complex using a modified version of a plasmid that contains all of the Bam complex genes under the control of the trc promoter (40). The plasmid we used contains an extra copy of the gene encoding BamB that compensates for a variable loss of the protein from the Bam complex during purification that has been observed previously (13, 14). After isolating the Bam complex by affinity purification, we verified that all of the subunits were present in roughly equal amounts, reconstituted the heterooligomer into preformed lipid vesicles of chosen compositions using rapid dilution, and pelleted the resulting proteoliposomes. Chemical structures of the lipids used for Bam complex reconstitutions and their phase transition temperatures (Tm) are shown in Fig. 1A. We used two different assays to evaluate the folding and orientation of the reconstituted Bam complex. In one assay, we exploited the observation that properly folded BamA, like many other folded OMPs, migrates more rapidly than its predicted molecular weight on SDS-PAGE in the absence of heat (16, 39, 45). The so-called “heat modifiability” of BamA is only observed on gels that are run at 4 °C (36, 39). In the second assay, we evaluated the orientation of the Bam complex using a tryptic digest. Previous work has shown that when BamA molecules insert into liposomes in an inside-out orientation (i.e. with their POTRA domains exposed on the surface), trypsin treatment generates a ∼43-kDa species (BamA*) that corresponds to the transmembrane β-barrel (36). We also expected that trypsin would digest Bam complex lipoproteins that are bound to surface-exposed POTRA domains. Presumably, only the fraction of the Bam complex that is in an inside-out orientation can interact productively with SurA and exogenously added OMP substrates.
Figure 1.

Folded state and orientation of the Bam complex following reconstitution into proteoliposomes that contain distinct phospholipids. A, chemical structures of the synthetic lipids used in this study and their phase transition temperatures (Tm). B, proteoliposomes that were generated using the purified Bam complex and the indicated lipids were untreated, heated in SDS sample buffer, or treated with trypsin and subjected to SDS-PAGE at 4 °C. Proteins were visualized by Coomassie Blue staining. A trypsin-protected species that probably corresponds to the β barrel domain of BamA (BamA*) is highlighted. The small BamE protein was difficult to visualize on some of the gels and is not shown here.
To validate our methodology, we first generated proteoliposomes that contain E. coli polar lipids. Nearly all of the BamA was heat-modifiable (Fig. 1B, top leftmost gel, left and middle lanes). Furthermore, trypsin treatment fully converted BamA to BamA* and led to complete digestion of the lipoproteins (Fig. 1B, top leftmost gel, right lane). The results indicate that the vast majority of the Bam complexes integrated into the vesicles in the desired orientation and contained a correctly folded BamA subunit. Although a priori the Bam complex would not be predicted to exhibit a preferred orientation in the absence of an electrostatic potential, the combined bulk of the soluble components (the BamA POTRA domains and associated lipoproteins) would give the detergent-solubilized Bam complex a marked asymmetry and might favor an inside-out orientation during the reconstitution procedure.
We next reconstituted the Bam complex into liposomes containing seven different synthetic phospholipids and phospholipid combinations. A series of lipids were chosen with the phosphatidylcholine (PC) headgroup (DLPC (12:0), DMPC (14:0), DPPC (16:0), and POPC (16:0, 18:1)) to probe the effect of membrane thickness and fluidity on Bam complex-catalyzed OMP folding (Fig. 1A). Although the E. coli OM does not contain a significant quantity of these lipids, we used PC as a model zwitterionic (“neutral”) headgroup. Bilayers that contain the widely used model lipid POPC have about the same fluidity as those produced by E. coli polar lipid extracts (Tm = −2 °C) and about the same hydrophobic width as the E. coli OM (29 Å) (46). DPPC has similar length acyl chains, but because it has only saturated C–C bonds, it forms gel phase bilayers at physiological temperatures (Tm = 41 °C) that mimic the rigidity of the OM (35). DLPC and DMPC produce bilayers of 22 and 26 Å, respectively, that are thinner than native E. coli membranes and are fluid at physiological temperatures (47). The remaining liposomes contained a mixture of POPC and lipids with different headgroups but the same fatty acid tails (16:0, 18:1). One type of vesicle contained 80% POPE, a lipid that harbors a zwitterionic headgroup (phosphatidylethanolamine; PE) that is highly abundant in bacterial membranes. The other vesicles contained 20% POPG, a lipid that harbors the anionic headgroup phosphatidylglycerol (PG), or 20% of the anionic lipid cardiolipin (CL; 18:1 acyl chains). Although the lipid composition of E. coli membranes varies with growth conditions (48), the amount of PE or PG headgroups was chosen to match the upper limit of their concentration in the inner leaflet of the E. coli OM (49, 50). The E. coli OM typically contains ≤7% CL (49), but we used a higher concentration to maximize the probability than any effect of the lipid could be observed in our assays. To test the boundaries of our reconstitution method further, we also created proteoliposomes that contain a 30%/70% (mol/mol) mixture of the non-phospholipids palmitic acid (PA) and cholesterol. This mixture has previously been shown to form stable vesicles (51).
Heat-modifiability and trypsin accessibility assays showed that proteoliposomes generated using all of the synthetic phospholipids and phospholipid mixtures as well as the non-phospholipid mixture contained high levels (>90%) of properly folded BamA and Bam complexes in an inside-out orientation (Fig. 1B and Fig. S1A). Trypsin treatment often generated a small amount of several ∼8–30-kDa polypeptides in addition to BamA*. Western blot analysis suggested that these fragments were also derived from BamA (Fig. S2 (A and B), pre-assay). None of them were membrane-protected fragments of BamC, a subunit that has been suggested to be exposed on the cell surface (52) (Fig. S2C, pre-assay). Although we successfully constructed homogeneous proteoliposomes using all of the selected lipids, we nevertheless observed some variability in the efficiency of Bam complex reconstitution and the fraction of BamA that was properly folded, particularly when reconstitution reactions were scaled down. Most notably, we found that thinner bilayers and bilayers containing pure PC headgroups typically yielded the highest percentage of membrane-embedded Bam complex. As expected, proteoliposomes that contained a comparatively large amount of improperly folded BamA were much less active in OMP assembly assays (Fig. S3). After interrogating the folding of BamA and the orientation of the Bam complex, we discarded all defective proteoliposome preparations.
The Bam complex catalyzes efficient folding of EspP into a wide variety of lipid bilayers
Having established a method to optimize the generation of proteoliposomes that contain the Bam complex embedded in a variety of lipids, we next examined the assembly of several model E. coli OMPs. In these assays, proteoliposomes (normalized by the concentration of the Bam complex) were mixed with an excess of SurA and a urea-denatured substrate. Initially, we examined the assembly of EspP, a member of the autotransporter family of OMPs (53). Like other autotransporters, EspP has a surface-exposed N-terminal “passenger domain” and a 12-stranded C-terminal β-barrel (54, 55). The two domains are connected by a short α-helical linker that traverses the β-barrel (54–56). After the β-barrel is assembled and the passenger domain is translocated across the OM, the linker is severed in an autocatalytic intrabarrel cleavage reaction (55, 56). The Bam complex appears to promote the secretion of the passenger domain as well as the membrane insertion of the β-barrel, and the assembly of EspP into proteoliposomes that contain the Bam complex has been reconstituted in vitro (40). The passenger domain is secreted in a C- to N-terminal direction (57, 58), and even large N-terminal deletions do not affect protein biogenesis (31, 59). Because we wished to focus on the assembly of the EspP β-barrel, we used a ∼35-kDa truncated version of EspP that lacks most of the passenger domain (EspPΔ5, residues 998–1300) in our experiments. Assembly of EspPΔ5 can be assessed by monitoring the appearance of a ∼30-kDa C-terminal fragment that corresponds to the cleaved β-barrel on a Western blot (40).
The Bam complex catalyzed the efficient assembly of EspPΔ5 into all of the proteoliposomes made with synthetic lipids. The folding efficiency (based on the percentage folded in 30 min at 30 °C) was somewhat variable, but it was significantly higher when we used proteoliposomes composed entirely of lipids with PC headgroups instead of E. coli polar lipids (Fig. 2). Interestingly, the thickness of the membrane, which has been reported to exert a large effect on the folding of β-barrel proteins into pure lipid bilayers (27, 33, 60), only slightly affected the assembly of EspPΔ5. Whereas on average slightly more EspPΔ5 assembled into the thinnest bilayers containing DLPC than thicker bilayers containing DMPC or POPC, the spread of the data suggests that the difference is not highly significant (Fig. 2B). Remarkably, EspPΔ5 folded into proteoliposomes prepared with the gel phase lipid DPPC only slightly less efficiently than proteoliposomes prepared with the corresponding fluid phase lipid POPC. This is a striking result given that the folding of OMPs into pure gel phase liposomes has not been observed (25, 28, 32). We also found that EspPΔ5 folded slightly less efficiently into proteoliposomes prepared with a mixture of POPC and POPE, POPG, or CL than those prepared with POPC alone (Fig. 2B). Although our results suggest that naturally occurring lipid headgroups exert a modest inhibitory effect on OMP folding mediated by the Bam complex, in contrast, the spontaneous folding of many β-barrel proteins into pure lipid bilayers is strongly inhibited (or completely blocked) by the high levels of PE that are found in native membranes (26). Surprisingly, EspPΔ5 also folded efficiently into non-phospholipid proteoliposomes prepared with a mixture of PA and cholesterol (Fig. S1B). The folding of the protein into this highly unnatural environment shows that interactions between OMPs and specific phospholipids are not required for Bam complex-catalyzed assembly. It should also be noted that when we treated proteoliposomes with trypsin after performing an assembly reaction, we did not observe a protected fragment of BamC that might result from the transfer of the lipoprotein across the lipid bilayer during activation of the Bam complex (Fig. S2C, post-assay) (52).
Figure 2.

The Bam complex catalyzes the efficient assembly of EspPΔ5 into proteoliposomes that vary in lipid composition. A, urea-denatured EspPΔ5 was incubated with SurA and proteoliposomes that contained the Bam complex and the indicated lipids for 30 min at 30 °C. The folding/self-cleavage of the protein was monitored by Western blotting using an antiserum raised against a C-terminal EspP peptide. Representative results are shown. B, quantitation of the efficiency of EspPΔ5 folding into proteoliposomes that contain different lipids. Folding assays that used each type of proteoliposome were repeated at least five times. A diamond denotes the mean of each data set, and boxes indicate the 25th to 75th percentiles. Vertical lines indicate data points that lie outside of these percentiles and show the full range of folding efficiencies.
To obtain further insight into the effect of lipids on Bam complex–mediated OMP folding, we evaluated the kinetics of EspPΔ5 assembly into the different proteoliposomes we generated. To compensate for differences in folding efficiency, we defined the highest fraction of EspPΔ5 assembled in each experiment as 1.0. We then fit the data in each experiment to a single exponential to determine a rate constant, k, and characteristic timescale (the time required for 50% assembly, or t½), as in previous studies on OMP folding (26, 40). Although this first-order kinetic model may not capture all details involved in the folding process (e.g. intermediate folding states), it provides a convenient way to quantitatively compare Bam complex–mediated OMP assembly under different conditions. The t½ of assembly into proteoliposomes prepared using all four of the PC headgroup lipids was ∼2 min, regardless of membrane thickness or fluidity (Fig. 3A). A similar t½ was reported when the assembly of EspPΔ5 into proteoliposomes that contain E. coli polar lipids was examined (40). Despite the slight inhibitory effect of naturally occurring lipids on the maximum level of EspPΔ5 assembly (Fig. 2B), the rate of assembly into proteoliposomes that contain a mixture of POPC and POPE, POPG, or CL was also ∼2 min (Fig. 3B). The kinetic data suggest that although the Bam complex does not completely overcome any thermodynamic influence that the lipid headgroup imposes on OMP folding, its catalytic action remains unaltered in different lipid contexts. To confirm that changes in folding kinetics can be observed in our assay, we examined the assembly of EspPΔ5 into proteoliposomes made with DMPC at 25 and 37 °C instead of 30 °C. Presumably due to a reduction in thermal fluctuations, the rate of folding was slowed more than 2-fold at the lower temperature (Fig. S4). The rate of folding, however, did not change at 37 °C, presumably because there is a temperature threshold above which thermal fluctuations no longer have an impact.
Figure 3.

The lipid composition of proteoliposomes does not affect the kinetics of Bam complex–mediated EspPΔ5 assembly. Urea-denatured EspPΔ5 was incubated with SurA and proteoliposomes that contained the Bam complex and the indicated lipids at 30 °C, and the folding/self-cleavage of the protein after 0–30 min was monitored as described in the legend to Fig. 2. The effect of varying the thickness/fluidity of the bilayer (A) or the lipid headgroup (B) was examined. The data were normalized to the maximum fraction of the protein that underwent self-cleavage during each assembly reaction, which was defined as 1.0. Average values from multiple experiments (symbols) and S.D. values (error bars) at each time point are shown. The rate constant (k) and time required to reach 50% maximal assembly (t½) for each reaction were calculated from single-exponential fits. To facilitate comparisons, the kinetics of assembly of EspPΔ5 into proteoliposomes that contained POPC are shown in both A and B.
To confirm that the assembly of EspPΔ5 was catalyzed by the Bam complex, we examined the folding of the protein into pure lipid vesicles. We did not detect any assembly of EspPΔ5 into LUVs composed of POPC within 1 h under our standard assay conditions (pH 8, 30 °C) or at pH 10 (Fig. S5). No assembly was observed even in the presence of SurA, which might prevent misfolding and thereby increase the time window during which the protein remains insertion-competent. In an attempt to replicate conditions that promote the spontaneous folding of OMPs (26), we also examined the assembly of EspPΔ5 into highly curved small unilamellar vesicles composed of DLPC at pH 10. Appreciable levels of assembly could only be observed after several hours, however, and required the presence of SurA (data not shown).
The kinetics of Bam complex–mediated OMP assembly are substrate-specific
We next wished to examine the efficiency and kinetics with which the Bam complex mediates the folding of other OMPs in different lipid environments. Considerable variation in the spontaneous folding of β-barrel proteins into pure lipid vesicles has been reported (26, 27), but no correlation between protein sequence or structure and folding efficiency has been established. Furthermore, the rate at which the Bam complex catalyzes the assembly of OMPs other than EspP has not been measured directly. An enzyme assay that was used to obtain an indirect estimate of the folding kinetics of OmpT, a 10-stranded β-barrel protease, suggests that t½ ≈ 8 min (16, 38, 40). It is unclear, however, if the results underestimate the rate of assembly due to a lag in the diffusion or hydrolysis of the fluorogenic peptide substrate or the need to add lipopolysaccharide, which partially inhibits Bam complex–mediated folding of OmpT (37).
In one set of experiments, we monitored the ability of the Bam complex to catalyze the folding of OmpA, a model protein that has been studied extensively both in vivo and in vitro (25, 36). OmpA is composed of an N-terminal 8-stranded β-barrel and an independently folded C-terminal periplasmic domain. The Bam complex has been shown to mediate the assembly of up to 50% of the denatured OmpA added to proteoliposomes produced using E. coli polar lipid extracts, but the folding of the protein does not require SurA (39). Although we confirmed that OmpA folds efficiently (and slightly faster) without SurA (Fig. S6), we included the chaperone in all of our experiments to match the conditions that we used to examine the assembly of other OMPs. To assess the folding of OmpA, we exploited the observation that the folded form of the protein migrates relatively rapidly on SDS-PAGE in the absence of heat (25, 33, 45). After incubating unfolded OmpA with proteoliposomes and SurA, the fraction of the protein that was folded was determined by Western blotting. Consistent with previous studies (61–63), high-molecular-weight polypeptides that presumably correspond to oligomeric or aggregated forms of the protein were also detected but were not included in our calculations. We found that OmpA assembled into each type of proteoliposome more efficiently than EspPΔ5; >90% assembly was observed in some experiments (Fig. 4). The assembly of OmpA, like that of EspPΔ5, was highest in PC-based proteoliposomes, and folding efficiency was independent of membrane thickness (Fig. 4B). The presence of POPE, POPG, or CL likewise inhibited OmpA assembly slightly. Regardless of the lipid composition, however, OmpA appeared to fold into all of the proteoliposomes we tested at least as efficiently as it folds into proteoliposomes made with E. coli polar lipid extracts (39).
Figure 4.
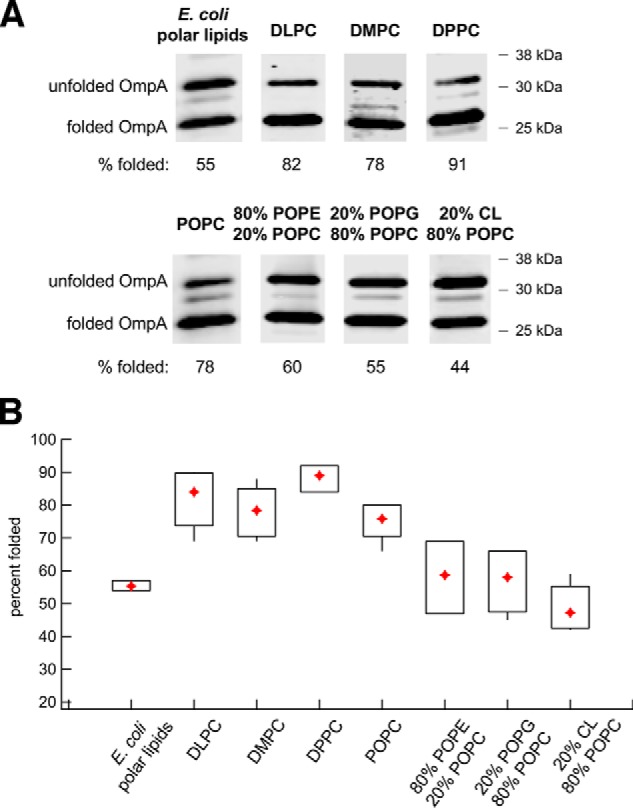
The Bam complex catalyzes the efficient assembly of OmpA into proteoliposomes that vary in lipid composition. A, urea-denatured OmpA was incubated with SurA and proteoliposomes that contained the Bam complex and the indicated lipids for 40 min at 30 °C. The folding of the protein was monitored by Western blotting using an anti-OmpA antiserum. Representative results are shown. B, quantitation of the efficiency of OmpA folding into proteoliposomes that contained different lipids. Folding assays that used each type of proteoliposome were repeated 3–6 times. A diamond denotes the mean of each data set, and boxes indicate the 25th to 75th percentiles. Vertical lines denote data points that lie outside these percentiles and show the full range of folding efficiencies.
Surprisingly, OmpA folded more slowly into a variety of proteoliposomes than EspPΔ5, although it assembled more efficiently and has a smaller β-barrel. The protein assembled into proteoliposomes that contained POPC, the gel-phase lipid DPPC, or mixtures of POPC and POPE, POPG, or CL with a t½ of ∼5 min (Fig. 5). The observation that OmpA folded more rapidly into proteoliposomes generated using the short chain lipids DLPC and DMPC (t½ = 2.0 and 3.3 min, respectively) suggests that membrane thickness does influence assembly kinetics. The range of assembly rates that we observed, however, is considerably smaller than the range that has been reported for the spontaneous folding of OmpA into pure lipid vesicles or vesicles that contain BamA alone (19, 33). Consistent with previous results, we found that OmpA assembled efficiently into pure POPC vesicles with a t½ of ∼30 min under our standard assay conditions in the presence or absence of SurA (Fig. S7A). In contrast, we did not observe significant assembly of OmpA into lipid vesicles that contained 80% POPE, 20% POPC within 90 min at either pH 8 or pH 10 even in the presence of SurA (Fig. S7B).
Figure 5.

Lipid headgroups modestly affect the kinetics of Bam complex–mediated OmpA assembly. Urea-denatured OmpA was incubated with SurA and proteoliposomes that contained the Bam complex and the indicated lipids at 30 °C, and the folding of the protein after 0–40 min was monitored as described in the legend to Fig. 4. The effect of varying the thickness/fluidity of the bilayer (A) or the lipid headgroup (B) was examined. The data were normalized to the maximum fraction of the protein that folded during each assembly reaction, which was defined as 1.0. Average values from multiple experiments (symbols) and S.D. values (error bars) at each time point are shown. The rate constant (k) and time required to reach 50% maximal assembly (t½) for each reaction were calculated from single-exponential fits. To facilitate comparisons, the kinetics of assembly of OmpA into proteoliposomes that contained POPC are shown in both A and B.
We next tested the assembly of the 14-stranded β-barrel protein OmpG by incubating the denatured protein with SurA and proteoliposomes containing the Bam complex. When SDS-PAGE was performed at 4 °C, a folded form of the protein that migrates slightly faster than the unfolded form was detected on Western blots (Fig. 6A). Although our anti-OmpG antiserum did not appear to recognize the folded form of the protein effectively in all experiments, we verified that the rapidly migrating species was integrated into proteoliposomes by demonstrating its resistance to proteinase K (PK) treatment and its heat-modifiability (Fig. S8A). OmpG folded into proteoliposomes that contain DLPC relatively slowly (t½ ≈ 11 min; Fig. 6B). Although we could not calculate a rate constant for the assembly of the protein into proteoliposomes that contain POPC, the observation that about half of the folding occurred between 10 and 15 min (Fig. 6A) suggests that the rate of assembly is similar.
Figure 6.

Bam complex–mediated assembly of OmpG is relatively slow. Urea-denatured OmpG was incubated with SurA and proteoliposomes that contained the Bam complex and either DLPC or POPC at 30 °C, and the folding of the protein after 0–60 min was monitored by Western blotting using an anti-OmpG antiserum. Representative results are shown in A. Molecular weight markers were run to the right of the 2.5-min time point on the top gel and have been deleted for clarity. The assembly of OmpG into proteoliposomes that contained DLPC was quantitated in B. The data were normalized to the maximum fraction of the protein that folded during each assembly reaction, which was defined as 1.0. Average values from multiple experiments (squares) and S.D. values (error bars) at each time point are shown. The time required to reach 50% maximal assembly (t½) was calculated from a single-exponential fit.
Taken together, the experiments described above indicate that the kinetics of Bam complex–mediated folding are substrate-specific. If proteoliposomes that contain a neutral, fluid bilayer of roughly physiological thickness (POPC) are used as a reference, EspPΔ5 is assembled ∼2.5 times faster than OmpA and ∼5 times faster than OmpG. Interestingly, the rate of assembly does not correlate with the number of β strands. EspPΔ5, the protein that folds the fastest, is unique in that it contains an α-helical segment embedded within the β-barrel that links it to the passenger domain (53, 55). Based on evidence that the linker is embedded inside a partially folded form of the EspP β-barrel in the periplasm and is essential for assembly in vivo (64, 65), we hypothesized that the α-helix nucleates the folding of the EspP barrel and enables it to fold significantly more rapidly than similar empty β-barrel proteins. To test this idea, we deleted the α-helix from EspPΔ5 and examined the assembly of the resulting EspP derivative (EspPΔ6, residues 1033–1300). EspPΔ6 does not undergo self-cleavage because it lacks the linker segment, but when SDS-PAGE is performed at 4 °C, a rapidly migrating polypeptide that corresponds to the folded form of the protein, based on its resistance to PK treatment and heat modifiability, can be detected (Fig. 7A and Fig. S8B). Consistent with our hypothesis, we found that EspPΔ5 folds into proteoliposomes that contain the Bam complex and POPC about 3 times faster than EspPΔ6 (Fig. 7B).
Figure 7.

Deletion of an embedded α-helix delays the assembly of the EspP β barrel. A, urea-denatured EspPΔ6 was incubated with SurA and proteoliposomes that contained the Bam complex and POPC at 30 °C, and the folding of the protein after 0–30 min was monitored by Western blotting as described in the legend to Fig. 2. Molecular weight markers were run to the right of the 0-min time point and have been deleted for clarity. B, quantitation of the results shown in A. The data were normalized to the maximum fraction of the protein that folded, which was defined as 1.0. The rate constant (k) and time required to reach 50% maximal assembly (t½) were calculated from a single-exponential fit. The assembly of EspPΔ5 into proteoliposomes that contain POPC (Fig. 3) is replotted for comparison.
Finally, we examined the effect of the periplasmic domain on OmpA folding. Previous work has shown that the deletion of this domain, which folds as an independent entity, does not significantly affect the assembly kinetics of the transmembrane β-barrel into pure PC lipid bilayers (34). As expected, we found that a truncated version of OmpA that contains only the β-barrel domain (TM-OmpA, residues 22–195) folds into proteoliposomes that contain the Bam complex and POPC at essentially the same rate as full-length OmpA (Fig. S6). Inconsistencies in the folding of TM-OmpA into other proteoliposomes that might be due to abnormal intraprotein interactions in the urea-denatured state (63) precluded a systematic study of lipid effects on kinetics. Nevertheless, the results suggest that, unlike the EspP α-helix, the OmpA periplasmic domain does not facilitate assembly of the covalently linked β-barrel domain.
Discussion
In this study, we present evidence that membrane lipids do not play a major role in Bam complex–mediated assembly of bacterial OMPs. We found that the Bam complex together with SurA catalyzed the assembly of a large fraction of two different model OMPs (EspPΔ5 and OmpA) into proteoliposomes that varied considerably in chemical composition, thickness, and fluidity at near physiological pH and timescales. The assembly of a third model protein that folded less efficiently (OmpG) also did not appear to be significantly affected by membrane thickness. Presumably, the differences in assembly efficiency that we observed were influenced by the ability of each OMP to remain assembly-competent following its dilution from urea. Although the presence of native lipid headgroups slightly affected the efficiency of assembly, we never observed the dramatic inhibitory effects that have been reported in studies that examined the spontaneous folding of OMPs into pure lipid vesicles (26, 34). Whereas the presence of a physiological concentration of PE decreased the efficiency of Bam complex–mediated folding of EspPΔ5 and OmpA no more than ∼1.5–2-fold, the same amount of PE completely blocks the assembly of many OMPs into LUVs (26). It is conceivable that the Bam complex simply accelerates the kinetics of OMP assembly and thereby prevents off-pathway reactions (e.g. aggregation) that account for the low efficiency of folding into LUVs. Our finding that the presence of SurA did not increase the efficiency of EspPΔ5 and OmpA assembly into LUVs (Figs. S5 and S7), however, suggests that prolonging the insertion competence of OMPs is not sufficient to enhance their folding in the absence of the Bam complex. The need to use non-physiological conditions, such as high pH, to favor folding over aggregation (26, 27) may at least partly account for the sensitivity of OMPs to the lipid environment seen in spontaneous folding experiments. Indeed, the net charge of OMPs at high pH may produce non-native folding intermediates and/or affect the interaction of the proteins with membrane lipids. Although we were able to examine OMP assembly under more physiological conditions, it should be noted that the proteoliposomes we generated do not reproduce the asymmetrical nature of the bacterial OM and that we cannot exclude the possibility that lipopolysaccharide located in the outer leaflet influences the fate of OMPs in vivo.
Our results are consistent with the notion that the Bam complex shields incoming OMPs from the membrane surface and largely offsets inhibitory effects imposed by native PE and other, predominantly anionic, lipid headgroups. Previous studies have shown that BamA alone accelerates the folding of OmpA and other empty β-barrel proteins, including OmpX and OmpLA, into LUVs that contain PG or a small amount of PE (20%), but only at high pH (34, 36). Furthermore, even in the presence of BamA, the assembly of OMPs into liposomes composed purely of neutral PC lipids was faster and more efficient, and BamA relieved the inhibitory effect of lipid headgroups on the assembly of some proteins more effectively than others (34). Thus, whereas BamA alone can catalyze limited OMP assembly, our results imply that the Bam complex lipoproteins play an important role in promoting folding in a native lipid context. Although evidence that the BamA POTRA domains interact with negatively charged lipids (21) and that BamE binds to PG (13, 67) suggests that the recruitment of specific lipids stabilizes lipoprotein binding or facilitates OMP folding, our finding that the lipid environment does not significantly affect OMP assembly challenges the hypothesis that the Bam complex functions by exploiting the anionic membrane surface. Likewise, our results indicate that CL, which appears to play a stimulatory role in the assembly of mitochondrial β-barrels (68), does not facilitate Bam complex–mediated folding. Given that the lipid composition of bacterial membranes is highly diverse (69), it might indeed be surprising if specific lipids were required for efficient Bam complex function. Finally, our results are inconsistent with the idea that the Bam complex localizes to specific regions of the OM to steer incoming OMPs to regions that have a favorable lipid composition.
Whereas the lipid composition of the proteoliposomes did not strongly influence folding kinetics, we observed considerable variation in the characteristic rate of assembly of each model protein we examined. Interestingly, there was no strict correlation between the number of β-strands in a given model protein and the kinetics of folding. The assembly of the 8-stranded OmpA β-barrel into “reference” bilayers composed of the neutral lipid POPC was consistently slower than the assembly of the 12-stranded EspPΔ5 β-barrel. This observation would seem to be compatible with a simple stepwise assembly model only if individual β-strands (or hairpins formed by two β-strands) are inserted into the lipid bilayer at significantly different rates. Of course, a wide variety of factors including the rate at which unfolded OMPs reach an insertion-competent conformation, the duration of interactions with SurA and the Bam complex, and differences in the mechanism of insertion, might affect assembly kinetics. The finding that the removal of the α-helix embedded inside the EspP β-barrel significantly slows assembly is consistent with other evidence (55, 64, 65) that this segment primes folding or at least enhances the acquisition of an insertion-competent state. Many large β-barrels (>18 strands) contain embedded segments that stabilize the protein in vivo, and it is conceivable that these segments likewise speed up an otherwise slow assembly process. Embedded segments might not only accelerate the folding of β-barrel structures, but may also reduce the magnitude of the defect in the lipid bilayer that the Bam complex needs to create to catalyze insertion. In this regard, it is striking that OmpA, but not EspPΔ5, was assembled ∼2-fold more rapidly into proteoliposomes made from short-chain lipids than those composed of long-chain lipids. This observation raises the possibility that the insertion of the unfolded OmpA β-barrel into unnaturally thin bilayers is faster because the Bam complex perturbs the membrane more readily but that the insertion of the partially folded EspPΔ5 β-barrel is less dependent on the extent of membrane distortion and is efficient regardless of the properties of the lipid bilayer.
Finally, we show that the purified Bam complex mediates the highly efficient assembly of OMPs into a wide variety of different lipid environments, including a non-phospholipid bilayer, in a robust and versatile in vitro assay. Because proteoliposomes that are generated using synthetic lipids are better defined than those generated using mixtures of polar lipids extracted from bacteria, in principle they should provide more reproducible results. Proteoliposomes that contain synthetic lipids may also have practical utility for experiments in which it is desirable or necessary to use bilayers that have a specific lipid composition to study the assembly of an OMP of interest. Furthermore, the ability to choose the membrane environment should facilitate examination of the hypothesis that the function of periplasmic chaperones, such as Skp and DegP, requires interactions with specific lipid headgroups (36, 70).
Experimental procedures
Plasmid construction
Plasmid pYG120 (pTRC-bamABCDE8His-bamB) and plasmids that encode His-tagged SurA and EspPΔ5 have been described (14, 40). To generate a plasmid that encodes OmpG (pJH207), the ompG gene was amplified by PCR using the oligonucleotides 5′-GAATGGCCTGCGCCATGGCCGAGGAAAGGAACG-3′ and 5′-GATAAAGTACTCGCGCCGGATCCGGCAA-3′ and genomic DNA from E. coli strain MC4100 as a template. The resulting PCR product was digested with NcoI and BamHI and cloned into the cognate sites of pET28b. To construct a plasmid that encodes EspPΔ6, a C-terminal fragment of espP was amplified by PCR using the oligonucleotides 5′-GCCTGAACAAACGTATGGGTCATATGCGTGATATC-3′ and 5′-CTCTCATCCGCCAAAACAGCCAAG-3′ and pRLS11 (71) as a template. The resulting PCR product was digested with NdeI and BamHI and cloned into the cognate sites of pET28b. Plasmids encoding OmpA and TM-OmpA were constructed using the Gibson assembly method (72). Oligonucleotide primers were designed using the NEBuilder web tool (nebuilder.neb.com).3 The OmpA insert was generated by PCR using the forward primer 5′-CTTTAAGAAGGAGATATACCATGGCTCCGAAAGATAACACCTG-3′, the reverse primer 5′-CGGAGCTCGAATTCGGATCCTTAAGCCTGCGGCTGAGT-3′, and MC4100 genomic DNA as a template. The TM-OmpA insert was generated using the same forward primer and the reverse primer 5′-CGGAGCTCGAATTCGGATCCTTATTCGCCCTGACCGAAACG-3′. pET28b digested with NcoI and BamHI and a 3-fold molar excess of each insert was then added to Gibson assembly master mix (New England Biolabs) and incubated at 50 °C for 1 h.
Expression and purification of BamABCDE
The Bam complex was expressed and purified by modifying a protocol described previously (40, 66). BL21(DE3) transformed with pYG120 was grown overnight at 37 °C in 10 ml of lysogeny broth (LB) containing 100 μg/ml ampicillin. Cells were then pelleted (2500 × g, 10 min, 4 °C), washed, and inoculated into 1 liter of fresh LB containing 100 μg/ml ampicillin. The culture was grown at 37 °C to OD600 = 0.8, at which point 0.4 mm isopropyl 1-thio-β-d-galactopyranoside was added. After 2.5 h, cells were harvested by centrifugation (4000 × g, 15 min, 4 °C). The cell pellet was then resuspended in 50 ml of cold PBS, pH 7.4 (KD Medical), and lysed immediately or flash-frozen in liquid N2 and stored at −80 °C. Cells were lysed at 4 °C using an Avestin Emulsiflex C3 at 15,000 p.s.i. or a Constant Systems cell disrupter at 30,000 p.s.i. The cell lysate was then clarified by centrifugation (6000 × g, 15 min, 4 °C), and the resulting supernatant was centrifuged in a Type70 Ti rotor (58,000 rpm, 40 min, 4 °C) to obtain cell membranes. The membrane pellet was resuspended in 5–10 ml of buffer A (PBS, 1% n-dodecyl-β-maltoside (DDM) (Anatrace), 37 mm imidazole), thoroughly dispersed using a Dounce homogenizer, and agitated at 4 °C for 2 h. Insoluble material was then pelleted in a Beckmann TLA100.4 rotor (70,000 rpm, 30 min, 4 °C), and soluble protein was added to 1 ml of Ni-NTA resin (Qiagen) equilibrated with buffer B (PBS, 0.03% DDM, 37 mm imidazole). After adding buffer B to increase the volume to 30 ml, the protein/Ni-NTA solution was incubated with shaking at 4 °C for 2 h. The Ni-NTA beads were pelleted (2000 × g, 2 min, 4 °C); washed four times with 10–15 bed volumes of PBS, 0.03% DDM, 50 mm imidazole; and poured into a column. The Bam complex was then eluted with PBS, 0.03% DDM, 500 mm imidazole in eight 500-μl fractions. The fractions that contained the most highly purified Bam complex proteins in stoichiometric ratios were then pooled and passed through a PD-10 desalting column (GE Healthcare) equilibrated with 20 mm Tris, pH 8.0, 0.03% DDM to remove the imidazole and exchange the buffer. The concentration of purified BamABCDE was estimated by measuring A280 (ϵ = 294,630 m−1 cm−1), and the protein was concentrated using a 4-ml Amicon 30,000-kDa cutoff filter (Millipore). The final protein concentration was verified using the Bio-Rad DC protein assay and subsequently adjusted to 20 μm. In some cases, the Bam complex was purified from up to four 1-liter cultures, and all of the volumes were scaled up accordingly.
Reconstitution of BamABCDE into synthetic lipid bilayers and analysis of proteoliposome quality
E. coli polar lipid extracts and the synthetic phospholipids used in this study were obtained from Avanti Polar Lipids in chloroform solution. PA and cholesterol were obtained from Sigma as dry powders and dissolved in chloroform before mixing in a 30%/70% (mol/mol) ratio (51). To generate lipid vesicles, an appropriate lipid or lipid mixture was first placed into a glass tube, and the solvent evaporated under a stream of N2. The lipids were then dried in a vacuum desiccator overnight and hydrated to a final concentration of 8 mg/ml. To obtain a homogeneous solution, lipids were incubated for 1 h at room temperature (except for DMPC and DPPC, which were incubated at 42 °C) with occasional vortexing. Hydrated lipids were kept above their transition temperatures during the remaining steps of the reconstitution. The lipids were then clarified by sonication in a Branson bath sonicator, centrifuged (3500 × g, 5 min, 4 °C) to remove aggregates, and extruded through 100-nm filters using an Avanti mini-extruder.
The Bam complex was reconstituted into lipid vesicles by rapid dilution essentially as described (38, 40, 66). Reconstitutions into most liposomes were performed at 4 °C, but reconstitutions into DMPC and DPPC liposomes were performed at 25 and 42 °C, respectively. Immediately before the reconstitution reactions, both the liposomes and the Bam complex in detergent solution were spun at 12,000 × g to remove any aggregates. Reconstitutions were performed using 0.2 mg/ml lipids and 1 μm BamABCDE. After the dilution step, the reconstitution reactions were incubated at the same temperature for 45 min with shaking and then centrifuged (3500 × g, 10 min) to remove aggregates. Subsequently, the proteoliposomes were pelleted in a TLA100.4 rotor (85,000 rpm, 65 min, 4 °C) and resuspended in 100–200 μl of 20 mm Tris, pH 8.0. The proteoliposome samples were flash-frozen in liquid N2 and stored at −80 °C.
The reconstitution efficiency (i.e. the fraction of the protein that was added to a reconstitution reaction that integrated into lipid vesicles) and the final concentration of the Bam complex in the proteoliposomes were determined by SDS-PAGE and Coomassie Blue staining. To evaluate the folding of BamA after reconstitution, proteoliposome aliquots that were heated in SDS-PAGE sample buffer at 95 °C for 5 min or unheated were run on 8–16% Tris minigels (Invitrogen) at 150 V for 110 min at 4 °C (39). The orientation of the Bam complex was determined by incubating proteoliposomes in 20 mm Tris, pH 8.0, with 0.2 mg/ml trypsin for 15 min at 37 °C (36). Trypsin-treated samples were heated at 95 °C for 5 min before SDS-PAGE. Western blotting was performed using rabbit polyclonal antisera generated against BamA and BamC to determine whether tryptic fragments were derived from those proteins. To make the anti-BamA antiserum, membranes were isolated from cells that overproduced BamA, and membrane proteins were resolved by SDS-PAGE. The band containing BamA was then excised and used for immunizations. To make the anti-BamC antiserum, the BamCDE subcomplex was first purified essentially as described (38), and the three proteins were resolved by SDS-PAGE. The band containing BamC was then excised and used for immunizations.
Expression and purification of SurA and OMPs
His-tagged SurA was expressed and purified as described (38, 40). Likewise, OMP substrates that lacked signal peptides were expressed and purified from inclusion bodies essentially as described (40) except that OmpA and TM-OmpA were produced from 500-ml cultures. In brief, OMP expression was induced when cultures reached A600 = 0.7 by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. After 3 h, cells were harvested, resuspended in 10 ml of cold PBS, and lysed as described above (see Bam complex purification). Inclusion bodies were obtained by subjecting cell lysates to centrifugation (6000 × g, 15 min, 4 °C). The inclusion bodies were then washed twice with PBS, resuspended in 3.5–5 ml of 8 m urea, and incubated at room temperature for 1 h with gentle rocking. Insoluble material was removed by centrifugation in a TLA100.4 rotor (26,000 rpm, 20 min, 4 °C). Protein concentrations were then determined using the Bio-Rad DC protein assay.
OMP assembly assays and data analysis
As described previously (40), OMP assembly reactions were prepared by sequentially adding 2 μm SurA, 0.2 μm OMP substrate (diluted from a 6 μm stock in 8 m urea), and 0.5 μm proteoliposomes containing the Bam complex to 20 mm Tris, pH 8.0. Proteoliposomes were briefly sonicated before use. Unless otherwise noted, assembly reactions were performed at 30 °C. Aliquots were removed at appropriate time points and placed on ice, and the reactions were quenched by the addition of an equal volume of 2× SDS-PAGE sample buffer. To examine the assembly of OMPs into pure lipid vesicles, LUVs consisting of POPC or 80% POPE, 20% POPC were extruded through 100-nm filters as described above and added to assembly reactions at a final concentration of 1.3 mg/ml. Although samples containing EspPΔ5 were heated at 95 °C for 5 min before SDS-PAGE, the other samples were not heated so that the heat-labile (“heat-modifiable”) folded forms of the OMPs could be detected. In experiments that examined the assembly of OmpG and EspPΔ6, SDS-PAGE was performed at 4 °C as described above. To verify membrane integration, OMP assembly reactions were performed for 30 min, and samples were then incubated with 4 μg/ml PK for 5 min on ice. Following SDS-PAGE, OMPs were visualized by Western blotting using an appropriate rabbit polyclonal antiserum. An antiserum generated against a C-terminal peptide of EspP (71) was used to detect EspPΔ5 and EspPΔ6. Full-length OmpA was detected using an antiserum obtained from P. C. Tai, and TM-OmpA was detected using an antiserum generated against the peptide NH2-CQWTNNIGDAHTIGTRPDNG-COOH. OmpG was detected using an antiserum generated against the full-length protein. A portion of the OmpG produced for assembly assays (see above) was subjected to SDS-PAGE, and a band containing the protein was excised and used for immunizations. An IRDye 680–conjugated goat anti-rabbit antiserum was used to visualize all antibody-antigen complexes using an Odyssey infrared imager (LI-COR).
To analyze the results, folded and unfolded forms of the OMPs that were detected on Western blots were quantified using ImageJ software. The quantification procedure was validated by verifying that multiple runs of the same sample gave similar results. The fraction folded was defined as the intensity of the folded band/(the intensity of the folded and unfolded bands). We note that although this formula does not account for possible differences in the efficiency with which the antisera bind to folded and unfolded forms of a given protein, it can still be used to compare the results of independent experiments. Kinetic folding assays were fit to a single-exponential model, and the values k and t½ (ln2/k) were determined using Igor Pro software. To compare experiments in which different folding efficiencies were observed, the values were normalized to the maximum fraction folded, which was determined by extrapolating the single-exponential fit to infinite time. The error bars shown on the kinetic folding assay curves were obtained by averaging data from a minimum of three experiments and calculating the S.D. for each time point.
Author contributions
S.H. and H.D.B. conceptualization; S.H. formal analysis; S.H. and H.D.B. validation; S.H. investigation; S.H. methodology; S.H. and H.D.B. writing-original draft; H.D.B. supervision; H.D.B. funding acquisition.
Supplementary Material
Acknowledgments
We thank Changjiang Dong for providing plasmid pYG120 and Janine Peterson for expressing and purifying urea-denatured OmpG and EspPΔ6. We also thank members of the Bernstein laboratory for providing helpful comments on the manuscript.
This work was supported by the Intramural Research Program of NIDDK, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- OM
- outer membrane
- OMP
- outer membrane protein
- Bam
- β-barrel assembly machinery
- POTRA domain
- polypeptide transport-associated domain
- LUV
- large unilamellar vesicle
- Tm
- phase transition temperature
- PC
- phosphatidylcholine
- DLPC
- dilauroyl-sn-phosphatidylcholine
- DMPC
- dimyristoyl-sn-phosphatidylcholine
- DPPC
- 1,2-dipalmitoyl-sn-phosphatidylcholine
- POPC
- 1-palmitoyl-2-oleoyl-sn-phosphatidylcholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-phosphatidylethanolamine
- POPG
- 1-palmitoyl-2-oleoyl-sn-phosphatidylglycerol
- PE
- phosphatidylethanolamine
- PG
- phosphatidylglycerol
- CL
- cardiolipin
- PA
- palmitic acid
- PK
- proteinase K
- DDM
- n-dodecyl-β-d-maltopyranoside
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Fairman J. W., Noinaj N., and Buchanan S. K. (2011) The structural biology of β-barrel membrane proteins: a summary of recent reports. Curr. Opin. Struct. Biol. 21, 523–531 10.1016/j.sbi.2011.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rollauer S. E., Sooreshjani M. A., Noinaj N., and Buchanan S. K. (2015) Outer membrane protein biogenesis in Gram-negative bacteria. Philos. Trans. R. Soc. B Biol. Sci. 370, 20150023 10.1098/rstb.2015.0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koebnik R., Locher K. P., and Van Gelder P. (2000) Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 37, 239–253 10.1046/j.1365-2958.2000.01983.x [DOI] [PubMed] [Google Scholar]
- 4. Wülfing C., and Plückthun A. (1994) Protein folding in the periplasm of Escherichia coli. Mol. Microbiol. 12, 685–692 10.1111/j.1365-2958.1994.tb01056.x [DOI] [PubMed] [Google Scholar]
- 5. Schäfer U., Beck K., and Müller M. (1999) Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J. Biol. Chem. 274, 24567–24574 10.1074/jbc.274.35.24567 [DOI] [PubMed] [Google Scholar]
- 6. Rouvière P. E., and Gross C. A. (1996) SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10, 3170–3182 10.1101/gad.10.24.3170 [DOI] [PubMed] [Google Scholar]
- 7. Misra R., CastilloKeller M., and Deng M. (2000) Overexpression of protease-deficient DegPS210A rescues the lethal phenotype of Escherichia coli OmpF assembly mutants in a degP background. J. Bacteriol. 182, 4882–4888 10.1128/JB.182.17.4882-4888.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T. J., and Kahne D. (2005) Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245 10.1016/j.cell.2005.02.015 [DOI] [PubMed] [Google Scholar]
- 9. Voulhoux R., Bos M. P., Geurtsen J., Mols M., and Tommassen J. (2003) Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265 10.1126/science.1078973 [DOI] [PubMed] [Google Scholar]
- 10. Sklar J. G., Wu T., Gronenberg L. S., Malinverni J. C., Kahne D., and Silhavy T. J. (2007) Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 104, 6400–6405 10.1073/pnas.0701579104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malinverni J. C., Werner J., Kim S., Sklar J. G., Kahne D., Misra R., and Silhavy T. J. (2006) YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol. Microbiol. 61, 151–164 10.1111/j.1365-2958.2006.05211.x [DOI] [PubMed] [Google Scholar]
- 12. Webb C. T., Heinz E., and Lithgow T. (2012) Evolution of the β-barrel assembly machinery. Trends Microbiol. 20, 612–620 10.1016/j.tim.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 13. Bakelar J., Buchanan S. K., and Noinaj N. (2016) The structure of the β-barrel assembly machinery complex. Science 351, 180–186 10.1126/science.aad3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gu Y., Li H., Dong H., Zeng Y., Zhang Z., Paterson N. G., Stansfeld P. J., Wang Z., Zhang Y., Wang W., and Dong C. (2016) Structural basis of outer membrane protein insertion by the BAM complex. Nature 531, 64–69 10.1038/nature17199 [DOI] [PubMed] [Google Scholar]
- 15. Han L., Zheng J., Wang Y., Yang X., Liu Y., Sun C., Cao B., Zhou H., Ni D., Lou J., Zhao Y., and Huang Y. (2016) Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat. Struct. Mol. Biol. 23, 192–196 10.1038/nsmb.3181 [DOI] [PubMed] [Google Scholar]
- 16. Iadanza M. G., Higgins A. J., Schiffrin B., Calabrese A. N., Brockwell D. J., Ashcroft A. E., Radford S. E., and Ranson N. A. (2016) Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat. Commun. 7, 12865 10.1038/ncomms12865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noinaj N., Kuszak A. J., Balusek C., Gumbart J. C., and Buchanan S. K. (2014) Lateral opening and exit pore formation are required for BamA function. Structure 22, 1055–1062 10.1016/j.str.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doerner P. A., and Sousa M. C. (2017) Extreme dynamics in the BamA β-barrel seam. Biochemistry 56, 3142–3149 10.1021/acs.biochem.7b00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schiffrin B., Calabrese A. N., Higgins A. J., Humes J. R., Ashcroft A. E., Kalli A. C., Brockwell D. J., and Radford S. E. (2017) Effects of periplasmic chaperones and membrane thickness on BamA-catalysed outer membrane protein folding. J. Mol. Biol. 429, 3776–3792 10.1016/j.jmb.2017.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noinaj N., Kuszak A. J., Gumbart J. C., Lukacik P., Chang H., Easley N. C., Lithgow T., and Buchanan S. K. (2013) Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501, 385–390 10.1038/nature12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fleming P. J., Patel D. S., Wu E. L., Qi Y., Yeom M. S., Sousa M. C., Fleming K. G., and Im W. (2016) BamA POTRA domain interacts with a native lipid membrane surface. Biophys. J. 110, 2698–2709 10.1016/j.bpj.2016.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinnige T., Weingarth M., Renault M., Baker L., Tommassen J., and Baldus M. (2014) Solid-state NMR studies of full-length BamA in lipid bilayers suggest limited overall POTRA mobility. J. Mol. Biol. 426, 2009–2021 10.1016/j.jmb.2014.02.007 [DOI] [PubMed] [Google Scholar]
- 23. Mahoney T. F., Ricci D. P., and Silhavy T. J. (2016) Classifying β-barrel assembly substrates by manipulating essential Bam complex members. J. Bacteriol. 198, 1984–1992 10.1128/JB.00263-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charlson E. S., Werner J. N., and Misra R. (2006) Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J. Bacteriol. 188, 7186–7194 10.1128/JB.00571-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Surrey T., and Jähnig F. (1992) Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 89, 7457–7461 10.1073/pnas.89.16.7457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgess N. K., Dao T. P., Stanley A. M., and Fleming K. G. (2008) β-Barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J. Biol. Chem. 283, 26748–26758 10.1074/jbc.M802754200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Otzen D. E., and Andersen K. K. (2013) Folding of outer membrane proteins. Arch. Biochem. Biophys. 531, 34–43 10.1016/j.abb.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 28. Fleming K. G. (2015) A combined kinetic push and thermodynamic pull as driving forces for outer membrane protein sorting and folding in bacteria. Philos. Trans. R. Soc. Biol. Sci. 370, 20150026 10.1098/rstb.2015.0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Freudl R., Schwarz H., Stierhof Y. D., Gamon K., Hindennach I., and Henning U. (1986) An outer membrane protein (OmpA) of Escherichia coli K-12 undergoes a conformational change during export. J. Biol. Chem. 261, 11355–11361 [PubMed] [Google Scholar]
- 30. Ureta A. R., Endres R. G., Wingreen N. S., and Silhavy T. J. (2007) Kinetic analysis of the assembly of the outer membrane protein LamB in Escherichia coli mutants each lacking a secretion or targeting factor in a different cellular compartment. J. Bacteriol. 189, 446–454 10.1128/JB.01103-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skillman K. M., Barnard T. J., Peterson J. H., Ghirlando R., and Bernstein H. D. (2005) Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol. Microbiol. 58, 945–958 10.1111/j.1365-2958.2005.04885.x [DOI] [PubMed] [Google Scholar]
- 32. Danoff E. J., and Fleming K. G. (2015) Membrane defects accelerate outer membrane β-barrel protein folding. Biochemistry 54, 97–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleinschmidt J. H., and Tamm L. K. (2002) Secondary and tertiary structure formation of the β-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J. Mol. Biol. 324, 319–330 10.1016/S0022-2836(02)01071-9 [DOI] [PubMed] [Google Scholar]
- 34. Gessmann D., Chung Y. H., Danoff E. J., Plummer A. M., Sandlin C. W., Zaccai N. R., and Fleming K. G. (2014) Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc. Natl. Acad. Sci. U.S.A. 111, 5878–5883 10.1073/pnas.1322473111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nikaido H. (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656 10.1128/MMBR.67.4.593-656.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patel G. J., and Kleinschmidt J. H. (2013) The lipid bilayer-inserted membrane protein BamA of Escherichia coli facilitates insertion and folding of outer membrane protein A from its complex with Skp. Biochemistry 52, 3974–3986 10.1021/bi400103t [DOI] [PubMed] [Google Scholar]
- 37. Hagan C. L., and Kahne D. (2011) The reconstituted Escherichia coli Bam complex catalyzes multiple rounds of β-barrel assembly. Biochemistry 50, 7444–7446 10.1021/bi2010784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hagan C. L., Kim S., and Kahne D. (2010) Reconstitution of outer membrane protein assembly from purified components. Science 328, 890–892 10.1126/science.1188919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hagan C. L., Westwood D. B., and Kahne D. (2013) Bam lipoproteins assemble BamA in vitro. Biochemistry 52, 6108–6113 10.1021/bi400865z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roman-Hernandez G., Peterson J. H., and Bernstein H. D. (2014) Reconstitution of bacterial autotransporter assembly using purified components. Elife 3, e04234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rassam P., Copeland N. A., Birkholz O., Tóth C., Chavent M., Duncan A. L., Cross S. J., Housden N. G., Kaminska R., Seger U., Quinn D. M., Garrod T. J., Sansom M. S., Piehler J., Baumann C. G., and Kleanthous C. (2015) Supramolecular assemblies underpin turnover of outer membrane proteins in bacteria. Nature 523, 333–336 10.1038/nature14461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleanthous C., Rassam P., and Baumann C. G. (2015) Protein–protein interactions and the spatiotemporal dynamics of bacterial outer membrane proteins. Curr. Opin. Struct. Biol. 35, 109–115 10.1016/j.sbi.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Epand R. M., and Epand R. F. (2009) Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta 1788, 289–294 10.1016/j.bbamem.2008.08.023 [DOI] [PubMed] [Google Scholar]
- 44. Vanounou S., Parola A. H., and Fishov I. (2003) Phosphatidylethanolamine and phosphatidylglycerol are segregated into different domains in bacterial membrane: a study with pyrene-labelled phospholipids. Mol. Microbiol. 49, 1067–1079 10.1046/j.1365-2958.2003.03614.x [DOI] [PubMed] [Google Scholar]
- 45. Noinaj N., Kuszak A. J., and Buchanan S. K. (2015) Heat modifiability of outer membrane proteins from Gram-negative bacteria. Methods Mol. Biol. 1329, 51–56 10.1007/978-1-4939-2871-2_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ellena J. F., Lackowicz P., Mongomery H., and Cafiso D. S. (2011) Membrane thickness varies around the circumference of the transmembrane protein BtuB. Biophys. J. 100, 1280–1287 10.1016/j.bpj.2011.01.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kučerka N., Nieh M.-P., and Katsaras J. (2011) Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 1808, 2761–2771 10.1016/j.bbamem.2011.07.022 [DOI] [PubMed] [Google Scholar]
- 48. Lugtenberg E. J., and Peters R. (1976) Distribution of lipids in cytoplasmic and outer membranes of Escherichia coli K12. Biochim. Biophys. Acta 441, 38–47 10.1016/0005-2760(76)90279-4 [DOI] [PubMed] [Google Scholar]
- 49. Morein S., Andersson A., Rilfors L., and Lindblom G. (1996) Wild-type Escherichia coli cells regulate the membrane lipid composition in a window between gel and non-lamellar structures. J. Biol. Chem. 271, 6801–6809 10.1074/jbc.271.12.6801 [DOI] [PubMed] [Google Scholar]
- 50. White D. A., Lennarz W. J., and Schnaitman C. A. (1972) Distribution of lipids in the wall and cytoplasmic membrane subfractions of the cell envelope of Escherichia coli. J. Bacteriol. 109, 686–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bastiat G., Oliger P., Karlsson G., Edwards K., and Lafleur M. (2007) Development of non-phospholipid liposomes containing a high cholesterol concentration. Langmuir 23, 7695–7699 10.1021/la700824m [DOI] [PubMed] [Google Scholar]
- 52. Webb C. T., Selkrig J., Perry A. J., Noinaj N., Buchanan S. K., and Lithgow T. (2012) Dynamic association of BAM complex modules includes surface exposure of the lipoprotein BamC. J. Mol. Biol. 422, 545–555 10.1016/j.jmb.2012.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dautin N., and Bernstein H. D. (2007) Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 61, 89–112 10.1146/annurev.micro.61.080706.093233 [DOI] [PubMed] [Google Scholar]
- 54. Barnard T. J., Gumbart J., Peterson J. H., Noinaj N., Easley N. C., Dautin N., Kuszak A. J., Tajkhorshid E., Bernstein H. D., and Buchanan S. K. (2012) Molecular basis for the activation of a catalytic asparagine residue in a self-cleaving bacterial autotransporter. J. Mol. Biol. 415, 128–142 10.1016/j.jmb.2011.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barnard T. J., Dautin N., Lukacik P., Bernstein H. D., and Buchanan S. K. (2007) Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat. Struct. Mol. Biol. 14, 1214–1220 10.1038/nsmb1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dautin N., Barnard T. J., Anderson D. E., and Bernstein H. D. (2007) Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 26, 1942–1952 10.1038/sj.emboj.7601638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Junker M., Besingi R. N., and Clark P. L. (2009) Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol. Microbiol. 71, 1323–1332 10.1111/j.1365-2958.2009.06607.x [DOI] [PubMed] [Google Scholar]
- 58. Ieva R., and Bernstein H. D. (2009) Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 19120–19125 10.1073/pnas.0907912106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pavlova O., Peterson J. H., Ieva R., and Bernstein H. D. (2013) Mechanistic link between β barrel assembly and the initiation of autotransporter secretion. Proc. Natl. Acad. Sci. U.S.A. 110, E938–E947 10.1073/pnas.1219076110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tamm L. K., Hong H., and Liang B. (2004) Folding and assembly of β-barrel membrane proteins. Biochim. Biophys. Acta 1666, 250–263 10.1016/j.bbamem.2004.06.011 [DOI] [PubMed] [Google Scholar]
- 61. Wang H., Andersen K. K., Vad B. S., and Otzen D. E. (2013) OmpA can form folded and unfolded oligomers. Biochim. Biophys. Acta 1834, 127–136 10.1016/j.bbapap.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 62. Danoff E. J., and Fleming K. G. (2011) The soluble, periplasmic domain of OmpA folds as an independent unit and displays chaperone activity by reducing the self-association propensity of the unfolded OmpA transmembrane β-barrel. Biophys. Chem. 159, 194–204 10.1016/j.bpc.2011.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Danoff E. J., and Fleming K. G. (2015) Aqueous, unfolded OmpA forms amyloid-like fibrils upon self-association. PLoS One 10, e0132301 10.1371/journal.pone.0132301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ieva R., Skillman K. M., and Bernstein H. D. (2008) Incorporation of a polypeptide segment into the β-domain pore during the assembly of a bacterial autotransporter. Mol. Microbiol. 67, 188–201 [DOI] [PubMed] [Google Scholar]
- 65. Peterson J. H., Tian P., Ieva R., Dautin N., and Bernstein H. D. (2010) Secretion of a bacterial virulence factor is driven by the folding of a C-terminal segment. Proc. Natl. Acad. Sci. U.S.A. 107, 17739–17744 10.1073/pnas.1009491107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roman-Hernandez G., and Bernstein H. D. (2015) An in vitro assay for outer membrane protein assembly by the BAM complex. Methods Mol. Biol. 1329, 203–213 10.1007/978-1-4939-2871-2_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Knowles T. J., Browning D. F., Jeeves M., Maderbocus R., Rajesh S., Sridhar P., Manoli E., Emery D., Sommer U., Spencer A., Leyton D. L., Squire D., Chaudhuri R. R., Viant M. R., Cunningham A. F., et al. (2011) Structure and function of BamE within the outer membrane and the β-barrel assembly machine. EMBO Rep. 12, 123–128 10.1038/embor.2010.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., and Pfanner N. (2009) Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 10.1016/j.cub.2009.10.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sohlenkamp C., and Geiger O. (2016) Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol. Rev. 40, 133–159 10.1093/femsre/fuv008 [DOI] [PubMed] [Google Scholar]
- 70. Skórko-Glonek J., Lipińska B., Krzewski K., Zolese G., Bertoli E., and Tanfani F. (1997) HtrA heat shock protease interacts with phospholipid membranes and undergoes conformational changes. J. Biol. Chem. 272, 8974–8982 10.1074/jbc.272.14.8974 [DOI] [PubMed] [Google Scholar]
- 71. 71 Szabady R. L., Peterson J. H., Skillman K. M., and Bernstein H. D. (2005) An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc. Natl. Acad. Sci. U.S.A. 102, 221–226 10.1073/pnas.0406055102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.