

Abstract
Conformational changes in proteins due to ligand binding are ubiquitous in biological processes and are integral to many biological systems. However, it is often challenging to link ligand-induced conformational changes to a resulting biological function because it is difficult to distinguish between the energetic components associated with ligand binding and those due to structural rearrangements. Here, we used a unique approach exploiting conformation-specific and regio-specific synthetic antibodies (sABs) to probe the energetic contributions of ligand binding to conformation changes. Using maltose-binding protein (MBP) as a model system, customized phage-display selections were performed to generate sABs that stabilize MBP in different conformational states, modulating ligand-binding affinity in competitive, allosteric, or peristeric manners. We determined that the binding of a closed conformation–specific sAB (sAB-11M) to MBP in the absence of maltose is entropically driven, providing new insight into designing antibody-stabilized protein interactions. Crystal structures of sABs bound to MBP, together with biophysical data, delineate the basis of free energy differences between different conformational states and confirm the use of the sABs as energy probes for dissecting enthalpic and entropic contributions to conformational transitions. Our work provides a foundation for investigating the energetic contributions of distinct conformational dynamics to specific biological outputs. We anticipate that our approach also may be valuable for analyzing the energy landscapes of regulatory proteins controlling biological responses to environmental changes.
Keywords: phage display, antibody engineering, crystal structure, epitope mapping, thermodynamics, allostery, conformation-specific synthetic antibodies, energy probes
Introduction
The functions of many types of biological processes are initiated through a ligand-binding event that induces programed conformational changes (1–3). However, assigning and quantifying the energetic forces that link specific conformational changes to a resulting function are only understood at a basic level. This is because it is difficult to distinguish between the energetic contributions derived from the ligand-binding event and energies originating from the molecular rearrangements encoded in the conformational changes. Further, since most protein molecules are inherently dynamic with characteristic distributions of different conformational forms, ligand binding may not necessarily induce a unique conformation but, rather, simply alter the distribution of the populations of the existing forms. Thus, to better understand these phenomena, we endeavored to develop a unique approach that would provide a more direct readout about how the thermodynamic relationships between ligand binding and conformational partitioning are coupled.
This approach involves using conformationally selective synthetic antibodies (sABs)5 generated by phage display as energetic probes to dissect the thermodynamic contributions of ligand-binding versus conformation changes. We used maltose-binding protein (MBP) as a model system, taking advantage of the ligand-induced conformational change from the open (apo) to the closed (maltose-bound) conformation (4). There have been studies using a variety of techniques aimed at gauging the energetics involved in conformational transition of MBP from open to closed state. These involve mutagenesis to generate variants of MBP either for attachment of molecular probes or hinge mutants having distribution of populations from open to partially closed hinge angles. The hinge mutants were used to assign cause and effect relationships produced through the different levels of closure of the binding pocket (5–8).
Although these studies have been informative, we believe that the use of conformation-selective sABs as energy probes provides a much more precise and quantitative approach to establish functional linkages between allostery and ligand binding in systems like MBP. By exploiting the power of phage display, we have generated and characterized a cohort of three classes of sABs: endosteric (binding to the maltose-binding pocket), allosteric (opposite to the maltose-binding pocket), and peristeric (close to the maltose-binding pocket) sABs of high affinity that specifically capture and stabilize distinct conformational states of wildtype MBP. One of the several advantages of using sABs over strategies involving mutations is that the characteristics of MBP can be modulated systematically without resorting to modifying the protein itself.
We show that conformationally selective sABs can significantly alter the conformational ensembles of the molecules they target to enhance or inhibit ligand binding in competitive and allosteric ways. For MBP, closed-specific sABs generated in the presence of maltose were found to enhance maltose binding by up to 100-fold, whereas open-specific sABs block maltose binding. We also utilized an “epitope-masking” strategy to identify the rare sAB candidates that bind to non-immunodominant epitopes. This resulted in an activator that enhances binding affinity of maltose 1000-fold in a peristeric manner. Interestingly, binding of a closed-specific sAB to MBP in the absence of maltose is entropically driven, a phenomenon rarely observed for antibody/antigen interactions. This provides insight into antibody-stabilized protein interaction design. Crystal structures of members of each of the three classes of sABs in complex with MBP, along with kinetic and thermodynamic data, provide quantitative information about the structural basis of the free energy differences between ligand free and bound states and demonstrate their usefulness as novel energy probes in establishing the interplay between enthalpic and entropic contributions to the transitions between different conformational states.
Results
Generation of conformation-specific binders
MBP is composed of two domains that are connected by a flexible linker. In the absence of maltose, MBP is in an open conformation providing unfettered access to the ligand. Upon the addition of maltose, the hinge between the two domains rearranges, effectively closing the maltose-binding pocket to encapsulate the ligand much like the closing of a clamshell. We used a phage-display strategy that exploits the structural differences between the open and closed state of MBP to generate sABs that preferentially bind to either state. To generate open and closed conformation–specific sABs, biopanning was carried out in the absence or presence of 1 mm maltose, respectively. Unique clones were identified (Table S1) by competitive phage ELISA followed by DNA sequencing. Conformational specificities of the binders were further evaluated by phage ELISA. The results showed that all of the binders obtained for the open form of MBP did not bind to MBP in the presence of 1 mm maltose (Fig. 1A). Similarly, the sABs generated against the closed form of MBP did not bind, or bound with significantly reduced affinity, to MBP in its open form (Fig. 1B). This selection strategy enabled us to obtain binders with exquisite conformational selectivity. For instance, sAB-11M generated against the closed form of MBP is able to discriminate between the different closure conformations of a set of hinge angle–altering mutants of MBP at different maltose concentrations (Fig. S1A).
Figure 1.

Characterization of conformation-specific sABs. Phage ELISA shows that sABs generated against the open form of MBP in the absence of maltose do not bind to the closed maltose-bound form of MBP (A) and sABs generated against the maltose-bound, closed form of MBP do not bind or bind with very reduced affinity to the open form of MBP (B). C, kinetics of sAB-7O binding to MBP at different maltose concentration. The kinetics is on-rate–dependent, and affinity decreases steadily with increasing maltose concentration. Interaction Map shows a homogeneous 1:1 interaction. Change in relative position of the heat map on the y axis (log ka) is clear due to a decrease in the on-rate with maltose concentration, whereas the position on the x axis (log kd) remains constant due to invariance in the off-rate. Error bars, S.E.
The binding kinetics of representative sABs were determined using surface plasmon resonance (SPR) (Table S1 and Fig. S1B). The eight closed-specific sABs overall were of higher affinity, and their dissociation constants were generally <10 nm. Without maltose, the dissociation constants (KD) of the eight open-specific sABs tested ranged from 1 to 50 nm. Affinities of the open conformation sABs depended on the maltose concentration. For example, sAB-7O binds MBP with KD of 1 nm in the absence of maltose and 152 nm in 1 μm maltose and does not bind in 1 mm maltose. With increased maltose concentration, the SPR sensograms displayed a pronounced delay in binding. Kinetic parameters at different maltose concentrations indicate that this decrease in affinity is due to a gradual decrease in on-rates (ka) with increasing maltose concentration, whereas the off-rates (kd) remained practically invariant (Table S2). To determine whether the interaction between sAB-7O and MBP under different maltose concentrations is a classical 1:1 binding event or a heterogeneous one composed of multiple interaction processes, we used Interaction Map® (IM) analysis that can resolve complex, parallel kinetics into individual events (9, 10). An IM analysis at individual maltose concentrations showed predominantly single peaks that demonstrate a clear 1:1 interaction (Fig. 1C). Higher maltose concentrations drive MBP toward the closed conformation, thus increasing the energetic cost for sAB-7O binding. The decrease in on-rate as a function of maltose concentration with no apparent effect on off-rate suggests a mechanism whereby the maltose has to be displaced before open-specific sABs can bind; however, once bound, the influence of maltose is decreased because accessibility to the binding pocket is limited.
Binding of open- and closed-specific sABs is mutually exclusive
To determine how many distinct epitopes there were for each class of these sABs, an epitope binning experiment was performed by phage ELISA. In the case of sABs targeting apo-MBP, competitive screening of individual clones was performed using sAB-7O, the sAB variant that was dominant in the post-selection phage pool. This competitive screening showed that all of the open-specific sABs shared a similar overall surface epitope (Fig. S2A). Likewise, the ELISA for the sABs specific for the closed form of MBP performed in the presence of sAB-11M (which dominated the phage pool targeting maltose-bound MBP) indicated only a single overlapping immunodominant surface epitope (Fig. S2B).
Because the sABs were generated for two different conformations of MBP, it was of interest to determine whether sABs from one selection could bind simultaneously with sABs from the other. To do this, 1 μm maltose was added to half-saturate the maltose-binding sites (Kd,mal ∼ 1 μm) of MBP, thus favoring neither conformation over the other. At 1 μm maltose, sAB-11M and sAB-7O bind MBP with affinities of 20 and 152 nm, respectively. However, their binding is mutually exclusive (Fig. 2A). In fact, a decrease in the ELISA signal from bound sAB-11M is observed upon titration of sAB-7O into MBP presaturated with sAB-11M (Fig. 2B). This observation can be explained either by competition for an overlapping epitope or a sAB-induced structural change in MBP that hinders the binding of sABs having the other conformation preference.
Figure 2.
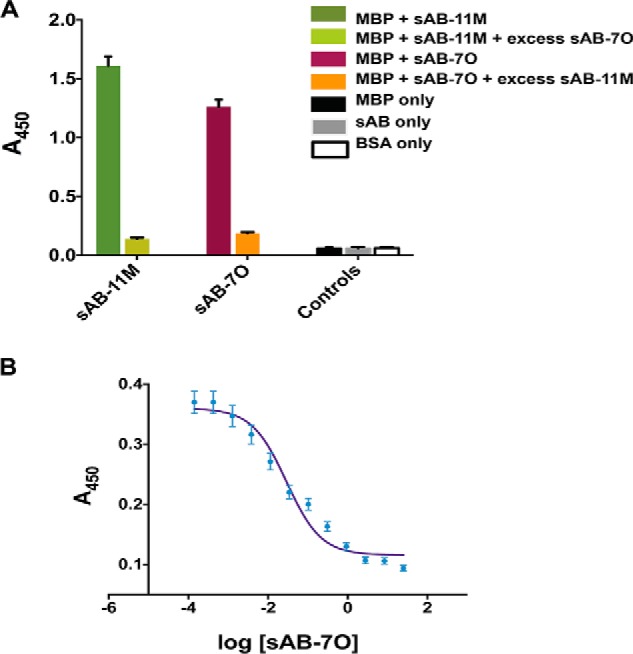
Closed and open conformation–specific sABs are competitive. A, single-point ELISA shows that the binding of sAB-7O and sAB-11M are exclusive. Neither of them binds in the presence of an excess of the other. B, competitive ELISA showing sAB-7O displacing His6-sAB-11M from MBP saturated with His6-sAB-11M. In both A and B, the sAB of interest was His-tagged, and the ELISA signal (A450) was monitored using anti-His antibody. Error bars, S.D.
Structures of the open- and closed-specific sAB·MBP complexes
The mutually exclusive binding results prompted us to perform a structural study to establish whether the conformation-specific sABs share an overlapping surface footprint or compete for different conformations of MBP. As sAB-7O and sAB-11M were the most dominant clones from the two selections, the structures of sAB-7O·MBP open-specific complex and the sAB-11M·MBP closed-specific complex were chosen as representatives to delineate the structural basis of their specificities.
Open-specific sAB-7O·MBP complex
The crystal structure of sAB-7O·MBP was determined at 2.1 Å (Table 1) and showed that sAB-7O binds to a large endosteric site on MBP using its heavy-chain CDR-H1, -H2, and -H3 and light chain CDR-L1 and -L3 residues (Fig. 3A). Approximately 1200 Å2 of surface area is buried in the interaction (heavy chain, 900 Å2; light chain, 300 Å2) (Fig. 3B). This large interface is stabilized by extensive hydrophobic, electrostatic, and hydrogen-bonding interactions. The CDR-H3 loop inserts into the maltose-binding pocket and interacts with a majority of the residues in the pocket involved in maltose binding (Fig. 3C). Notably, the aromatic side chains of Tyr-106H3 and Tyr-108H3 are involved in π-stacking interactions with Trp-62 and Trp-230 of MBP. The sugar rings and hydroxyl groups of maltose are emulated by the aromatic ring of tyrosines and the backbone carbonyls in CDR-H3, resulting in similarities of the hydrophobic and polar contacts with the residues involved in maltose binding (Fig. 3D). This molecular mimicry (11) is probably the reason behind the strong bias of the sABs targeting the maltose-binding pocket as the immunodominant epitope. Another contributing factor is the biased design of the sAB library toward tyrosine and tryptophan residues (12) that can form favorable interactions with the fairly hydrophobic environment in the binding pocket.
Table 1.
Data collection and refinement statistics
Values in parentheses are for the highest-resolution shell.
| sAB-7O·MBP | sAB-11M·MBP | sAB-P1·MBP | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P21 | C2 |
| Cell dimensions | |||
| a, b, c (Å) | 76.8, 120.8, 193.6 | 98.4, 87.6, 114.7 | 217.2, 42.4, 200.7 |
| α, β, γ (degrees) | 90.0, 90.0, 90.0 | 90.0, 114.3, 90.0 | 90.0, 90.1, 90.0 |
| Wavelength (Å) | 0.9792 | 0.9791 | 0.9791 |
| Resolution (Å) | 19.81–2.15 (2.20–2.15) | 19.96–1.95 (2.0–1.95) | 19.94–2.60 (2.70–2.60) |
| Rmerge (%) | 10.4 (89.0) | 7.6 (89.6) | 20.0 (138.0) |
| CC½ (%) | 99.6 (60.4) | 99.7 (64.2) | 99.1 (63.1) |
| I/σ(I) | 10.2 (1.6) | 9.9 (1.8) | 8.4 (1.7) |
| Completeness (%) | 99.2 (100) | 97.5 (98.2) | 99.5 (100) |
| Redundancy | 4.0 (4.1) | 3.3 (3.4) | 6.5 (7.0) |
| Refinement | |||
| Resolution (Å) | 19.81–2.15 | 19.96–1.95 | 19.94–2.60 |
| No. of reflections | 97,669 | 126,099 | 57,135 |
| Rwork/Rfree | 0.21/0.25 | 0.20/0.23 | 0.21/0.26 |
| No. of atoms | |||
| Protein | 12,262 | 12,525 | 12,489 |
| Ligands/ions | 13 | 93 | 139 |
| Waters | 981 | 1162 | 221 |
| B-factors | |||
| Protein | 38.2 | 38.8 | 52.8 |
| Ligands/ions | 58.2 | 54.0 | 65.7 |
| Waters | 40.0 | 41.3 | 43.2 |
| Root mean square deviations | |||
| Bond lengths (Å) | 0.003 | 0.006 | 0.003 |
| Bond angles (degrees) | 0.592 | 0.769 | 0.543 |
| Ramachandran plot statistics | |||
| Favored (%) | 96.7 | 98.1 | 96.9 |
| Allowed (%) | 3.2 | 1.9 | 3.0 |
| Outliers (%) | 0.1 | 0.0 | 0.1 |
| PDB code | 5BK1 | 5BJZ | 5BK2 |
Figure 3.

Structures of open-specific sAB-7O·MBP complex. A, sAB-7O binds to MBP in open conformation by the light chain CDRs (L1 and L3) and heavy chain CDRs (H1, H2, and H3). B, open-book view of the interface between sAB-7O and MBP. Residues in the interface are colored according to their percentage of reduction in accessible surface area upon complex formation (yellow, 10–49%; orange, 50–70%; red, >70%). C, CDR-H3 residues of sAB-7O (green) interact with the residues in the maltose-binding pocket of MBP (salmon). D, residues of MBP (salmon) involved in maltose binding. Selected hydrogen-bonding interactions with maltose (ball and stick) are highlighted. Most of the maltose-binding residues in MBP are involved in interaction with CDR-H3 residues of sAB-7O as seen in C. Sugar rings and hydroxyl groups of maltose are mimicked by the aromatic ring of tyrosine and backbone carbonyls in CDR-H3 residues, resulting in strong bias of the maltose binding pocket as the sole immunodominant epitope.
Closed-specific sAB-11M·MBP complex
The crystal structure of sAB-11M·MBP in the presence of maltose at 2.1 Å (Table 1) indicated that sAB-11M binds to the hinge region of MBP. This region is on the opposite face of the molecule from the maltose-binding pocket (Fig. 4A). The interface between sAB-11M and MBP buries ∼1330 Å2 of solvent-accessible surface area almost equally distributed between the two chains, ∼680 Å2 by the CDR-H2 and -H3 and ∼660 Å2 by the light chain (Fig. 4B). Residues 110–114 of CDR-H3 occupy a cleft formed between the helices comprising residues 90–98 and 314–327 of MBP that is only exposed in the closed conformation. CDR-H3 residues 103–109 stabilize the loop that is disordered in a previously reported sAB·MBP structure (13) (Fig. S3).
Figure 4.

Structures of closed-specific sAB-11M·MBP complex. A, sAB-11M binds to the hinge region in the maltose (red sticks)-bound conformation of MBP on the side opposite to the maltose-binding pocket. B, detailed picture of sAB-11M residues (shown in sticks) interacting with MBP. CDR-L1, -L2, and -L3 and LC scaffold residues are colored marine, and CDR-H1, -H2, and -H3 are colored green. MBP is color-coded as in Fig. 3B. C, superposition of structures of sAB-7O-MBP structure (salmon) with sAB-11M-MBP (slate) shows that sAB-7O and sAB-11M bind on opposite sides of MBP in open and closed states, respectively.
The structures of sAB-7O·MBP and sAB-11M·MBP reveal that the two immunodominant binding surfaces reside on opposite faces of MBP (Fig. 4C), demonstrating that their mutual exclusivity is a conformational selectivity, not a spatial phenomenon. Upon binding to MBP, sAB-11M shifts the MBP conformation to the closed form, preventing sAB-7O from binding to the endosteric epitope that is occluded in the closed form of the protein. Similarly, sAB-7O binds to the maltose-binding pocket of MBP driving the equilibrium to the open form altering the epitope required for sAB-11M.
sAB-induced conformational modulation is reflected in ligand binding affinity
To determine the influence of the conformational specific sABs on the binding thermodynamics between maltose and MBP, a series of isothermal calorimetry (ITC) experiments were performed. Based on ITC, the maltose/wildtype MBP binding event is entropically driven with an equilibrium dissociation constant of ∼830 nm (Fig. 5A), which is in good agreement with previous results (14). In the presence of excess sAB-7O, no appreciable maltose binding to MBP was observed (Fig. 5B). This can be attributed to sAB-7O binding the endosteric epitope of MBP with sufficient affinity to effectively outcompete maltose binding. Conversely, MBP in the presence of a 5-fold excess of sAB-11M increases the affinity for maltose by 120-fold (Fig. 5C). Thus, assuming the enhanced affinity is due to the conformational modulation of MBP by sAB-11M, this provides a unique opportunity to decouple the conformational energetics from the energetics stemming from the direct effects of maltose interacting with the binding pocket. Consequently, we sought to dissect the underlying enthalpic and entropic contributions to the sAB-driven shift in ligand affinity.
Figure 5.

Thermodynamics of maltose and sABs binding to MBP. Shown are ITC titration curves of maltose binding to MBP (A), MBP with a 5-fold excess of sAB-7O (B), and MBP with 5-fold excess of sAB-11M (C). D, thermodynamic cycle of maltose binding to MBP with and without sAB-11M.
The binding thermodynamics in the presence of the sABs contain two overlapping components: (i) the component due to MBP-maltose interaction alone and (ii) that due to conformational bias induced by the sAB and its interaction with MBP. A thermodynamic cycle was generated (Fig. 5D), which consists of four MBP states: (i) open, (ii) maltose-bound (closed), (iii) sAB-11M–bound (closed) without maltose, and (iv) sAB-11M with maltose (closed). Using ITC, the binding thermodynamics (ΔG0, ΔH0, and −TΔS0) were determined at 25 °C for maltose binding to MBP and the MBP·sAB-11M complex, as well as sAB-11M binding to the maltose·MBP complex (Table 2). Although the remaining interaction, between sAB-11M and MBP, possessed negligible heats of binding, the three experimentally accessible equilibria provided a route to determine the binding thermodynamics of the sAB-11M/MBP interaction through the thermodynamic cycle (i.e. ΔH0sAB = ΔH0mal + ΔH0sABC − ΔH0malC).
Table 2.
Thermodynamic parameters from ITC experiments at 25 °C
| Titration | ΔH0 | −TΔS0 | ΔG0 | KD = 1/KB |
|---|---|---|---|---|
| kcal mol−1 | kcal mol−1 | kcal mol−1 | nm | |
| Maltose: MBP | 2.1 ± 0.1 | −10.4 ± 0.1 | −8.3 ± 0.1 | 830.0 |
| MBP: sAB-11M in 1 mm maltose | −14 ± 2 | 2 ± 2 | −11.4 ± 0.1 | 4.0 |
| Maltose: MBP in 5-fold molar excess of sAB-11M | −8.9 ± 0.8 | −2 ± 1 | −11.1 ± 0.2 | 7.1 |
| MBP: sAB11M(calculated) | −3 ± 2 | −5.6 ± 2 | −8.6 ± 0.2 | 470.0 |
| MBP: sAB-P1 in 1 mm maltose | −16.8 ± 0.5 | 5.8 ± 0.7 | −11.0 ± 0.2 | 9.0 |
| Maltose: MBP in 5-fold molar excess of sAB-P1 | −14 ± 2 | 1 ± 2 | −12.6 ± 0.1 | 0.6 |
The maltose/MBP binding equilibrium (Kmal) was determined to be a highly entropically driven event. Due to the complex nature of the binding equilibrium, it is not possible to decouple the energetics of the two major apparent events, intramolecular changes within MBP and the formation of new intermolecular maltose/MBP interactions, without the ability to isolate each event. However, analyzing the thermodynamic profile of sAB-11M binding provides a route to “capture” a conformation of MBP that is similar to its closed form in the absence of maltose. This allows the isolation of maltose/MBP binding presumably without major changes in MBP's conformation. In the presence of excess sAB-11M, the maltose-binding equilibrium constant (KmalC) exhibited an approximately 120-fold enhancement (ΔΔG0 = −2.8 kcal/mol). This result is due to the sAB-11M biasing the conformational state to the closed form, thereby significantly reducing the energetic penalty going from the open to closed state during maltose binding. Interestingly, the thermodynamic profile for the maltose-binding step (KmalC) changes to a predominantly enthalpically driven event. The difference in the changes in enthalpies and entropies between maltose binding to the closed versus open MBP states are striking: ΔΔH0 = −11.0 kcal/mol and −TΔΔS0 =8.4 kcal/mol, which suggests that any entropic gain from solvent release from the maltose-binding pocket has already been realized through the binding of sAB-11M. Moreover, the maltose-binding site in the closed state of MBP is predicted to be structurally aligned to maximize interactions with maltose, consistent with the more favorable enthalpy for maltose binding to the sAB-11M–stabilized closed state of MBP versus MBP alone.
Notably, the sAB-11M/MBP binding interaction in the absence of maltose (KsAB) displays an atypical antibody/antigen thermodynamic profile (ΔH0 = −1.4 kcal/mol, −TΔS0 = −7.3 kcal/mol), where sAB-11M binds to a higher-energy closed conformation of MBP without maltose. The sAB-11M/MBP interaction is entropy-driven, whereas most antibody/antigen interactions are enthalpy-driven binding events (15). As a comparison, the binding equilibrium for sAB-11M and MBP in the presence of 1 mm maltose (KsABC) possessed a typical antibody/antigen profile (ΔH0 = −12.4 kcal/mol, −TΔS0 = 0.9 kcal/mol). Nevertheless, the entropy-driven sAB-11M/MBP binding event is consistent with the observed thermodynamic model for maltose binding discussed above, whereby the trapping of the closed MBP state upon sAB-11M binding includes a significantly favorable entropic component, which is attributable to the intramolecular closing of MBP and subsequent solvent release.
Maltose concentration does not alter the binding hot spot in the closed-form sABs
Our interpretation of the thermodynamic coupling data using the differences observed between sAB-11M binding in the presence and absence of maltose requires that the binding interactions remain identical (i.e. if sAB-11M binds using one set of interactions in the presence of maltose and a different set in its absence, invoking thermodynamic coupling between states is incorrect). Therefore, to establish its validity, we compared the energy epitopes of the binding interface of sAB-11M at 1 μm and 1 mm maltose, the idea being that if the sAB/MBP interactions were affected by a constant ratio, the interactions across the interface were preserved. To establish this, four individual residues in the paratope were identified from the structure of sAB-11M–bound MBP that are likely to be hot spots for MBP binding and mutated to alanine. SPR was used to determine the binding affinity between MBP and these four alanine mutants of sAB-11M at the different maltose concentrations (Fig. S4). In all of the cases, we observed that ΔΔG0 (1 mm versus 1 μm maltose) of the mutants are virtually identical to that of WT sAB-11M (Table 3). This is a strong indication that the epitope/paratope interactions stabilizing the interface of the MBP·sAB-11M complex are conserved along the entire pathway of conformational change. Furthermore, this shows that the changes in affinities in sAB binding between the different conformational states provide direct readout of the energies associated with the transitions between those states.
Table 3.
Kinetic parameters of binding of WT sAB-11M and Ala mutants with WT MBP
| Variants of sAB-11M | KD in 1 μm maltose | KD in 1 mm maltose | ΔΔG0 |
|---|---|---|---|
| nm | nm | kcal mol−1 | |
| WT | 20.0 | 1.5 | −1.5 |
| Y57A | 104.5 | 7.6 | −1.5 |
| R67A | 305.0 | 20.0 | −1.6 |
| W112A | 350.0 | 29.4 | −1.5 |
| P113A | 715.0 | 63.0 | −1.4 |
Directing sABs to other epitopes on MBP
Selections in the presence or absence of maltose produced two distinct immunodominant populations of sABs: the closed-specific sABs binding to the hinge region, promoting maltose binding in an allosteric manner and the open-specific variants to an endosteric epitope that comprises residues of the maltose-binding pocket. Although it is not unusual for conformation specific sABs to display immunodominant preferences, we had nevertheless anticipated that some open-specific sABs would bind to the external hinge region in its open conformation. Likewise, it seemed plausible that some of the closed-specific sABs could bind across the closed face of the binding pocket, effectively trapping the maltose inside. We realized that such sABs might have existed in the original pools, but perhaps at such a low abundance that they would not have been identified under the stringent selection conditions favoring the high-affinity binders. To enhance the probability of finding such rare binding epitopes, we undertook an “epitope exclusion” strategy during selection. This eliminates accessibility to the immediate vicinity of the “immunodominant hot spots” and thus can lead to the identification of a pool of rare binders that target other less immunogenic surfaces.
To obtain sABs that bind away from the maltose-binding pocket in the open conformation of MBP, we used 5 μm sAB-7O to block the region around the endosteric site during biopanning. The selection resulted in the generation of three sABs (A5, B10, and D9). Of these, only sAB-B10 bound to MBP with reasonable affinity as judged by competitive phage ELISA. However, sAB-B10 is conformationally nonselective, as it binds to the open and closed forms of MBP with similar KD values of 8 and 30 nm, respectively. Presumably, it binds to a region that does not undergo reorganization between the open and closed states. Based on size-exclusion chromatography–multiangle static light scattering and SPR experiments, sAB-B10 binds to an epitope distinct from sAB-7O and sAB-11M (Fig. S5). Alanine scanning of the CDRs shows that the majority of the interactions were contributed by the aromatic and the hydrophobic amino acid residues (Fig. S6). Overall, attempts to find an open-specific sAB whose footprint included the hinge region were unsuccessful. A plausible explanation could be that the intrinsic dynamics of the open form of MBP presumably involves a degree of conformational heterogeneity of the hinge itself. Whereas this might have some effect, sAB-7O should have stabilized MBP in its open conformation and greatly limited its dynamic fluctuations. This suggests that the conformation and local environment of the hinge region itself in the open conformation is not conducive to sAB binding even under the strong selection pressure during biopanning.
To obtain sABs binding to epitopes other than the hinge region of MBP in the closed conformation, 5 μm sAB-11M was used during biopanning to block the footprint defined by the immunodominant hinge epitope, generating four sABs that bound to MBP with high affinity in the presence of maltose. Epitope binning experiments by SPR showed that as expected, these sABs bound to unique epitopes different from where sAB-11M binds (data not shown). Three of these were found to be conformationally agnostic. However, sAB-P1 was found to have the desired property of being highly specific for the closed conformation of MBP and does not bind to the open form. To investigate the thermodynamic properties of this sAB, ITC experiments were performed showing that the affinity of maltose toward MBP is enhanced more than 1000-fold in the presence of an excess of sAB-P1 with ΔΔG0 = −4.1 kcal/mol (Table 2 and Fig. S7). Interestingly, such a potent activator of maltose binding turned out to be a potential inhibitor of maltose transport by the Escherichia coli maltose transporter, MalFGK2, across the periplasm (Fig. S8). Because sAB-P1 binds to an epitope different from the sABs that enhance maltose binding and maltose transport across periplasm under low-maltose conditions (13) in an allosteric manner, we hypothesized that the mechanism of operation by sAB-P1 is presumably different.
Structure of the sAB-P1·MBP complex
The structure of the sAB-P1·MBP complex shows that sAB-P1 binds to the closed conformation of MBP across the face of the closed binding pocket, effectively trapping the maltose inside (Fig. 6A). Unlike sAB-11M, which binds to the allosteric hinge region, sAB-P1 binds to the peristeric site of MBP with its epitope spanning across the two lobes that form the entrance to the maltose-binding pocket (Fig. 6B). A short α-helix comprising residues 64–73 on one lobe of MBP and the long helix formed by residues 334–352 on its other lobe, form part of the extended interface between sAB-P1 and MBP (Fig. 6C). Light-chain CDR residues interact with the residues 341–350 on the long helix, whereas heavy chain CDRs do so with the MBP residues 66–72 contained in the short helix and residues 334–342 on the long helix. These two helices have a remarkably different relative orientation in the open and closed forms of MBP, thereby imparting conformational specificity of sAB-P1 (Fig. 6D). The structure of the sAB-P1·MBP complex when superposed with that of MBP-bound E. coli MalFGK2 (PDB code 3PUZ) shows that sAB-P1 has a significant overlapping epitope with MalF (Fig. S9). Thus, sAB-P1 precludes the association of MalF with MBP by partially blocking its epitope. The high-affinity interaction between the sAB-P1 and MBP outcompetes the modest affinity of the protein with the transporter, thereby inhibiting maltose transport across the periplasm.
Figure 6.

Structural features of sAB-P1·MBP complex. A, sAB-P1 binds to the closed conformation of MBP interacting across the face of the binding pocket with the maltose trapped inside. B, superposition of MBP bound to sAB-P1 (green) with that of sAB-11M (slate) shows that sAB-P1 binds to the peristeric site of MBP in the closed form opposite to sAB-11M. C, interactions of the HC (magenta) and LC (blue) CDRs of sAB-P1 with the two helices (64–73 and 334–352) of MBP (green) at the peristeric interface. D, the relative orientations of the helices comprising residues 64–73 and 341–350 are different in open (salmon) and closed (green) forms of MBP. These helices in the closed conformation interact extensively with sAB-P1 (C), thereby imparting conformational specificity.
In comparison with sAB-11M, sAB-P1 provides additional stabilization of the maltose-bound state by ∼1.5 kcal/mol. Notably, the enhanced binding of maltose afforded by the binding of sAB-P1 is even more enthalpically favored, relative to the sAB-11M–stabilized MBP (Table 2). This suggests that the close proximity of sAB-P1 to the maltose-binding pocket brings the two lobes of MBP together to stabilize the closed-form state, and the sAB-driven stabilization of MBP's maltose binding–competent state is sensitive to the molecular details of the epitope of interaction.
Discussion
Conformational changes in response to ligand binding are ubiquitous among biological processes. The energetics governing ligand binding involve a complex mixture of both direct, short-range atomic interactions and indirect, long-range forces controlling conformational changes. However, comprehensive analyses of the roles of these factors have been frustrated by a lack of adequate tools and methodologies that could distinguish between the contributions of the individual types of forces. In this regard, exploiting the power of phage display, we have developed a versatile set of affinity reagents that can be used to investigate the energetics of these complex biological phenomena by exploiting the ability of these reagents to trap functionally important conformations. These reagents serve as highly informative energy probes that can experimentally elucidate the thermodynamic forces that partition protein conformational ensembles, thus providing unique insights into the functional linkages between conformational transitions and ligand binding.
We interpret the thermodynamic data to indicate that closed-form sABs can stabilize the ligand-bound conformation of MBP, thereby enhancing the maltose-MBP binding event between 2 and 4 kcal/mol. Interestingly, whereas the ΔΔG0 values across the thermodynamic cycle are comparable (ΔG0 ∼ 3 kcal/mol) for either maltose- or sAB-based stabilization of MBP, the individual entropic and enthalpic contributions to each step differ significantly. The thermodynamic parameters suggest that closed-specific sAB-11M “primes” the conformation of MBP to facilitate better intermolecular interactions that are otherwise less favorable due to the poor intramolecular interactions within the maltose-binding pocket. It is noted that the intramolecular forces of the unfavorable closing of MBP from the open to the closed state (ΔG0 ∼ 3 kcal/mol) are dominated by an unfavorable enthalpy over favorable entropy. This sAB-assisted affinity modulation provides insight into protein interaction design, where optimal high affinity may be obtained by generating a binding partner that locks the protein in a binding-competent state, thereby minimizing energetic penalties due to conformational changes upon binding.
It has been shown that allosteric closed-specific sABs induce positive cooperativity in maltose binding to MBP (13). A notable observation from our data is that what drives this linkage is not simply the binding affinity of the sAB, but also the particular binding epitope relative to an allosterically sensitive region of the MBP molecule. In other words, two sABs can have binding footprints that overlap significantly but can have quite different allosteric coupling properties. This suggests that there are allosteric hot spots much akin to energetic binding hot spots, which are characterized by having a relatively small proportion of the interactions within the binding interface contributing a disproportionally high amount to the overall binding energy. This is elaborated even at the level of individual interactions, where the pattern of the contributions of allosteric hotspot residues is independent of maltose concentration. However, the concentration range over which this is true depends on the binding affinity of the sAB to the closed conformation. Because the energetic cost to induce the closed conformation in an allosteric manner is relatively constant, higher-affinity sABs can contribute ∼3 kcal/mol in their binding without completely losing their binding capacity. Lower-affinity sABs or sABs that bind outside the allosteric hot spot will have less impact on ligand binding.
The ability to affect binding of the ligand systematically by modulating forces that shift the conformational equilibrium between functional states has many potential applications. Previous work has shown how sABs can drive a target protein to adopt a specific form (on versus off), effectively enhancing or diminishing its activity (16–18). In this study, we have shown that sABs of high affinities against a well-characterized protein that undergoes ligand-dependent conformational change can modulate functional outcomes and therefore be used uniquely to provide quantitative energetic readouts of conformational transitions. We have used the ligand to shift the conformational equilibrium between the open and closed forms and further utilized an “epitope masking” strategy to direct the sAB binding to a specific epitope on the protein away from the immunodominant hot spot.
The open conformation-specific sABs bind MBP in the ligand-binding pocket, thereby inhibiting maltose binding competitively. Among the closed conformation–specific sABs, those binding to the hinge region of MBP enhance the affinity of maltose in an allosteric manner, whereas those binding to the peristeric epitope (close to the maltose-binding pocket) stabilize the closed conformation of MBP by “stapling” the two lobes of MBP together with the maltose trapped inside, thereby increasing the affinity toward maltose. Notably, with a large number of regulatory proteins that exhibit functionally relevant conformational change, a similar strategy may be used to generate sABs of customized functions by targeting and trapping unique protein states of specific biological outputs, thus opening opportunities to dissect the energetic contributions of different conformational states. Therefore, this study provides a framework for investigating the energetic contributions of conformational dynamics between different forms of the same protein to its function under different conditions. We believe an expansion of the use of this methodology will prove valuable for the comprehensive analyses of the underlying energy landscapes employed by regulatory and signaling proteins in promoting biological response to environmental changes.
Materials and methods
Preparation of proteins and library sorting
The constructs used for the studies are as follows: (i) CBPH6-MBP, MBP that has a calmodulin-binding peptide (CBP) followed by a His6 tag at the C terminus (19); (ii) pHFT2-MBP, which has a His10, FLAG tag, and tobacco etch virus site at the N terminus (20); and (iii) pHBT-CAM and pHBT-MBP, which adds a His6 and a biotin tag at the N terminus of the construct (21). CBPH6-MBP and pHFT2-MBP were expressed in E. coli BL21(DE3), and pHBT-CAM and pHBT-MBP were expressed in E. coli BL21 (DE3)/pBirA that co-expresses E. coli biotin ligase BirA for in vivo biotinylation of the target proteins. All of the constructs were overexpressed with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h. For in vivo biotinylation, 50 μm d-biotin was added to the media.
The proteins were purified by nickel-nitrilotriacetic acid chromatography followed by size-exclusion chromatography (SEC) on a 16/60 Superdex 75 column (GE Healthcare). For biophysical studies on MBP, an additional step of pH- or urea-induced unfolding followed by refolding or extensive dialysis at high salt (7) was performed to remove contaminating oligosaccharides, which often co-purify with MBP.
Library sorting was performed at room temperature following published procedures (19). Briefly, streptavidin-coated magnetic beads were precoated with Avi-CaM to prepare the CaM-coated magnetic beads. These CaM-coated beads were used in sorting to capture CBP-tagged MBP. All of the buffers used in sorting, except the elution buffer, were supplemented with 1 mm CaCl2. To eliminate the potential CaM binders from the amplified phage pool, the phage library was negatively selected against 250 μl of streptavidin-coated magnetic beads coated with 200 nm Avi-CaM from the very first round before the phages were added to the target. 1 ml of phage library (12) containing 1012–1013 virions was added to the beads and incubated for 30 min. The resuspended beads containing bound virions after extensive washing were used to infect freshly grown log-phase E. coli XL1-Blue cells. Phages were amplified overnight in 2YT media with 50 μg/ml Amp and 109 plaque-forming units/ml M13-KO7 helper phage. To increase the stringency of selection, three additional rounds of sorting were performed by decreasing the target concentration in each round (second round, 50 nm; third round, 10 nm; fourth round, 10 nm), using the amplified pool of virions of the preceding round as the input. Sorting from the second to fourth rounds was done on a Kingfisher instrument. From the second to the fourth round, the targets were premixed with the amplified phage pool, and then CaM coated beads were added to the mixture. From the second round onward, 10 mm EDTA was used to elute the bound phage by disrupting the interaction of CBP-tagged targets with CaM on the beads.
To obtain conformation-specific (closed) sABs for MBP, sorting buffer was supplemented with 1 mm maltose in all rounds. sABs specific for the open conformation of MBP were obtained without any maltose in the selection conditions.
Epitope-specific sABs were obtained by an “epitope exclusion strategy.” Before sorting, the target was preincubated with 5 μm sAB-11M or sAB-7O to block the immunodominant epitope in the presence or absence of maltose, respectively. Additionally, 5 μm masking sAB was added to all of the buffers during each of the four rounds. This ensures that the epitope is masked and that all binders specific for the masking sABs are washed away. The enrichment ratio was determined as the number of phage from the MBP-containing selection divided by the number of phage from the control selection, which did not contain immobilized MBP. Enrichment ratios in all of the cases were >100 after the fourth round of selection.
Single-point competitive phage ELISA
A single-point competitive phage ELISA was used to rapidly estimate the affinities of the obtained sABs in phage format (16). Colonies of E. coli XL1-Blue harboring phagemids were used to inoculate 500 μl of 2YT medium supplemented with 100 μg/ml ampicillin and M13-KO7 helper phage. The cultures were grown at 37 °C for 16–20 h at 280 rpm in a 96-deep-well block plate. Culture supernatants containing sAB-phage were diluted 10-fold in PBST buffer (PBS buffer containing 0.05% (v/v) Tween 20) with or without 50 nm target proteins as soluble competitor. After 15 min of incubation at room temperature, the mixtures were transferred to ELISA plates containing immobilized MBP. The plates were incubated with the phage/competitor mixture for another 15 min and then washed with PBST. The washed ELISA plates were incubated with HRP-conjugated anti-M13 mouse monoclonal antibody (1:5000 dilution in PBST buffer) for 30 min. The plates were again washed with PBST, developed with TMB substrate, and quenched with 1.0 m H3PO4, and absorbance (A450) was determined at 450 nm. For each clone, the ratio of A450 in the presence of competitor to that in its absence gives the fraction of sAB-phage uncomplexed with soluble competitor. A plot of the signal intensities (y axis) without any competitor as a function of this ratio (x axis) gives an estimate of the affinities of the binders. For further characterization, clones with high y values and low x values were chosen. To determine conformational specificity of the sABs against the closed form of MBP, all of the buffers used in the ELISA were supplemented with 1 mm maltose, whereas open-specific sABs were identified by ELISA in absence of maltose in the buffers. Clones that showed differential binding under these two conditions with high competition ratios were identified as conformation-specific and sequenced.
Epitope binning experiments
To determine how many distinct surface epitopes there are for each class of conformation-specific sAB, an epitope binning experiment was performed. This analysis is straightforward and can be done rapidly using a standard phage ELISA. The assay determines how much a purified sAB is able to compete with the binding of another sAB displayed on phage using phage ELISA. Binding of the individual sABs (in phage format) to the immobilized target is compared with the binding when a 1 μm concentration of a potentially competing sAB (purified protein) is present in solution. Thus, during epitope binning experiments of the open and closed form of MBP, Avi-MBP was preincubated with 1 μm sAB-7O and sAB-11M, respectively, for 15 min at room temperature before the addition of the phage displaying a different sAB. The culture supernatants of the phage displaying the sABs were diluted 10-fold in buffers containing a 1 μm concentration of the respective purified sAB to make sure that the epitopes are bound to the competing sAB during phage binding. As before, ELISAs involving the closed form of MBP were done in the presence of 1 mm maltose in all of the buffers.
To monitor whether each class of sABs bind in the presence of the other, protein ELISA was performed with purified sABs, which were cloned in expression vector pRH2.2, expressed in E. coli BL21 (gold) cells, and purified by protein A followed by ion-exchange chromatography on AKTA-Explorer. Both sAB-7O and sAB-11M were expressed and purified as His-tagged and non-His-tagged proteins. To verify whether sAB-11M binds to MBP in the presence of sAB-7O, MBP was preincubated with a 15 μm concentration of the non-His-tagged sAB-7O before binding His-tagged sAB-11M. Similarly, the binding of sAB-7O in the presence of sAB-11M was monitored by preincubation with 2 μm non-His-tagged sAB-11M. The secondary antibody used in this experiment was HRP-conjugated anti-His6 antibody (Ab1187), and the signal intensities at 450 nm in the absence and presence of the competing sAB were compared. For the competitive ELISA, non-His-tagged sAB-7O was serially diluted (2-fold in each well) starting at 25 μm and incubated with the MBP presaturated with 2 μm His-tagged sAB-11M. The drop in the signal intensity at 450 nm indicates displacement of the His-tagged sAB by the competing non-His-tagged sAB. Notably, all of the ELISA experiments were done in the presence of 1 μm maltose.
Binding kinetics by SPR and Interaction Map calculations
Interaction analyses between MBP and sABs were performed at 20 °C using a BIACORE 3000 (GE Healthcare). Pure, monodispersed His10-tagged MBPs were immobilized onto a nitrilotriacetic acid sensor chip via the N-terminal His10 tag. For each assay, 2-fold dilution series of sABs were injected at a flow rate of 30 μl/min. All conditions were tested for at least five different sAB concentrations. Sensograms were double-referenced using blank channel and buffer injections. Data processing and kinetic analysis were performed using BiaEvaluation software (GE Healthcare). To determine kinetic rate constants, all data sets were fit to a 1:1 interaction model. To measure the affinity of the sABs for the closed form of MBP, the running buffer was supplemented with 1 mm maltose, and all of the dilutions of sample were prepared in the running buffer with maltose. The kinetic experiments of alanine mutants of sAB-11M were performed at 1 μm and 1 mm maltose. Difference in change of Gibbs free energy (ΔΔG0) was calculated from the dissociation constants (KD) at two maltose concentrations.
To study the effect of maltose on binding of sAB-7O to MBP, kinetic experiments were performed without maltose, at 50 nm, 100 nm, 250 nm, 500 nm, and 1 μm maltose. For IM calculation, SPR data were loaded to TraceDrawer® software version 1.7 to prepare all data for Interaction Map® calculations by the Ridgeview Server Software (Ridgeview Diagnostics AB, Uppsala, Sweden). Interaction Map has been developed to decompose kinetic binding curves into the contributions of parallel interactions, based on a two-dimensional distribution of kinetic parameters, and is capable of separating the signals from multiple parallel 1:1 interactions using a distribution-based fitting approach (9). Under the assumption that each interaction contributing to the complex interaction can be described as a 1:1 interaction, the measured binding curve will be a sum of all such interactions. The measured curve is approximated with a sum of a range of binding curves, each representing a 1:1 interaction with a unique combination of association rate kon (ka) and dissociation rate koff (kd) and consequently an equilibrium dissociation constant KD = koff/kon. IM identifies the most important interaction processes based on the weight factor for each peak (9). Each circular IM peak on the heat map represents a homogenous 1:1 interaction.
Isothermal titration calorimetry
All ITC experiments were conducted using a Microcal VP-ITC titration calorimeter and were performed at 25 °C. Before each titration, protein samples were dialyzed overnight against PBS (20 mm sodium phosphate, 150 mm NaCl, pH 7.4). Maltose samples were prepared using dialysis buffer. Maltose/MBP titration experiments contained MBP concentrations between 50 and 100 μm, whereas maltose concentrations in the syringe ranged from 500 to 1000 μm. For experiments titrating maltose into MBP·sAB complex, a 5:1 molar ratio of sAB/MBP was present in the sample cell, and maltose concentrations were 10-fold higher than the concentration of MBP in the syringe. The experiments titrating sAB into the maltose·MBP complex were prepared by dialyzing MBP and sABs overnight against PBS, 1 mm maltose, pH 7.4. Typically, concentrations of sABs between 5.0 and 10 μm were used, and the concentration of the maltose in the syringe ranged from 50 to 100 μm. The binding parameters (K, ΔH0, ΔS0, and n) were determined using the single-site binding model using the ITC add-in in Origin version 7 (MicroCal LLC).
Crystallization and structure determination of MBP·sAB complexes
Crystal structures of MBP in complex with endosteric sAB-7O, allosteric sAB-11M, and peristeric sAB-P1 were determined. For this, His10-MBP was purified by nickel-nitrilotriacetic acid. The His tag was cleaved subsequently by tobacco etch virus protease. The protein was further purified on a nickel column to eliminate the tag and the protease from the tag-free protein. The flow-through fractions from the subtractive purification step were concentrated and further purified to monodispersity by SEC using a 16/60 Superdex 200 column on AKTA explorer. The sAB·MBP complexes were formed by incubating monodispersed MBP with a 2-fold molar excess of purified sAB-7O, sAB-11M, and sAB-P1. The 1:1 complex was purified from excess sAB by SEC using a 16/60 Superdex 200 column equilibrated with 10 mm HEPES, pH 7.5, 100 mm NaCl. For complex formation with sAB-11M and sAB-P1, 1 mm maltose was supplemented in the buffer. Crystallization trials were set up with the concentrated, monodispersed 1:1 MBP·sAB complexes using PEG-ION suite from Hampton Research and NEXTAL Protein Complex Suite from Qiagen at 19 °C. Small crystals for all of the three complexes appeared at multiple conditions in the screens. These initial conditions were optimized by varying the pH, ionic strength, precipitant, and protein concentration. The crystallization conditions of the crystals from which diffraction data were collected were as follows: (i) sAB-7O·MBP, 0.2 m NaCl, 0.1 m sodium/potassium phosphate, pH 6.4, and 24% PEG 1000; (ii) sAB-11M·MBP, 0.1 m HEPES, pH 7.0, and 15% PEG 4000; (iii) sAB-P1·MBP, 0.2 m KSCN and 18% PEG 3350. Crystals of MBP·sAB-7O, MBP·sAB-11M, and MBP·sAB-P1 were cryoprotected in respective reservoir solutions containing 5, 20, and 15% glycerol, respectively, and flash-frozen in liquid nitrogen. Data were collected at 24-ID-E and C at the Advanced Photon Source and processed with XDS (22). Initial phases were determined in Phaser (23) by molecular replacement using previously determined crystal structures of MBP (PDB code 1OMP (4) (open form) and 1ANF (24) (closed form)) and sAB (PDB code 3PGF) (13) without CDRs as models. Pieces of variable and the constant domains of sABs were used as different ensembles during molecular replacement. The initial promising solution obtained from Phaser was subjected to iterative cycles of refinement in Phenix (25) alternated with manual model building in Coot (26). After the preliminary refinement of the polypeptide chain, the ligands were placed unambiguously according to the difference electron maps calculated at this stage followed by modeling the solvent molecules, considering their geometrical constraints as well. The progress of the refinement was monitored by a steady decrease and convergence of R and Rfree values. The stereochemical quality of the model was validated using Molprobity (27). The data collection and refinement statistics are shown in Table 1. Solvent-accessible surface areas were calculated using PISA (28), and the figures were created in PyMOL (29).
Purification of E. coli transporter MalFGK2 and reconstitution in nanodiscs
MalF, MalG, and MalK proteins were co-purified as a complex (MalFGK2) by IMAC on Talon resin (Clontech) using the His6 tag on MalK followed by SEC (30). The complex was incorporated in nanodiscs using E3D1 as the membrane scaffold protein and soy lipids (Avanti) (31). The optimal molar ratio of the components used in the reconstitution of the protein in nanodiscs is a 1:5:250 ratio of MalFGK2/membrane scaffold protein/lipids. The empty nanodiscs were separated from the loaded ones by IMAC following published protocols (32, 33). The transporter was further purified by SEC on a 10/300 S200 column to obtain monodispersed sample for the activity assay.
NADH-coupled ATPase activity assay
Maltose transport across the membrane by the ABC transporter MALFGK2 is coupled with the hydrolysis of ATP. The extent of ATP hydrolysis is a direct measure of the efficiency of the transporter in transporting maltose across the membrane, and any factor modulating the transport process affects the rate of ATP hydrolysis as well. Thus, to study the effect of sAB-P1 on maltose transport, the extent of ATP hydrolysis was monitored at 22 °C by determining the rate of conversion of NADH to NAD at 340 nm in a coupled assay (30). Typically, 1 μg of transporter was added to the assay mixture containing 50 mm Hepes/KOH, pH 8.0, 10 mm MgCl2, 4 mm phosphoenolpyruvate, 60 μg/ml pyruvate kinase, 32 μg/ml lactate dehydrogenase, 0.3 mm NADH, 1.5 mm ATP, and 150 μm maltose in the presence or absence of 2.5 μm MBP. The rate of ATP hydrolysis was monitored for 10 min. To determine the effect of sAB-P1 on the ATP hydrolysis, 7.5 μm (3-fold molar excess of MBP) sAB-P1 was added to the assay mixture and incubated for 5 min before adding the transporter.
Author contributions
S. M., J. R. H., S. S. R., and A. A. K.: conceptualization; S. M., D. H. G., J. R. H., S. S. R., M. N. L., M. M., S. S. K., and A. A. K.: formal analysis; A. A. K.: supervision; S. M., D. H. G., J. R. H., S. S. R., M. N. L., M. M., and A. A. K.: validation; S. M., D. H. G., J. R. H., S. S. R., M. N. L., M. M., S. S. K., and A. A. K.: investigation; S. M., J. R. H., M. M., and A. A. K.: methodology; S. M., J. R. H., S. S. R., and A. A. K.: writing original draft; A. A. K.: project administration; S. M., J. R. H., S. S. R., M. N. L., M. M., S. S. K., and A. A. K.: writing, review, and editing; A. A. K.: funding acquisition.
Supplementary Material
Acknowledgments
We thank Satchal K. Erramilli for kind help with the ATPase assay and Engin Ozkan, Pawel Dominik, Mateusz Lugowski, Marcin Paduch, and Katarzyna Radziwon for helpful discussions. Assistance of staff from 24-ID beamline, Advanced Photon Source, in diffraction data collection is duly acknowledged.
This work was supported by the Chicago Biomedical Consortium and the National Institutes of Health Grants GM117372 and GM094588 (to A. A. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S9.
The atomic coordinates and structure factors (codes 5BK1, 5BK2, and 5BJZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- sAB
- synthetic antibody
- MBP
- maltose-binding protein
- SPR
- surface plasmon resonance
- IM
- Interaction Map®
- CDR
- complementarity-determining region
- ITC
- isothermal calorimetry
- PDB
- Protein Data Bank
- CBP
- calmodulin-binding peptide
- CaM
- calmodulin
- SEC
- size-exclusion chromatography.
References
- 1. Aaronson D. S., and Horvath C. M. (2002) A road map for those who know JAK-STAT. Science 296, 1653–1655 10.1126/science.1071545 [DOI] [PubMed] [Google Scholar]
- 2. Oehler S., Eismann E. R., Krämer H., and Müller-Hill B. (1990) The three operators of the lac operon cooperate in repression. EMBO J. 9, 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yonetani T., Park S. I., Tsuneshige A., Imai K., and Kanaori K. (2002) Global allostery model of hemoglobin: modulation of O2 affinity, cooperativity, and Bohr effect by heterotropic allosteric effectors. J. Biol. Chem. 277, 34508–34520 10.1074/jbc.M203135200 [DOI] [PubMed] [Google Scholar]
- 4. Sharff A. J., Rodseth L. E., Spurlino J. C., and Quiocho F. A. (1992) Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry 31, 10657–10663 10.1021/bi00159a003 [DOI] [PubMed] [Google Scholar]
- 5. Millet O., Hudson R. P., and Kay L. E. (2003) The energetic cost of domain reorientation in maltose-binding protein as studied by NMR and fluorescence spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 100, 12700–12705 10.1073/pnas.2134311100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim E., Lee S., Jeon A., Choi J. M., Lee H.-S., Hohng S., and Kim H.-S. (2013) A single-molecule dissection of ligand binding to a protein with intrinsic dynamics. Nat. Chem. Biol. 9, 313–318 10.1038/nchembio.1213 [DOI] [PubMed] [Google Scholar]
- 7. Marvin J. S., and Hellinga H. W. (2001) Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 8, 795–798 10.1038/nsb0901-795 [DOI] [PubMed] [Google Scholar]
- 8. Telmer P. G., and Shilton B. H. (2003) Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants. J. Biol. Chem. 278, 34555–34567 10.1074/jbc.M301004200 [DOI] [PubMed] [Google Scholar]
- 9. Altschuh D., Björkelund H., Strandgård J., Choulier L., Malmqvist M., and Andersson K. (2012) Deciphering complex protein interaction kinetics using Interaction Map. Biochem. Biophys. Res. Commun. 428, 74–79 10.1016/j.bbrc.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 10. Peess C., von Proff L., Goller S., Andersson K., Gerg M., Malmqvist M., Bossenmaier B., and Schräml M. (2015) Deciphering the stepwise binding mode of HRG1β to HER3 by surface plasmon resonance and interaction map. PLoS One 10, e0116870 10.1371/journal.pone.0116870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koide A., Gilbreth R. N., Esaki K., Tereshko V., and Koide S. (2007) High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. U.S.A. 104, 6632–6637 10.1073/pnas.0700149104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller K. R., Koide A., Leung B., Fitzsimmons J., Yoder B., Yuan H., Jay M., Sidhu S. S., Koide S., and Collins E. J. (2012) T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS One 7, e43746 10.1371/journal.pone.0043746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rizk S. S., Paduch M., Heithaus J. H., Duguid E. M., Sandstrom A., and Kossiakoff A. A. (2011) Allosteric control of ligand-binding affinity using engineered conformation-specific effector proteins. Nat. Struct. Mol. Biol. 18, 437–442 10.1038/nsmb.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomson J., Liu Y., Sturtevant J. M., and Quiocho F. A. (1998) A thermodynamic study of the binding of linear and cyclic oligosaccharides to the maltodextrin-binding protein of Escherichia coli. Biophys. Chem. 70, 101–108 10.1016/S0301-4622(97)00113-0 [DOI] [PubMed] [Google Scholar]
- 15. Stites W. E. (1997) Protein-protein interactions: interface structure, binding thermodynamics, and mutational analysis. Chem. Rev. 97, 1233–1250 10.1021/cr960387h [DOI] [PubMed] [Google Scholar]
- 16. Paduch M., Koide A., Uysal S., Rizk S. S., Koide S., and Kossiakoff A. A. (2013) Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 60, 3–14 10.1016/j.ymeth.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rizk S. S., Kouadio J. L., Szymborska A., Duguid E. M., Mukherjee S., Zheng J., Clevenger C. V., and Kossiakoff A. A. (2015) Engineering synthetic antibody binders for allosteric inhibition of prolactin receptor signaling. Cell Commun. Signal. 13, 1 10.1186/s12964-014-0080-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rizk S. S., Mukherjee S., Koide A., Koide S., and Kossiakoff A. A. (2017) Targeted rescue of cancer-associated IDH1 mutant activity using an engineered synthetic antibody. Sci. Rep. 7, 556 10.1038/s41598-017-00728-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukherjee S., Ura M., Hoey R. J., and Kossiakoff A. A. (2015) A new versatile immobilization tag based on the ultra high affinity and reversibility of the calmodulin-calmodulin binding peptide interaction. J. Mol. Biol. 427, 2707–2725 10.1016/j.jmb.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang J., Koide A., Nettle K. W., Greene G. L., and Koide S. (2006) Conformation-specific affinity purification of proteins using engineered binding proteins: application to the estrogen receptor. Protein Expr. Purif. 47, 348–354 10.1016/j.pep.2005.10.021 [DOI] [PubMed] [Google Scholar]
- 21. Sha F., Gencer E. B., Georgeon S., Koide A., Yasui N., Koide S., and Hantschel O. (2013) Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc. Natl. Acad. Sci. U.S.A. 110, 14924–14929 10.1073/pnas.1303640110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 10.1107/S0021889893005588 [DOI] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quiocho F. A., Spurlino J. C., and Rodseth L. E. (1997) Extensive features of tight oligosaccharide binding revealed in high-resolution structuresof the maltodextrin transport/chemosensory receptor. Structure 5, 997–1015 10.1016/S0969-2126(97)00253-0 [DOI] [PubMed] [Google Scholar]
- 25. Echols N., Grosse-Kunstleve R. W., Afonine P. V., Bunkóczi G., Chen V. B., Headd J. J., McCoy A. J., Moriarty N. W., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Adams P. D. (2012) Graphical tools for macromolecular crystallography in PHENIX. J. Appl. Crystallogr. 45, 581–586 10.1107/S0021889812017293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 29. DeLano W. L. (2017) The PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC, New York [Google Scholar]
- 30. Orelle C., Ayvaz T., Everly R. M., Klug C. S., and Davidson A. L. (2008) Both maltose-binding protein and ATP are required for nucleotide-binding domain closure in the intact maltose ABC transporter. Proc. Natl. Acad. Sci. U.S.A. 105, 12837–12842 10.1073/pnas.0803799105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alvarez F. J., Orelle C., and Davidson A. L. (2010) Functional reconstitution of an ABC transporter in nanodiscs for use in electron paramagnetic resonance spectroscopy. J. Am. Chem. Soc. 132, 9513–9515 10.1021/ja104047c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dominik P. K., and Kossiakoff A. A. (2015) Phage display selections for affinity reagents to membrane proteins in nanodiscs. Methods Enzymol. 557, 219–245 10.1016/bs.mie.2014.12.032 [DOI] [PubMed] [Google Scholar]
- 33. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 10.1016/S0076-6879(09)64011-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.