

Abstract
Rhodanese domains are structural modules present in the sulfurtransferase superfamily. These domains can exist as single units, in tandem repeats, or fused to domains with other activities. Despite their prevalence across species, the specific physiological roles of most sulfurtransferases are not known. Mammalian rhodanese and mercaptopyruvate sulfurtransferase are perhaps the best-studied members of this protein superfamily and are involved in hydrogen sulfide metabolism. The relatively unstudied human thiosulfate sulfurtransferase-like domain–containing 1 (TSTD1) protein, a single-domain cytoplasmic sulfurtransferase, was also postulated to play a role in the sulfide oxidation pathway using thiosulfate to form glutathione persulfide, for subsequent processing in the mitochondrial matrix. Prior kinetic analysis of TSTD1 was performed at pH 9.2, raising questions about relevance and the proposed model for TSTD1 function. In this study, we report a 1.04 Å resolution crystal structure of human TSTD1, which displays an exposed active site that is distinct from that of rhodanese and mercaptopyruvate sulfurtransferase. Kinetic studies with a combination of sulfur donors and acceptors reveal that TSTD1 exhibits a low Km for thioredoxin as a sulfane sulfur acceptor and that it utilizes thiosulfate inefficiently as a sulfur donor. The active site exposure and its interaction with thioredoxin suggest that TSTD1 might play a role in sulfide-based signaling. The apical localization of TSTD1 in human colonic crypts, which interfaces with sulfide-releasing microbes, and the overexpression of TSTD1 in colon cancer provide potentially intriguing clues as to its role in sulfide metabolism.
Keywords: hydrogen sulfide, post-translational modification (PTM), enzyme kinetics, crystal structure, sulfur, sulfur transferase
Introduction
Hydrogen sulfide (H2S) is an important signaling molecule with effects on multiple physiological processes including neuromodulation, inflammation and cardiac function (1–7). Maintaining healthy levels of H2S in mammalian cells requires tight control of its biosynthesis and its catabolism (8, 9). H2S is produced endogenously by two enzymes in the transsulfuration pathway, cystathionine β-synthase and γ-cystathionase, as well as by mercaptopyruvate sulfurtransferase (MST)3 (10–13). H2S is oxidized via the sulfide oxidation pathway to generate the end products thiosulfate and sulfate via the concerted action of the mitochondrial enzymes sulfide quinone oxidoreductase, persulfide dioxygenase, rhodanese, and sulfite oxidase (14–16).
Protein persulfidation, a posttranslational modification of cysteine residues, is postulated to be a major mechanism by which H2S signals (17). However, the mechanism by which target proteins acquire this modification and the sulfur source(s) used in persulfidation reactions are unclear (18). Sulfurtransferases like rhodanese or the single rhodanese domain–containing TSTD1 could potentially play a role in protein persulfidation because of their capacity to form an active-site cysteine persulfide during their reaction cycle (18).
The rhodanese superfamily members are distinguished by a structural module with an α/β topology in which α-helices surround a central five-stranded β-sheet core (19). At least three variations of the rhodanese domain are found in family members (20). In some members, only a single rhodanese domain is present, such as in the bacterial GlpE (21) and human TSTD1 (22). Alternatively, the rhodanese domain can be present in a tandem repeat, as in rhodanese and MST, where the active sites are located in a cleft between the N- and C-terminal domains (14, 15). Finally, rhodanese domains are fused to other protein domains as in Cdc25 phosphatase (20) or in the PRF and CstB proteins (23, 24).
Sulfurtransferases are ubiquitously distributed enzymes that function in a range of pathways, including sulfur metabolism, iron-sulfur cluster formation, selenium metabolism, and cyanide detoxification (13, 14, 25–28). In humans, there are five sulfurtransferases, including mitochondrial rhodanese and MST, which is found in the cytoplasmic and mitochondrial compartments (29, 30). Three isoforms of TSTD, 1, 2, and 3, are annotated in the human genome database. Of these, TSTD3 lacks a catalytic cysteine and is not predicted to function as a stand-alone sulfurtransferase. TSTD2 has the conserved active site cysteine, but its function is not known. TSTD1 (previously referred to as KAT) is found in the cytoplasm, in the vicinity of the nuclear membrane (31).
Rhodanese was initially described as a thiosulfate:cyanide sulfurtransferase (32–34). The catalytic cycle of sulfurtransferases involves two half-reactions. In the first, the sulfur is transferred from a donor to an active site cysteine to form a cysteine persulfide (Cys-SSH) intermediate. In the second half-reaction, the outer sulfur from the Cys-SSH intermediate is transferred to a thiophilic acceptor to regenerate the resting enzyme (34, 35). Human rhodanese preferentially catalyzes sulfur transfer from GSSH to sulfite, forming thiosulfate and GSH (Reaction 1), and exhibits a >2 × 105-fold preference for this over the reverse reaction (14, 15).
Residues on an active-site loop containing the cysteine at which the Cys-SSH intermediate is formed are predicted to confer substrate specificity (Fig. 1A). In rhodanese, the positive charges at the proximal arginine and lysine residues in the CRKGVT motif are predicted to interact with the negative charges on the substrate (e.g. oxygen atoms in the case of thiosulfate). The uncharged serine residue in the corresponding MST motif, CGSGVT, participates in a hydrogen-bonding network with two other arginines (at positions 188 and 197) that includes the carboxyl groups of pyruvate (13, 36). In fact, the dual replacement of Arg-248 and Lys-249 in rhodanese with Gly and Ser, respectively, to mimic the MST active site increased the Km value for thiosulfate (∼17-fold) and decreased the kcat for rhodanese 6-fold (36).
Figure 1.
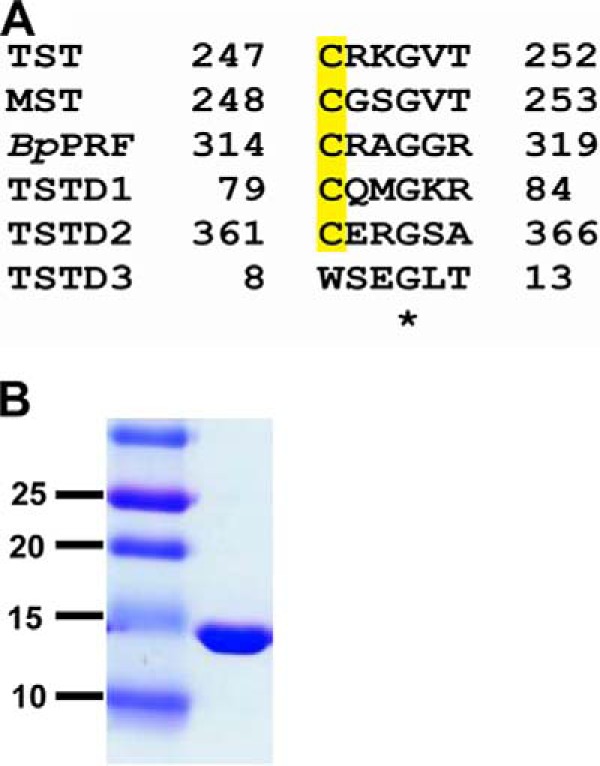
Rhodanese (TST) sequence comparison and purity of recombinant human TSTD1. A, sequence alignment of active-site sequences in the human sulfurtransferases: TST, MST, B. phytofirmans PRF (BpPRF); and TSTD1, TSTD2, and TSTD3. The conserved active-site cysteine is highlighted in yellow. B, recombinant human TSTD1 (20 μg) was estimated to be >95% pure by SDS-PAGE analysis and Coomassie Blue staining. Molecular mass markers (in kDa) are shown on the left.
The structure of the Burkholderia phytofirmans PRF, a persulfide dioxgenase-rhodanese fusion protein, displays a unique β-hairpin extension in its single rhodanese domain (24). The sequence motif in the active-site loop is CRAGGR. PRF preferentially catalyzes a sulfur transfer reaction from thiosulfate to GSH, forming sulfite and GSSH (i.e. in the direction opposite to that preferred by rhodanese) (Reaction 1). GSSH is a substrate for the fused persulfide dioxygenase domain in PRF. This difference in reaction specificity between rhodanese and the single-rhodanese domain containing PRF raises the question as to whether it is more broadly relevant to other single versus double rhodanese domain-containing proteins.
Whereas MST and rhodanese have well-described roles in sulfide biogenesis and oxidation pathways (13, 14), the role of TSTD1 is poorly characterized. Recombinant human TSTD1 utilizes thiosulfate as a sulfur donor, although the kinetics of this reaction were only characterized at the alkaline pH of 9.2 (22). Based on this activity, the involvement of TSTD1 in the sulfide oxidation pathway was proposed, although the remaining enzymes in this pathway exist inside the mitochondrion.
In this study, we report a high-resolution crystal structure and steady-state kinetic characterization of TSTD1 at a physiologically relevant pH. These studies suggest that TSTD1 is more likely to play a role in sulfide signaling than in the sulfide oxidation pathway.
Results
Expression and purification of TSTD1
A synthetic cDNA codon-optimized for Escherichia coli expression of human TSTD1 was obtained from GenScript. The recombinant protein was purified as described previously (22) and obtained in a yield of 20 mg/liter of culture. TSTD1 was judged to be >95% pure by SDS-PAGE analysis (Fig. 1B). TSTD1 eluted from a Superdex 200 column with an apparent molecular mass of ∼12 kDa, consistent with being a monomer in solution (not shown).
Crystal structure of TSTD1
The crystal structure of TSTD1 was obtained at 1.04 Å resolution by single-wavelength anomalous diffraction phasing with selenomethionine (Table 1). The final model contains two chains in the asymmetric unit. Each molecule consists of a five-stranded parallel β-sheet core framed by six α-helices (Fig. 2A). Two of these β-strands are notably short, consisting of only two residues each. Comparison of TSTD1 with the Saccharomyces cerevisiae homolog RDL1 shows that they display similar folds (Fig. 2B). TSTD1 also shares a similar fold with other rhodanese domain–containing proteins. However, some minor differences are observed. TSTD1 displays shorter helices, namely α2, α4, and α6, compared with RDL1 (Fig. 2, A and B) (19). Another point of difference is the presence of an additional α-helix, α3, in TSTD1, which is absent in RLD1.
Table 1.
X-ray data collection and refinement statistics
| TSTD1 | SeMet TSTD1 | |||
|---|---|---|---|---|
| Data collection | ||||
| Space group | P1 | P1 | ||
| Cell dimensions | ||||
| a, b, c (Å) | 37.9, 40.2, 47.2 | 38.1, 40.5, 47.5 | ||
| α, β, γ (degrees) | 113, 90, 117 | 113, 90, 117 | ||
| X-ray source | APS 23ID-B | APS 23ID-B | ||
| Wavelength (Å) | 0.7293 | 0.9793 | ||
| dmin (Å) | 1.04 (1.08 to 1.04)a | 1.26 (1.31 to 1.26)a | ||
| R-merge | 0.030 (0.698) | 0.055 (1.562) | ||
| Average I/σ(I) | 24.2 (1.7) | 18.31 (0.3) | ||
| Completeness (%) | 95.7 (89.7) | 81.08 (20.82) | ||
| Multiplicity | 7.0 (6.3) | 3.3 (1.9)b | ||
| Total observations | 703,631 (59,675) | 162,761 (3068)b | ||
| CC½ | 1 (0.908) | 0.998(0.257) | ||
| CC* | 1 (0.976) | 1 (0.64) | ||
| Refinement | ||||
| Data range (Å) | 23.28–1.04 | |||
| Reflections used in refinement | 100,881 (9498) | |||
| Rwork/Rfree (%) | 13.0/14.4 | |||
| Number of non-hydrogen atoms | 2494 | |||
| Protein | 1947 | |||
| Water | 547 | |||
| Amino acid residues | 227 | |||
| Deviation from ideality | ||||
| Bond lengths (Å) | 0.006 | |||
| Bond angles (degrees) | 0.98 | |||
| Average B-factor | 18.98 | |||
| Macromolecules | 15.51 | |||
| Solvent | 31.31 | |||
| Ramachandran plot (%) | ||||
| Favored | 97.3 | |||
| Allowed | 2.7 | |||
| Outliers | 0 | |||
| PDB code | 6BEV | |||
a Values in parentheses pertain to the outermost shell of data.
b Friedel pairs are kept separate for the anomalous wavelength data.
Figure 2.
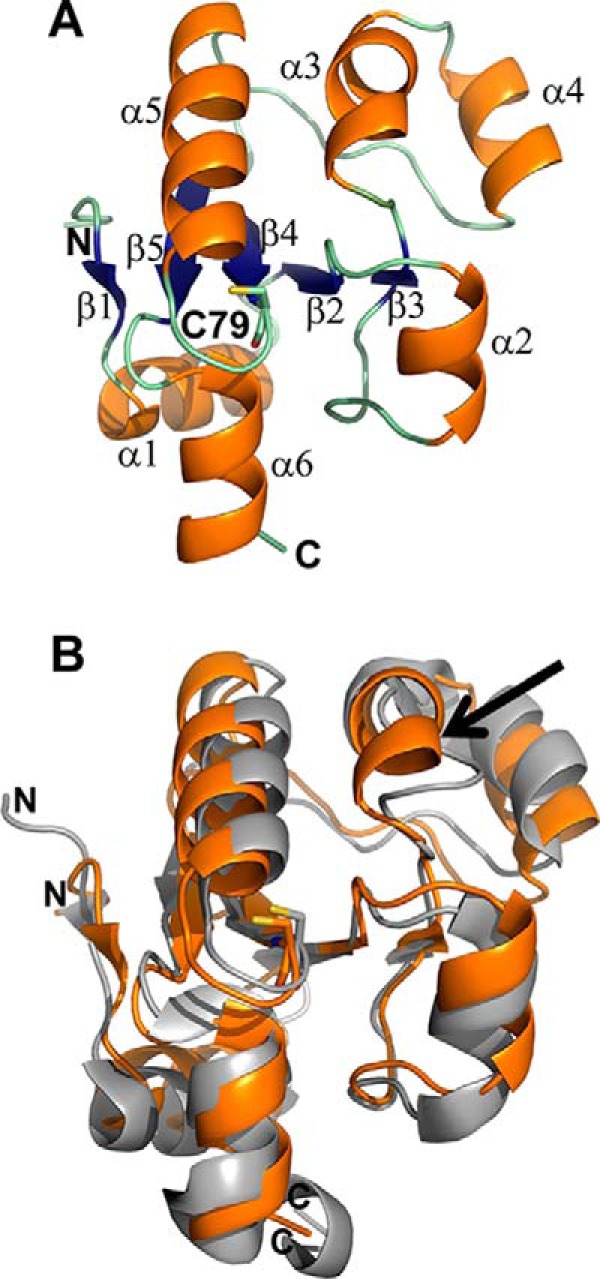
Crystal structure of TSTD1. The X-ray crystal structure of TSTD1 was solved at 1.04 Å resolution by SAD phasing with SeMet. A, the structure of the TSTD1 monomer consists of a five-stranded parallel β-sheet core (blue) surrounded by six α-helices (orange; α1–6). The active-site cysteine, Cys-79, is shown in a stick representation. B, comparison of TSTD1 with RDL1. Structural overlay of TSTD1 (orange) with the S. cerevisiae homolog RDL1 (PDB code 3D1P; gray). The active-site cysteine residues are shown in stick representation. The presence of an additional α-helix, α3, in TSTD1 is indicated by the arrow.
The active-site cysteine residue (Cys-79) in TSTD1 is located at the bottom of a shallow pocket. In contrast, in rhodanese and MST, the corresponding cysteines are sequestered in deep clefts (Fig. 3A). The architecture of the TSTD1 active-site loop is superficially similar to that of rhodanese and MST (Fig. 3B). Specifically, the orientations of the conserved cysteine and the backbone amide groups are similar in the three proteins. However, a closer inspection reveals that substitutions in the loop residues result in stereoelectronic differences between the three sulfurtransferases and probably reflect differences in their substrate preferences (Fig. 3B). In TSTD1, polar but uncharged residues Gln-80 and Met-81 replace the bulky and positively charged Arg-248 and Lys-249 in bovine rhodanese and Gly-249 and Ser-250 in human MST. The positively charged and bulky residues Lys-83 and Arg-84 in TSTD1 replace valine and threonine residues found in rhodanese and MST in the corresponding positions.
Figure 3.
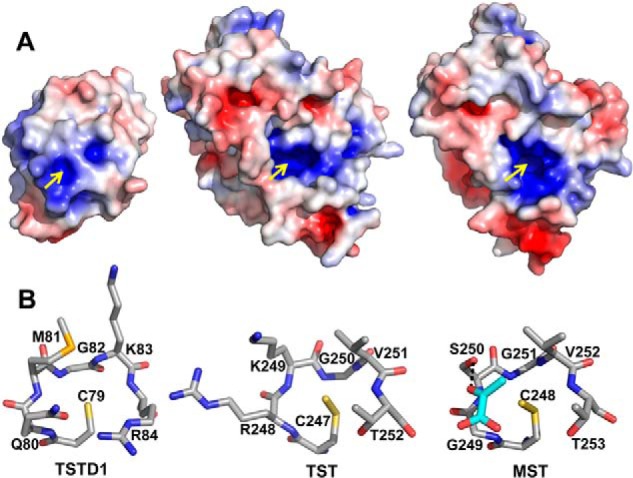
Comparison of TSTD1 with rhodanese. A, surface electrostatic potential representation of TSTD1 highlighting its exposed active-site pocket, bovine rhodanese (PDB code 1RHD), and MST (PDB code 4JGT). Positive and negative electrostatic potentials are shown in blue and red, respectively, in the range of ±5 kT/e. The yellow arrows point to the active-site cysteines. B, structural comparison of the rhodanese active-site loops of TSTD1, bovine rhodanese, and MST. Pyruvate bound in the active site of MST is shown in cyan.
Thiosulfate:cyanide sulfurtransferase activity
The capacity of TSTD1 to detoxify cyanide via sulfur transfer from thiosulfate was assessed. From the dependence of the reaction rate on substrate concentration, Km values of 0.27 ± 0.02 mm for cyanide and 22 ± 3 mm for thiosulfate were obtained (Fig. 4 (A and B) and Table 2). The specific activity of the protein at saturating substrate concentrations was 2.6 ± 0.1 μmol min−1 mg−1 protein at 37 °C, corresponding to a kcat of 0.52 s−1.
Figure 4.
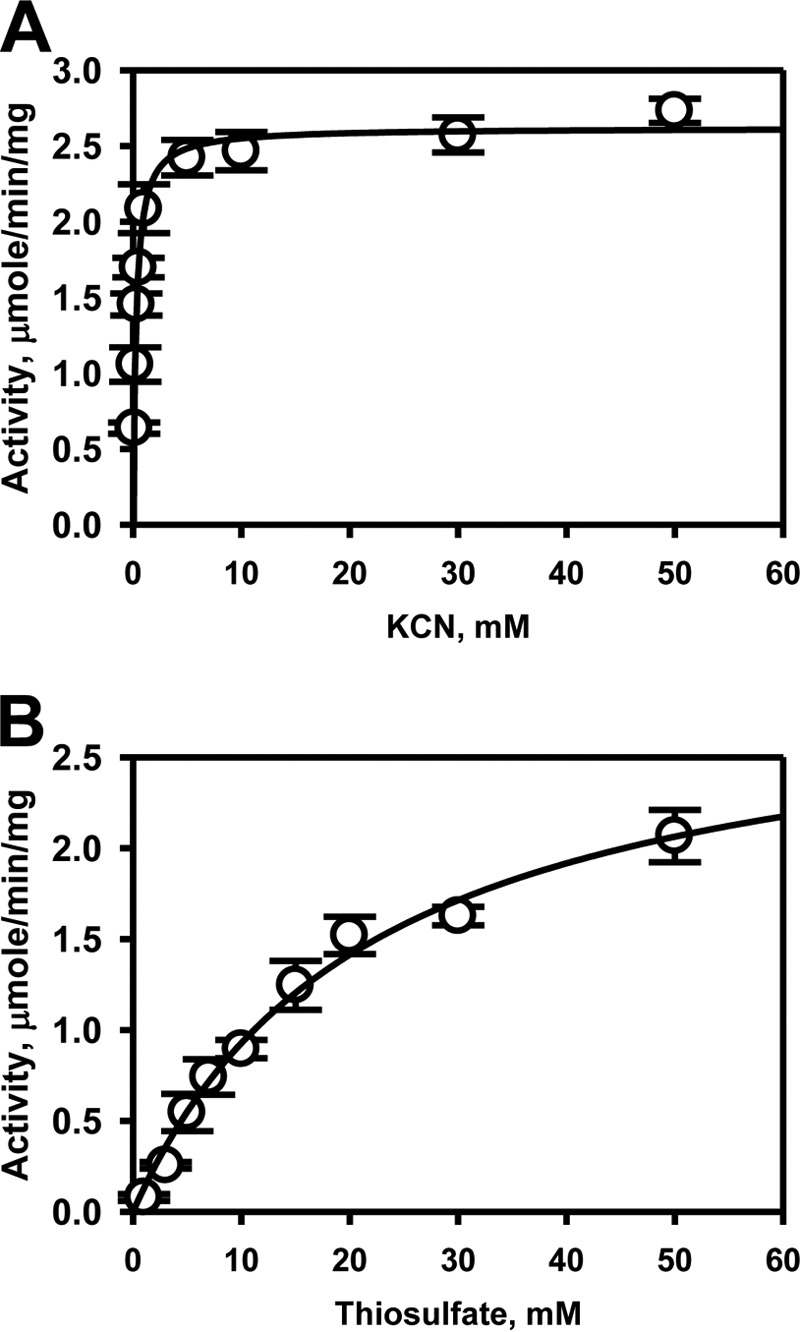
Kinetic analysis of TSTD1 thiosulfate:cyanide sulfurtransferase activity. The dependence of the reaction velocity on cyanide concentration (0.02–50 mm) in the presence of 50 mm thiosulfate (A) and on thiosulfate concentrations (1–50 mm) in the presence of 30 mm cyanide (B) were determined. The data were fitted with the Michaelis–Menten equation. Data represent the mean ± S.D. (error bars) of three independent experiments performed in 300 mm HEPES, pH 7.4, 150 mm NaCl.
Table 2.
Kinetic parameters for the sulfurtransferase activity of TSTD1, rhodanese, and MST
ND, not detected; n, Hill coefficient; S2O32− and SO32−, thiosulfate and sulfite, respectively.
| Donor | Acceptor | Km donor | Km acceptor | kcat | n | kcat/Km donor | kcat/Km acceptor |
|---|---|---|---|---|---|---|---|
| mm | mm | s−1 | m−1 s−1 | m−1 s−1 | |||
| TSTD1 | |||||||
| S2O32− | KCN | 22 ± 3 | 0.27 ± 0.02 | 0.52 | 24 | 1.94 × 103 | |
| S2O32− | GSH | 17 ± 1 | 11 ± 1 | 0.432 | 1.8 ± 0.2 | 25 | 39 |
| GSSH | SO32− | ND | ND | ND | ND | ND | |
| S2O32− | Cys | 14 ± 2 | 13.7 ± 1.9 | 0.7 | 1.9 ± 0.1 | 50 | 51 |
| S2O32− | Hcy | 18 ± 1 | 10.7 ± 0.4 | 0.61 | 2.0 ± 0.1 | 34 | 57 |
| S2O32− | Trx | 22 ± 3 | 0.017 ± 0.002 | 0.116 | 5 | 6.8× 103 | |
| Rhodanesea | |||||||
| S2O32− | KCN | 39.5 ± 2.5 | 29 ± 4 | 910 | 23 × 103 | 31 × 103 | |
| S2O32− | GSH | 0.34 ± 0.05 | 21.0 ± 0.4 | 0.67 | 2.3 ± 0.2 | 2.0 × 103 | 0.03 × 103 |
| GSSH | SO32− | 0.450 ± 0.004 | 0.06 ± 0.01 | 389 | 0.86 × 106 | 6.5 × 106 | |
| MSTa | |||||||
| 3MP | KCN | 0.35 ± 0.06 | 6 ± 1 | 2.4 | 6.8 × 103 | 0.4 × 103 | |
| 3MP | GSH | (20 ± 0.4)× 10−3 | 28 ± 2 | 0.3 | 15 × 103 | 0.01× 103 | |
| 3MP | Trx | 0.35 ± 0.06 | (2.5 ± 0.4) × 10−3 | 1.3 | 3.7 × 103 | 520× 103 |
Thiosulfate:thiol sulfurtransferase activity
The efficiency of TSTD1 in catalyzing sulfur transfer from thiosulfate to the low-molecular weight thiol acceptors, GSH, l-cysteine, and l-homocysteine, was assessed (Fig. 5 and Table 2). The Km values for the thiol acceptors ranged from 11 ± 1 mm (GSH) to 13.7 ± 1.9 mm (cysteine), whereas the specific activity ranged from 2.0 to 3.5 μmol/min/mg of protein at 37 °C (Fig. 5, A, C, and E). Substrate inhibition with thiosulfate was not observed with TSTD1 (Fig. 5, B, D, and F), as seen previously with rhodanese (14). The Km for thiosulfate ranged from 14 to 18 mm in the presence of varying acceptors.
Figure 5.

Kinetic analysis of TSTD1 thiosulfate:thiol sulfurtransferase activity. TSTD1 sulfurtransferase activity was determined in the presence of 50 mm thiosulfate and varying concentrations of GSH (A), cysteine (C), or homocysteine (E). Dependence of the reaction velocity on thiosulfate (1–50 mm) was determined in the presence of 50 mm GSH (B), 50 mm cysteine (D), or 50 mm homocysteine (F), as described under “Experimental Procedures.” The data were fitted with the Michaelis–Menten or Hill equations and represent the mean ± S.D. (error bars) of three independent experiments performed in 300 mm HEPES, pH 7.4, 150 mm NaCl. Hill coefficients for the thiol acceptors GSH, cysteine, and homocysteine are 1.8 ± 0.2, 1.9 ± 0.1, and 2.0 ± 0.1, respectively.
Thiosulfate:thioredoxin sulfurtransferase activity
Unlike rhodanese, TSTD1 can utilize thioredoxin as an acceptor in the presence of thioredoxin reductase and NADPH (Fig. 6 and Table 2). Whereas the Km for thiosulfate (Km = 22 ± 3 mm) is high as in the assays described above, the Km for thioredoxin is low (Km = 17 ± 2 μm). The kcat/Km(Trx) is ∼175-fold greater than kcat/Km(GSH) (Table 2).
Figure 6.
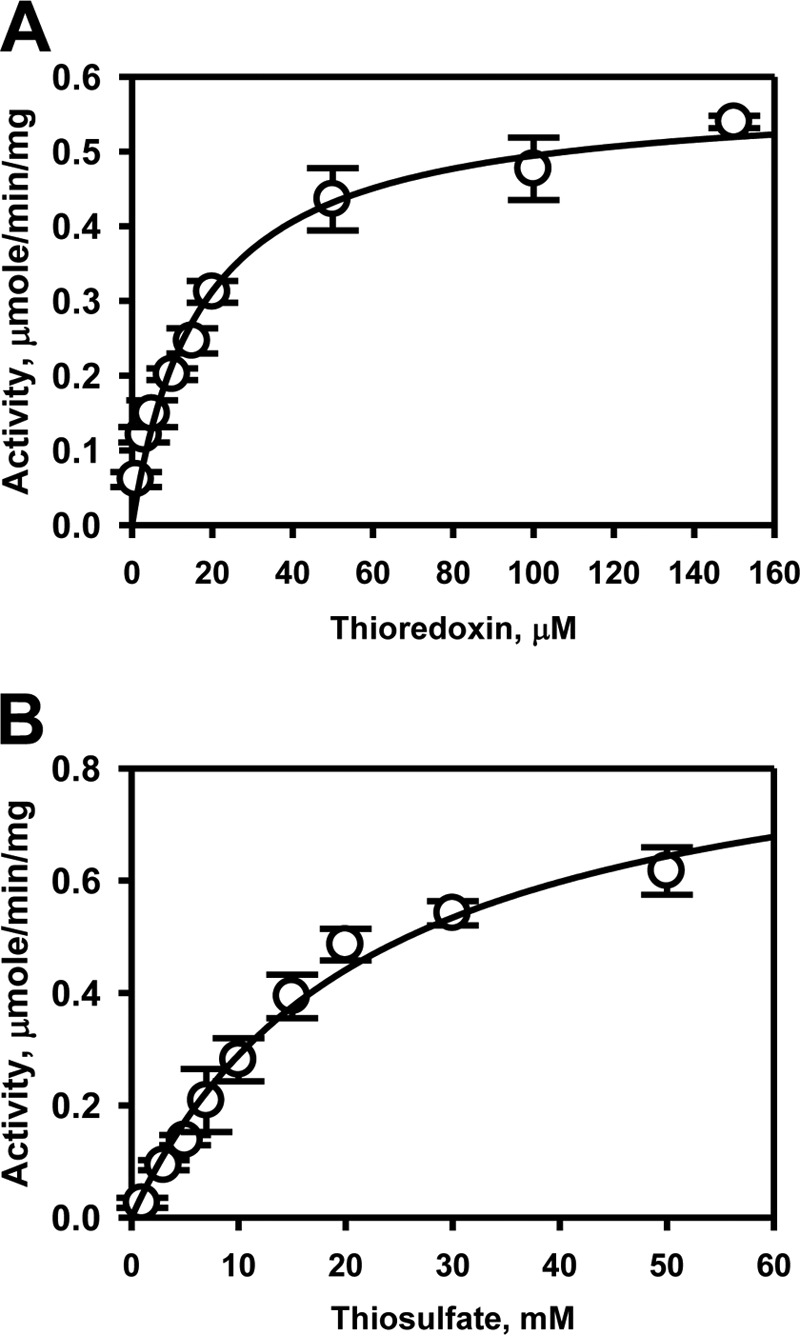
Kinetic analysis of TSTD1 thiosulfate-thioredoxin sulfur transfer activity. TSTD1 sulfur transfer activity to thioredoxin was determined in the presence of 50 mm thiosulfate and varying concentrations of thioredoxin (A) and 150 μm thioredoxin and varying concentrations of thiosulfate (B). Reactions were performed as described under “Experimental Procedures” and fitted with the Michaelis–Menten equation. Data represent the mean ± S.D. (error bars) of three independent experiments performed in 300 mm HEPES, pH 7.4, 150 mm NaCl.
Persulfide transfer to thioredoxin
To validate transfer of the sulfane sulfur from TSTD1 to thioredoxin, the biotin thiol assay (BTA) was used. This assay distinguishes between persulfides and other oxidative cysteine modifications, such as sulfenic acid (37). For this, wildtype thioredoxin and two cysteine mutants (i.e. the catalytic cysteine C32A and the resolving cysteine C35S) were used (Fig. 7A). Biotin-maleimide alkylation of thiols and persulfides led to immobilization of the proteins on a streptavidin column. DTT treatment reduced the mixed disulfide bonds in the (initially) persulfide-containing proteins and led to their elution from the column (Fig. 7B). Western blot analysis revealed the presence of wildtype thioredoxin and the C35S mutant but not of the nucleophilic C32A mutant (Fig. 7C). These results are consistent with TSTD1-dependent persulfidation of thioredoxin at the nucleophilic Cys-32 residue. In the absence of DTT, none of the thioredoxins eluted from the column.
Figure 7.
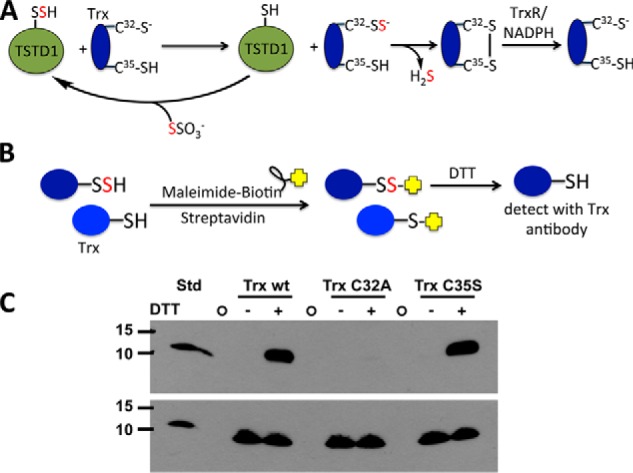
Mechanism of TSTD1-dependent persulfidation of thioredoxin and its detection by the biotin thiol assay. A, mechanism of sulfur transfer from thiosulfate to thioredoxin (Trx) via TSTD1. Cys-32 is the nucleophilic cysteine, and Cys-35 is the resolving cysteine. Oxidized thioredoxin is recycled by thioredoxin reductase (TrxR) and NADPH. B, strategy for detection of persulfidated thioredoxin using the biotin thiol assay. Reactive and accessible persulfide and thiol groups on thioredoxin are alkylated by maleimide biotin and immobilized on a streptavidin column. Following DTT treatment, only thioredoxin, which had a persulfide group, is released from the column and can be detected by immunoblotting. C, Western blot analysis of TSTD1-dependent persulfidation of thioredoxin from the biotin thiol assay. A representative Western blot is shown in which elution of wildtype thioredoxin (Trx wt) and the C35S but not the C32A mutant was detected following DTT treatment of the loaded streptavidin column. The bottom panel represents an equal loading control for the presence of thioredoxin in each sample before loading on the streptavidin column. The first lane contains purified wildtype thioredoxin (Trx wt STD; 35 ng). The circles represent empty lanes. The position of the molecular mass markers (in kDa) is shown on the left.
GSSH:sulfite sulfurtransferase activity
Because the preferred sulfur transfer reaction catalyzed by rhodanese is from GSSH to thiosulfate, the efficacy of TSTD1 in this reaction was assayed. However, unlike human rhodanese, TSTD1-catalyzed sulfur transfer from GSSH to sulfite was undetectable under steady-state turnover conditions (Table 2). To investigate whether product inhibition could account for the observed behavior of TSTD1 in this assay, the activity was measured under single-turnover conditions (Fig. 8A). Under these conditions, sulfite consumption was stoichiometric within experimental error, with thiosulfate formation. To further investigate product inhibition, the single-turnover experiment was repeated in the presence of 2× or 5× thiosulfate. Under these conditions, sulfite consumption was inhibited (Fig. 8B).
Figure 8.
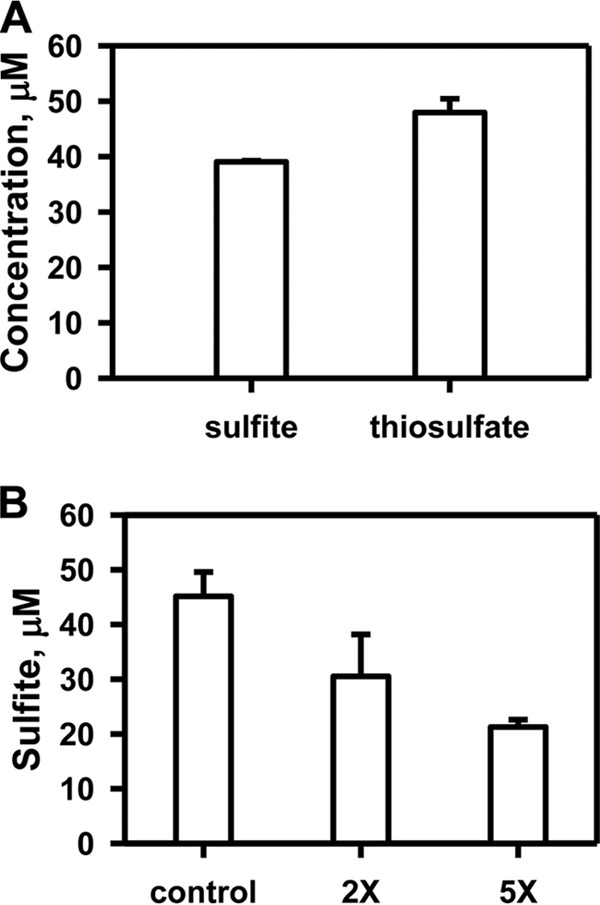
Product inhibition of TSTD1 in the GSSH-sulfite sulfur transfer reaction. A, analysis of TSTD1 reaction components under single-turnover conditions in the presence of 50 μm GSSH, 50 μm sulfite, and 50 μm enzyme. B, substrate consumption by TSTD1 under single-turnover reaction was performed in the presence of 50 μm each of GSSH, sulfite, and TSTD1 and 0 μm (control), 100 μm (2×), or 250 μm (5×) thiosulfate. Data are representative of three independent experiments each performed in duplicate in 100 mm HEPES, pH 7.4, 150 mm NaCl. Error bars, S.D.
Immunohistochemical localization of TSTD1 in colon
Since the colon is routinely exposed to high concentrations (1–2.4 mm) of sulfide produced by gut microbiota (38), we sought to examine the localization of TSTD1 in this tissue. TSTD1 antibody was validated using purified recombinant TSTD1 protein and verified in murine liver and kidney lysates (data not shown). Staining for TSTD1 was observed in colon epithelium, with stronger staining seen at the luminal surface region compared with the crypt base region (Fig. 9A). Increased staining for TSTD1 was observed in seven colon cancer tissues compared with normal colon epithelium (Fig. 9, B and C). Overall, the H-scores were significantly higher (p < 0.001) in colon cancer versus normal colon epithelium (Fig. S1).
Figure 9.

Immunohistochemical localization of TSTD1 in human colon. TSTD1 was localized in normal epithelium (A) and in colorectal cancer tissue (B) at ×10 magnification. C, close-up of the area marked in B shown at a magnification of ×40.
Discussion
The sulfurtransferase superfamily is enigmatic. For instance, 18 sulfurtransferases are annotated in the Arabidopsis thaliana genome, but their specific functions are largely unknown (39). In mammals, the roles of the two sulfurtransferases, MST and rhodanese, involved in sulfide metabolism, are relatively well-studied. Although the role of rhodanese was originally described in cyanide detoxification, its high Km for cyanide (29 mm) makes the physiological relevance of this reaction dubious. Instead, it is more likely that rhodanese catalyzes sulfur transfer from GSSH to sulfite-forming GSH and thiosulfate and is a component of the mitochondrial sulfide oxidation pathway (14). MST transfers sulfur from mercaptopyruvate to thioredoxin, generating H2S (13). The potential for MST and rhodanese to transfer sulfur to other protein acceptors has not been rigorously evaluated.
Like rhodanese and MST, TSTD1 also catalyzes sulfur transfer to cyanide. It exhibits a significantly lower Km for cyanide (0.27 mm) in comparison with rhodanese (29 mm) and MST (6 mm). The catalytic efficiency (kcat/Km) of TSTD1 for sulfur transfer from thiosulfate to cyanide is ∼16-fold lower than for rhodanese but ∼5-fold higher than for MST (for sulfur transfer from mercaptopyruvate to cyanide) (13, 14). These kinetic data beg the question as to whether TSTD1 could be more proficient in detoxifying cyanide with a different (i.e. preferred) sulfur donor, whose identity remains to be established.
Because the physiologically relevant reaction catalyzed by TSTD1 is unknown, we have examined its activity with a variety of substrates. Kinetic analysis of TSTD1 at the physiologically relevant pH of 7.4 reveals that it can catalyze a variety of sulfur transfer reactions, albeit with low efficiency. Of the sulfur acceptors tested, human TSTD1 exhibits the lowest Km for thioredoxin (18 μm), which is ∼650–1000-fold lower than for the physiologically relevant low-molecular weight thiol acceptors (Table 2). We note that the Km for GSH, although high (11 ± 1 mm), is within a physiologically relevant concentration range for this thiol, which varies from 1 to 10 mm in tissues (40, 41). Thioredoxin is also reported to be an efficient acceptor (Km = 34 μm) for the E. coli single-rhodanese domain sulfurtransferase GlpE in the presence of thiosulfate as the sulfur donor (42). Rhodanese from Azotobacter vinelandii reportedly uses thioredoxin as a sulfur acceptor with 2-propenyl thiosulfate, an organosulfur compound extracted from garlic (43), as a sulfur donor. However, we were unable to detect activity of human rhodanese with thioredoxin and thiosulfate as donor (data not shown). BTA analysis of thiosulfate-dependent persulfidation catalyzed by TSTD1 demonstrated that persulfidation of thioredoxin requires the catalytic cysteine, Cys-32. Mutation of the resolving Cys-35 in the same CXXC motif did not affect persulfide transfer to thioredoxin (Fig. 7C).
In the colon, thiosulfate is the major product of the sulfide oxidation pathway (44). The efficiency of thiosulfate production by rhodanese from GSSH and sulfite (kcat/Km(GSSH) = ∼1 × 106 m−1 s−1) is consistent with its role in the sulfide oxidation pathway (14). Unlike rhodanese, TSTD1 does not catalyze detectable sulfur transfer from GSSH (Table 2), which is due, in part, to product inhibition by thiosulfate (Fig. 8B). This product inhibition behavior of TSTD1 is similar to that of the PRF protein, which has a single rhodanese sulfurtransferase domain fused to a persulfide dioxygenase (24). In contrast, the Staphylococcus aureus CstB, which has a persulfide dioxygenase fused to a tandem repeat of rhodanese domains, catalyzes sulfur transfer from coenzyme A persulfide or bacillithiol persulfide to sulfite as an acceptor (23).
Even in the opposite direction (i.e. sulfur transfer from thiosulfate to GSH), TSTD1 is an inefficient catalyst in comparison with rhodanese. Although the specific activity and Km value for GSH are not dramatically different in TSTD1 compared with rhodanese (Table 2), the Km value of TSTD1 for thiosulfate (13 mm) is 38-fold higher than for rhodanese (14). These kinetic data suggest that TSTD1 is unlikely to play a role in the sulfide oxidation pathway using thiosulfate as a sulfur donor as previously postulated (Fig. 10A).
Figure 10.
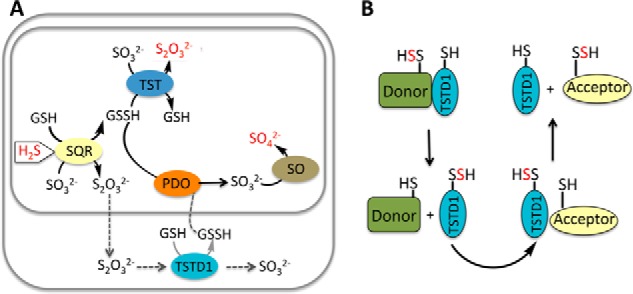
Alternative proposed roles for TSTD1. A, in one model of the mitochondrial sulfide oxidation pathway denoted by gray arrows, the product of sulfide quinone oxidoreductase (SQR) is thiosulfate (S2O32−), which is further processed in the cytoplasm by TSTD1. The product of this reaction is GSSH, which must go across the mitochondrial membranes to be completely oxidized to sulfate. This pathway is considered to be unlikely because the major product of the SQR reaction at physiologically relevant sulfite and glutathione concentrations is predicted to be GSSH. In the mitochondrially contained sulfide oxidation pathway, H2S is progressively oxidized by SQR, persulfide dioxygenase (PDO), TST, and sulfite oxidase (SO) to give thiosulfate and sulfate, which are the major products of H2S oxidation. The input (H2S) and outputs (S2O32−, SO42−) of the sulfide oxidation pathway are highlighted in red. B, based on the structure of TSTD1 and its ability to transfer sulfane sulfur to thioredoxin, we propose that TSTD1 functions in sulfur relay shuttles between donor and acceptor proteins. Alternatively, thioredoxin could potentially function as a persulfide donor and TSTD1 as an intermediary carrier for sulfane sulfur donation to acceptors.
Our conclusion that the sulfur donor for TSTD1 is unlikely to be among the ones that we and others have previously tested is supported by its crystal structure. Comparison of the active-site loop residues in TSTD1 versus rhodanese and MST (Fig. 3B) reveals significant stereoelectronic differences. Arg-248 and Lys-249, which are postulated to play a role in substrate and Cys-SSH stabilization in rhodanese, are replaced by Gln-80 and Met-81 in TSTD1, whereas similarly charged residues (Lys-83 and Arg-84) in TSTD1 are located at the opposite end of the active-site loop within an ∼5-Å distance from the active-site cysteine. Ser-250 in MST, which is postulated to play a role in substrate binding, is replaced by Met-81 in the TSTD1 active site.
Comparison of the electrostatic potential surfaces of rhodanese, MST, and TSTD1 reveals no notable differences (Fig. 3A). Neutral and positively charged residues frame the active-site entrance in each protein. However, whereas the active site of TSTD1 is relatively surface-exposed, it resides in a deep cleft in rhodanese and MST, between the two rhodanese domains (Fig. 3, A and B). The differences in the access to and geometry of the active-site pockets probably reflect differences in the substrate specificities for these three sulfurtransferases. For instance, in rhodanese, which can catalyze many of the same reactions as TSTD1, the deeply set active-site pocket probably promotes interactions with low-molecular weight substrates like GSSH, whereas in TSTD1, the shallow active-site pocket might promote interactions with larger substrates, namely proteins. In addition, the sequestered active sites in rhodanese and MST probably contribute to stabilizing the Cys-SSH intermediate, which has been seen in their crystal structures (13, 45).
Thioredoxin is postulated to play a role in H2S signaling, and inhibition of thioredoxin reductase is associated with increased intracellular persulfide levels (46, 47). The concentration of thioredoxin is reportedly ∼8–10 μm in liver (48), supporting the plausibility of its interaction with TSTD1. In principle, persulfidated thioredoxin can serve as a donor to TSTD1, whereas we have shown that the thiol form can serve as a sulfur acceptor. We speculate that other single rhodanese domain–containing proteins like TSTD1 might also have relatively accessible active sites and serve as intermediaries in sulfur transfer reactions involving other proteins (Fig. 10B). The single-domain rhodanese proteins GlpE, ThiI, and RhdA also exhibit sulfur transfer activity with thiosulfate as a donor (42, 49, 50). For example, ThiI reportedly carries sulfur between IscS and tRNA. Using [35S]cysteine, radiolabel transfer from IscS to Tus was detected (51, 52). Indeed, IscS transfers sulfane sulfur to TusA, which is then sequentially transferred to TusB/TusC/TusD/TusE and then to tRNA (51–53). The possible involvement of TSTD1 in a persulfide relay mechanism remains to be elucidated.
TSTD1 is expressed in several human tissues, including kidney, liver, skeletal muscle, heart, colon, thymus, spleen, placenta, and lung (31). The TSTD1 protein is reportedly overexpressed in breast, colon, and lung carcinoma cell lines (31). The colon is a unique compartment in which host cells are exposed to high concentrations of microbially derived sulfide and where thiosulfate rather than sulfate is the predominant sulfide oxidation product (44). As part of a larger focus on colon sulfide homeostasis in our laboratory, we examined the localization of TSTD1 in human colon. Interestingly, TSTD1 expression is higher at the apices of colonic crypts (i.e. at the host-gut microbiota interface) (Fig. 9A). A significant increase in protein expression and diffuse localization of TSTD1 were observed in cross-sections from cancerous colon tissue (H-score, p < 0.001) (Fig. 9 (B and C) and Fig. S1).
In summary, although the physiological role of TSTD1 is not known, our kinetic studies at a physiologically relevant pH make its postulated role in thiosulfate utilization unlikely. Instead, the crystal structure of the protein and its low Km for thioredoxin suggest a role in persulfide transfer involving protein partners.
Experimental procedures
Materials
Sodium sulfide, sodium sulfite, sodium thiosulfate, GSH, oxidized glutathione, l-homocysteine, and l-cysteine were purchased from Sigma.
Expression and purification of TSTD1
A synthetic gene encoding human TSTD1, which was codon-optimized for expression in E. coli, was purchased from GeneScript (Piscataway, NJ). The cDNA was inserted into the pET-28b vector using a 5′-NdeI and a 3′-SacI site. BL21 (DE3) One Shot chemically competent E. coli cells (Invitrogen) were transformed with the TSTD1 expression plasmid. A 200-ml culture in Luria–Bertani medium supplemented with 50 μg ml−1 kanamycin was grown overnight at 37 °C and used to inoculate a 6 × 1-liter culture in the same medium. Cultures were grown at 28 °C, and expression was induced with isopropyl-β-d-thiogalactopyranoside (100 μm) when the optical density at 600 nm reached 0.5. At the time of induction, the temperature was reduced to 16 °C, and growth was continued for an additional 12 h. Cells were harvested by centrifugation at 2683 × g for 20 min at 4 °C.
For selenomethionine TSTD1 expression, a 1-liter culture in Terrific broth medium was grown overnight at 37 °C and used to inoculate a 10 × 1-liter culture in minimal medium supplemented with 50 mg/liter selenomethionine to a final absorbance at 600 nm of 0.2. Cultures were grown at 28 °C, and expression was induced with isopropyl-β-d-thiogalactopyranoside (200 μm) when the absorbance at 600 nm reached 0.4. Growth was continued for an additional 16 h at 28 °C. Cells were harvested by centrifugation at 10,800 × g for 20 min at 4 °C.
TSTD1 was purified as described previously with the following modifications (22). The cell pellet from a 6-liter culture was resuspended in 150 ml of 50 mm Tris buffer, pH 8, containing 0.15 m NaCl (Buffer A), one tablet of protease inhibitor mixture (Roche Applied Science), DNase (3 mg), soybean trypsin inhibitor (2 mg), aprotinin (3 mg), phenylmethanesulfonyl fluoride (4 mg), tosyllysine chloromethyl ketone (0.45 mg), and MgSO4 (5 mm final concentration) were added to the cell suspension and stirred at 4 °C for 60 min followed by sonication on ice with the following pulse sequence: 30-s burst, 1-min rest for a total burst time of 10 min at a power output setting of 6. The sonicate was centrifuged at 38,758 × g for 1 h at 4 °C. The resulting supernatant was loaded onto a 20-ml nickel-nitrilotriacetic acid column equilibrated with Buffer A containing imidazole (final concentration 8 mm). The column was washed with 500 ml of Buffer A containing 8 mm imidazole buffer. TSTD1 was eluted from the column with a linear gradient ranging from 8 to 250 mm imidazole in Buffer A. Fractions containing the TSTD1 protein were pooled, concentrated, and dialyzed overnight against 4 liters of 50 mm Tris, pH 8. TSTD1 was then loaded on to a 5-ml Mono-Q column equilibrated with 50 mm Tris, pH 8. TSTD1 was eluted from the column with a linear gradient ranging from 0 to 1 m NaCl in 50 mm Tris, pH 8. Fractions containing TSTD1 were pooled, concentrated, and stored at −80 °C. Protein concentration was determined using Bradford reagent (Bio-Rad) with BSA as a standard. The SeMet TSTD1 was purified using the same protocol.
Thiosulfate:cyanide sulfurtransferase activity assay
The cyanide detoxification activity of TSTD1 was measured by a colorimetric assay as described previously with the following modifications (26). The reaction was performed at 37 °C and contained thiosulfate (1–50 mm), potassium cyanide (0.02–50 mm), and 5–10 μg of TSTD1 in 300 mm HEPES, pH 7.4, containing 150 mm NaCl (330-μl final volume). After a 20-min incubation, the reaction was quenched with 100 μl of 15% (w/v) formaldehyde, followed by the addition of a ferric nitrate solution (165 mm ferric nitrate nonahydrate, 13.3% (v/v) nitric acid) (500 μl) to form a ferric thiocyanate complex. Thiocyanate concentrations were determined from the absorbance at 460 nm (54). Kinetic data were fitted using SigmaPlot software (Systat Software, San Jose, CA).
Thiosulfate:thiol sulfurtransferase activity assay
The TSTD1 sulfur transfer activities from thiosulfate to various thiol acceptors were monitored as described previously by detecting H2S using the lead acetate assay (14). The reactions were performed at 37 °C as described previously and contained 1–50 mm thiosulfate and either 1–50 mm GSH, l-cysteine, or l-homocysteine. Reactions were initiated by the addition of 5–10 μg of enzyme. The reactions were monitored by the formation of lead sulfide at 390 nm, and H2S concentrations were calculated using an extinction coefficient of 5500 m−1 cm−1 (14, 54).
Thiosulfate:thioredoxin sulfurtransferase activity assay
Steady-state kinetic parameters for TSTD1 were determined in the presence of thioredoxin/thioredoxin reductase and thiosulfate. The reaction mixture contained 300 mm HEPES buffer, pH 7.4, 0.2 mm NADPH, 3.5 μm thioredoxin reductase, thioredoxin (0–150 μm), and thiosulfate (0–50 mm) as described previously (13). The persulfide formed on thioredoxin is spontaneously displaced by the resolving cysteine, releasing H2S and forming a disulfide, which is subsequently reduced by NADPH/thioredoxin reductase (Fig. 7B). The initial rate of NADPH oxidation was monitored at 340 nm, and an extinction coefficient of 6220 m−1 cm−1 was used to determine the concentration of product formed.
Persulfide analysis on thioredoxin using the BTA assay
Persulfide transfer from TSTD1 to thioredoxin was assessed using a modified BTA as described previously (37). Briefly, 120 μg of TSTD1 was preincubated with 10 mm thiosulfate for 5 min in 1 ml of 100 mm HEPES buffer, pH 7.4, at 37 °C. Then excess thiosulfate was removed using a 10 K Amicon concentrator (Millipore), and TSTD1 was incubated with 150 μg of thioredoxin (wildtype, C32A, or C35S) in 1 ml of 100 mm Tris, pH 8, for 5 min at 37 °C. The reaction mixture was incubated with 50 μm NM-biotin (EZ-Link Maleimide-PEG2-Biotin, Thermo Scientific) for 30 min with gentle mixing at room temperature and subsequently precipitated by the addition of 4 volumes of cold acetone. After incubation for 1 h at −20 °C, the samples were centrifuged at 17,000 × g for 10 min, and the precipitated pellets were suspended in 0.1% SDS, 150 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, pH 8, and gently mixed with 300 μl of streptavidin-agarose resin (Thermo Scientific) overnight at 4 °C. 50 μl of each sample was removed for use as equal loading controls. The resin was washed with 10 volumes of buffer (0.1% SDS, 600 mm NaCl, 1 mm EDTA, 1% Triton X-100, 50 mm Tris-HCl, pH 8) and incubated with 500 μl of the elution buffer (150 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, pH 8) with or without DTT (30 mm) for 30 min at room temperature. The eluted protein was concentrated using 10K Amicon concentrators (Millipore) and analyzed by Western blotting. The eluted proteins were separated by SDS-PAGE, transferred to polyvinylidene fluoride membranes, and probed with a rabbit monoclonal anti-thioredoxin antibody (Abcam; ab133524, lot GR91668-12; 1:5000 dilution).
GSSH:sulfite sulfurtransferase activity assay
The standard reaction was performed at 37 °C with a mixture containing GSSH (0.05–10 mm), sulfite (0.05–10 mm), and enzyme (5–45 μg) in 100 mm HEPES buffer, pH 7.4, 150 mm NaCl (200-μl final volume) as described previously (14, 54). The reaction was initiated by the addition of enzyme. After 30 min of incubation, monobromobimane was added to a final concentration of 8 mm and incubated at room temperature for 15 min. Samples containing millimolar concentrations of substrate were diluted 2–10-fold (final volume 200 μl) before the addition of monobromobimane. The derivatization reaction was terminated by the addition of 100 μl of 0.2 m sodium citrate, pH 2.0. Production of thiosulfate from GSSH and sulfite was monitored by HPLC. Bimane adducts of GSSH, Na2S, GSH, sulfite, and thiosulfate were prepared in 20 mm sodium phosphate, pH 7.4. Each standard (final concentration 250 μm) was incubated with a final concentration of 3 mm monobromobimane (200-μl final volume) for 10 min at 22 °C.
The derivatized samples were centrifuged at 16,000 × g for 10 min at 4 °C, and the supernatants were separated on a 4.6 × 150-cm C18 reverse-phase HPLC column (3 μm packing, SunFire). The column was equilibrated with the following solution: 100% solvent A (897.5 ml of water, 100 ml of methanol, and 2.5 ml of acetic acid) and 20% solvent B (97.5 ml of water, 900 ml of methanol, and 2.5 ml of acetic acid). The flow rate was 0.75 min/ml. Samples (50 μl) were injected onto the column and resolved using the following gradient of solvent B: 20% from 0 to 5 min, 20–45% from 5 to 10 min, 45% isocratic from 10 to 15 min, 45–50% from 15 to 25 min, 50% isocratic from 25 to 28 min, 50–100% from 28 to 30 min, 100% isocratic from 30 to 39 min, 100–20% from 39 to 42 min, 20% isocratic from 42 to 48 min. Fluorescence of the bimane adducts was detected using 340-nm excitation and 450-nm emission. Thiosulfate and sulfite concentrations were determined using a standard curve.
Protein crystallization
TSTD1 (native and SeMet) was crystallized by the sitting-drop vapor diffusion method from a 0.75 μl/0.75 μl mixture of protein stock (10 mg/ml in 50 mm Tris, pH 8, 0.25 m NaCl) and well solution. Initial crystals appeared in 2 days at 20 °C using a well solution of 0.1 m sodium cacodylate, pH 6.5, 0.1 m sodium citrate. Optimal crystals were achieved by microseeding into 0.09 m sodium cacodylate, pH 6.5, 0.77 m sodium citrate. Rhombohedron-shaped crystals were cryoprotected by passing harvested crystals through 2 μl of 0.08 m sodium cacodylate, 1.38 m sodium citrate, followed by flash cooling in liquid nitrogen.
Data collection and crystal structure determination
Diffraction data were collected at the Advanced Photon Source (APS, Argonne National Laboratory) on the GM/CA beamline 23ID-B (Table 1). For the wildtype and SeMet TSTD1 crystals, data were collected to 1.04 and 1.1 Å, respectively. All data were processed using XDS (55). The TSTD1 structure was determined by single-wavelength anomalous diffraction using Phenix to locate the selenium atoms and to determine the initial phases. Phenix AUTOBUILD was used to build ∼50% of the initial model. Model building and refinement was performed with native data using Coot (56) and Phenix Refine (57). The structure was validated using MolProbity (58). Electron density is complete for all TSTD1 residues excepting two N-terminal residues (Met-1 and Ala-2) and the N-terminal histidine tag. Figures were prepared using PyMOL (59–61).
Immunohistochemistry
Immunostaining of paraffin-embedded, formalin-fixed colon tissue sections was performed as described previously using the ABC technique (62). Briefly, seven microsections of formalin-fixed and paraffin-embedded tissues were mounted on slides. The slides were then deparaffinized in xylene and rehydrated into distilled H2O through incubation in graded alcohols. The antigen retrieval process was pursued by microwaving the slides in citrate buffer (pH 6.0; Biogenex, San Ramon, CA) for 10 min. Incubation with hydrogen peroxide (6%) in methanol was used to quench endogenous tissue peroxidase activity. The sections were post-fixed in 10% buffered formalin, washed, and blocked with 1.5% rabbit serum for 1 h. Sections were then incubated with a goat anti-TSTD1 antibody (Santa Cruz Biotechnology, Inc., sc-324457, lot C1411) overnight at 4 °C. Slides were washed in PBS and then incubated with a biotinylated anti-goat secondary antibody for 30 min at room temperature. Antigen-antibody complexes were detected with the avidin-biotin peroxidase method, using the chromogenic substrate 3,3′-diaminobenzidine (Vectastain ABC kit, catalogue no. PK-6105; Vector Laboratories, Burlingame, CA). Immunostained sections were lightly counterstained with hematoxylin and examined by light microscopy. The tumor tissues were staged according to the pTNM pathological classification (63).
The stained tissues were evaluated by a semiquantitative analysis to assign an H-score, which was calculated based on the staining intensity and the percentage of stained cells with a specific magnitude of intensity. The H-score was calculated using the equation, H-score = ∑Pi(i), where i represents the intensity of staining (0, 1, 2, and 3 represent no, weak, intermediate, and strong staining) and Pi is the percentage of stained cells with intensities varying from 0 to 100%. The H-score value ranged from 0 to 300, where 0 is complete negative staining and 300 is complete positive staining (64, 65). The criteria used to evaluate staining intensity are shown in Fig. S1.
Author contributions
M.L., N.M., and R.B. conceptualization; M.L., N.M., D.L.A., and N.S. data curation; M.L., N.M., D.L.A., N.S., E.R.F., J.L.S., and R.B. formal analysis; M.L., N.M., D.L.A., and N.S. investigation; M.L. and N.M. methodology; M.L., N.M., and R.B. writing-original draft; M.L., N.M., D.L.A., N.S., E.R.F., J.L.S., and R.B. writing-review and editing; E.R.F., J.L.S., and R.B. resources; E.R.F., J.L.S., and R.B. supervision; R.B. funding acquisition.
Supplementary Material
Acknowledgment
We thank Dr. Pramod Yadav (University of Michigan) for generating the cysteine mutants of thioredoxin used in this study.
This work was supported in part by National Institutes of Health (NIH) Grant GM112455 (to R. B.) and NIGMS, NIH, Training Grant T32GM008353 (to N. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 6BEV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Fig. S1.
- MST
- mercaptopyruvate sulfurtransferase
- TSTD1
- thiosulfate sulfurtransferase-like domain–containing 1 protein
- TST
- rhodanese
- Cys-SSH
- cysteine persulfide
- BTA
- biotin thiol assay
- SeMet
- selenomethionine
- SQR
- sulfide quinone oxidoreductase.
References
- 1. Abe K., and Kimura H. (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sodha N. R., Clements R. T., Feng J., Liu Y., Bianchi C., Horvath E. M., Szabo C., Stahl G. L., and Sellke F. W. (2009) Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J. Thorac. Cardiovasc. Surg. 138, 977–984 10.1016/j.jtcvs.2008.08.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ha C., Tian S., Sun K., Wang D., Lv J., and Wang Y. (2015) Hydrogen sulfide attenuates IL-1β-induced inflammatory signaling and dysfunction of osteoarthritic chondrocytes. Int. J. Mol. Med. 35, 1657–1666 10.3892/ijmm.2015.2183 [DOI] [PubMed] [Google Scholar]
- 4. Calvert J. W., Jha S., Gundewar S., Elrod J. W., Ramachandran A., Pattillo C. B., Kevil C. G., and Lefer D. J. (2009) Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374 10.1161/CIRCRESAHA.109.199919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., and Lefer D. J. (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565 10.1073/pnas.0705891104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., and Wang R. (2008) H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 10.1126/science.1162667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kabil O., Motl N., and Banerjee R. (2014) H2S and its role in redox signaling. Biochim. Biophys. Acta 1844, 1355–1366 10.1016/j.bbapap.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kabil O., and Banerjee R. (2014) Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox. Signal. 20, 770–782 10.1089/ars.2013.5339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kabil O., and Banerjee R. (2010) Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 10.1074/jbc.R110.128363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh S., and Banerjee R. (2011) PLP-dependent H2S biogenesis. Biochim. Biophys. Acta 1814, 1518–1527 10.1016/j.bbapap.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabil O., Vitvitsky V., Xie P., and Banerjee R. (2011) The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox. Signal. 15, 363–372 10.1089/ars.2010.3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., and Banerjee R. (2009) H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612 10.1074/jbc.M808026200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yadav P. K., Yamada K., Chiku T., Koutmos M., and Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 10.1074/jbc.M113.466177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 10.1074/jbc.M114.602664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Libiad M., Sriraman A., and Banerjee R. (2015) Polymorphic variants of human rhodanese exhibit differences in thermal stability and sulfur transfer kinetics. J. Biol. Chem. 290, 23579–23588 10.1074/jbc.M115.675694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 10.1111/j.1742-4658.2008.06482.x [DOI] [PubMed] [Google Scholar]
- 17. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 10.1038/nchembio.1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ploegman J. H., Drent G., Kalk K. H., Hol W. G., Heinrikson R. L., Keim P., Weng L., and Russell J. (1978) The covalent and tertiary structure of bovine liver rhodanese. Nature 273, 124–129 10.1038/273124a0 [DOI] [PubMed] [Google Scholar]
- 20. Bordo D., and Bork P. (2002) The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 3, 741–746 10.1093/embo-reports/kvf150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spallarossa A., Forlani F., Carpen A., Armirotti A., Pagani S., Bolognesi M., and Bordo D. (2004) The “rhodanese” fold and catalytic mechanism of 3-mercaptopyruvate sulfurtransferases: crystal structure of SseA from Escherichia coli. J. Mol. Biol. 335, 583–593 10.1016/j.jmb.2003.10.072 [DOI] [PubMed] [Google Scholar]
- 22. Melideo S. L., Jackson M. R., and Jorns M. S. (2014) Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53, 4739–4753 10.1021/bi500650h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen J., Keithly M. E., Armstrong R. N., Higgins K. A., Edmonds K. A., and Giedroc D. P. (2015) Staphylococcus aureus CstB is a novel multidomain persulfide dioxygenase-sulfurtransferase involved in hydrogen sulfide detoxification. Biochemistry 54, 4542–4554 10.1021/acs.biochem.5b00584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Motl N., Skiba M. A., Kabil O., Smith J. L., and Banerjee R. (2017) Structural and biochemical analyses indicate that a bacterial persulfide dioxygenase-rhodanese fusion protein functions in sulfur assimilation. J. Biol. Chem. 292, 14026–14038 10.1074/jbc.M117.790170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cipollone R., Ascenzi P., and Visca P. (2007) Common themes and variations in the rhodanese superfamily. IUBMB Life 59, 51–59 10.1080/15216540701206859 [DOI] [PubMed] [Google Scholar]
- 26. Sörbo B. H. (1953) Rhodanese. Acta Chem. Scand. 7, 1137–1145 10.3891/acta.chem.scand.07-1137 [DOI] [Google Scholar]
- 27. Pagani S., Bonomi F., and Cerletti P. (1984) Enzymic synthesis of the iron-sulfur cluster of spinach ferredoxin. Eur. J. Biochem. 142, 361–366 10.1111/j.1432-1033.1984.tb08295.x [DOI] [PubMed] [Google Scholar]
- 28. Ogasawara Y., Lacourciere G., and Stadtman T. C. (2001) Formation of a selenium-substituted rhodanese by reaction with selenite and glutathione: possible role of a protein perselenide in a selenium delivery system. Proc. Natl. Acad. Sci. U.S.A. 98, 9494–9498 10.1073/pnas.171320998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smirnov A., Comte C., Mager-Heckel A. M., Addis V., Krasheninnikov I. A., Martin R. P., Entelis N., and Tarassov I. (2010) Mitochondrial enzyme rhodanese is essential for 5 S ribosomal RNA import into human mitochondria. J. Biol. Chem. 285, 30792–30803 10.1074/jbc.M110.151183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagahara N., Ito T., and Minami M. (1999) Mercaptopyruvate sulfurtransferase as a defense against cyanide toxication: molecular properties and mode of detoxification. Histol. Histopathol. 14, 1277–1286 [DOI] [PubMed] [Google Scholar]
- 31. Wenzel K., Felix S. B., Flachmeier C., Heere P., Schulze W., Grunewald I., Pankow H., Hewelt A., Scherneck S., Bauer D., and Hoehe M. R. (2003) Identification and characterization of KAT, a novel gene preferentially expressed in several human cancer cell lines. Biol. Chem. 384, 763–775 [DOI] [PubMed] [Google Scholar]
- 32. Clemedson C. J., Hultman H. I., and Sorbo B. (1954) The antidote effect of some sulfur compounds and rhodanese in experimental cyanide poisoning. Acta Physiol. Scand. 32, 245–251 10.1111/j.1748-1716.1954.tb01171.x [DOI] [PubMed] [Google Scholar]
- 33. Westley J. (1981) Thiosulfate: cyanide sulfurtransferase (rhodanese). Methods Enzymol. 77, 285–291 10.1016/S0076-6879(81)77039-3 [DOI] [PubMed] [Google Scholar]
- 34. Leininger K. R., and Westley J. (1968) The mechanism of the rhodanese-catalyzed thiosulfate-cyanide reaction. Thermodynamic and activation parameters. J. Biol. Chem. 243, 1892–1899 [PubMed] [Google Scholar]
- 35. Jarabak R., and Westley J. (1974) Human liver rhodanese: nonlinear kinetic behavior in a double displacement mechanism. Biochemistry 13, 3233–3236 10.1021/bi00713a006 [DOI] [PubMed] [Google Scholar]
- 36. Nagahara N., Okazaki T., and Nishino T. (1995) Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese: striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 270, 16230–16235 10.1074/jbc.270.27.16230 [DOI] [PubMed] [Google Scholar]
- 37. Gao X. H., Krokowski D., Guan B. J., Bederman I., Majumder M., Parisien M., Diatchenko L., Kabil O., Willard B., Banerjee R., Wang B., Bebek G., Evans C. R., Fox P. L., Gerson S. L., et al. (2015) Quantitative HS-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 4, e10067 10.7554/eLife.10067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Macfarlane G. T., Gibson G. R., and Cummings J. H. (1992) Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 72, 57–64 10.1111/j.1365-2672.1992.tb04882.x [DOI] [PubMed] [Google Scholar]
- 39. Bauer M., and Papenbrock J. (2002) Identification and characterization of single-domain thiosulfate sulfurtransferases from Arabidopsis thaliana. FEBS Lett. 532, 427–431 10.1016/S0014-5793(02)03723-7 [DOI] [PubMed] [Google Scholar]
- 40. Vitvitsky V., Dayal S., Stabler S., Zhou Y., Wang H., Lentz S. R., and Banerjee R. (2004) Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R39–R46 10.1152/ajpregu.00036.2004 [DOI] [PubMed] [Google Scholar]
- 41. Vitvitsky V., Martinov M., Ataullakhanov F., Miller R. A., and Banerjee R. (2013) Sulfur-based redox alterations in long-lived Snell dwarf mice. Mech. Ageing Dev. 134, 321–330 10.1016/j.mad.2013.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ray W. K., Zeng G., Potters M. B., Mansuri A. M., and Larson T. J. (2000) Characterization of a 12-kilodalton rhodanese encoded by glpE of Escherichia coli and its interaction with thioredoxin. J. Bacteriol. 182, 2277–2284 10.1128/JB.182.8.2277-2284.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabelli R., Iorio E., De Martino A., Podo F., Ricci A., Viticchiè G., Rotilio G., Paci M., and Melino S. (2008) Rhodanese-thioredoxin system and allyl sulfur compounds. FEBS J. 275, 3884–3899 10.1111/j.1742-4658.2008.06535.x [DOI] [PubMed] [Google Scholar]
- 44. Furne J., Springfield J., Koenig T., DeMaster E., and Levitt M. D. (2001) Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem. Pharmacol. 62, 255–259 10.1016/S0006-2952(01)00657-8 [DOI] [PubMed] [Google Scholar]
- 45. Ploegman J. H., Drent G., Kalk K. H., and Hol W. G. (1978) Structure of bovine liver rhodanese. I. Structure determination at 2.5 Å resolution and a comparison of the conformation and sequence of its two domains. J. Mol. Biol. 123, 557–594 10.1016/0022-2836(78)90207-3 [DOI] [PubMed] [Google Scholar]
- 46. Dóka É., Pader I., Bíró A., Johansson K., Cheng Q., Ballagó K., Prigge J. R., Pastor-Flores D., Dick T. P., Schmidt E. E., Arnér E. S., and Nagy P. (2016) A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2, e1500968 10.1126/sciadv.1500968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wedmann R., Onderka C., Wei S., Szijártó I. A., Miljkovic J. L., Mitrovic A., Lange M., Savitsky S., Yadav P. K., Torregrossa R., Harrer E. G., Harrer T., Ishii I., Gollasch M., Wood M. E., et al. (2016) Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 7, 3414–3426 10.1039/C5SC04818D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Demarquoy J., Fairand A., Vaillant R., and Gautier C. (1991) Development and hormonal control of thioredoxin and the thioredoxin-reductase system in the rat liver during the perinatal period. Experientia 47, 497–500 10.1007/BF01959954 [DOI] [PubMed] [Google Scholar]
- 49. Palenchar P. M., Buck C. J., Cheng H., Larson T. J., and Mueller E. G. (2000) Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J. Biol. Chem. 275, 8283–8286 10.1074/jbc.275.12.8283 [DOI] [PubMed] [Google Scholar]
- 50. Colnaghi R., Pagani S., Kennedy C., and Drummond M. (1996) Cloning, sequence analysis and overexpression of the rhodanese gene of Azotobacter vinelandii. Eur. J. Biochem. 236, 240–248 10.1111/j.1432-1033.1996.00240.x [DOI] [PubMed] [Google Scholar]
- 51. Ikeuchi Y., Shigi N., Kato J., Nishimura A., and Suzuki T. (2006) Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol. Cell 21, 97–108 10.1016/j.molcel.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 52. Kambampati R., and Lauhon C. T. (2003) MnmA and IscS are required for in vitro 2-thiouridine biosynthesis in Escherichia coli. Biochemistry 42, 1109–1117 10.1021/bi026536+ [DOI] [PubMed] [Google Scholar]
- 53. Mueller E. G. (2006) Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2, 185–194 10.1038/nchembio779 [DOI] [PubMed] [Google Scholar]
- 54. Banerjee R., Chiku T., Kabil O., Libiad M., Motl N., and Yadav P. K. (2015) Assay methods for H2S biogenesis and catabolism enzymes. Methods Enzymol. 554, 189–200 10.1016/bs.mie.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 57. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., and Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 10.1093/nar/gkm276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ramsden J. D., Cocks H. C., Shams M., Nijjar S., Watkinson J. C., Sheppard M. C., Ahmed A., and Eggo M. C. (2001) Tie-2 is expressed on thyroid follicular cells, is increased in goiter, and is regulated by thyrotropin through cyclic adenosine 3′,5′-monophosphate. J. Clin. Endocrinol. Metab. 86, 2709–2716 10.1210/jc.86.6.2709 [DOI] [PubMed] [Google Scholar]
- 63. Edge S. B., Byrd D. R., Compton C. C., Fritz A. G., Greene F. L., and Trotti A. (2010) AJCC Cancer Staging Manual, 7th Ed., Springer, New York [Google Scholar]
- 64. Godbole G. B., Modi D. N., and Puri C. P. (2007) Regulation of homeobox A10 expression in the primate endometrium by progesterone and embryonic stimuli. Reproduction 134, 513–523 10.1530/REP-07-0234 [DOI] [PubMed] [Google Scholar]
- 65. Jiang J., Jin M. S., Kong F., Cao D., Ma H. X., Jia Z., Wang Y. P., Suo J., and Cao X. (2013) Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS One 8, e81799 10.1371/journal.pone.0081799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.