

Abstract
Acrolein, an α,β-unsaturated aldehyde, is generated in vivo as the end product of lipid peroxidation and from metabolic oxidation of polyamines, and it is a ubiquitous environmental pollutant. The reaction of acrolein with the N2 of guanine in DNA leads to the formation of γ-hydroxy-1-N2-propano-2′ deoxyguanosine (γ-HOPdG), which can exist in DNA in a ring-closed or a ring-opened form. Here, we identified the translesion synthesis (TLS) DNA polymerases (Pols) that conduct replication through the permanently ring-opened reduced form of γ-HOPdG ((r) γ-HOPdG) and show that replication through this adduct is mediated via Rev1/Polη-, Polι/Polκ-, and Polθ-dependent pathways, respectively. Based on biochemical and structural studies, we propose a role for Rev1 and Polι in inserting a nucleotide (nt) opposite the adduct and for Pols η and κ in extending synthesis from the inserted nt in the respective TLS pathway. Based on genetic analyses and biochemical studies with Polθ, we infer a role for Polθ at both the nt insertion and extension steps of TLS. Whereas purified Rev1 and Polθ primarily incorporate a C opposite (r) γ-HOPdG, Polι incorporates a C or a T opposite the adduct; nevertheless, TLS mediated by the Polι-dependent pathway as well as by other pathways occurs in a predominantly error-free manner in human cells. We discuss the implications of these observations for the mechanisms that could affect the efficiency and fidelity of TLS Pols.
Keywords: DNA damage, DNA damage response, DNA polymerase, DNA replication, DNA repair, gamma-HOPdG, (r) gamma-HOPdG phosphoramidite synthesis, translesion synthesis in human cells
Introduction
Acrolein, an α,β-unsaturated aldehyde, is a ubiquitous environmental pollutant formed by incomplete combustion of organic materials, and it occurs in the environment as a component of tobacco smoke and automobile exhaust. Moreover, acrolein is generated endogenously as the end product of lipid peroxidation and during the metabolic oxidation of polyamines (1–5). Acrolein adducts have been detected in DNA from a variety of tissues in rats, mice, and humans, indicating that this DNA adduct is generated in vivo from cellular reactions (1–3, 6, 7).
The reaction of acrolein with the N2 of guanine in DNA followed by ring closure results in the formation of the cyclic adduct γ-hydroxy-1,N2-propano-2′-deoxyguanosine (γ-HOPdG).2 γ-HOPdG can exist in DNA in a ring-closed or ring-opened form (8–10). γ-HOPdG presents a strong block to synthesis by replicative DNA polymerases, and it is also inhibitory to synthesis by yeast and human Polη, particularly at the nucleotide (nt) incorporation step (11, 12). DNA synthesis opposite γ-HOPdG, however, can be mediated by the sequential action of Pols ι and κ, in which Polι incorporates an nt opposite γ-HOPdG and Polκ performs the subsequent extension step (12). In the presence of a reducing agent, γ-HOPdG can be trapped as the N2-(3-hydroxypropyl) 2′-deoxyguanosine adduct, which permanently stays in the ring-opened configuration (Fig. 1A). We refer to this reduced ring-opened form of γ-HOPdG as (r) γ-HOPdG. Biochemical studies with (r) γ-HOPdG have indicated that both yeast and human Polη can carry out proficient synthesis opposite this adduct by inserting the correct nt and by extending synthesis (11, 13); Polκ also performs proficient TLS opposite this adduct by inserting the correct nt and by extending synthesis (13). Polι incorporates an nt opposite (r) γ-HOPdG; however, it incorporates a T with only an ∼3-fold reduced catalytic efficiency as compared with the correct C (13).
Figure 1.

Assay for TLS opposite (r) γ-HOPdG. A, structure of reduced γ-HOPdG. B, the target 16-mer sequence containing (r) γ-HOPdG nt at G* is shown on top, and the lacz′ sequence in the leading strand in the pBS vector containing the adduct at G* is shown below. The (r) γ-HOPdG–containing DNA strand is in frame, and it carries the Kan+ gene. TLS through the adduct generates Kan+ blue colonies.
In this study, we identify the TLS Pols required for replicating through the (r) γ-HOPdG adduct in human cells and show that TLS opposite this adduct is mediated via three independent pathways that involve Rev1 and Polη in one pathway, Polι and Polκ in another pathway, and Polθ in the third pathway, and TLS by all of these pathways is mediated in a predominantly error-free manner. We discuss the possible implications of these observations for TLS opposite (r) γ-HOPdG and other minor groove DNA lesions.
Results
Synthesis of (r) γ-HOPdG phosphoramidite
The reduced form of γ-HOPdG needed for these studies was introduced into synthetic DNA sequence during automated solid-phase DNA synthesis via a suitably protected phosphoramidite form of N2-hydroxypropanol-2′-deoxyguanosine. Previously, the reduced form of γ-HOPdG has been generated by treatment of γ-HOPdG-adducted DNA with sodium borohydride (11). Here, we describe a new method for the direct synthesis of the reduced form of γ-HOPdG rather than having to first synthesize γ-HOPdG and then converting it to the reduced form. However, site-specific direct alkylation of the N2 position of 2′-deoxyguanosine in DNA is particularly difficult because of multiple competing reactions (14). The method chosen here, although similar to γ-HOPdG modifications synthesized earlier by others (8–10), provides a more direct route for forming site-specific N2-(r) γ-HOPdG adduct during solid-phase DNA synthesis. This method is described in detail under “Experimental Procedures.” Briefly, based on the earlier method of Hofmann et al. (15) using 2′-deoxy-4-desmethylwyosine for direct N2-alkylation and conversion to N2-alkylated 2′-deoxyguanosine, this synthesis method was modified for alkylation with (3-bromopropoxy)-t-butyldimethylsilyl ether. t-butyldimethylsilyl-protected N2-(r) γ-HOPdG nucleoside was then converted to t-butyldimethylsilyl-N2-(r) γ-HOPdG phosphoramidite for use in solid-phase DNA synthesis. The t-butyldimethylsilyl protecting group of the propano ether side chain is stable to deprotection conditions during DNA synthesis and is easily converted to the free alcohol by t-butylammonium fluoride treatment before final reverse-phase HPLC purification of the (r) γ-HOPdG DNA sequence.
TLS Pols required for replicating through the (r) γ-HOPdG adduct in human cells
To identify the TLS Pols required for replicating through the (r) γ-HOPdG adduct (Fig. 1A), we examined the effects of siRNA depletions of TLS Pols on the frequency of TLS opposite this lesion carried on the template for leading strand replication in the SV40-based duplex plasmid (Fig. 1B). In this plasmid, bidirectional replication initiates from a replication origin, and TLS through the DNA lesion generates Kan+ blue colonies (16, 17). The frequency of Kan+ blue colonies among total Kan+ colonies gives a highly reliable and repeatable estimate of TLS frequencies (16–18).
In normal human fibroblasts treated with control (NC) siRNA, TLS occurs with a frequency of ∼35%, and siRNA depletion of the Rev3 catalytic or Rev7 accessory subunit of Polζ confers no reduction in TLS frequencies, indicating that Polζ plays no role in TLS opposite (r) γ-HOPdG (Table 1). siRNA depletion of Polη, Polι, Polκ, or Polθ reduced the TLS frequency to ∼22%, and siRNA depletion of Rev1 reduced the TLS frequency to ∼11% (Table 1). To determine which of the Pols function together in the same TLS pathway and which Pols function in different pathways, we examined the effects of their simultaneous depletion on TLS frequency. Our observation that co-depletion of Pols ι and κ conferred a similar reduction in TLS frequency as observed upon their individual depletion indicated that these Pols function together in the same TLS pathway (Table 1). Co-depletion of Polη with Polι or with Polκ, however, led to a further reduction in TLS frequency to ∼12% compared with that seen upon their individual depletion (∼22%), indicating that Polη functions in a TLS pathway independently of Pols ι and κ (Table 1).
Table 1.
Effects of siRNA knockdowns of TLS polymerases on the replicative bypass of the (r) γ-HOPdG lesion carried on the leading strand template in human fibroblasts
| siRNA | No. of Kan+ colonies | No. of blue colonies among Kan+ | TLS |
|---|---|---|---|
| % | |||
| NC | 669 | 235 | 35.1 |
| Rev3 | 642 | 248 | 38.6 |
| Rev7 | 486 | 185 | 38.1 |
| Polη | 548 | 130 | 23.7 |
| Polι | 623 | 141 | 22.6 |
| Polκ | 523 | 106 | 20.3 |
| Polθ | 547 | 123 | 22.5 |
| Rev1 | 412 | 46 | 11.2 |
| Polι + Polκ | 396 | 96 | 24.2 |
| Polη + Polι | 308 | 39 | 12.7 |
| Polη + Polκ | 326 | 41 | 12.6 |
| Polη + Polθ | 278 | 32 | 11.5 |
| Polι + Polθ | 317 | 36 | 11.4 |
| Polκ + Polθ | 302 | 36 | 11.9 |
To determine whether Polθ functions together with Polη or whether it functions in an independent pathway, we examined the effects of co-depletion of Pols η and θ. Our observation that co-depletion of these Pols confers a greater reduction in TLS frequency than that seen upon their individual depletion indicated that Polη and Polθ function in different TLS pathways (Table 1). To verify that Polι/Polκ-mediated TLS operates independently of Polθ, we examined the effects of co-depletion of Polι or Polκ with Polθ. The observation that co-depletion of these Pols causes a greater reduction in TLS frequencies than that observed upon their individual depletion (Table 1) indicated that Polι/Polκ and Polθ function in different TLS pathways. From these observations, we infer that TLS opposite the (r) γ-HOPdG adduct is mediated by three independent pathways dependent on Polι/Polκ, Polη, and Polθ, respectively.
To further verify this inference, we examined the effects of depletion of TLS Pols on the frequency of TLS opposite (r) γ-HOPdG in human XPV fibroblasts defective in Polη (Table 2). In XPV cells treated with control siRNA, TLS opposite (r) γ-HOPdG occurs with a frequency of ∼25%. As expected from the involvement of Polη, Pols ι/κ, and Polθ, in three independent pathways, respectively, TLS is reduced to ∼12% upon depletion of Polι, Polκ, or Polθ or upon co-depletion of Pols ι and κ, and co-depletion of Polι or Polκ with Polθ reduced TLS frequencies to ∼5% (Table 2). The residual level of TLS that remains in XPV cells co-depleted for Polι or Polκ with Polθ probably results from the low levels of TLS Pols (∼2–3%) that persist in siRNA-treated cells (18, 19) (data not shown). Regardless of this consideration, the requirement of Polι/Polκ and Polθ for TLS through (r) γ-HOPdG in XPV cells provides further confirmatory evidence for the involvement of three independent pathways for replicating through this minor groove DNA adduct (Fig. 2).
Table 2.
Effects of siRNA knockdowns of TLS polymerases on the replicative bypass of the (r) γ-HOPdG lesion carried on the leading strand template in XPV human fibroblasts
| siRNA | No. of Kan+ colonies | No. of blue colonies among Kan+ | TLS |
|---|---|---|---|
| % | |||
| NC | 238 | 60 | 25.2 |
| Polι | 272 | 33 | 12.1 |
| Polκ | 286 | 30 | 10.5 |
| Polθ | 236 | 34 | 12.8 |
| Polι + Polκ | 245 | 30 | 12.2 |
| Polι + Polθ | 204 | 10 | 4.9 |
| Polκ + Polθ | 196 | 9 | 4.6 |
Figure 2.
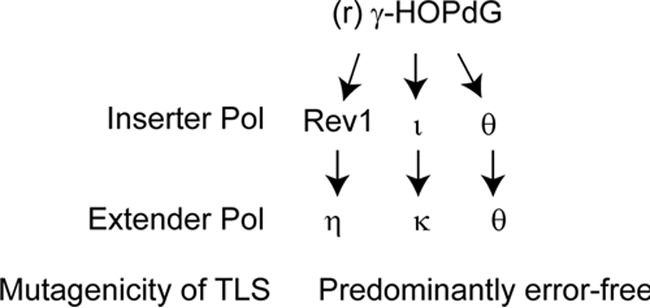
Pathways for replicating through the (r) γ-HOPdG adduct in human cells. Replication through the (r) γ-HOPdG adduct is mediated by three independent pathways in a highly error-free manner. See “Discussion” for roles of the TLS Pols in these pathways.
Catalytic and non-catalytic roles of Rev1 in TLS opposite (r) γ-HOPdG in human cells
We have shown previously that Rev1 promotes replication through UV lesions together with Pols η, ι, and κ (18). To verify that for TLS opposite (r) γ-HOPdG also, Rev1 functions together with Y-family Pols, we analyzed the effects of simultaneous depletion of Rev1 with Polη, Polι, or Polκ on TLS frequencies. TLS frequency in Rev1-depleted cells is reduced to ∼11%, a level similar to that in cells co-depleted for Polη with Polι or for Polη with Polκ (Table 1). Our finding that simultaneous depletion of Rev1 with Polη, Polι, or Polκ confers no further reduction in TLS frequency than that observed upon Rev1 depletion alone indicates that Rev1 functions together with Pols η, ι, and κ for TLS opposite (r) γ-HOPdG (Table 3). Because co-depletion of Rev1 with Polθ leads to a much greater reduction in TLS frequency than observed upon Rev1 depletion, Polθ-mediated TLS occurs independently of Rev1 (Table 3).
Table 3.
Effects of co-depletion of Rev1 with Y-family Pols or with Polθ on TLS opposite (r) γ-HOPdG carried on the leading stand template in human fibroblasts
| siRNA | No. of Kan+ colonies | No. of blue colonies among Kan+ | TLS |
|---|---|---|---|
| % | |||
| NC | 669 | 235 | 35.1 |
| Rev1 | 412 | 46 | 11.2 |
| Rev1 + Polη | 381 | 43 | 11.3 |
| Rev1 + Polι | 394 | 45 | 11.4 |
| Rev1 + Polκ | 426 | 47 | 11.0 |
| Rev1 + Polθ | 468 | 20 | 4.3 |
Among the TLS Pols, Rev1 is highly specialized for incorporating a C opposite template G (20, 21). Crystal structures of yeast and human Rev1 have shown that template G is evicted from the DNA helix into a solvent-filled cavity, and an Arg residue in Rev1 forms hydrogen bonds with the incoming C (22, 23). This protein-template–directed mechanism of nt incorporation is highly suited to allow Rev1 to insert a C opposite the minor groove N2-dG adducts. Therefore, to confirm that Rev1 DNA polymerase activity was required for TLS opposite (r) γ-HOPdG, we expressed the 3× FLAG-siRNA-sensitive wildtype Rev1 (Fig. 3A) or the siRNA-resistant (siR) form of either the wildtype or the catalytic mutant D570A/E571A Rev1 in human cells (Fig. 3B) and examined the effects of this mutation on TLS opposite (r) γ-HOPdG. In Rev1 siRNA-treated cells expressing 3× FLAG-wildtype Rev1, Rev1 was efficiently depleted (Fig. 3A), and the siRNA-resistant form of wildtype Rev1 or catalytically inactive (AA) Rev1 was not affected (Fig. 3B). As shown in Table 4, TLS in human fibroblast cells treated with Rev1 siRNA and carrying the vector control occurs with a frequency of ∼12%, and TLS occurs with a similar frequency in cells expressing the siRNA-sensitive wildtype Rev1 protein. By contrast, in cells expressing the siRNA-resistant WT Rev1, TLS is restored near to WT levels (∼32%), indicating that expression of WT Rev1 complements the TLS deficiency caused by Rev1 depletion. Expression of the siRNA-resistant Rev1 D570A/E571A catalytic mutant in cells from which genomic Rev1 has been depleted, however, reduced the TLS frequency to ∼22%, which suggested that in addition to its non-catalytic role in TLS in Polη and Polι/Polκ pathways, Rev1 DNA polymerase activity is required for TLS opposite (r) γ-HOPdG. To determine whether Rev1 DNA polymerase activity contributes to TLS in the Polη-dependent pathway, we expressed the siRNA-resistant Rev1 catalytic mutant in cells from which both Rev1 and Polη have been depleted. Our observation that TLS frequencies remain the same in Polη-proficient or -deficient cells (∼22%) expressing the Rev1 catalytic mutant indicates that Rev1 polymerase activity functions in TLS opposite (r) γ-HOPdG in conjunction with Polη (Table 4). Altogether, the genetic data in Tables 1–4 support the conclusion that replication through the (r) γ-HOPdG adduct is mediated via three different pathways in which Rev1 and Polη function in one pathway, Polι and Polκ function in another pathway, and Polθ mediates the third pathway (Fig. 2).
Figure 3.

Stable expression of wildtype Rev1 and catalytic mutant of Rev1 in human fibroblasts. Normal human fibroblasts (GM637) expressing 3× FLAG-siRNA-sensitive WT Rev1 (A) or 3× FLAG-siRNA-resistant (siR) WT Rev1 or 3× FLAG-siR D570A/E571A (AA) mutant Rev1 (B) were treated with siRNA for 48 h. The efficiencies of Rev1 depletion and Rev1 expression were determined by Western blot analysis with anti-FLAG antibody. Actin was used as the loading control.
Table 4.
Effect of siRNA knockdown of Rev1 alone or together with Polη on TLS opposite (r) γ-HOPdG present on the leading strand template in wildtype human fibroblasts carrying a vector expressing the siRNA-resistant (siR) form of wildtype or catalytically inactive Rev1
| siRNA | Vector-expressing | No. of Kan+ colonies | No. of blue colonies among Kan+ | TLS |
|---|---|---|---|---|
| % | ||||
| Rev1 | Vector control | 325 | 39 | 12.0 |
| Rev1 | FLAG-WT-Rev1 | 384 | 45 | 11.7 |
| Rev1 | FLAG-WT-siR-Rev1 | 326 | 104 | 31.9 |
| Rev1 | FLAG-D570A/E571A-siR-Rev1 | 358 | 79 | 22.1 |
| Polη + Rev1 | FLAG-WT-Rev1 | 348 | 40 | 11.5 |
| Polη + Rev1 | FLAG-WT-siR-Rev1 | 316 | 75 | 23.7 |
| Polη + Rev1 | FLAG-D570A/E571A-siR-Rev1 | 372 | 81 | 21.8 |
Low mutagenicity of TLS opposite (r) γ-HOPdG
We find that TLS opposite (r) γ-HOPdG occurs in a highly error-free manner, as only ∼1% of TLS products harbor mutations. Among the ∼400 TLS products sequenced from cells treated with control siRNA, we observed four mutations wherein an A or T was incorporated opposite the lesion site (Table 5). TLS opposite (r) γ-HOPdG incurred the same low mutagenicity regardless of whether Polη, Polι, Polκ, or Polθ was depleted, indicating that all of these Pols function in a predominantly error-free manner opposite this DNA lesion (Table 5).
Table 5.
Effects of siRNA knockdowns of TLS polymerases on the frequencies of nucleotides inserted opposite (r) γ-HOPdG carried on the leading strand template in human fibroblasts
| siRNA | No. of kan+ blue colonies sequenced | Nucleotide inserted |
Mutation frequency | |||
|---|---|---|---|---|---|---|
| A | G | C | T | |||
| % | ||||||
| NC siRNA | 386 | 2 | 0 | 382 | 2 | 1.0 |
| Polη | 384 | 2 | 0 | 381 | 1 | 0.8 |
| Polι | 288 | 2 | 0 | 284 | 2 | 1.4 |
| Polκ | 196 | 1 | 0 | 194 | 1 | 1.0 |
| Polθ | 196 | 0 | 0 | 194 | 2 | 1.0 |
Roles of TLS Pols in DNA synthesis opposite (r) γ-HOPdG
Rev1 inserts a C opposite the ring-closed form of γ-HOPdG with the same catalytic efficiency and fidelity as it inserts a C opposite undamaged G (24). As expected, in the presence of all four dNTPs, Rev1 inserts a C opposite (r) γ-HOPdG and does not carry out extension of synthesis from the inserted nt (data not shown). For the Rev1 pathway, based on this biochemical observation, genetic data shown in Table 4, and structural observations (22, 23), we suggest a role for Rev1 at the nt insertion step and for Polη at the extension step of TLS opposite (r) γ-HOPdG. For the Polι/Polκ pathway, we have previously reported biochemical evidence indicating that Polι incorporates a C or a T opposite this lesion (13). Based on biochemical (13) and structural considerations (25, 26), we suggest a role for Polι at the nt insertion step of TLS opposite (r) γ-HOPdG and for Polκ in extending synthesis from the nt inserted by Polι opposite the lesion site (27). By contrast to the highly error-prone role of Polι in incorporating a T nt opposite (r) γ-HOPdG with only an ∼3-fold reduced catalytic efficiency compared with a C (13), Polθ primarily incorporates a C opposite (r) γ-HOPdG (Fig. 4). Although Polθ can carry out the subsequent extension of synthesis, it is considerably blocked in extending synthesis from the nt inserted opposite the adduct (Fig. 4).
Figure 4.
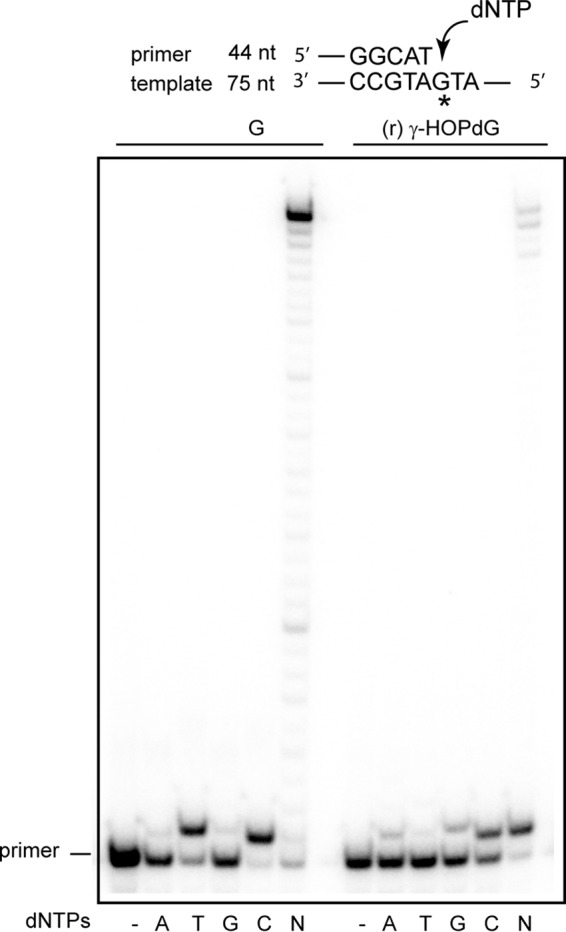
Deoxynucleotide incorporation opposite undamaged G and (r) γ-HOPdG by Polθ. Polθ (1 nm) was incubated with DNA substrate (10 nm) and 10 μm each of a single deoxynucleotide dATP, dTTP, dGTP, or dCTP, or all four deoxynucleotides (N) for 10 min at 37 °C. DNA substrate is shown above the gel where an asterisk denotes an undamaged G or an (r) γ-HOPdG template base.
Discussion
Our genetic observations indicating that replication through the (r) γ-HOPdG adduct is mediated by three independent pathways, composed of Rev1/Polη, Polι/Polκ, and Polθ, respectively (Fig. 2), differ strikingly from the roles predicted for TLS Pols from biochemical studies. Thus, from the proficient ability of Polη and Polκ to perform TLS opposite (r) γ-HOPdG by both inserting a correct nt opposite the adduct and then by extending synthesis from the inserted nt, one would have expected these two Pols to conduct replication through the adduct via two independent pathways (11, 13). However, rather than acting by themselves in their respective pathway, we find that in human cells TLS opposite (r) γ-HOPdG is performed by Rev1 and Polη in one pathway and by Polι and Polκ in the other pathway. To explain this discordance in the roles for TLS Pols indicated from biochemical studies versus those inferred from genetic studies in human cells, we suggest that replication through DNA lesions in human cells is performed by TLS Pols as components of multiprotein assemblies and that the proficiency and fidelity of TLS Pols are modulated in these assemblies.
Roles of TLS Pols in Rev1/Polη and Polι/Polκ pathways
Rev1/Polη pathway
The protein-template–directed mechanism of nt incorporation provides Rev1 the ability to insert a C opposite N2-dG adducts that protrude into the DNA minor groove (22, 23). We find that in the presence of all four dNTPs, Rev1 selectively incorporates a C opposite (r) γ-HOPdG, but it fails to extend synthesis from the inserted nt (data not shown). Based on the ability of Rev1 for nt insertion and on the ability of Polη to extend synthesis from the inserted nt, we suggest that TLS in the Rev1/Polη pathway is mediated by the sequential action of Rev1 and Polη, in which following the incorporation of a C opposite (r) γ-HOPdG by Rev1, Polη would extend synthesis from the inserted nt. As judged from the proficient ability of Rev1 for incorporating a C and of Polη for extending synthesis from it, Rev1/Polη-mediated TLS opposite (r) γ-HOPdG would occur in an error-free manner in human cells.
Polι/Polκ pathway
Based on biochemical and structural considerations, we suggest a role for Polι at the nt incorporation step and for Polκ at the extension step of TLS. Polι differs greatly from other DNA Pols in the ways it synthesizes DNA opposite template purines and pyrimidines (28–31). Polι incorporates nt with a much higher catalytic efficiency and fidelity opposite template purines than opposite template pyrimidines, and among template purines, Polι shows a higher efficiency and fidelity opposite template A than opposite template G (28–31). Polι incorporates nt opposite purine templates by pushing the template A or G into a syn conformation, which then forms a Hoogsteen base pair with the incoming T or C, respectively (25, 26, 32). The ability of Polι to push the (r) γ-HOPdG adduct into a syn conformation would allow the adduct to form a Hoogsteen base pair with the correct C or the incorrect T nt, and our previous biochemical studies with Polι have shown that it proficiently incorporates a C or a T opposite (r) γ-HOPdG (13). Biochemical and structural studies have shown that Polκ is highly adapted for extending synthesis opposite from minor groove DNA lesions (13, 27); hence, Polκ could extend synthesis from the nt inserted by Polι.
The propensity of Polι for incorporating a T opposite (r) γ-HOPdG (13) would suggest that TLS mediated by the sequential action of Pols ι and κ would occur in a highly error-prone manner. However, TLS opposite this adduct by this pathway as well as by the other two pathways operates in a predominantly error-free manner in human cells. Thus, the inference derived from biochemical studies for the highly error-prone role of Polι in TLS opposite (r) γ-HOPdG does not extend to TLS that occurs in human cells.
Non-catalytic role of Rev1 in TLS
Previously, we showed that for TLS opposite UV-induced CPDs and 6-4 photoproducts, Rev1 functions as an indispensable scaffolding component of Y-family Pols η, ι, and κ and that it does not function together with Polζ (18). For TLS opposite (r) γ-HOPdG, we find that Rev1 functions together with Y-family Pols η, ι, and κ and not with Polθ. Presumably, Rev1 bound to Polη, Polι, or Polκ plays a crucial role in the formation of multiprotein assembly. It would be of great interest to identify the components of such assemblies, to determine the roles they play in the efficiency and fidelity of TLS Pols opposite DNA lesions, and to see whether the composition of multiprotein assemblies for Y-family Pols and for other TLS Pols differs for different types of DNA lesions. For elucidating the highly error-free role of Polι in TLS opposite (r) γ-HOPdG, it would be important to decipher how Rev1 together with other components of the multiprotein assembly modulates the Polι active site opposite this lesion such that its proficiency for the insertion of the incorrect nt T is greatly reduced and its ability to insert the correct nt C is highly enhanced.
Possible roles of multiprotein assemblies in activation or inhibition of TLS Pols in the Rev1/Polη and Polι/Polκ pathways
We surmise that in the Rev1/Polη and Polι/Polκ pathways, in which Rev1 and Polι would act at the nt incorporation step of TLS and Polη and Polκ would carry out the extension step in the respective pathway, the proficient ability of Polη or Polκ to insert an nt opposite (r) γ-HOPdG would be inhibited in the multiprotein ensemble of these Pols. Such inhibitory effects of multiprotein assemblies on TLS Pols, such as Polη and Polκ, which can replicate through (r) γ-HOPdG with a high catalytic efficiency and fidelity (11, 13), raise the intriguing possibility that multiprotein assemblies of TLS Pols have evolved to become highly specialized for replicating through particular types of DNA lesions. Thus, for replication through the large variety of minor groove N2-dG adducts, the Y-family Pols may employ an identical or a very similar multiprotein assembly in which the various components coordinate TLS in a highly efficient and relatively error-free manner.
Role of Polθ in TLS
By contrast to the requirement of different inserter and extender Pols for TLS mediated by Rev1/Polη and Polι/Polκ opposite (r) γ-HOPdG, our genetic studies indicate that Polθ would act at both the nt insertion and the subsequent extension step of TLS opposite this DNA lesion. How could Polθ incorporate the correct nt opposite this minor groove adduct? One possibility is that in the Polθ active site, the N2-dG adduct stays in the anti conformation and forms a Watson–Crick base pair with the incoming C, and Polθ then extends synthesis from this base pair. An alternative possibility is that this minor groove DNA adduct is accommodated in a syn conformation in the Polθ active site, and it forms a Hoogsteen base pair with the incoming C residue. Such a mode of accommodating minor groove DNA lesions in its active site could provide Polθ a greater latitude in its ability to replicate through the diverse array of DNA adducts that form at the highly reactive N2 group of a deoxyguanine.
The high fidelity of TLS in human cells
In human cells, TLS opposite (r) γ-HOPdG occurs with a high fidelity, generating only ∼1% of mutations, similar to that observed for TLS opposite DNA lesions, such as cis-syn TT dimer (16), 6-4 TT photoproduct (17), thymine glycol (33), N1-methyl adenine (34), and N3-methyl adenine (35), where at most ∼1–2% of the TLS products harbor mutations. This is despite the fact that TLS Pols synthesize DNA opposite DNA lesions with a poor fidelity. For example, although Polι incorporates a T opposite the (r) γ-HOPdG adduct with only a somewhat reduced catalytic efficiency as compared with a C (13), TLS opposite this adduct occurs in a predominantly error-free manner in human cells. The evidence for predominantly error-free TLS opposite a number of DNA lesions in human cells strongly suggests that TLS mechanisms have been adapted to act in highly specialized and predominantly error-free ways.
Experimental procedures
Synthesis of (r) γ-HOPdG phosphoramidite
All solvents and reagents were purchased from either Fisher or Sigma-Aldrich with the exception of bromine, which was purchased from Fluka. Anhydrous solvents were additionally treated with molecular sieves and measured to be less than 50 ppm water content by Karl–Fischer titration before use.
All NMR data were obtained on a Bruker Ultrashield 300, Avance II, 300 MHz. Positive-mode mass spectroscopy data were obtained on a Sciex 5800 MALDI TOF/TOF.
The method of Hofmann et al. (15) via 2′-deoxy-4-desmethylwyosine as the protected intermediate is preferred for the direct alkylation of the N2 position of guanosine by 3-bromo-1-(t-butyldimethylsilyl)-propanoether. For this synthesis, bromoacetone was freshly prepared using the method of Levene et al. (36). 3-Bromo-1-(t-butyldimethylsilyl)-propanoether was prepared as described (37). Synthesis of (r) γ-HOPdG phosphoramidite comprised the six steps, 1–6, described below and outlined in Fig. 5.
Figure 5.

Synthesis of (r) γ-HOPdG phosphoramidite. Synthesis of (r) γ-HOPdG phosphoramidite comprised six steps beginning with protection of the N1-N2 positions of deoxyguanosine (1) with bromoacetone to give 2′-deoxy-4-desmethylwyosine (2). Alkylation of 2 with (3-bromopropoxy)-t-butyldimethylsilyl ether gave 3-(t-butyldimethylsilyloxy)propyl-2′-deoxy-4-desmethylwyosine (3). Protection of the 5′-hydroxyl group of 3 using dimethoxytrityl chloride gave 5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxy-4-desmethylwyosine (4). Deprotection of the N1-N2 positions of 4 gave 5′-DMT-(3(t-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine (5). Phosphoramidite (6) was prepared using the bis-N,N,N′,N′-diisopropyl-β-cyanoethyl phosphitylation method.
Bromoacetone (1)
Following the method of Levene (36), acetone (50 ml, 0.676 mol) was added to a three-neck flask containing water (160 ml, 8.855 mol). Acetic acid (38 ml, 0.661 mol) was added, and the mixture was heated to 70–80 °C. Bromine (37.4 ml, 0.730 mol) was added dropwise over 1–1.5 h. When the solution was completely decolorized, it was diluted with cold water (80 ml). The mixture was cooled to 10 °C and neutralized with sodium carbonate (which turns yellow/orange; there are two layers). The oil was separated and dried with anhydrous calcium chloride. The product was distilled using a house vacuum; the fraction at 65–70 °C (brown) was the main fraction (25.88 g). 1H NMR showed mostly product, with some dibromo side products present. A second distillation gave pure product as a pale yellow liquid (22.54 g, 24.34%). 1H NMR (CDCl3), δ (ppm): 3.87 (CH2); 2.34 (CH3).
2′-Deoxy-4-desmethylwyosine (2)
Using the protection method of Hofmann et al. (15), 2′-deoxyguanosine monohydrate (1) (10 g, 35.1 mmol) was coevaporated with 20 ml of anhydrous pyridine twice and then dissolved in anhydrous dimethyl sulfoxide (120 ml) under argon with stirring. Sodium hydride (60% suspension) (1.472 g, 36.8 mmol) was added, and the mixture was stirred at room temperature under argon for 1 h. Bromoacetone (5.04 g, 36.8 mmol) was added, and stirring continued under argon for 1 h (the solution immediately turned brown). Ammonium hydroxide (60 ml, 889 mmol) was added, and the mixture was stirred an additional 2 h at room temperature and then evaporated to ∼60 ml. Acetone (60 ml) was added, and the mixture was poured into a solution of acetone (800 ml) and ether (200 ml). The mixture was stirred at 0 °C for 3 h (monitored by TLC using 10% MeOH/CH2Cl2, and developed using 1% anisaldehyde/ethanol spray with heating). When the reaction showed no further progress, the mixture was transferred to a 2-liter separatory funnel, and the layers were separated. The resulting lower layer of oil was washed further with ether (2 × 500 ml), separated, and evaporated. Water (900 ml) was added, and the crude product was preabsorbed onto silica gel (10 g) by evaporation followed by coevaporation three times with 200 ml of ethanol. The 2′-deoxy-4-desmethylwyosine absorbed silica was added to a silica gel column (200 g) and eluted using a step gradient elution of 100% CH2Cl2 to 90% CH2Cl2/MeOH to yield pure product (6.62 g, 21.68 mmol, 61.9% yield). 1H NMR (DMSO-d6) δ (ppm): 2.23 (d, J = 1.2 Hz, 3H, CH3); 2.57 (m, 2H, H2′); 3.53 (m, 2H, H5′); 3.83 (m, 1H, H4′); 4.35 (m, 1H, H3′); 4.93 (t, J = 5.4 Hz, 1H, 5′-OH); 5.27 (dd, J = 4.2, 2.1 Hz, 1H, 3′-OH); 6.21 (t, J = 6 Hz, 1H, H1′); 7.30 (s, 1H, H9); 8.07 (s, 1H, H2); 12.30 (s, 1H, NH). ES-MS+ 306.0 g/mol, exact mass = 305.11 g/mol.
(3-Bromopropoxy)-t-butyldimethylsilyl ether
Following the preparation method of Choi et al. (37), fresh 3-bromo-1-propanol (2 ml, 22.12 mmol) was dissolved in dry CH2Cl2 (8 ml). Triethylamine (3.39 ml, 24.33 mmol) and 4-(dimethylamino) pyridine (0.29 g, 2.433 mmol) were added under argon with stirring. The solution was cooled to 0 °C under argon, and t-butyldimethylsilyl chloride (3.33 g, 22.12 mmol) was added. The solution was allowed to come to room temperature while stirring for 3 h under argon (monitored by TLC using 15% ethyl acetate/hexane with anisaldehyde detection). The reaction was quenched with water (5 ml) and allowed to stir for 15 min. The crude product was extracted from the aqueous solution with CH2Cl2, (3 × 10 ml). The combined organic extracts were dried over Na2SO4, filtered, and evaporated. Crude (3-bromopropoxy)-t-butyldimethylsilyl ether was purified on a silica gel column using 15% ethyl acetate/hexane to yield 4.52 g, 17.85 mmol, 81% yield, as a yellow oil. 1H NMR (CDCl3) δ (ppm): 0.089 (s, 3H, SiCH3); 0.094 (s, 3H, SiCH3); 0.91 (s, 9H, t-Bu); 2.06 (m, 2H, BrCH2CH2CH2O); 3.53 (t, J = 5.7Hz, 2H, BrCH2CH2CH2O); 3.75 (t, J = 5.7 Hz, 2H, BrCH2CH2CH2O).
3-(t-Butyldimethylsilyloxy)propyl-2′-deoxy-4-desmethylwyosine (3)
Using the alykylation method of Hofmann et al. (15), 2′-deoxy-4-desmethylwyosine (1 g, 3.28 mmol) was dissolved in anhydrous N,N-dimethylformamide (13 ml) under an argon atmosphere. Anhydrous potassium carbonate (0.475 g, 3.44 mmol) and (3-bromopropoxy)-t-butyldimethylsilyl ether (2.074 g, 8.19 mmol) were added. The mixture was stirred at 37 °C for 19 h (turned dark reddish brown) and followed by TLC (10% MeOH/CH2Cl2, anisaldehyde detection). A second addition of (3-bromopropoxy)-t-butyldimethylsilyl ether (0.83 g, 3.28 mmol) was added, and the reaction was stirred at room temperature for an additional 65 h (followed by TLC using 15% MeOH/CHCl3). The reaction mixture was filtered through celite, and the celite pad was washed with additional warm dimethylformamide until the washings showed no UV absorbance at 254 nm. The filtrates and washings were combined and evaporated to give crude product that was purified on a silica gel column using an elution gradient of chloroform with 0.2% triethylamine to 30% methanol/chloroform with 0.2% triethylamine to give pure 3-(t-butyldimethylsilyloxy)propyl-2′-deoxy-4-desmethylwyosine (550 mg, 1.152 mmol, 35.2% yield). 1H NMR (CDCl3) δ (ppm): .03 (s, 6H, Si-CH3); .87 (s, 9H, t-Bu); 1.96 (m, 2H, NHCH2CH2CH2O); 2.33 (s, 3H, CH3); 2.44 (m, 1H, H-2′); 2.81 (m, 1H, H-2′); 3.55 (m, 2H, NHCH2CH2CH2O); 3.64 (m, 2H, H-5′); 3.71 (m, 2H, NHCH2CH2CH2O); 3.87 (m, 1H, H4′); 4.15 (m, 2H, NHCH2CH2CH2O); 4.77 (m, 1H, H-3′); 6.37 (t, J = 6.6 Hz, 1H, H-1′); 7.32 (s, 1H, H9); 8.04 (s, 1H, H-2). ES-MS+: 478.2/mol, exact mass = 477.24 g/mol.
5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxy-4-desmethylwyosine (4)
3-(t-Butyldimethylsilyloxy)propyl-2′-deoxy-4-desmethylwyosine (700 mg, 1.47 mmol, from two prepared batches) was coevaporated twice with anhydrous pyridine (5 ml) and then redissolved in anhydrous pyridine (15 ml) under argon with stirring. Dimethoxytrityl chloride (0.546 g, 1.61 mmol) was added, and the reaction was stirred at room temperature for 3 h under argon (monitored by TLC 10% MeOH/CH2Cl2 containing 0.1% triethylamine, anisaldehyde detection). A second addition of fresh dimethoxytrityl chloride (0.25 g, 0.74 mmol) was added, and after an additional 1-h reaction, TLC showed completion. The reaction was evaporated of solvent and redissolved in 15 ml of CH2Cl2. The organic solution was washed with saturated NaHCO3 (10 ml), followed by water (10 ml), and the organic layer was dried over Na2SO4, filtered, and evaporated. The 5′-DMT-(3-(tert-butyldimethylsilyloxy)propyl)-2′-deoxy-4-desmethylwyosine was purified on a silica gel column using a step gradient of CH2Cl2 with 0.1% triethylamine to 10% MeOH/CH2Cl2 with 0.1% triethylamine to give pure product (0.794 g, 1.01 mmol, 68.7% yield). 1H NMR (CDCl3) δ (ppm): 0.08 (s, 6H, SiCH3); 0.92 (s, 9H, t-Bu); 1.86 (m, 2H, NHCH2CH2CH2O); 2.45 (s, 3H, CH3); 2.5 (m, 1H, H-2′); 2.68 (m, 1H, H-2′); 3.29 (m, 1H, NHCH2CH2CH2O); 3.4 (m, 1H, NHCH2CH2CH2O); 3.62 (t, J = 5.7 Hz, 2H, H5′); 3.72 (m, 1H, H4′); 3.77 (s, 6H, OCH3); 4.1 (m, 2H, NHCH2CH2CH2O); 4.64 (m, 1H, H-3′); 6.39(t, J = 6.3 Hz, 1H, H-1′); 6.78 (dd, J = 2.4, 4.5 Hz, 4H, DMT-meta H's); 7.26 (m, 7H, DMT-ortho H's, Bz and H9); 7.38 (m, 2H, DMT-ortho H's); 7.7 (s, 1H, H-2). ES-MS+: 780.2 g/mol, exact mass = 779.37 g/mol.
5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine (5)
Deprotection was performed following the methods of Casale and McLaughlin (38), Hofmann et al. (15), and Boryski and Ueda (39). 5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxy-4-desmethylwyosine (0.79 g, 0.99 mmol) was dissolved in a mixture of THF (27 ml) and 0.2 m aqueous potassium acetate, pH 5.7 (11 ml). N-Bromosuccinimide (0.216 g, 1.19 mmol) was added, and the mixture was stirred at room temperature for 40 min. (the solution turns light blue and then pale yellow as the reaction proceeds). Ammonium hydroxide (5.7 ml) was added, and stirring was continued for 30 min (monitored by TLC using 5% MeOH/CH2Cl2 with 0.1% triethylamine). When the reaction was complete, solvent was evaporated, and the crude product was purified by silica gel column using a step gradient of 100% CH2Cl2 to 5% MeOH/CH2Cl2 with a 1% solution of triethylamine in H2O to give pure 5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine (226 mg, 0.305 mmol, 30% yield). 1H NMR (CDCl3) δ (ppm): 0.010 (s, 6H, Si-CH3); 0.85 (s, 9H, t-Bu); 1.78 (m, (2H, NHCH2CH2CH2O); 2.51 (m, 1H, H2′); 2.70 (m, 1H, H2′); 3.33 (m, 2H, NHCH2CH2CH2O); 3.61 (t, J = 5.7 Hz, 2H, H5′); 3.67 (m, 1H, H4′); 3.70 (s, 6H, OCH3); 4.08 (m, 2H, NHCH2CH2CH2O); 4.68 (m, 1H, H3′); 6.23 (t, J = 6 Hz, 1H, H1′), 6.73 (m, 4H, DMT meta H's); 7.16 (m, 7H, DMT-ortho H's and Benzyl H's); 7.33 (m, 2H, DMT-ortho H's); 7.64 (s, 1H, H2) ES-MS+: 742.2 g/mol; exact mass 741.36 g/mol.
DMT-5-(3-(tert-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine-3′-(N,N,-diisopropyl-β-cyanoethyl-phosphoramidite, [(r) γ-HOPdG phosphoramidite] (6)
5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine (0.30 g, 0.40 mmol) was dissolved in dry methylene chloride (11 ml) in an argon atmosphere in a sealed round-bottom flask with rubber stopper. 2-Cyanoethyl-N,N,N′,N′-tetraisopropylphosphine (0.177 ml, 0.55 mmol) and tetrazole/THF solution (1.2 ml, 0.51 mmol tetrazole) were added simultaneously via syringes under argon. The solution was stirred at room temperature for 1 h (monitored by TLC using prerun ethyl acetate, 0.1% triethylamine TLC plates, dried, spotted, and eluted with the same solvent and detected with anisaldehyde). A second addition of 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphine (0.128 ml, 0.40 mmol) and tetrazole solution (0.938 ml, 0.40 mmol) were added, again simultaneously with stirring, and the reaction was continued for 30 min. The reaction was diluted with CH2Cl2 (11 ml), and the product was washed with 5% aqueous sodium bicarbonate (10 ml) followed by washing with an aqueous brine solution (10 ml). The organic layer was separated, dried over sodium sulfate, and filtered. Triethylamine (20 μl) was added to the filtrate, and the solution was evaporated. The crude 5′-DMT-(3-(t-butyldimethylsilyloxy)propyl)-2′-deoxyguanosine phosphoramidite mixture was purified on a silica gel column using a step gradient of 100% ethyl acetate with 0.1% triethylamine to 10% MeOH/ethyl acetate with 0.1% triethylamine as eluant. TLC using prerun ethyl acetate, 0.1% triethylamine TLC plates, dried, spotted, and eluted with the same solvent gave two isomeric phosphoramidite products that were detected with anisaldehyde. Dittmer reagent was utilized to detect any excess 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphine agent separated from the product isomers. The phosphoramidite products were combined to yield 120 mg (0.127 mmol, 47.2% yield) 1H NMR (CD3CN) δ (ppm): 0.04 (s, 6H, SiCH3); 0.89 (s, 9H, t-Bu); 1.20 (m, 12H, NCH(CH3)2); 1.74 (m, 2H, NHCH2CH2CH2O); 2.52 (m, 3H, H2′ and CH2CH2CN); 2.63 (t, J = 6 Hz, 2H, NCH(CH3)2); 2.94 (m, 1H, H2′); 3.30 (m, 4H, H5′ and NHCH2CH2CH2O); 3.67 (m, 4H, CH2CH2CN and NHCH2CH2CH2O); 3.73 (s, 6H, OCH3); 4.16 m, 1H, H4′); 4.74 (m, 1H, H3′); 6.21 (t, J = 6.6 Hz, 1H, H1′); 6.78 (m, 4H, DMT meta H's); 7.24 (m, 7H, DMT ortho H's and bz H's); 7.39 (m, 2H, DMT ortho H's); 7.66 (s, 1H, H2). 31P NMR (CD3CN) δ (ppm): 148.79; 148.88.
Oligonucleotide syntheses
Oligonucleotides containing (r) γ-HOPdG were synthesized on a model 8909 Expedite DNA synthesizer using standard DNA synthesis chemistry. The (r) γ-HOPdG phosphoramidite was incorporated using an offline coupling method to conserve reagent. The oligonucleotides were deprotected using standard concentration ammonia deprotection, with an additional deprotection step using 0.1 m t-butylammonium fluoride, THF for removal of the t-butyldimethylsilyl chloride group from the propanohydroxyl side chain. Oligonucleotides were purified and analyzed by reverse-phase HPLC on a Beckman System Gold HPLC. Purified oligonucleotides were analyzed and confirmed by MALDI-MS on a Bruker Autoflex MALDI mass spectrophotometer.
Construction of plasmid vectors containing (r) γ-HOPdG
The in-frame target sequence of the lacz′ gene containing (r) γ-HOPdG is shown in Fig. 1B. Because the lacz′ sequence in the (r) γ-HOPdG–containing DNA strand is in-frame, it encodes functional β-gal; the opposite DNA strand harbors an SpeI restriction site containing a +1 frameshift, which makes it non-functional for β-gal. The (r) γ-HOPdG–containing strand carries the kanamycin gene (Kan+), whereas the other DNA strand has the kan− gene (Fig. 1B). The detailed methods for the construction of lesion-containing SV40-based duplex plasmids have been published previously (16, 17).
Assays for translesion synthesis and mutation analyses of TLS products in human cells
The detailed methods for TLS assays have been described previously (16, 17). Briefly, human fibroblast GM637 cells are transfected with the particular siRNA, and after 48 h of incubation, the target vector DNA and siRNA (second transfection) are co-transfected. After 30 h of incubation, plasmid DNA is transfected into E. coli XL1-Blue supercompetent cells (Stratagene), and cells are plated on LB/Kan plates containing isopropyl-1-thio-β-d-galactopyranoside (GenDEPOT) and 100 μg/ml X-Gal (GenDEPOT). TLS frequency is determined from the number of blue colonies among total colonies growing on LB/Kan plates, and mutation frequencies and mutational changes are analyzed by DNA sequencing.
DNA polymerase assays
DNA substrates consisted of a radiolabeled oligonucleotide primer annealed to a 75-nt oligonucleotide DNA template by heating a mixture of primer/template at a 1:1.5 molar ratio to 95 °C and allowing it to cool to room temperature for several hours. The template 75-mer oligonucleotide contained the sequence 5′-AGC AAG TCA CCA ATG TCT AAG AGT TCG TAT GAT GCC TAC ACT GGA GTA CCG GAG CAT CGT CGT GAC TGG GAA AAC-3′ and was either undamaged G or harbored an (r) γ-HOPdG at the underlined position. For examining the incorporation of dATP, dTTP, dCTP, or dGTP nucleotides individually or of all four dNTPs, a 44-mer primer, 5′-GTT TTC CCA GTC ACG ACG ATG CTC CGG TAC TCC AGT GTA GGC AT-3′ was annealed to the abovementioned 75-mer template.
The standard DNA polymerase reaction (5 μl) contained 25 mm Tris·HCl (pH 7.5), 5 mm MgCl2, 1 mm dithiothreitol, 100 μg/ml BSA, 10% glycerol, 10 nm DNA substrate, and 1 nm Polθ. For nucleotide incorporation assays, 10 μm dATP, dTTP, dCTP, or dGTP (Roche Applied Science) were used, and for examining synthesis through the (r) γ-HOPdG lesion, all four dNTPs (10 μm each) were used. Reactions were carried out for 10 min at 37 °C. Reaction products were resolved on a 12% polyacrylamide gel containing 8 m urea and analyzed by a PhosphorImager.
Author contributions
J. -H. Y. and R. P. H. data curation; J. -H. Y., R. P. H., S. P., and L. P. formal analysis; J. -H. Y., R. P. H., J. P., J. R. C., S. P., and L. P. investigation; J. -H. Y., R. P. H., L. C. H., and J. R. C. methodology; R. P. H., S. P., and L. P. funding acquisition; R. P. H., S. P., and L. P. writing-original draft; S. P. and L. P. conceptualization; S. P. and L. P. supervision; S. P. and L. P. validation; S. P. and L. P. project administration; S. P. and L. P. writing-review and editing.
This work was supported by National Institutes of Health (NIH) Grants ES022948 and ES020833 and in part by NIEHS, NIH, Center Grant P30 ES06676. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- γ-HOPdG
- γ-hydroxy-1,N2-propano-2′-deoxyguanosine
- (r) γ-HOPdG
- permanently ring-opened reduced form of γ-HOPdG
- nt
- nucleotide(s)
- NC
- negative control
- TLC
- thin layer chromatography
- THF
- tetrahydrofuran
- DMT
- dimethoxytrityl
- Pol
- polymerase
- TLS
- translesion synthesis
- siR
- siRNA-resistant.
References
- 1. Chung F.-L., Chen H.-J. C., and Nath R. G. (1996) Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 17, 2105–2111 10.1093/carcin/17.10.2105 [DOI] [PubMed] [Google Scholar]
- 2. Chung F.-L., Nath R. G., Nagao M., Nishikawa A., Zhou G.-D., and Randerath K. (1999) Endogenous formation and significance of 1,N2-propanodeoxyguanosine adducts. Mutat. Res. 424, 71–81 10.1016/S0027-5107(99)00009-3 [DOI] [PubMed] [Google Scholar]
- 3. Chung F.-L., Zhang L., Ocando J. E., and Nath R. G. (1999) Role of 1,N2-propanodeoxygunosine adducts as endogenous DNA lesions in rodents and humans. IARC Sci. Publ. 45–54 [PubMed] [Google Scholar]
- 4. Esterbauer H., Schaur R. J., and Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonadehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 10.1016/0891-5849(91)90192-6 [DOI] [PubMed] [Google Scholar]
- 5. Vaca C. E., Wilhelm J., and Harms-Ringdahl M. (1988) Interaction of lipid peroxidation products with DNA: a review. Mutat. Res. 195, 137–149 10.1016/0165-1110(88)90022-X [DOI] [PubMed] [Google Scholar]
- 6. Nath R. G., and Chung F.-L. (1994) Detection of exocyclic 1,N2-propanodeoxyguanosine adducts as common DNA lesions in rodents and humans. Proc. Natl. Acad. Sci. U.S.A. 91, 7491–7495 10.1073/pnas.91.16.7491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nath R. G., Ocando J. E., and Chung F.-L. (1996) Detection of 1,N2-propanodeoxyguanosine adducts as potential endogenous DNA lesions in rodent and human tissues. Cancer Res. 56, 452–456 [PubMed] [Google Scholar]
- 8. de los Santos C., Zaliznyak T., and Johnson F. (2001) NMR characterization of a DNA duplex containing the major acrolein-derived deoxyguanosine adduct γ-OH-1,-N2-propano-2′-deoxyguanosine. J. Biol. Chem. 276, 9077–9082 10.1074/jbc.M009028200 [DOI] [PubMed] [Google Scholar]
- 9. Kim H.-Y. H., Voehler M., Harris T. M., and Stone M. P. (2002) Detection of an interchain carbinolamine cross-link formed in a CpG sequence by the acrolein DNA adduct γ-OH-1,N2-propano-2′-deoxyguanosine. J. Am. Chem. Soc. 124, 9324–9325 10.1021/ja020333r [DOI] [PubMed] [Google Scholar]
- 10. Kozekov I. D., Nechev L. V., Moseley M. S., Harris C. M., Rizzo C. J., Stone M. P., and Harris T. M. (2003) DNA interchain cross-links formed by acrolein and crotonaldehyde. J. Am. Chem. Soc. 125, 50–61 10.1021/ja020778f [DOI] [PubMed] [Google Scholar]
- 11. Minko I. G., Washington M. T., Kanuri M., Prakash L., Prakash S., and Lloyd R. S. (2003) Translesion synthesis past acrolein-derived DNA adduct, γ-hydroxypropanodeoxyguanosine, by yeast and human DNA polymerase η. J. Biol. Chem. 278, 784–790 10.1074/jbc.M207774200 [DOI] [PubMed] [Google Scholar]
- 12. Washington M. T., Minko I. G., Johnson R. E., Wolfle W. T., Harris T. M., Lloyd R. S., Prakash S., and Prakash L. (2004) Efficient and error-free replication past a minor groove DNA adduct by the sequential action of human DNA polymerases ι and κ. Mol. Cell. Biol. 24, 5687–5693 10.1128/MCB.24.13.5687-5693.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wolfle W. T., Johnson R. E., Minko I. G., Lloyd R. S., Prakash S., and Prakash L. (2005) Human DNA polymerase ι promotes replication through a ring-closed minor-groove adduct that adopts a syn conformation in DNA. Mol. Cell. Biol. 25, 8748–8754 10.1128/MCB.25.19.8748-8754.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nechev L. V., Harris C. M., and Harris T. M. (2000) Synthesis of nucleosides and oligonucleotides containing adducts of acrolein and vinyl chloride. Chem. Res. Toxicol. 13, 421–429 10.1021/tx990167+ [DOI] [PubMed] [Google Scholar]
- 15. Hofmann T., Zweig K., and Engels J. W. (2005) A new synthetic approach for the synthesis of N2-modified guanosines. Synthesis 2005, 1797–1800 10.1055/s-2005-869905 [DOI] [Google Scholar]
- 16. Yoon J.-H., Prakash L., and Prakash S. (2009) Highly error-free role of DNA polymerase η in the replicative bypass of UV induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U.S.A. 106, 18219–18224 10.1073/pnas.0910121106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoon J.-H., Prakash L., and Prakash S. (2010) Error-free replicative bypass of (6-4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev. 24, 123–128 10.1101/gad.1872810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoon J. H., Park J., Conde J., Wakamiya M., Prakash L., and Prakash S. (2015) Rev1 promotes replication through UV lesions in conjunction with DNA polymerases η, ι, and κ but not DNA polymerase ζ. Genes Dev. 29, 2588–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoon J. H., Roy Choudhury J., Park J., Prakash S., and Prakash L. (2014) A role for DNA polymerase θ in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem. 289, 13177–13185 10.1074/jbc.M114.556977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haracska L., Prakash S., and Prakash L. (2002) Yeast Rev1 protein is a G template-specific DNA polymerase. J. Biol. Chem. 277, 15546–15551 10.1074/jbc.M112146200 [DOI] [PubMed] [Google Scholar]
- 21. Nelson J. R., Lawrence C. W., and Hinkle D. C. (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature 382, 729–731 10.1038/382729a0 [DOI] [PubMed] [Google Scholar]
- 22. Nair D. T., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2005) Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309, 2219–2222 10.1126/science.1116336 [DOI] [PubMed] [Google Scholar]
- 23. Swan M. K., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2009) Structure of the human REV1-DNA-dNTP ternary complex. J. Mol. Biol. 390, 699–709 10.1016/j.jmb.2009.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nair D. T., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2008) Protein-template directed synthesis across an acrolein-derived DNA adduct by yeast Rev1 DNA polymerase. Structure 16, 239–245 10.1016/j.str.2007.12.009 [DOI] [PubMed] [Google Scholar]
- 25. Nair D. T., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2005) Human DNA polymerase ι incorporates dCTP opposite template G via a G.C+ Hoogsteen base pair. Structure 13, 1569–1577 10.1016/j.str.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 26. Nair D. T., Johnson R. E., Prakash S., Prakash L., and Aggarwal A. K. (2006) An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-ι active site. Structure 14, 749–755 10.1016/j.str.2006.01.010 [DOI] [PubMed] [Google Scholar]
- 27. Lone S., Townson S. A., Uljon S. N., Johnson R. E., Brahma A., Nair D. T., Prakash S., Prakash L., and Aggarwal A. K. (2007) Human DNA polymerase κ encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell 25, 601–614 10.1016/j.molcel.2007.01.018 [DOI] [PubMed] [Google Scholar]
- 28. Haracska L., Johnson R. E., Unk I., Phillips B. B., Hurwitz J., Prakash L., and Prakash S. (2001) Targeting of human DNA polymerase ι to the replication machinery via interaction with PCNA. Proc. Natl. Acad. Sci. U.S.A. 98, 14256–14261 10.1073/pnas.261560798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson R. E., Washington M. T., Haracska L., Prakash S., and Prakash L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406, 1015–1019 10.1038/35023030 [DOI] [PubMed] [Google Scholar]
- 30. Tissier A., McDonald J. P., Frank E. G., and Woodgate R. (2000) Polι, a remarkably error-prone human DNA polymerase. Genes Dev. 14, 1642–1650 [PMC free article] [PubMed] [Google Scholar]
- 31. Washington M. T., Johnson R. E., Prakash L., and Prakash S. (2004) Human DNA polymerase ι utilizes different nucleotide incorporation mechanisms dependent upon the template base. Mol. Cell. Biol. 24, 936–943 10.1128/MCB.24.2.936-943.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nair D. T., Johnson R. E., Prakash S., Prakash L., and Aggarwal A. K. (2004) Replication by human DNA polymerase ι occurs via Hoogsteen base-pairing. Nature 430, 377–380 10.1038/nature02692 [DOI] [PubMed] [Google Scholar]
- 33. Yoon J.-H., Bhatia G., Prakash S., and Prakash L. (2010) Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 14116–14121 10.1073/pnas.1007795107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Conde J., Yoon J. H., Roy Choudhury J., Prakash L., and Prakash S. (2015) Genetic control of replication through N1-methyladenine in human cells. J. Biol. Chem. 290, 29794–29800 10.1074/jbc.M115.693010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoon J. H., Roy Choudhury J., Park J., Prakash S., and Prakash L. (2017) Translesion synthesis DNA polymerases promote error-free replication through the minor-groove DNA adduct 3-deaza-3-methyladenine. J. Biol. Chem. 292, 18682–18688 10.1074/jbc.M117.808659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levene P. A. (1930) Bromoacetone. Org. Synth. Coll. 10, 12 10.15227/orgsyn.010.0012 [DOI] [Google Scholar]
- 37. Choi J. S., Kang C. W., Jung K., Yang J. W., Kim Y. G., and Han H. (2004) Synthesis of DNA triangles with vertexes of bis(terpyridine)iron(II) complexes. J. Am. Chem. Soc. 126, 8606–8607 10.1021/ja048537q [DOI] [PubMed] [Google Scholar]
- 38. Casale R., and McLaughlin L. W. (1990) Synthesis and properties of an oligodeoxynucleotide containing a polycyclic aromatic hydrocarbon site specifically bound to the N2 amino group of a 2′-deoxyguanosine residue. J. Am. Chem. Soc. 112, 5264–5271 10.1021/ja00169a039 [DOI] [Google Scholar]
- 39. Boryski J., and Ueda T. (1985) A new simple synthesis of N-2-methylguanosine and its analogues via derivatives of 4-desmethylwyosine. Nucleosides Nucleotides 4, 595–606 10.1080/07328318508081892 [DOI] [Google Scholar]