

Abstract
The microtubule-associated protein tau forms insoluble, amyloid-type aggregates in various dementias, most notably Alzheimer's disease. Cellular chaperone proteins play important roles in maintaining protein solubility and preventing aggregation in the crowded cellular environment. Although tau is known to interact with numerous chaperones, it remains unclear how these chaperones function mechanistically to prevent tau aggregation and how chaperones from different classes compare in terms of mechanism. Here, we focused on the small heat shock protein HspB1 (also known as Hsp27) and the constitutive chaperone Hsc70 (also known as HspA8) and report how each chaperone interacts with tau to prevent its fibril formation. Using fluorescence and NMR spectroscopy, we show that the two chaperones inhibit tau fibril formation by distinct mechanisms. HspB1 delayed tau fibril formation by weakly interacting with early species in the aggregation process, whereas Hsc70 was highly efficient at preventing tau fibril elongation, possibly by capping the ends of tau fibrils. Both chaperones recognized aggregation-prone motifs within the microtubule-binding repeat region of tau. However, HspB1 binding remained transient in both aggregation-promoting and non-aggregating conditions, whereas Hsc70 binding was significantly tighter under aggregation-promoting conditions. These differences highlight the fact that chaperones from different families play distinct but complementary roles in the prevention of pathological protein aggregation.
Keywords: 70 kilodalton heat shock protein (Hsp70), aggregation, amyloid, chaperone, small heat shock protein (sHsp), tau
Introduction
Tau is an intrinsically disordered microtubule-associated protein involved in numerous neurodegenerative diseases collectively termed tauopathies. Tau is highly expressed in neurons and plays roles in microtubule stability, axonal transport, and neurite outgrowth (1–3). Tauopathies are characterized by the deposition of tau in insoluble amyloid-type fibrillar aggregates (4), and based on disease-associated tau mutations, this aggregation appears to play a causative role in at least some of these neurodegenerative diseases (5). Although the mechanism of tau toxicity is subject to some debate, increasing evidence suggests that a prefibrillar oligomeric state of tau is the primary toxic species responsible for neurodegeneration (6, 7), similar to models proposed for many other amyloid-forming proteins. According to this model, tau dissociates from microtubules and forms oligomers with the potential to disrupt cellular membranes and impair synaptic and mitochondrial function before ultimately forming relatively inert amyloid fibrils in what may or may not be a protective mechanism to remove toxic oligomers (6–8). How cells control the relative populations of tau monomers, oligomers, and fibrils, and protect against associated toxicity, remains poorly understood.
Fibril formation is primarily driven by the microtubule-binding repeat region of tau, which consists of three or four 31–32-residue pseudorepeats (Fig. 1) involved in microtubule binding (9, 10). The tau gene is alternatively spliced, with the ratio of three-repeat to four-repeat isoforms varying based on normal aging (4) or in disease states (11). Two six-residue motifs at the start of the second and third repeats, 275VQIINK280 and 306VQIVYK311 (also called PHF6* and PHF6), are believed to be the primary drivers of aggregation (9, 12, 13). A recent cryo-electron microscopy study of AD patient-derived fibrils revealed that residues 306–378 form a highly structured core in two distinct fibril morphologies from a single patient's brain (14). These core residues consist of the third and fourth repeats as well as 10 additional C-terminal residues. Whether such morphologies are common among all AD patients or whether alternate fibril morphologies exist in AD or in other tauopathies remains an open question.
Figure 1.

Domain organization of tau, HspB1, and Hsc70. Top, full-length tau2N4R is natively unstructured and consists of four domains. The tau4RD construct used in this work contains residues 244–372, which include the four microtubule-binding repeats. The sequences 275VQIINK280 and 306VQIVYK311 at the beginning of the second and third repeats drive tau fibril formation. Shaded areas, regions that can be alternatively spliced. Middle, HspB1 contains three domains. The core ACD has a β-sandwich fold and forms a stable dimer. The CTR is intrinsically disordered and contains an IXI motif that interacts with the ACD in the context of large oligomers. The NTR is partially disordered and involved in oligomerization, and it contains three serine residues that are phosphorylated in response to cellular stress. The “D3” and “dimer” constructs, described under “Results,” affect the oligomerization state of HspB1. Bottom, Hsc70 contains two domains: an N-terminal nucleotide binding domain with ATPase activity and a C-terminal substrate binding domain that interacts with client proteins.
Whereas the precise mechanism of tau fibril formation is unclear, it appears that tau monomers oligomerize and form elongation-competent species, which then elongate to form mature fibrils, with the formation of elongation-competent species the rate-determining step (15–17). Each stage of the fibril formation process is probably targeted by multiple cellular players, and a better understanding of how the cell works to counteract this process could inform future pharmacological strategies.
The cellular chaperone network is responsible for maintaining protein solubility, promoting proper folding, and preventing aberrant aggregation. Age-linked protein aggregation diseases, including Alzheimer's disease (AD)3 and other tauopathies, may represent an eventual saturation of the chaperone machinery's ability to prevent aggregation (18). Individuals carrying disease-associated mutations in the tau gene that enhance aggregation propensity only develop symptoms later in life (19), suggesting that neuronal cells have the machinery to maintain aggregation-prone tau in a soluble form for years. Interactions between tau and a number of chaperones have been documented, indicating that multiple players within the chaperone network are involved in this process (20–25). However, the mechanisms by which these chaperones interact with tau and their effects on tau fibril formation remain unclear. A better biochemical and mechanistic understanding of how chaperone proteins interact with tau will provide insight into the cellular defense mechanism against tauopathies. This work focuses on the chaperones HspB1 and Hsc70, which both interact with tau in ways that are probably relevant to disease pathology (discussed below). These chaperones belong to distinct families, and their juxtaposition provides an example of how different components of the chaperone network may complement each other in the prevention of pathological tau fibril formation.
HspB1 is a member of the small heat shock proteins (sHSPs), a family of ATP-independent chaperones that act as “holdases” to prevent protein aggregation but lack enzymatic activity (26). Like many members of the sHSP family, HspB1 forms large, polydisperse, highly dynamic oligomers ranging in size from dimers to 30-mers (27), governed by a network of interactions between the α-crystallin domain (ACD), N-terminal region (NTR), and C-terminal region (CTR) (28, 29). Due in part to experimental limitations in dealing with dynamic, polydisperse systems, little is known about how HspB1 recognizes clients and functions as a holdase. HspB1 co-localizes with tau fibrils in tauopathy brains and is pulled down by phosphorylated tau (30, 31), suggesting an important interaction between HspB1 and tau in disease states. A tauopathy mouse model exhibits deficits in long-term potentiation that are rescued by HspB1 overexpression (24). This indicates that HspB1 can alter the physiological outcome of aberrant tau aggregation, but the underlying mechanism remains unclear.
Hsc70 is a constitutively expressed member of the Hsp70 family of chaperones, which are “refoldases” that use the energy of ATP hydrolysis to help misfolded proteins refold to their native states. It contains an N-terminal nucleotide binding domain and a C-terminal substrate binding domain and interacts with Hsp40 family co-chaperones and nucleotide exchange factors to accomplish protein refolding and ATP hydrolysis (32). However, ATP-independent activity has also been reported in Hsp70-family chaperones (33–35). Hsc70 binds to the two aggregation-prone motifs in tau and has been proposed as a pharmacological target (22, 23, 36). Its overexpression leads to an increase in the concentration of tau in cell culture models, whereas an Hsc70 inhibitor causes a decrease in tau concentration in cell cultures and rescues long-term potentiation deficits in tauopathy model mouse brain slices (22, 36). Hsc70 may also play roles in enhancing tau's association with microtubules (23) and in modulating the extracellular release of tau (37). Although it is unclear how Hsc70 affects tau fibril formation, it has been shown to block the formation of fibrils by another amyloid-prone protein, α-synuclein, and even disaggregate preformed α-synuclein fibrils when combined with co-chaperones from the Hsp40 and Hsp110 families (33, 38). Its paralog, Hsp70, has been shown to inhibit tau fibril formation by interacting with tau oligomers (21, 39). It remains to be determined whether these activities are generally applicable to interactions between members of the Hsp70 family and amyloid-prone proteins and which, if any, apply to the interaction between Hsc70 and tau.
Here we compare the effects of Hsc70 and HspB1 on tau self-assembly to explore how different components of the cellular chaperone network interact with tau and impact fibril formation. Using fluorescence spectroscopy, EM, and time-dependent NMR spectroscopy, we study how each chaperone alters the kinetics of tau aggregation and how this is achieved mechanistically. We also compare the binding and recognition of tau by each chaperone under stable and aggregation-promoting conditions using NMR and fluorescence correlation spectroscopy (FCS). These experiments reveal that Hsc70 and HspB1 have profoundly different mechanisms of inhibition, suggesting that they have evolved complementary functions within the context of the larger chaperone network. This would enable the cell to avoid redundancy while targeting distinct stages of the tau fibril formation process with different chaperones.
Results
Hsc70 and HspB1 inhibit tau fibril formation in distinct ways
We focus primarily on a tau construct containing the four microtubule-binding repeats (tau4RD, also referred to in the literature as K18). This sequence, consisting of residues 244–372, forms the core of tau fibrils (13) and contains the two 6-residue motifs that are highly aggregation-prone in isolation and in the context of full-length protein (9). Tau4RD aggregates more readily in vitro than full-length tau and is more amenable to study by NMR spectroscopy. As with other amyloidogenic proteins, environmental factors such as temperature, ionic strength, shaking, the amount and nature of aggregation-inducers such as heparin, and even the type of reaction vessel can dramatically affect tau4RD aggregation. We therefore explicitly note these experimental parameters where relevant.
We also used selected constructs (Fig. 1, middle) to tune the activity and oligomeric state of HspB1 to explore the effects of well-known modifications on its activity and to enable a broader range of spectroscopic techniques. Wild-type HspB1 forms large and polydisperse oligomers, driven by at least three types of interactions: ACD-ACD, ACD-CTR, and NTR-NTR (Fig. S1a) (28, 29). Mutation of three serine residues in the NTR (Ser-15, Ser-78, and Ser-82) to aspartate mimics stress-induced phosphorylation and alters NTR-NTR interactions, yielding a construct termed HspB1D3 that forms much smaller oligomers than HspB1WT (27, 40). Further mutation of the IXI motif in the CTR to GXG, residues 179ITIPV183 in HspB1 to GTGPG, disfavors interactions between the ACD and the CTR, similar to the effects of high temperature (41, 42). We combined the D3 and GXG mutations to generate a construct, denoted HspB1dimer, that forms a well-defined, monodisperse ∼45-kDa dimer (Fig. S1, b and c) and retains the local secondary structure of the ACD (Fig. S1d). Crucially, HspB1dimer displays much lower background thioflavin T (ThT) fluorescence than either HspB1D3 or HspB1WT (Fig. S1e). ThT is a popular reporter of amyloid formation but is neither perfectly specific nor selective for amyloid structure (43). Indeed, ThT fluoresces strongly in the presence of (presumably non-amyloid) HspB1 oligomers but not dimers. HspB1dimer therefore enables mechanistic experiments based on ThT fluorescence that are impossible with HspB1WT or HspB1D3.
In a ThT-monitored assay, tau4RD shows the sigmoidal fluorescence kinetics expected of unseeded amyloid formation, with an initial lag phase leading into a rapid elongation phase followed by an asymptotic approach to a plateau. HspB1dimer caused a delay in the onset of tau4RD aggregation and in the transition midpoint (t50) in a dose-dependent manner without substantially altering the magnitude of the final ThT fluorescence signal (Fig. 2a). Hsc70 was a much more potent, substoichiometric inhibitor of this process, measurably decreasing the final plateau intensity in a dose-dependent manner even when present at a molar ratio as low as 1:100 (Fig. 2b). However, in contrast to HspB1dimer, Hsc70 does not substantially alter the t50 of the tau aggregation reactions.
Figure 2.

Effects of Hsc70 and HspB1 on tau4RD fibril formation. a, ThT fluorescence traces over the course of a representative fibril formation reaction. Tau4RD (3.5 μm) fibrillizes over the course of 10 h following the addition of the aggregation inducer heparin (6 μm, average molecular mass 3 kDa) and follows expected unseeded amyloid formation kinetics. HspB1dimer delays the onset of tau4RD fibril formation in a dose-dependent manner but does not alter the final ThT fluorescence signal. Each trace is the average of 6 wells in a 96-well plate. For clarity, error bands (S.E.) are shown only for the highest and lowest chaperone concentrations. b, Hsc70 is a highly potent substoichiometric inhibitor and decreases the extent of fibril formation in a dose-dependent manner. c, representative negative stain electron micrographs of end products of ThT fibril formation reactions. Tau4RD forms long fibrils in the absence of chaperone and is still able to form long, fully developed fibrils in the presence of HspB1dimer. It forms smaller oligomeric species in the presence of Hsc70, indicating that Hsc70 holds much of the protein in a non-fibrillar state.
Hsc70's activity toward tau4RD appears to be nucleotide-independent. In the presence of 1 mm ATP, Hsc70 had a similar effect on tau4RD aggregation, decreasing the plateau intensity without substantially altering the t50 (Fig. S2a). Although it is somewhat less potent under these conditions, it retains substoichiometric activity and presumably acts through a similar mechanism.
We verified the ability of HspB1 and Hsc70 to inhibit the aggregation of the longest full-length tau isoform, tau2N4R, to confirm that the chaperone activities we observe are generalizable to interactions with cellular tau. The tau4RD construct lacks six residues (positions 373–378) that were recently shown to form part of the ordered core of AD patient–derived fibrils (14), potentially raising questions about the relevance of this well-studied construct. As with tau4RD, we found that HspB1 predominantly affects the lag phase of tau2N4R aggregation, whereas Hsc70 predominantly affects the plateau (Fig. S2, b and c). This indicates that the interactions of these chaperones with tau4RD and tau2N4R are mechanistically similar and that chaperone/tau4RD studies can shed light on chaperone interactions with full-length tau isoforms. We focus on the widely studied tau4RD construct for the remainder of our studies, as the slower aggregation kinetics and larger molecular weight of tau2N4R make mechanistic and spectroscopic studies challenging.
Negative stain EM of the end products of these reactions (Fig. 2c) shows that tau4RD forms long fibrils in the absence of chaperone or in the presence of HspB1dimer. This indicates that HspB1dimer only delays fibril formation rather than preventing it. In contrast, Hsc70-containing reactions yield increasing amounts of small oligomeric species (as compared with long fibrils), indicating that Hsc70 holds much of the tau4RD in an oligomeric state and can prevent elongation of these species into mature fibrils. This confirms that the reduction in ThT fluorescence in reactions containing Hsc70 does indeed correspond to a decrease in the amount of fibrillar material, as opposed to morphological differences that could alter ThT fluorescence. Together, these results demonstrate that, although both chaperones have inhibitory effects on tau aggregation, they are very different in mechanism and outcome.
HspB1 and Hsc70 have distinct mechanisms of inhibition
To better understand the mechanisms by which each chaperone alters tau4RD aggregation, we used one-dimensional NMR spectroscopy to study the interactions of HspB1 and Hsc70 with prefibrillar, ThT-negative species. Large, slowly tumbling species have a weaker signal than smaller species, so the intensity of an NMR spectrum is dependent upon the oligomeric state of protein in solution. We expect monomeric tau4RD to have a strong signal, small oligomers to have a weak signal, and large oligomeric and fibrillar tau4RD to be essentially invisible. A high intensity resonance in the methyl region at 0.77 ppm provides a good read-out for the intensity of the protein signal that is well-separated from buffer components or heparin (Fig. S3a). We used 13C-labeled tau4RD and unlabeled chaperones and collected 13C-edited spectra to observe tau4RD specifically (Fig. S3, b and c). Loss of peak intensity over time reports on the disappearance of soluble, monomeric tau4RD and on binding to heparin and chaperones. We normalized all data relative to the intensity of the first spectrum after the addition of heparin, which is slightly lower than that of free tau4RD. Under these conditions, tau4RD again displayed sigmoidal aggregation kinetics, with a final intensity <20% of the first heparin-bound spectrum (Fig. 3, a and b). Note that these experiments used a different sample concentration and reaction vessel than the ThT experiments and are not shaken, making direct comparison between the two types of experiments impossible. However, an evaluation of the effects of HspB1 and Hsc70 is still informative.
Figure 3.

Mechanisms of chaperone inhibition. a, intensity from 1D NMR spectra monitoring the disappearance of small, soluble tau4RD species during aggregation. In the absence of chaperone (gray circles), 50 μm tau4RD loses some intensity following the addition of 120 μm heparin (drop between first and second data points) and then loses intensity in a sigmoidal manner as it becomes incorporated in NMR-invisible fibrils. HspB1dimer slows this intensity loss in a dose-dependent manner (blue squares and triangles). b, Hsc70 is inhibitory even at a 1:50 molar ratio, making it a highly potent substoichiometric inhibitor. At a 1:1 ratio, it causes a rapid decrease in intensity, probably due to enhanced binding in the presence of heparin, followed by a plateau. c, SDS-PAGE analysis of end products of 1D NMR experiments. End products were centrifuged, and total end products (T), supernatant (S), and pellet (P) were run on a gel. In the absence of chaperone and in the presence of 1 molar eq of HspB1dimer, most of the tau4RD ends up in the insoluble fraction. In the presence of 1 molar eq of Hsc70, almost all of the tau4RD remains soluble. This indicates that the rapid intensity loss in the presence of Hsc70 is due to binding rather than aggregation. d, ThT fluorescence traces over the course of a tau4RD fibril formation reaction. HspB1dimer (1 molar eq) was added to 3.5 μm tau4RD either at the beginning of the reaction (dark blue) or 0.5, 1, or 2.5 h after the reaction had been initiated by the addition of 6 μm heparin (lighter blues). HspB1dimer was still effective at delaying aggregation when added during the lag phase of the reaction but was no longer effective when added during the elongation phase of the reaction. Adding buffer instead of chaperone at any time point did not alter aggregation kinetics (gray curves). Curves are averages of six replicates. For clarity, error bands (S.E.) are shown only for the 0 h experiments. e, Hsc70 (0.05 molar eq) was added to 3.5 μm tau4RD either at the beginning of the reaction (dark red) or 0.5, 1, or 2.5 h after the reaction had been started by the addition of 6 μm heparin (lighter reds). The addition of Hsc70 during the lag phase did not alter its effect on the aggregation kinetics, and addition during the elongation phase prevented any further increase in ThT fluorescence. Adding buffer instead of chaperone at any time point did not alter aggregation kinetics (gray curves). Curves are averages of four or six replicates.
HspB1dimer slowed the intensity loss of tau4RD in a dose-dependent manner (Fig. 3a). In the presence of 1 molar eq of HspB1dimer, ∼70% of the original intensity of tau4RD was lost after 18 h, and in the presence of 4 molar eq, only about one-third of the intensity was lost. Notably, in unedited spectra, which contain signal from both tau4RD and HspB1dimer, this intensity loss was more gradual than in the 13C-edited spectra that contain signal from only tau4RD (Fig. S3d). This means that HspB1dimer is losing signal intensity at a slower rate than tau4RD, remaining predominantly soluble, whereas tau4RD forms large, NMR-invisible species.
Hsc70 is much more potent and has distinct effects (Fig. 3b); in the presence of just 1 μm Hsc70 ( molar eq), tau4RD intensity decreases by only ∼50%, confirming the highly effective substoichiometric nature of its inhibition. In the presence of 1 molar eq of Hsc70, the tau4RD intensity dropped quickly in the 1–2 h following the addition of heparin and then plateaued at ∼70% of the initial intensity. We believe that this drop in intensity is due to binding by Hsc70 rather than fibril formation, as the formation of a larger complex with Hsc70 would also result in a loss of NMR signal intensity for tau4RD. Indeed, when the product of this reaction was pelleted and the supernatant and pellet fractions were analyzed by SDS-PAGE, nearly all of the protein remained in the supernatant, indicating that it remained soluble over the course of the 18-h experiment (Fig. 3c). This is in contrast to reactions containing tau4RD or tau4RD + HspB1dimer, in which most of the tau4RD is in the pellet. Additionally, unedited 1D spectra lose intensity more quickly than 13C-edited spectra, indicating that Hsc70 is losing intensity at an even higher proportion than tau4RD during this process, probably due to binding. Therefore, we conclude that Hsc70 binds aggregation-prone tau4RD early in the aggregation reaction and is able to maintain its solubility over the course of 18 h. By contrast, HspB1 does not form a tight complex with tau4RD during aggregation but instead interacts with it transiently and is less effective at maintaining its solubility than Hsc70.
To better understand the mechanisms of inhibition, we added each chaperone to tau fibril formation reactions at varying time points after initiating fibril formation with heparin. HspB1dimer was able to delay fibril formation when it was added at the beginning of a reaction or during the lag phase but had no effect on fibril formation when it was added after the start of the elongation phase (Fig. 3d). This indicates that HspB1 acts early in the fibril formation process and interacts with prefibrillar species. By contrast, Hsc70 is an effective inhibitor regardless of when it is added during the course of a fibril formation reaction. It has the same effect on fibril formation when it is added at the beginning of a reaction or during the lag phase. When added during the elongation phase, it blocks any subsequent increase in ThT fluorescence and thus halts further fibril formation (Fig. 3e). Taken together with the 1D NMR results, this reveals important differences in how the two chaperones function mechanistically. HspB1 acts early to delay aggregation but is ineffective at blocking elongation of pre-existing fibrils, whereas Hsc70 is effective during all stages of the aggregation process, forming a tight initial complex with aggregation-prone tau and blocking elongation of existing tau fibrils.
HspB1 and Hsc70 both recognize aggregation-prone regions of tau4RD, but with differences in affinity
2D NMR spectroscopy was used to obtain a better structural understanding of tau-chaperone interactions. Despite initial challenges due to the poor chemical shift dispersion of the disordered tau spectrum, recent advances have enabled the complete backbone resonance assignment of the tau4RD and tau2N4R spectra (10, 44–47). Previously published assignments (47) were transferred to our 15N HSQC spectrum of tau4RD, using HNCACB and CBCA(CO)NH spectra for reference and to resolve ambiguities. Our spectrum was highly similar to that previously published, with a few exceptions in the histidine- and lysine-rich regions at the end of each microtubule-binding repeat. These peaks shifted with slight changes in pH or with EDTA (data not shown), suggesting that these peaks are highly sensitive to pH and to the presence of metals in water and that the observed discrepancies are probably due to small differences in buffer conditions.
The binding sites of each chaperone were mapped onto the sequence of tau4RD by recording spectra of 15N-labeled tau4RD in the presence of unlabeled chaperone proteins. HspB1dimer causes significant broadening in peaks corresponding to the 306VQIVYK311 motif, and it causes less pronounced broadening in 275VQIINK280 peaks (Fig. 4a). Hsc70 causes significant broadening in peaks corresponding to both aggregation-prone motifs (Fig. 4b), the same regions affected in full-length tau (22). This indicates that the two chaperones recognize similar regions in tau under non-aggregation-promoting conditions. Notably, the region of tau4RD recognized by HspB1dimer contains the sequence VXI, which is similar to the well-known IXI recognition motif for sHSP binding. Valine can be tolerated in place of isoleucine in such sequences (41), so we predict that HspB1 binds this region of tau in the β4–β8 groove of the ACD.
Figure 4.
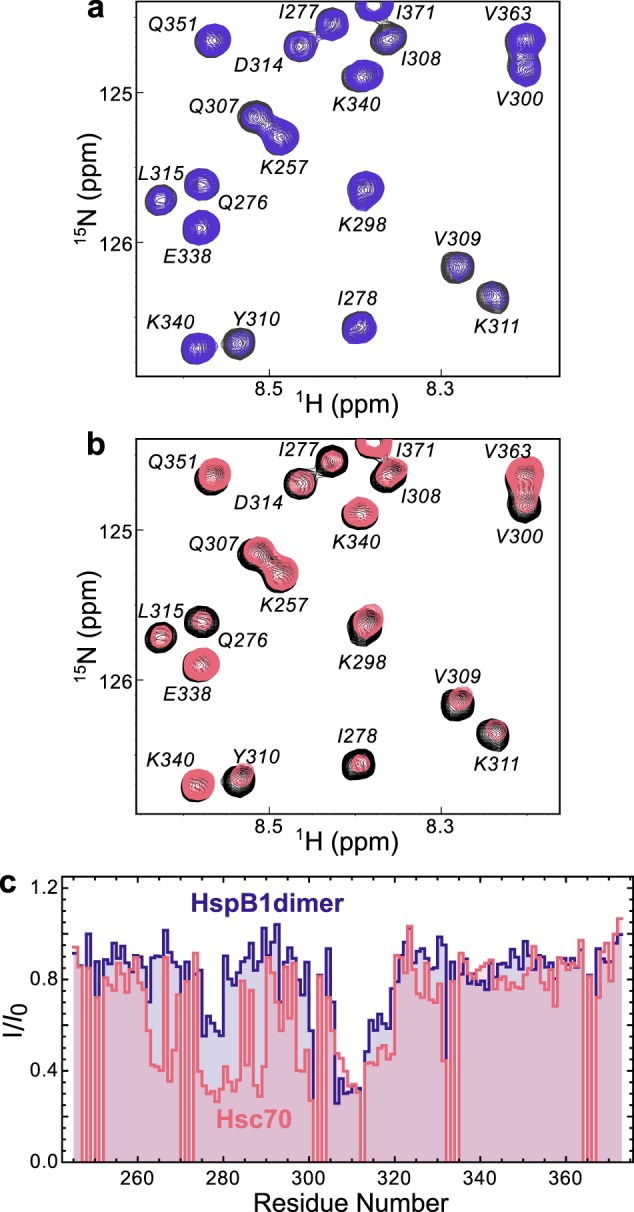
Chaperone binding sites. a, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 2 molar eq of HspB1dimer (blue). b, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 2 molar eq of Hsc70 (red). c, peak broadening from a and b, quantified by dividing the intensity of a peak in the bound spectrum (I) by the intensity of the same peak in the unbound spectrum (I0). Broadening due to HspB1dimer maps primarily to the aggregation-prone motif in the third repeat of tau4RD (306VQIVYK311), whereas broadening due to Hsc70 maps to the aggregation-prone motifs at the start of both the second and third repeats of tau4RD (275VQIINK280 and 306VQIVYK311).
Because a variety of factors can affect NMR peak width, we turned to FCS as an orthogonal, direct probe of binding affinity. FCS can measure changes in hydrodynamic radius (Rh) of a fluorescently labeled protein in the presence of binding partners. We used site-directed mutagenesis to selectively label tau4RD with Alexa Fluor 488 (A488) at residue 2 and measured Rh of the resulting tau4RD∼A488 construct in the presence of increasing concentrations of chaperones (Fig. 5). HspB1dimer had a minor effect on tau4RD∼A488 Rh, increasing it only by ∼25% when present at 500 μm. This subtle change in Rh increased without saturation over HspB1dimer concentrations. The small apparent change in Rh and lack of a plateau could be due to a relatively compact HspB1dimer–tau4RD complex and/or one or more weak binding modes. It is worth noting that HspB1dimer contains two putative binding sites, one within each protomer, and NMR results suggest that it can bind to two distinct sites in tau4RD with different affinity (Fig. 4b). These factors make it impossible to definitively determine a KD for the HspB1dimer–tau4RD interaction by FCS. By contrast, Hsc70 caused a saturable ∼1.7-fold increase in Rh, suggesting the formation of an Hsc70–tau4RD∼A488 complex with an apparent KD of 99 ± 28 μm.
Figure 5.
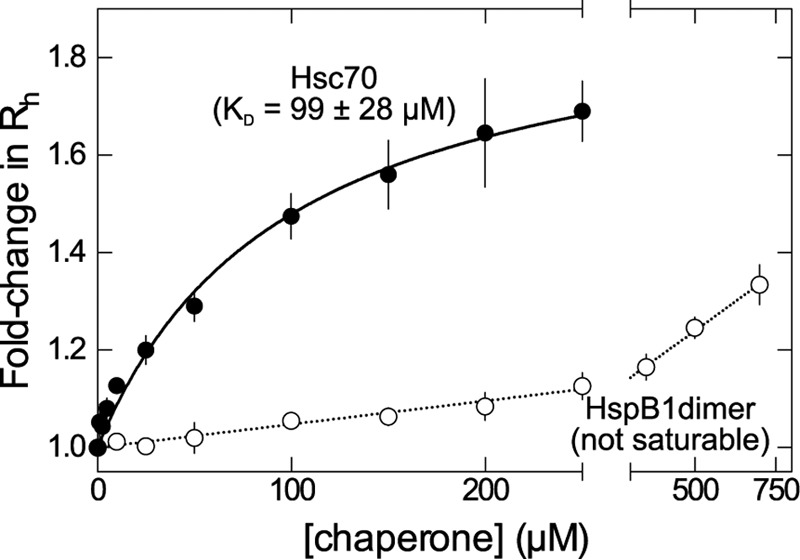
Chaperone binding affinities. FCS-derived changes in hydrodynamic radius of tau4RD∼A488, measured in the presence of increasing concentrations of Hsc70 (solid circles) or HspB1dimer (empty circles). The Hsc70 data are well fit by a one-site binding model with KD = 99 ± 28 μm, whereas HspB1dimer does not appear to saturate up to a chaperone concentration of 750 μm. Error bars, S.E.
Hsc70 binding is enhanced under aggregation-promoting conditions
Because the interactions between these chaperones and tau are most relevant under aggregation-promoting conditions, we used NMR to explore how the aggregation inducer heparin modulates tau-chaperone interactions. Heparin caused peaks in the tau4RD 15N HSQC spectrum to shift dramatically, most notably those in the region corresponding to the aggregation-prone motif 275VQIINK280 (Fig. 6, a and b), in agreement with previous reports (10, 48). Under the conditions used here, binding was nearly saturated, and the peaks represent a heparin-bound state of tau (Fig. S4a). When HspB1dimer was added along with heparin, the peaks still shifted as they do in the presence of heparin alone and still broadened as they do in the presence of HspB1dimer alone. However, these effects were purely additive; there did not appear to be any additional change beyond what we saw when adding HspB1dimer or heparin to tau4RD separately (Fig. 6c). By contrast, when Hsc70 was added in the presence of heparin, significantly more broadening was observed than in the heparin-free Hsc70-bound spectrum (Fig. 6 (d and e) and Fig. S4b). This is presumably due to the formation of a three-component complex in which Hsc70 has enhanced affinity for heparin-bound tau. The difference between HspB1dimer and Hsc70 is well illustrated by the peaks corresponding to 306VQIVYK311. These peaks were broadened to a similar degree by either chaperone in the absence of heparin, but in the presence of heparin, they were significantly broader with Hsc70 than with HspB1dimer (Fig. 6e). These observations suggest that Hsc70 binds more tightly to aggregation-prone heparin-bound tau4RD, whereas HspB1dimer displays no such selectivity. This agrees well with 1D NMR results, which show that Hsc70 causes a rapid loss of intensity from the tau4RD spectrum following the addition of heparin, whereas HspB1dimer does not detectably affect the earliest time points relative to the tau4RD alone curve (Fig. 3, a and b). Hsc70's preference for aggregation-prone states may explain why it is so much more potent an aggregation inhibitor than HspB1dimer.
Figure 6.

Effects of heparin on chaperone binding. a, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 2.4 molar eq of heparin (gray). b, chemical-shift perturbations (CSP) of each peak in A plotted as a function of residue number. The highest chemical-shift perturbations map to the aggregation-prone motif at the start of the second repeat of tau4RD, 275VQIINK280. c, 15N HSQC TROSY of 15N-labeled tau4RD alone (black), in the presence of 2.4 molar eq of heparin (gray), in the presence of 2 molar eq of HspB1dimer (blue), and in the presence of both 2.4 eq of heparin and 2 eq of HspB1dimer (yellow). HspB1dimer binding is largely unchanged by the presence of heparin. d, 15N HSQC TROSY of 15N-labeled tau4RD alone (black), in the presence of 2.4 molar eq of heparin (gray), in the presence of 2 molar eq of Hsc70 (red), and in the presence of both 2.4 eq of heparin and 2 eq of Hsc70 (yellow). Hsc70 binding is enhanced by the presence of heparin, as indicated by the enhanced broadening in the yellow spectrum relative to the red. e, comparison of residues Val-309, Tyr-310, and Lys-311 in the HspB1dimer-bound and Hsc70-bound spectra in the presence of heparin. Although HspB1dimer and Hsc70 cause a similar amount of broadening in these peaks in the absence of heparin, in the presence of heparin, Hsc70 causes significantly more broadening in these peaks than HspB1dimer, indicating that heparin enhances Hsc70 binding in a way that it does not for HspB1dimer.
HspB1WT and HspB1D3 behave similarly to HspB1dimer
Finally, we studied the interactions of tau4RD with HspB1WT and HspB1D3. Whereas HspB1dimer is more experimentally tractable due to its defined, monodisperse dimeric state, HspB1WT and HspB1D3 are more physiologically relevant and can still be studied using a subset of the techniques available to us. By 1D NMR, HspB1D3 and HspB1WT inhibited tau4RD aggregation similarly to HspB1dimer (Fig. 7 (a and b) and Fig. S3), transiently interacting with tau4RD and slowing the loss of monomer signal in a dose-dependent manner. Interestingly, it seems that HspB1dimer is less effective than WT or D3 at an equimolar ratio (50 μm chaperone) but more effective in 4-fold excess (200 μm chaperone). The oligomeric states of HspB1D3 and WT probably vary over this concentration range, a phenomenon that alters their chaperone activity in complicated ways (27, 40).
Figure 7.

Effects of HspB1WT and HspB1D3 on tau4RD. a, intensity from 1D NMR spectra monitoring the disappearance of small, soluble tau4RD species during aggregation in the presence of HspB1WT. HspB1WT (green squares and triangles) slows this intensity loss in a manner similar to HspB1dimer (Fig. 3a). b, intensity from 1D NMR spectra monitoring tau4RD aggregation in the presence of HspB1D3 (teal squares and triangles). HspB1D3 slows intensity loss in a manner similar to HspB1dimer and WT. c, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 4 molar eq of HspB1WT (green). There is very little significant broadening in the spectrum with HspB1WT, suggesting that binding is very weak. d, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 4 molar eq of HspB1D3 (teal). There is slight broadening in peaks corresponding to 306VQIVYK311, indicating some binding. e, 15N HSQC TROSY of 15N-labeled tau4RD alone (black) and in the presence of 4 molar eq of HspB1dimer (blue). Peak broadening is much more substantial, indicating that HspB1WT and D3 both bind more weakly than HspB1dimer. f, quantification of tau 4RD peak intensity loss due to HspB1WT, HspB1D3, and HspB1dimer binding.
HspB1WT did not significantly perturb the 15N HSQC spectrum of tau4RD, even in 4-fold excess, indicating that its binding is very weak and difficult to detect. HspB1D3 caused some broadening in the spectrum of tau4RD, particularly in peaks corresponding to 306VQIVYK311, but less strongly than HspB1dimer, suggesting intermediate affinity (Fig. 7, c–f). Changes in binding affinity between HspB1WT, HspB1D3, and HspB1dimer could be due to differences in the oligomeric state of these constructs, structural changes due to the D3 mutations that enhance affinity for tau4RD, and/or increased availability of the HspB1 β4-β8 groove due to the GXG mutations. Notably, these differences in affinity do not appear to correlate with inhibition of tau4RD fibril formation in 1D NMR experiments, and additional work is needed to elucidate how inhibition of tau fibril formation relates to HspB1 structure and oligomeric state. Nevertheless, it is clear that HspB1 interacts with tau in a way that is similar across constructs and mechanistically distinct from the interactions between Hsc70 and tau.
Discussion
Molecular chaperones play important roles in maintaining protein solubility and preventing pathological aggregation. The several families of molecular chaperones, each with multiple isoforms, comprise an intricate functional network in the cell. The disparate mechanisms by which individual chaperones function are not fully understood. We have characterized the interactions between the amyloid-prone protein tau and two molecular chaperones from different families: HspB1, a ubiquitously expressed sHSP, and Hsc70, a constitutively expressed member of the Hsp70 family. This work shows that the two chaperones interact with distinct tau species and have profoundly different effects on the kinetics of tau aggregation, suggesting that they play complementary roles within the context of the cellular chaperone network.
HspB1 binds tau fairly weakly, and this binding is not enhanced under aggregation-promoting conditions. Rather, HspB1 interacts transiently with early species of tau in a manner that delays but does not prevent fibril formation (Fig. 8). Such transient interactions have been observed in many sHSP/client pairings and may be a common mechanism of sHSP “holdase” activity (49, 50). Little is known about how HspB1 interacts with client proteins structurally, in large part due to experimental limitations in dealing with dynamic, polydisperse systems. The client binding sites of HspB1 have not been mapped to the residue level, to our knowledge, although studies indicate an important role of the NTR in binding at least some clients (51). Two binding sites have been mapped on another ubiquitously expressed human sHSP, HspB5: one in the NTR and one in the ACD (52). We hypothesize that HspB1 binds the VXI motif in tau in the β4–β8 groove of the ACD, in accord with previous work on HspB5/client binding (41, 52) and HspB1 ACD-CTR interactions (29). We find no correlation between the oligomeric state of HspB1 and its chaperone activity toward tau. Whereas the oligomeric state does correlate with binding affinity in this work, it is unclear which factors cause this; the D3 and GXG mutations could alter affinity or chaperone activity independently of their effects on oligomer size.
Figure 8.
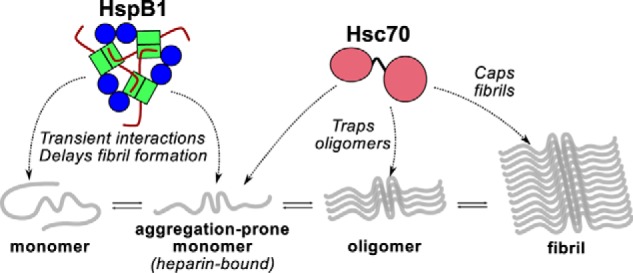
Model of chaperone effects on tau aggregation. HspB1 and Hsc70 interact with different species along the tau amyloid formation pathway. HspB1 interacts with early species and delays the formation of tau oligomers and fibrils (1D NMR and ThT results). Hsc70 has enhanced affinity for aggregation-prone, heparin-bound tau (1D and 2D NMR results); can hold tau in an oligomeric state (EM results); and can prevent fibril elongation (ThT results).
Hsc70 also has fairly low affinity for tau under non-aggregating conditions, with a KD around 100 μm. However, this binding is enhanced in the presence of the aggregation inducer heparin, enabling Hsc70 to form a tight complex with aggregation-prone tau. It is a highly potent inhibitor and is effective even when present at a 1:50 molar ratio, and it can arrest fibril formation when it is added to an aggregation reaction at any time point. Hsc70 is fully capable of binding monomeric tau4RD, but its substoichiometric potency in fibril formation reactions and ability to block elongation even late in a reaction suggest that it can also interact with larger oligomeric or fibrillar species. Our data are consistent with a model wherein Hsc70 caps the ends of fibrils and elongation-competent oligomers and prevents elongation of these species (Fig. 8). Notably, Hsp70 also has been shown to interact with tau oligomers (39), suggesting that this may be a common mode of action for members of the Hsp70 family. Hsp70 family chaperones have also been reported to display similarly ATP-independent, substoichiometric inhibition of α-synuclein amyloid formation, again by interacting with both monomeric and fibrillar states (33, 35). It should be noted that our experiments studied the activity of Hsc70 in the absence of co-chaperones from the Hsp40 family or nucleotide-exchange factors. How these co-chaperones engage with Hsc70 and tau remains to be seen. Another open question is whether Hsc70's activity would be beneficial to the cell by preventing fibril formation or detrimental by causing the accumulation of toxic oligomeric species.
Recent cryo-EM structures of AD patient-derived tau fibrils have provided valuable new insight into at least two tau fibril morphologies found in vivo (14). Additionally, proteolysis assays indicate that patient-derived fibrils from different tauopathies have distinct morphologies (53). An understanding of the diversity of tau fibril morphologies thus remains an important open question. Notably, the Fitzpatrick et al. (14) cryo-EM structures indicate that residues 373–378 form part of the fibril core in both morphologies, but these residues are absent from the tau4RD construct used in this paper and many others. It is unclear whether or how these residues affect the aggregation mechanism, as compared with canonical aggregation drivers, such as 306VQIVYK311 (9). We therefore compared chaperone effects on fibril formation by tau4RD and full-length tau2N4R.
HspB1 and Hsc70 have similar effects on the fibril formation kinetics of tau4RD and tau2N4R, indicating that they interact with the full-length protein in a manner similar to the shorter construct that lacks residues 373–378. NMR studies of Hsc70 binding to tau2N4R show that Hsc70 causes slight broadening in peaks corresponding to residues 375–380 of tau2N4R, although this broadening is not as strong as that of the 275VQIINK280 and 306VQIVYK311 motifs (22, 23). Because we found similar Hsc70 chaperone activity against tau4RD, which does not contain these residues, and tau2N4R, which does, we believe that this interaction does not significantly alter Hsc70's mechanism of action. Because no structural studies involving full-length tau and HspB1 have been conducted, it remains to be seen whether HspB1 interacts with regions of full-length tau in addition to those observed here using the tau4RD construct. HspB1dimer was a somewhat more potent inhibitor of tau2N4R fibril formation than of tau4RD fibril formation. Whether this is due to slower aggregation kinetics of tau2N4R, altered binding of HspB1 to tau2N4R, or a different factor is unknown.
The disparate effects of HspB1 and Hsc70 on tau fibril formation point to interesting ways different members of the cellular chaperone network complement each other in the prevention of protein aggregation. The sHSPs have been described as “first responders” to cell stress events (54), a descriptor that fits the activity of HspB1 described here well. It is easy to imagine a scenario in which HspB1 interacts with tau during early stages of fibril formation, delaying the formation of toxic oligomers, and Hsc70 interacts with oligomers and later species in the fibril formation process. HspB1 is phosphorylated and up-regulated in response to stress, whereas Hsc70 is constitutively expressed. The load of aggregation-prone proteins increases during stress events, and enhanced activity of HspB1 during these events could relieve some of the pressure on Hsc70 by delaying their aggregation until Hsc70 is better able to handle the increased workload. Few studies have examined the interactions between human Hsc70 and HspB1, although recent work suggests that the two proteins may be physically linked through interactions with the nucleotide-exchange factor BAG3 (55). There is evidence that Escherichia coli sHSPs work with the E. coli Hsp70 homolog, DnaK, in preventing protein aggregation (56, 57). Additional work is needed to elucidate how HspB1 and Hsc70 function in the context of the full human chaperone network and how this relates to pathological protein aggregation in diseases such as tauopathies. However, our work points to distinct but complementary roles that Hsc70 and HspB1 could play in the prevention of tau fibril formation by engaging distinct species along the amyloid formation pathway.
Experimental procedures
Protein expression and purification
Plasmids containing the tau4RD or tau2N4R genes, each with a His-tag and TEV cleavage site, were gifts from the Rhoades laboratory at the University of Pennsylvania. A pMCSG7 vector containing His-tagged Hsc70 (HSPA8) with a TEV cleavage site was a gift from the Southworth laboratory at the University of Michigan. Untagged HspB1 constructs were expressed in pET23a (WT) and pET151D (D3 and dimer) vectors. All recombinant proteins were expressed and purified from E. coli strain BL-21 (DE3). Unlabeled tau4RD, tau2N4R, and HspB1 were expressed in 1-liter cultures in Luria-Bertani medium, and unlabeled Hsc70 was expressed in Terrific Broth. Isotopically labeled tau4RD was expressed in 1 liter of MOPS minimal medium supplemented with [13C]glucose, [15N]ammonium chloride, or both. All were incubated at 37 °C until reaching an optical density of 0.6. Tau4RD and tau2N4R expression was induced with 0.4 mm IPTG and grown overnight at 16 °C. HspB1 expression was induced with 1 mm IPTG and grown at 22 °C, and Hsc70 expression was induced with 0.2 mm IPTG and grown at 28 °C.
Hsc70 was purified from a cell pellet derived from 1 liter of culture, resuspended in 30 ml of 50 mm Tris buffer, pH 8.0, with 500 mm NaCl and 10 mm imidazole. Phenylmethylsulfonyl fluoride, 1× Halt protease inhibitor mixture (Thermo Scientific), DNase, and RNase were added, and sample was lysed using a French press and then pelleted by centrifugation at 4100 rpm for 1 h. Supernatant was filtered with a 0.45-μm syringe filter and then passed over a 5-ml Ni2+ affinity column (GE Healthcare). His-tagged Hsc70 was eluted with buffer containing 250 mm imidazole and then dialyzed overnight back into buffer containing 10 mm imidazole in the presence of ∼1 μm TEV protease (expressed from Addgene plasmid 8830, a gift from David Waugh) to cleave the His tag. Dialyzed sample was run over a Ni2+ column to separate cleaved protein from uncleaved protein, cleaved His tag, and TEV protease. The flow-through was concentrated with a 10,000 molecular weight cutoff Amicon concentrator and then buffer-exchanged into 25 mm Tris buffer, pH 8.0, with 1 mm EDTA. Sample was loaded onto a 5-ml HiTrap Q HP column (GE Healthcare) and eluted in a 25-ml gradient to buffer containing 500 mm NaCl. Purity was assessed by SDS-PAGE, and pooled fractions were concentrated to 2–3 ml. Sample was passed over a Superdex 200 10/300 GL column (GE Healthcare) in 500-μl batches. Fractions were analyzed by SDS-PAGE, pooled based on purity, concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C. Tau4RD and tau2N4R constructs were purified as described previously (16) in a protocol that is very similar to that for Hsc70 but omits the anion-exchange chromatography step. HspB1 was purified as described previously (58).
Analytical SEC
All samples were prepared in pH 7.5 buffer containing 50 mm sodium phosphate, 100 mm sodium chloride, and 0.5 mm EDTA and allowed to equilibrate at room temperature for at least 3 h. For SEC, 100-μl samples of 50 μm HspB1 were run over a Superose 6 10/300 mm column (GE Healthcare) equilibrated in identical buffer as the samples, at room temperature. For SEC-multi-angle static light scattering, 25-μl samples of 100 μm HspB1 were equilibrated at room temperature for at least 3 h before being placed in a 4 °C autosampler. Samples were run over a Superose 6 Increase 3.2/300-mm column at room temperature, equilibrated in identical buffer. Chromatograms were collected on a GE AKTA Pure system coupled to a Wyatt miniDAWN TREOS and Optilab T-rEX differential refractive index detector. Molar mass was calculated from the Raleigh ratio based on multiangle (static) light scattering and protein concentration from the change in refractive index (dn/dc = 0.185). Analysis was performed using Wyatt ASTRA VI software, and curves were calibrated with apoferritin.
Circular dichroism
CD spectra of HspB1 and its mutants were collected on a Jasco J-1500 CD spectrometer. Samples contained 10 μm protein in 25 mm sodium phosphate buffer with 50 mm sodium chloride and 0.25 mm EDTA at pH 7.5. Samples were equilibrated at room temperature for at least 3 h before measurement and measured in a 0.1-cm cuvette. Measurements were taken at 20 °C from 260 to 190 nm in continuous scanning mode with a scanning rate of 50 nm/min, data integration time of 2 s, and bandwidth of 1 nm. Data presented are an average of three scans, and data are truncated at a wavelength resulting in a high-tension value at or above the recommended 800-V cutoff.
ThT fluorescence spectra
Samples were prepared containing 5 or 20 μm HspB1 in 50 mm sodium phosphate buffer (pH 7.5) containing 100 mm sodium chloride and 0.5 mm EDTA. ThT was added to a concentration of 75 μm, and samples were covered in foil and incubated at room temperature for 3 h. Samples were incubated at 37 °C immediately before fluorescence measurements, which were also recorded at 37 °C. Using a Horiba Fluorolog 3 fluorimeter, samples were excited at 440 nm (1.2-nm slit width), and emission spectra from 450 to 550 nm (5-nm slit width) were collected.
ThT kinetic assays
Tau4RD, ThT, and chaperone were added to wells in a 96-well black flat-bottom non-binding plate (Corning) in buffer containing 30 mm sodium phosphate, 20 mm sodium chloride, 5 mm dithiothreitol, and 0.5 mm EDTA at pH 7.4. Immediately before beginning the reads, 100 μl of heparin (MP Biomedicals, average molecular mass 3 kDa) solution was added to each well, bringing the total volume to 200 μl and the concentrations to 3.5 μm tau4RD, 6 μm heparin, and 75 μm ThT. Experiments done in the presence of ATP also included 1 mm ATP, 1 mm MgCl2, and 1 mm KCl. Solutions containing heparin and tau4RD + ThT were filtered with a 0.22-μm syringe filter immediately before the addition to the plate. Solutions containing chaperone were centrifuged at 21,100 × g for 10 min prior to addition. The plate was covered with clear polyolefin sealing tape (Thermo Scientific) to prevent evaporation and placed in a Biotek Synergy HTX multimode plate reader. Samples were held at 37 °C, and fluorescence was measured every 2.5 min using 440- and 485-nm filters for excitation and emission, respectively. Immediately before each fluorescence read, the plate was shaken linearly at 1096 cpm for 1 min. Delayed addition experiments were programmed to pause at appropriate time points to enable the addition of 10 μl of chaperone solution or buffer to the appropriate wells. All other conditions were the same. Experiments with full-length tau were conducted under similar conditions, except that they used 5 μm tau2N4R, were shaken continuously, and were read at 10-min intervals.
Electron microscopy
300-Mesh carbon-coated copper grids (Electron Microscopy Services) were glow-discharged and spotted with 3 μl of the end products of tau4RD ThT assays. Samples adsorbed for 60 s and then were blotted by filter paper, rinsed with filtered milliQ water and blotted twice, and floated on a drop of NanoW stain (NanoProbes) for 60 s. Stain was blotted with filter paper, and grids were allowed to dry on filter paper and then stored in a container at room temperature. Grids were visualized on a Morgagni electron microscope with a 100-kV beam.
1D NMR assays
50 μm samples of 13C-labeled tau4RD were prepared with or without chaperone in pH 7.4 buffer containing 25 mm sodium phosphate, 10 mm sodium chloride, 5 mm dithiothreitol, 0.5 mm EDTA, and 10% D2O. Initial unedited 1D watergate and 13C-edited spectra were collected on a 500-MHz Bruker magnet at 37 °C. 13C-Edited 1D spectra are the first slice of a 13C HSQC. The sample was then removed, heparin was added to a final concentration of 120 μm, and a series of alternating unedited and 13C-edited spectra were collected over the course of 18 h. At the end of the experiment, sample was removed from the NMR tube, and a 100-μl aliquot was centrifuged at 21,100 × g for 60 min. Supernatant was removed, and 100 μl of buffer was added to wash the pellet. Sample was centrifuged at 21,100 × g for 15 min, supernatant was removed, and pellet was resuspended in 100 μl of buffer. Samples from the total reaction, the supernatant, and the pellet were analyzed by SDS-PAGE. NMR spectra were processed using NMR pipe. For each spectrum, the intensity of a peak in the methyl region at 0.77 ppm in the unedited and 0.89 ppm in the edited spectra was measured. The intensity of this peak was normalized to the intensity of the same peak in the first spectrum following heparin addition, and relative intensities were plotted as a function of time.
Fluorescence correlation spectroscopy
A construct of tau4RD with both native cysteines mutated to serine and an additional cysteine incorporated as the second residue was created using the Phusion site-directed mutagenesis kit (Thermo Scientific). Alexa Fluor 488 C5 maleimide (Life Technologies) was conjugated to this cysteine residue by incubating 100 μm purified protein with ∼3-fold excess dye at room temperature for 2 h. Labeled protein was separated from unlabeled protein and free dye on a BDS Hypersil C18 column run with a water/acetonitrile gradient in the presence of 0.05% TFA on a Dionex Ultimate 3000 HPLC. Fractions containing labeled protein were collected, and solvent was evaporated. Dried labeled protein was stored at −20 °C and resuspended in buffer before use. Aliquots of resuspended protein were flash-frozen in liquid nitrogen and stored at −80 °C until immediately before use. 50-μl samples containing chaperone protein and 2 mg/ml BSA were prepared in pH 7.4 buffer containing 25 mm sodium phosphate, 100 mm sodium chloride, 2 mm dithiothreitol, and 0.5 mm EDTA. For Hsc70 binding experiments, 1 μl of labeled tau4RD was added to the sample 5 min before recording FCS measurements. For HspB1dimer binding experiments, tau4RD was added, and samples were incubated at room temperature for 3–5 h before FCS measurements. The diffusion of each sample was measured on a Zeiss Axio Observer D1 confocal microscope equipped with a Picoquant PDL 828 Sepia II pulsed laser driver, HydraHarp 400 detection electronics, and a Picoquant τ-SPAD single photon counting module. Laser excitation was at 485 nm, and emitted light was collected through a 488-nm dichroic mirror and 535/70 emission filter. Seven consecutive 1-min measurements were recorded, and autocorrelation curves for each measurement were calculated using SymphoTime 64 software. Each curve was fit using GraphPad Prism to the following equation for free diffusion, where N is the average number of particles in the focal volume, τD is the diffusion time of each particle, and s (the ratio of the radial to axial dimension of the focal volume) is fixed to 0.2 based on calibration measurements.
| (1) |
Each curve was normalized to the value of N determined by this fitting, and the seven curves for each condition were averaged. Because Rh ∝ τD, all diffusion times were normalized to that of free tau4RD, and the relative -fold change was fit to the following one-site model,
| (2) |
where Rhnorm is the normalized Rh at a given chaperone concentration, ΔRh,∞norm is the maximal -fold change in Rh, [hsp] is the concentration of chaperone, and KD is the dissociation constant.
2D NMR spectroscopy
NMR samples contained 100 or 200 μm labeled tau4RD protein in pH 7.4 buffer containing 25 mm sodium phosphate, 100 mm sodium chloride, 2 mm DTT, and 0.5 mm EDTA. Spectra were collected at 10 °C on a Bruker Avance III 800-MHz magnet equipped with a triple-resonance, z-gradient cryoprobe. HNCACB and CBCA(CO)NH spectra were used to transfer published assignments (47) to the 15N TROSY HSQC spectrum of tau4RD. Data were processed using NMRpipe and analyzed with NMRViewJ. Peak broadening was quantified by dividing the intensity of each peak in the bound spectrum by the intensity of the corresponding peak in the unbound spectrum. Chemical shift perturbations were quantified with the following equation, where ΔHN is the amide proton chemical shift difference and ΔN is the 15N backbone amide chemical shift difference.
| (3) |
Author contributions
H. E. R. B., A. F. C., R. E. K., and A. N. designed the research. H. E. R. B. conducted the fibril formation, EM, FCS, and NMR experiments. A. F. C. designed and characterized the HspB1dimer construct. H. E. R. B. and A. N. analyzed data. H. E. R. B, A. F. C., R. E. K., and A. N. wrote the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants 5T32GM008268 (to H. E. R. B. and A. F. C.) and 2R01EY017370 (to R. E. K.); the University of Washington Royalty Research Fund (to A. N.); and the Brady Fund for Natural Products Research (to A. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- AD
- Alzheimer's disease
- ACD
- α-crystallin domain
- NTR
- N-terminal region
- CTR
- C-terminal region
- ThT
- thioflavin T
- tau4RD
- 4-repeat domain of tau
- tau2N4R
- full-length tau
- FCS
- fluorescence correlation spectroscopy
- sHSP
- small heat shock protein
- HSQC
- heteronuclear single quantum coherence
- A488
- Alexa Fluor 488
- TEV
- tobacco etch virus
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- SEC
- size-exclusion chromatography
- TROSY
- transverse relaxation optimized spectroscopy.
References
- 1. Drubin D. G., and Kirschner M. W. (1986) Tau protein function in living cells. J. Cell Biol. 103, 2739–2746 10.1083/jcb.103.6.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stamer K., Vogel R., Thies E., Mandelkow E., and Mandelkow E. M. (2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063 10.1083/jcb.200108057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dawson H. N., Ferreira A., Eyster M. V., Ghoshal N., Binder L. I., and Vitek M. P. (2001) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 4. Goedert M., Spillantini M. G., Jakes R., Rutherford D., and Crowther R. A. (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- 5. Wolfe M. S. (2009) Tau mutations in neurodegenerative diseases. J. Biol. Chem. 284, 6021–6025 10.1074/jbc.R800013200 [DOI] [PubMed] [Google Scholar]
- 6. Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Clos A. L., Jackson G. R., and Kayed R. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 39 10.1186/1750-1326-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., et al. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flach K., Hilbrich I., Schiffmann A., Gärtner U., Krüger M., Leonhardt M., Waschipky H., Wick L., Arendt T., and Holzer M. (2012) Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 287, 43223–43233 10.1074/jbc.M112.396176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., and Mandelkow E. (2000) Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 10.1073/pnas.97.10.5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mukrasch M. D., Biernat J., von Bergen M., Griesinger C., Mandelkow E., and Zweckstetter M. (2005) Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986 10.1074/jbc.M501565200 [DOI] [PubMed] [Google Scholar]
- 11. D'Souza I., Poorkaj P., Hong M., Nochlin D., Lee V. M.-Y., Bird T. D., and Schellenberg G. D. (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. U.S.A. 96, 5598–5603 10.1073/pnas.96.10.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. von Bergen M., Barghorn S., Li L., Marx A., Biernat J., Mandelkow E. M., and Mandelkow E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J. Biol. Chem. 276, 48165–48174 10.1074/jbc.M105196200 [DOI] [PubMed] [Google Scholar]
- 13. Daebel V., Chinnathambi S., Biernat J., Schwalbe M., Habenstein B., Loquet A., Akoury E., Tepper K., Müller H., Baldus M., Griesinger C., Zweckstetter M., Mandelkow E., Vijayan V., Lange A., et al. (2012) β-sheet core of tau paired helical filaments revealed by solid-state NMR. J. Am. Chem. Soc. 134, 13982–13989 10.1021/ja305470p [DOI] [PubMed] [Google Scholar]
- 14. Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., Crowther R. A., Ghetti B., Goedert M., and Scheres S. H. W. (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547, 185–190 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shammas S. L., Garcia G. A., Kumar S., Kjaergaard M., Horrocks M. H., Shivji N., Mandelkow E., Knowles T. P. J., Mandelkow E., and Klenerman D. (2015) A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat. Commun. 6, 7025 10.1038/ncomms8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakatani-Webster E., and Nath A. (2017) Inferring mechanistic parameters from amyloid formation kinetics by approximate Bayesian computation. Biophys. J. 112, 868–880 10.1016/j.bpj.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carlson S. W., Branden M., Voss K., Sun Q., Rankin C. A., and Gamblin T. C. (2007) A complex mechanism for inducer mediated tau polymerization. Biochemistry 46, 8838–8849 10.1021/bi700403a [DOI] [PubMed] [Google Scholar]
- 18. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 19. Wszolek Z. K., Tsuboi Y., Ghetti B., Pickering-Brown S., Baba Y., and Cheshire W. P. (2006) Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis. 1, 30 10.1186/1750-1172-1-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karagöz G. E., Duarte A. M. S., Akoury E., Ippel H., Biernat J., Morán Luengo T., Radli M., Didenko T., Nordhues B. A., Veprintsev D. B., Dickey C. A., Mandelkow E., Zweckstetter M., Boelens R., Madl T., and Rüdiger S. G. D. (2014) Hsp90-tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974 10.1016/j.cell.2014.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voss K., Combs B., Patterson K. R., Binder L. I., and Gamblin T. C. (2012) Hsp70 alters tau function and aggregation in an isoform specific manner. Biochemistry 51, 888–898 10.1021/bi2018078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jinwal U. K., Akoury E., Abisambra J. F., O'Leary J. C. 3rd, Thompson A. D., Blair L. J., Jin Y., Bacon J., Nordhues B. A., Cockman M., Zhang J., Li P., Zhang B., Borysov S., Uversky V. N., Biernat J., Mandelkow E., Gestwicki J. E., Zweckstetter M., and Dickey C. A. (2013) Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 27, 1450–1459 10.1096/fj.12-220889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fontaine S. N., Martin M. D., Akoury E., Assimon V. A., Borysov S., Nordhues B. A., Sabbagh J. J., Cockman M., Gestwicki J. E., Zweckstetter M., and Dickey C. A. (2015) The active Hsc70/tau complex can be exploited to enhance tau turnover without damaging microtubule dynamics. Hum. Mol. Genet. 24, 3971–3981 10.1093/hmg/ddv135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abisambra J. F., Blair L. J., Hill S. E., Jones J. R., Kraft C., Rogers J., Koren J. 3rd, Jinwal U. K., Lawson L., Johnson A. G., Wilcock D., O'Leary J. C., Jansen-West K., Muschol M., Golde T. E., Weeber E. J., Banko J., and Dickey C. A. (2010) Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 30, 15374–15382 10.1523/JNEUROSCI.3155-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu L.-R., Liu X.-L., Chen J., and Liang Y. (2013) Protein disulfide isomerase interacts with tau protein and inhibits its fibrillization. PLoS One 8, e76657 10.1371/journal.pone.0076657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Treweek T. M., Meehan S., Ecroyd H., and Carver J. A. (2015) Small heat-shock proteins: important players in regulating cellular proteostasis. Cell Mol. Life Sci. 72, 429–451 10.1007/s00018-014-1754-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jovcevski B., Kelly M. A., Rote A. P., Berg T., Gastall H. Y., Benesch J. L. P., Aquilina J. A., and Ecroyd H. (2015) Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195 10.1016/j.chembiol.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 28. Lelj-Garolla B., and Mauk A. G. (2012) Roles of the N- and C-terminal sequences in Hsp27 self-association and chaperone activity. Protein Sci. 21, 122–133 10.1002/pro.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hochberg G. K. A., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., Collier M. P., Stroud J., Carver J. A., Baldwin A. J., Robinson C. V., Eisenberg D. S., Benesch J. L. P., and Laganowsky A. (2014) The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U.S.A. 111, E1562–E1570 10.1073/pnas.1322673111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nemes Z., Devreese B., Steinert P. M., Van Beeumen J., and Fésüs L. (2004) Cross-linking of ubiquitin, HSP27, parkin, and α-synuclein by γ-glutamyl-ϵ-lysine bonds in Alzheimer's neurofibrillary tangles. FASEB J. 18, 1135–1137 [DOI] [PubMed] [Google Scholar]
- 31. Shimura H., Miura-Shimura Y., and Kosik K. S. (2004) Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J. Biol. Chem. 279, 17957–17962 10.1074/jbc.M400351200 [DOI] [PubMed] [Google Scholar]
- 32. Stricher F., Macri C., Ruff M., and Muller S. (2013) HSPA8/HSC70 chaperone protein. Autophagy 9, 1937–1954 10.4161/auto.26448 [DOI] [PubMed] [Google Scholar]
- 33. Chaari A., Eliezer D., and Ladjimi M. (2016) The C-terminal α-helices of mammalian Hsc70 play a critical role in the stabilization of α-synuclein binding and inhibition of aggregation. Int. J. Biol. Macromol. 83, 433–441 10.1016/j.ijbiomac.2015.10.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rao J. L., Reddy P. S., Mishra R. N., Gupta D., Sahal D., Tuteja N., Sopory S. K., and Reddy M. K. (2010) Thermo and pH stable ATP-independent chaperone activity of heat-inducible Hsp70 from Pennisetum glaucum. Plant Signal. Behav. 5, 110–121 10.4161/psb.5.2.10547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aprile F. A., Arosio P., Fusco G., Chen S. W., Kumita J. R., Dhulesia A., Tortora P., Knowles T. P. J., Vendruscolo M., Dobson C. M., and Cremades N. (2017) Inhibition of α-synuclein fibril elongation by Hsp70 is governed by a kinetic binding competition between α-synuclein species. Biochemistry 56, 1177–1180 10.1021/acs.biochem.6b01178 [DOI] [PubMed] [Google Scholar]
- 36. Abisambra J., Jinwal U. K., Miyata Y., Rogers J., Blair L., Li X., Seguin S. P., Wang L., Jin Y., Bacon J., Brady S., Cockman M., Guidi C., Zhang J., Koren J., et al. (2013) Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry. 74, 367–374 10.1016/j.biopsych.2013.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fontaine S. N., Zheng D., Sabbagh J. J., Martin M. D., Chaput D., Darling A., Trotter J. H., Stothert A. R., Nordhues B. A., Lussier A., Baker J., Shelton L., Kahn M., Blair L. J., Stevens S. M. Jr., and Dickey C. A. (2016) DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549 10.15252/embj.201593489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao X., Carroni M., Nussbaum-Krammer C., Mogk A., Nillegoda N. B., Szlachcic A., Guilbride D. L., Saibil H. R., Mayer M. P., and Bukau B. (2015) Human Hsp70 disaggregase reverses Parkinson's-linked α-synuclein amyloid fibrils. Mol. Cell 59, 781–793 10.1016/j.molcel.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patterson K. R., Ward S. M., Combs B., Voss K., Kanaan N. M., Morfini G., Brady S. T., Gamblin T. C., and Binder L. I. (2011) Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 50, 10300–10310 10.1021/bi2009147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rogalla T., Ehrnsperger M., Preville X., Kotlyarov A., Lutsch G., Ducasse C., Paul C., Wieske M., Arrigo A.-P., Buchner J., and Gaestel M. (1999) Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J. Biol. Chem. 274, 18947–18956 10.1074/jbc.274.27.18947 [DOI] [PubMed] [Google Scholar]
- 41. Delbecq S. P., Jehle S., and Klevit R. (2012) Binding determinants of the small heat shock protein, αB-crystallin: recognition of the “IxI” motif. EMBO J. 31, 4587–4594 10.1038/emboj.2012.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baldwin A. J., Walsh P., Hansen D. F., Hilton G. R., Benesch J. L. P., Sharpe S., and Kay L. E. (2012) Probing dynamic conformations of the high-molecular-weight αB-crystallin heat shock protein ensemble by NMR spectroscopy. J. Am. Chem. Soc. 134, 15343–15350 10.1021/ja307874r [DOI] [PubMed] [Google Scholar]
- 43. Biancalana M., and Koide S. (2010) Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 10.1016/j.bbapap.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smet C., Leroy A., Sillen A., Wieruszeski J. M., Landrieu I., and Lippens G. (2004) Accepting its random coil nature allows a partial NMR assignment of the neuronal Tau protein. ChemBioChem 5, 1639–1646 10.1002/cbic.200400145 [DOI] [PubMed] [Google Scholar]
- 45. Mukrasch M. D., Bibow S., Korukottu J., Jeganathan S., Biernat J., Griesinger C., Mandelkow E., and Zweckstetter M. (2009) Structural polymorphism of 441-residue tau at single residue resolution. PLos Biol. 7, e34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Narayanan R. L., Dürr U. H. N., Bibow S., Biernat J., Mandelkow E., and Zweckstetter M. (2010) Automatic assignment of the intrinsically disordered protein Tau with 441-residues. J. Am. Chem. Soc. 132, 11906–11907 10.1021/ja105657f [DOI] [PubMed] [Google Scholar]
- 47. Barré P., and Eliezer D. (2013) Structural transitions in tau K18 on micelle binding suggest a hierarchy in the efficacy of individual microtubule-binding repeats in filament nucleation. Protein Sci. 22, 1037–1048 10.1002/pro.2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao J., Huvent I., Lippens G., Eliezer D., Zhang A., Li Q., Tessier P., Linhardt R. J., Zhang F., and Wang C. (2017) Glycan determinants of heparin-tau interaction. Biophys. J. 112, 921–932 10.1016/j.bpj.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cox D., Selig E., Griffin M. D. W., Carver J. A., and Ecroyd H. (2016) Small heat-shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 291, 22618–22629 10.1074/jbc.M116.739250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rajagopal P., Tse E., Borst A. J., Delbecq S. P., Shi L., Southworth D. R., and Klevit R. E. (2015) A conserved histidine modulates HSPB5 structure to trigger chaperone activity in response to stress-related acidosis. Elife 4, 10.7554/eLife.07304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McDonald E. T., Bortolus M., Koteiche H. A., and Mchaourab H. S. (2012) Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 51, 1257–1268 10.1021/bi2017624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mainz A., Peschek J., Stavropoulou M., Back K. C., Bardiaux B., Asami S., Prade E., Peters C., Weinkauf S., Buchner J., and Reif B. (2015) The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905 [DOI] [PubMed] [Google Scholar]
- 53. Taniguchi-Watanabe S., Arai T., Kametani F., Nonaka T., Masuda-Suzukake M., Tarutani A., Murayama S., Saito Y., Arima K., Yoshida M., Akiyama H., Robinson A., Mann D. M. A., Iwatsubo T., and Hasegawa M. (2016) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 131, 267–280 10.1007/s00401-015-1503-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Basha E., O'Neill H., and Vierling E. (2012) Small heat shock proteins and α-crystallins: Dynamic proteins with flexible functions. Trends Biochem. Sci. 37, 106–117 10.1016/j.tibs.2011.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rauch J. N., Tse E., Freilich R., Mok S.-A., Makley L. N., Southworth D. R., and Gestwicki J. E. (2017) BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J. Mol. Biol. 429, 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Veinger L., Diamant S., Buchner J., and Goloubinoff P. (1998) The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network. J. Biol. Chem. 273, 11032–11037 10.1074/jbc.273.18.11032 [DOI] [PubMed] [Google Scholar]
- 57. Żwirowski S., Kłosowska A., Obuchowski I., Nillegoda N. B., Piróg A., Ziȩtkiewicz S., Bukau B., Mogk A. and Liberek K. (2017) Hsp70 displaces small heat shock proteins from aggregates to initiate protein refolding. EMBO J. 36, 783–796 10.15252/embj.201593378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clouser A. F., and Klevit R. E. (2017) pH-dependent structural modulation is conserved in the human small heat shock protein HSBP1. Cell Stress Chaperones 22, 569–575 10.1007/s12192-017-0783-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.