

Abstract
Discoidin domain receptor 1 (DDR1) is a collagen receptor that mediates cell communication with the extracellular matrix (ECM). Aberrant expression and activity of DDR1 in tumor cells are known to promote tumor growth. Although elevated DDR1 levels in the stroma of breast tumors are associated with poor patient outcome, a causal role for tumor-extrinsic DDR1 in cancer promotion remains unclear. Here we report that murine mammary tumor cells transplanted to syngeneic recipient mice in which Ddr1 has been knocked out (KO) grow less robustly than in WT mice. We also found that the tumor-associated stroma in Ddr1-KO mice exhibits reduced collagen deposition compared with the WT controls, supporting a role for stromal DDR1 in ECM remodeling of the tumor microenvironment. Furthermore, the stromal–vascular fraction (SVF) of Ddr1 knockout adipose tissue, which contains committed adipose stem/progenitor cells and preadipocytes, was impaired in its ability to stimulate tumor cell migration and invasion. Cytokine array–based screening identified interleukin 6 (IL-6) as a cytokine secreted by the SVF in a DDR1-dependent manner. SVF-produced IL-6 is important for SVF-stimulated tumor cell invasion in vitro, and, using antibody-based neutralization, we show that tumor promotion by IL-6 in vivo requires DDR1. In conclusion, our work demonstrates a previously unrecognized function of DDR1 in promoting tumor growth.
Keywords: breast cancer, cancer biology, cell invasion, tumor cell biology, cytokine, extracellular matrix, tumor microenvironment, adipose stromal cells, DDR1, discoidin domain receptor 1, IL-6, mammary tumor, stromal–vascular fraction
Introduction
Excessive adiposity has been linked to increased breast cancer recurrence and mortality in both pre- and postmenopausal women(1–6). The underlying mechanisms of adiposity-associated cancer burden are likely multifactorial, including elevated production of hormones, cytokines, reactive oxygen species, and extracellular matrix (ECM).6 Obesity is also associated with altered adipose tissue homeostasis and metabolic reprogramming. These changes in adipose tissue could collectively impact tumor progression through both systemic and paracrine mechanisms. Given the abundance of tumor-surrounding adipocytes in breast tissue, communication between tumor and mature adipocytes has naturally been the primary focus of mechanistic studies of obesity-related cancer burden (7–9). However, it is increasingly evident that, in addition to mature adipocytes, altered abundance and properties of human adipose stem/stromal cells (ASCs) and elevated fibrosis contribute to tissue remodeling associated with tumor-associated adipose tissue (2, 10–12). For example, we and others have shown previously that human ASCs are a significant source of local estrogens that stimulate ERα+ breast tumor growth (11, 13–15).
Discoidin domain receptor 1 (DDR1) is a cell surface tyrosine kinase receptor that binds to and is activated by collagens (16–19). DDR1 is predominantly expressed in normal epithelial cells, and its aberrant expression is associated with multiple solid cancer types. For these reasons, the current literature on DDR1 function in cancer biology has exclusively been focused on its activity in tumor cells (20–22). However, comparative gene expression profiling shows that stromal DDR1 expression in invasive breast cancer is significantly elevated versus normal breast stroma (23) (6.4-fold, p = 1 × 10−15), suggesting a possible DDR1 function in stromal cells during cancer progression. In support, we previously reported a DDR1-dependent signaling pathway that regulates adipose production of estrogens in human ASC cultured in vitro (15). Furthermore, we found that the DDR1 function in human ASCs is not shared by other collagen receptors, including integrins or DDR2 (15), indicating a uniquely important role of DDR1 in regulating endocrine/paracrine ASC functions. Despite these lines of emerging evidence, there is a lack of in vivo evidence that definitively establishes a causal relationship between stromal DDR1 and cancer progression.
In this work, we utilized a Ddr1 knockout (KO) mouse model and syngeneic mouse mammary tumor cells to examine the role of host DDR1 in mammary tumor progression. To complement in vivo tumor studies, we assessed in vitro the tumor cell–promoting ability of the stromal–vascular fraction (SVF) of mouse adipose tissue, which is enriched with multipotent stem/progenitor cells and functionally similar to human ASCs (24). We conducted comprehensive cytokine profiling to identify the adipose stroma–secreted cytokine IL-6 as an important mediator of stromal DDR1 function in tumor pathogenesis. For the first time, our data provide compelling mechanistic insight into the role of stromal DDR1 in breast tumor growth in vivo.
Results
Genetic ablation of host Ddr1 blunts mammary tumor growth
To interrogate the role of host DDR1 in cancer progression, we used a previously established Ddr1 whole-body KO mouse model on the C57BL/6 genetic background (25). We first confirmed DDR1 protein expression in WT mouse SVF and its depletion in the counterpart from homozygous KO mice (lanes 2 and 3, Fig. 1a). The two Ddr1-dependent protein bands in WT mouse stroma are likely DDR1 isoforms because of alternative splicing (18). As a positive control for DDR1, we used primary ASCs isolated from human breast tissue (lane 1, Fig. 1a). For the in vivo tumor study, we orthotopically injected two syngeneic murine mammary tumor cell lines, AT-3 and M-Wnt, into 8- to 10-week-old female Ddr1 WT or homozygous KO recipient mice. To avoid potential animal cage–based variation, pairs of WT and KO mice from the same litter were used in tumor and cell culture experiments throughout our study. No significant body weight difference was observed between the WT and KO cohorts at the time of tumor study (data not shown).
Figure 1.
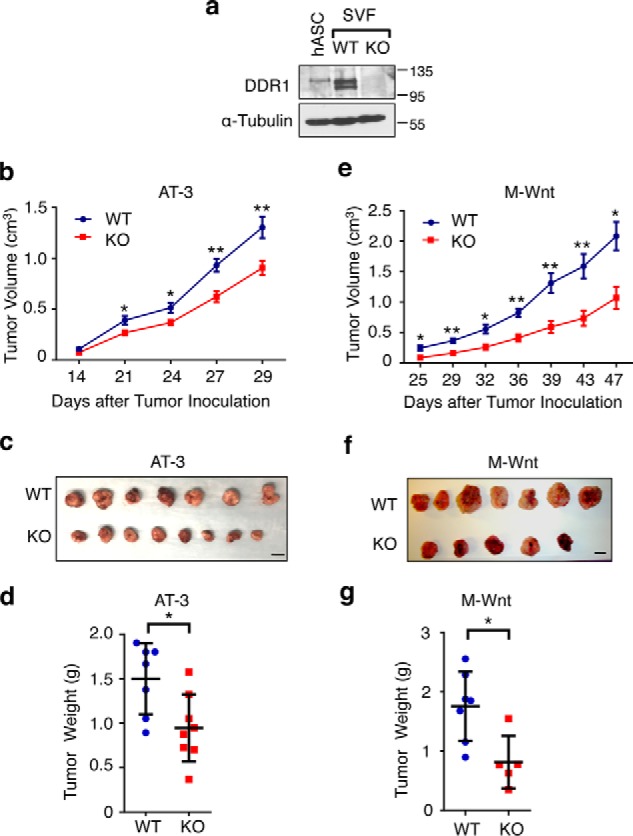
Host DDR1 promotes mammary tumor growth in syngeneic mouse models. a, immunoblot assessing DDR1 protein expression in primary human ASCs (hASC), WT mouse SVF, and littermate KO SVF. b–d, tumor volume (b), size (c), and weight (d) of AT-3 mammary tumor cells in Ddr1 WT (n = 7) and KO (n = 8) mice. e–g, tumor volume (e), size (f), and weight (g) of M-Wnt mammary tumor cells in Ddr1 WT (n = 7) and KO (n = 5) mice. Scale bars = 1 cm. Data are represented as mean ± S.D. *, p < 0.05; **, p < 0.01.
The AT-3 cell line was derived from an murine mammary tumor virus–PyMT (polyoma middle T) transgenic mouse mammary tumor (26, whereas M-Wnt was established from an murine mammary tumor virus–Wnt-1 transgenic mouse mammary tumor (27). We chose these two tumor cell lines because both are syngeneic with C57BL/6 mice and have been used as models for triple-negative breast cancer (28–31). Tumor sizes were assessed by caliper measurement over a period of 4–7 weeks, and tumors were weighed upon harvest. In both AT-3 (Fig. 1, b–d) and M-Wnt (Fig. 1, e–g) syngeneic tumor models, tumors grew more robustly in WT mice versus Ddr1 KO counterparts. Because DDR1 in tumor cells is also known to promote tumor progression, we examined DDR1 expression in tumors from WT and Ddr1 KO hosts. DDR1 protein levels in KO hosts were not lower than those in WT counterparts (Fig. S1a), further supporting tumor-extrinsic activity of host DDR1 in promoting tumor growth in vivo. Based on Ki67 and phospho-histone H3 staining, we did not find any significant difference in tumor cell proliferation between the WT and KO cohorts (Fig. S1, b and c). However, tumors from Ddr1 KO hosts displayed elevated apoptosis, as measured by TUNEL (Fig. S2a). The same tumors from KO hosts also expressed less Ctnnb1 (β-catenin) and Cdh2 (N-cadherin) but more Cdh1 (E-cadherin) versus those tumors in WT hosts (Fig. S2b), suggesting reduced epithelial–mesenchymal transition for tumors in Ddr1 KO hosts.
Host DDR1–dependent ECM remodeling in the tumor microenvironment
Under various physiopathological conditions, such as hypertensive nephropathy, collagen-triggered DDR1 activation is known to induce an inflammatory response, which, in turn, leads to excessive collagen synthesis and exaggerated fibrosis (19). To determine whether a similar DDR1-dependent positive feedback loop occurred in the mammary tumor microenvironment, we first conducted intratumoral collagen histochemistry with picrosirius red (PSR) on M-Wnt tumors harvested from Ddr1 WT and KO mice. Intratumoral PSR staining intensity was significantly reduced in tumors from Ddr1 KO mice versus their WT counterparts (Fig. 2, a and b), consistent with reduced collagen in Ddr1 KO mice. In further support, Col1a1 mRNA levels were markedly dampened in M-Wnt tumors in Ddr1 KO mice (Fig. 2c). Expression of Col3a1 and α-smooth muscle actin (α-SMA), another hallmark for accumulation of tumor-associated stromal cells (32), followed the same trend, but it was not statistically significant (Fig. 2c). Taken together, our findings support the notion that host DDR1 contributes to matrix remodeling in the tumor microenvironment.
Figure 2.
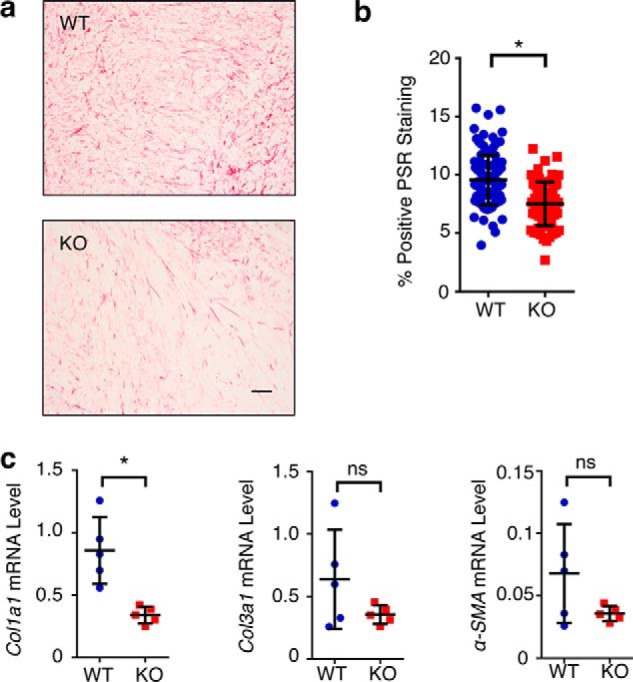
Host DDR1 affects ECM remodeling in the tumor microenvironment. a, PSR staining of tumor (M-Wnt) from Ddr1 WT and KO mice. b, quantification of PSR staining. c, RT-PCR of ECM markers in tumor (M-Wnt) from Ddr1 WT and KO mice. Scale bar = 50 μm. Data are represented as mean ± S.D. *, p < 0.05; ns, not significant. The same numbers of mice as shown in Fig. 1 was used here.
Mouse SVF stimulates tumor cell migration and invasion in a stromal DDR1-dependent manner
Because we previously showed that DDR1 is important for the paracrine action of primary human ASCs cultured in vitro (15), we asked whether host mouse DDR1 played a similar role in the current animal models. Upon isolation of SVF from WT and Ddr1 KO mice, we first verified DDR1 depletion in KO-SVF by immunoblotting (Fig. 3a). An MTT assay indicated no appreciable difference in cell proliferation between WT- and KO-SVF populations (Fig. 3b). Using a Boyden chamber–based co-culture system, we found that medium conditioned by WT-SVF significantly stimulated both migration (Fig. S3) and invasion (Fig. 3, c and d) of both M-Wnt and AT-3 murine mammary tumor cells. In stark contrast, medium conditioned with Ddr1 KO-SVF had a substantially impaired ability to stimulate tumor cell migration (Fig. S1) and invasion (Fig. 3, c and d). Consistent with the in vivo finding of elevated collagen deposition in tumors from the WT host versus the Ddr1 KO host (Fig. 2), medium conditioned with WT-SVF stimulated Col1a1 mRNA expression in tumor cells to a greater extent than that conditioned with KO-SVF (Fig. 3e). Thus, the in vitro system with SVF-conditioned medium recapitulates the observed DDR1 effect on the tumor microenvironment in the syngeneic mouse tumor models.
Figure 3.
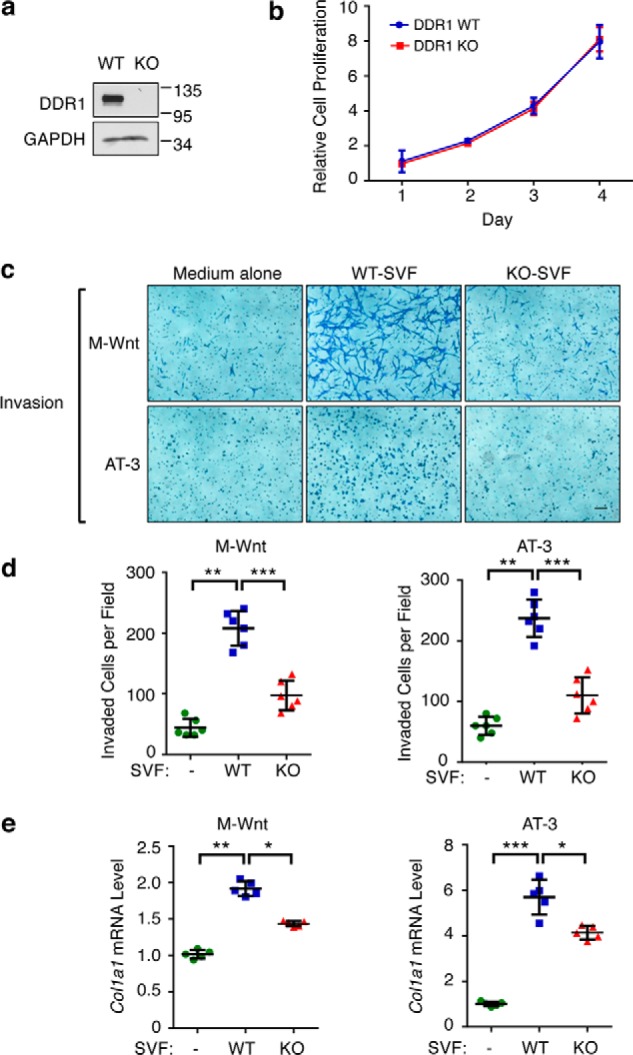
Stromal DDR1 promotes tumor cell migration and invasion. a, DDR1 protein expression in Ddr1 WT- and KO-SVF. GAPDH is the loading control. b, MTT assay assessing proliferation of primary SVF from Ddr1 KO and littermate WT controls. c, representative images of invasion of M-Wnt (top panels) and AT-3 (bottom panels) after 20 h of co-culture with medium alone or WT/KO-SVF. d, quantification of invaded cells by cell counting. e, RT-qPCR was used to assess mRNA levels of Col1a1 in M-Wnt or AT-3 cells following co-culture with medium alone or WT/KO SVF. Scale bar = 50 μm. Data represent mean ± S.D. of four littermate WT/KO pairs. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine the durability of the SVF effect on tumor cells, M-Wnt and AT-3 tumor cells were retrieved from the exposure to SVF-conditioned medium and assessed alone for their invasive behaviors in the absence of the conditioned medium. As shown in Fig. S4, a and b, tumor cells that had been exposed to medium conditioned with WT-SVF retained the more aggressive invasive behavior versus their counterparts exposed to either medium alone or medium conditioned with KO-SVF. Of note, neither WT nor KO-SVF-conditioned medium had any effect on the proliferation of M-Wnt or AT-3 tumor cells (Fig. S4, c and d). Thus, our findings clearly indicate that DDR1 in mouse SVF confers to tumor cells a prolonged invasive phenotype.
Stromal DDR1 promotes tumor cell invasion by regulating SVF-secreted IL-6
To identify the DDR1-dependent, SVF-secreted factor(s) that promote tumor cell invasion, we conducted a cytokine screen using a commercial array consisting of 111 cytokines and chemokines. SVF-conditioned medium from two independent pairs of WT and Ddr1 KO mice was used for probing the cytokine/chemokine array. The levels of several cancer-related, SVF-secreted factors were reduced in conditioned medium from KO-SVF versus WT-SVF, including IL-6, IL-11, CCL17, and vascular endothelial growth factor (Fig. 4a and data not shown). Because our previously published work showed that human ASC-secreted IL-6 contributes to the tumor-promoting action of ASC (33), we chose to focus on the functionality of the differential levels of IL-6 between mouse WT- and KO-SVF. We first used RT-qPCR (Fig. 4b) and ELISA (Fig. 4c) to confirm reduced IL-6 mRNA and protein levels, respectively, in KO-SVF versus WT-SVF in multiple independent pairs of WT and KO samples. Consistent with our published results for human ASCs (33), mRNA and protein expression of Cofilin, an upstream regulator of IL-6 production in the human stromal compartment, was also substantially reduced in mouse KO-SVF as compared with WT-SVF (Fig. 4, d–f).
Figure 4.

Identification of DDR1-dependent cytokines in SVF. a, representative images of the membrane-based antibody array using cell-free supernatant from WT- and KO-SVF. The boxed dots are a reference protein (blue) and IL-6 (red). b, mRNA levels of IL-6 in WT/KO-SVF by RT-qPCR. c, measurement of secreted IL-6 in cell-free supernatant from WT/KO-SVF by ELISA. d, mRNA levels of Cofilin in WT/KO-SVF by RT-qPCR. e, protein levels of DDR1 and Cofilin in three pairs of WT/KO SVF. GAPDH was a loading control. f, quantification of Cofilin protein level normalized by GAPDH. All mRNA analyses and ELISA were done with four pairs of WT and KO. Data represent mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether DDR1-dependent IL-6 secretion contributes to SVF-promoted tumor cell invasion, we used an IL-6–neutralizing antibody to reduce IL-6 activity in WT conditioned medium. Pretreatment with the anti-IL-6 antibody obliterated the difference in invasion stimulation between WT and KO-SVF (Fig. 5, a and b), clearly indicating that IL-6 is an important mediator of the DDR1-dependent effect on tumor cell invasion. In a reciprocal experiment, addition of recombinant IL-6 reduced the difference in stimulation of tumor invasion between WT- and KO-SVF from 1.7- to 1.4-fold (Fig. 5, c and d). Because exogenous IL-6 did not completely eliminate the difference between WT- and KO-SVF, we infer from this result that additional DDR1-regulated, SVF-secreted factors besides IL-6 likely play roles in mediating host DDR1 signaling.
Figure 5.
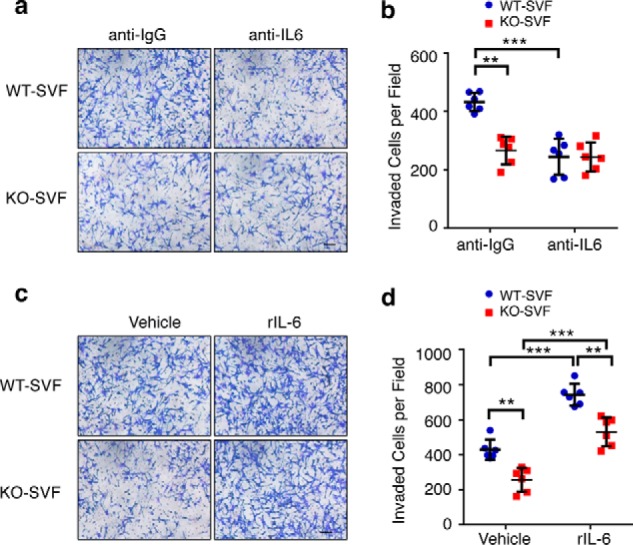
DDR1-dependent stromal IL-6 secretion is important for SVF to promote tumor cell invasion. a, representative images of M-Wnt cell invasion after 20 h of co-culture with WT/KO-SVF in the presence of anti-IL-6 or anti-IgG (1 μg/ml). b, quantification of the cell invasion result in a. c, representative images of M-Wnt cell invasion in the co-culture system in the presence or absence of recombinant IL-6 (10 ng/ml). d, quantification of the cell invasion result shown in c. Data represent mean ± S.D. of four littermate WT/KO pairs. **, p < 0.01; ***, p < 0.001.
IL-6 stimulation of mammary tumor growth in vivo is host DDR1–dependent
To interrogate the in vivo functional relationship between DDR1 and IL-6 in cancer progression, we systemically administered the anti-IL-6–neutralizing antibody in M-Wnt–bearing WT and Ddr1 KO mice (34). Tumor sizes were monitored by caliper for a 5-week period following tumor transplantation (Fig. 6a), and tumors were weighed upon harvest at the end point (Fig. 6, b and c). Consistent with the in vitro co-culture findings (Fig. 5, a and b), IL-6 neutralization in WT mice significantly mitigated tumor growth (compare the first and third columns in Fig. 6c). Notably, the same antibody treatment in Ddr1 KO mice did not lead to any further reduction in tumor growth (compare the second and fourth columns in Fig. 6c). Taken together with the findings from the above in vitro cytokine experiments, these in vivo results strongly suggest that host DDR1 is an important upstream regulator of IL-6 production and its tumor-promoting function.
Figure 6.

IL-6 stimulation of mammary tumor growth in vivo is host DDR1–dependent. a, growth curves of M-Wnt tumors in the WT/KO host treated with either anti-IL-6 or anti-IgG antibodies, each at 100 μg per mouse every 3 days starting 2 days before tumor inoculation. b, tumor images upon harvest. Scale bar = 1 cm. c, tumor weight at the final time point. Data represent mean ± S.D. of 7 tumors from each group. d, a model diagram showing the cross-talk between the ECM, ASCs, and tumor cells. Data are represented as mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Discussion
To date, published studies of DDR1 in cancer have been limited to its action in tumor cells (20, 21). Using genetically engineered mice and syngeneic tumor models, we demonstrate the importance of host DDR1 in tumor growth, significantly extending the current understanding of the DDR1 tumor-promoting function. We further show that DDR1 in adipose tissue-derived SVF is a previously unappreciated regulator of stromal IL-6 secretion that affects tumor cell migration and invasion. Notably, we provide compelling in vivo evidence for a functional link between host DDR1 and the tumor-promoting activity of IL-6. Given the cell surface localization of DDR1 and its innate tyrosine kinase activity, pharmacologic abrogation of host DDR1 function in tumor progression could mitigate cancer burden.
Emerging evidence indicates that adipose tissue–derived stem/progenitor cells are a significant source of stroma-secreted factors that have a profound impact on tissue regeneration as well as pathogenesis, including in cancer (2, 35, 36). Obesity is a well-known factor associated with poor prognosis for multiple cancer types, including breast cancer. Furthermore, obesity-associated fibrosis is an increasingly recognized hallmark of adipose dysfunction that is tightly associated with other adiposity-related changes such as inflammation (37). As a known collagen receptor, DDR1 is a key player in a collagen-initiated positive feedback loop that ultimately results in excessive ECM accumulation in various non-cancer disease models (19). Our in vitro and in vivo data suggest that the same positive feedback loop most likely also manifests in the mammary tumor microenvironment. In this regard, DDR1-dependent secretion of various inflammatory factors, including those detected in our cytokine assay, could serve dual functions: they promote tumor growth and, at the same time, exacerbate obesity-associated ECM remodeling and inflammation. We propose that stromal DDR1 is part of a signaling network that links the ECM, stromal cells, and tumor cells in the same tumor microenvironment (see the model in Fig. 6d). Although our current work used mice receiving a normal (not a high-fat) diet for studying primary tumor growth, it will be important to investigate the role of host DDR1 in other aspects of cancer biology, including obesity-associated tumor progression and metastasis.
The tumor-promoting functions of IL-6 in both tumor and host cells have been well documented as important in inflammation and breast cancer (33, 34, 38–45). In further support, high circulating IL-6 levels are associated with poor prognosis in breast cancer patients (46). Although our in vitro co-culture experiments focused on IL-6 secretion by adipose SVF, multiple cell types, including tumor and other stromal cells, most likely contribute to IL-6 levels in the tumor microenvironment and in the circulation. However, it is also worth noting that genetic ablation of host Ddr1 in our syngeneic mouse models completely eliminates the effect of IL-6–neutralizing antibody on tumor growth. Furthermore, neither AT-3 nor M-Wnt mammary tumor cells express appreciable amounts of IL-6 (Fig. S5). We therefore favor the possibility that stromal IL-6 predominantly contributes to the tumor-promoting activity of IL-6 observed in our study and that this adipose stromal pool of IL-6 is under the tight control of SVF DDR1. In support, it has been reported previously that mouse preadipocytes express significantly higher levels of IL-6 than mature adipocytes (47).
Our work does not exclude the possible involvement of other DDR1-dependent, SVF-secreted factors besides IL-6 that could also contribute to tumor promotion. Consistent with this possibility, Ddr1 KO–conditioned medium still exhibited lower invasion-promoting activity than WT-condition medium, even in the presence of an excessive amount of recombinant IL-6 (Fig. 5, c and d; 10 ng/ml recombinant IL-6 versus 0.7 and 0.2 ng/ml for endogenous IL-6 in WT- and KO-SVF, respectively). Further, with IL-6–neutralizing antibody, tumor growth in Ddr1 KO mice was still slower versus WT counterparts. Although this difference could be due to incomplete IL-6 neutralization in vivo, an alternative explanation is that host DDR1 regulates expression of additional tumor-promoting factors besides IL-6. Potential candidates for future investigation include cytokines and chemokines that displayed differential levels between WT and Ddr1 KO-derived SVF. Although the current whole-body Ddr1 KO animal model allows us to distinguish DDR1 in tumor versus host cells, DDR1 from multiple host cell and tissue types besides adipose tissue–derived stromal cells could also influence tumor growth. There is currently no suitable Cre-lox system in vivo to create adipose SVF–specific gene KO in mice. More sophisticated tissue-specific mouse models could shed light on this question.
This study of syngeneic mouse tumor models significantly extends our previously published work using cultured primary human ASCs (15, 33). Although human samples obviously bear more clinical relevance, the use of genetically engineered mouse models circumvents individual-based variation associated with clinical cohorts. Furthermore, compared with xenograft tumor models, the syngeneic tumor models used in this work ensure an immune-competent host environment that more faithfully recapitulates the tumor microenvironment in humans. It is satisfying that both lines of complementary investigation in mouse and human systems clearly point to an unequivocal role of a conserved DDR1-dependent signaling pathway that dictates production of a tumor-promoting secretome in breast cancer.
Materials and methods
Primary cells isolation and culture
Primary SVF was isolated from mouse inguinal fat pads using standard procedures (48). Briefly, harvested fat tissue was digested for 15–18 h on a rotating shaker at 37 °C using Dulbecco's modified Eagle's medium/F12 (Stem Cell Technologies, catalog no. 36254) supplemented with 10% gentle collagenase/hyaluronidase (Stem Cell Technologies, catalog no. 07912) and 1% penicillin/streptomycin plus 2% fetal bovine serum (FBS). Debris was removed by a 70-μm cell strainer (BD Biosciences, catalog no. 352350), followed by centrifugation at 100 × g for 5 min at 4 °C. Red blood cells were removed by lysis using ammonium chloride solution (Stem Cell Technologies, catalog no. 07800) at room temperature for 5 min, followed by centrifugation at 600 × g for 5 min at 4 °C. Cell pellets were washed and resuspended in SVF culture medium (Dulbecco's modified Eagle's medium/F12 supplemented with 10% FBS and 1% penicillin/streptomycin).
In vivo tumor study and IL-6 antibody neutralization
Ddr1+/− mice on the pure C57BL/6 background were bred to generate Ddr1+/+ (WT) and Ddr1−/− (KO) littermates, which were used in all in vitro and in vivo experiments. Murine mammary tumor cells (M-Wnt and AT-3) were propagated up to 70% confluency, harvested by trypsinization (0.05% trypsin-EDTA), washed, and resuspended in PBS. Cells were injected into the mouse mammary gland fat pad using 1 × 105 cells/100 μl for M-Wnt and 2 × 105 cells/100 μl for AT-3 per injection. Tumor volume was measured by caliper at the indicated time points, and tumor growth was measured for 4 to 7 weeks. Tumor weight was measured at the time of termination. In vivo IL-6 neutralization was performed by intraperitoneal injection of IL-6–neutralizing antibody (BD Biosciences, clone MP5-20F3, catalog no. 554398) or isotype control anti-IgG (BD Biosciences, clone R3-34, catalog no. 554682) at 100 μg per mouse every 3 days starting 2 days before tumor inoculation. All animal experiments were performed after obtaining approval from the University of Texas Health San Antonio Institutional Animal Care and Use Committee. All methods were carried out in accordance with the Institutional Animal Care and Use Committee–approved guidelines.
Picrosirius red staining
Tumors harvested from mice were fixed in 10% neutral-buffered formalin, dehydrated, embedded in paraffin, and sectioned at 3-μm thickness. The picrosirius red staining was performed as described by the manufacturer using 0.2% phosphomolybdic acid (Electron Microscopy Sciences, catalog no. RT-26357-01), Sirius red 0.1% in saturated picric acid (Electron Microscopy Sciences, catalog no. RT-26357-02), and 0.01 n hydrochloric acid (Electron Microscopy Sciences, catalog no. RT-26357-03). The intensity of positive staining was estimated by measuring the optical density = log [IB/IO], where IB is the average intensity in the background, and IO is the average intensity of the stained area. The percent positive staining was calculated by taking the ratio of area of pixels stained to the total area of pixels in the background. A minimum of 10 fields per tumor section were measured and averaged by ImageJ.
Cell migration and invasion assay
Tumor cells grown to 80% confluency were harvested by trypsinization, washed with PBS twice, resuspended in culture medium without FBS, and co-cultured with but physically separated from either Ddr1 WT-SVF or KO-SVF in a Transwell system. Briefly, 5 × 104 tumor cells were seeded in the top chamber. The bottom chamber was filled with medium with 1% FBS alone or medium with 1% FBS plus 3 × 104 WT-SVF or KO-SVF. After 12 h of co-culture at 37 °C, unmigrated cells on the upper side of the insert were gently removed with a cotton swab. Migrated cells on the undersurface of the inserts were stained by crystal violet. Six fields per insert were counted under an optical microscope.
For the cell invasion assay, the inserts were overlaid with ice-cold Matrigel Basement Membrane Matrix Growth Factor Reduced (Corning, catalog no. 354483) at 5 mg/ml with 50 μl Matrigel/well. The Matrigel was allowed to settle for 30 min at 37 °C. 5 × 104 tumor cells were seeded on the top chamber, and either medium alone or SVF was seeded in the bottom chamber. After 20 h, invaded cells were stained by crystal violet and counted by optical microscopy as described above for the migration assay. The invasion assay in Fig. 5 was conducted using 10 ng/ml recombinant mouse IL-6 (R&D Systems, catalog no. 406-ML-005) supplementation or 1 μg/ml anti-IL6–neutralizing antibody (R&D Systems, catalog no. MAB406) in the bottom chamber with either WT- or KO-SVF. For the experiment shown in Fig. S2, a and b, tumor cells were removed from the top chamber of the inserts (0.4-μm pore, Millipore, catalog no. MCHT12H48) after 3-day incubation with either medium alone or SVF seeded in the bottom chamber. Tumor cells were then examined in the Boyden chamber assay for cell invasion with 10% FBS-containing medium in the bottom chamber without SVF-conditioned medium.
RT-qPCR
RNA samples were reverse-transcribed using the ImProm-II reverse transcription system (Promega, catalog no. A3800). Real-time PCR was set up using Luminaris Color High Green High ROX qPCR Master Mix (Thermo Fisher Scientific, catalog no. K0364) and run in an Applied Biosystems 7900HT work station equipped with SDS 2.4 software. All primers used for RT-PCR were designed using Primer-3 software (Sigma-Aldrich). The primer sequences were as follows: Col1a1-F, GCTCCTCTTAGGGGCCACT; Col1a1-R, ATTGGGGACCCTTAGGCCAT; Col3a1-F, CTGTAACATGGAAACTGGGGAAA; Col3a1-R, CCATAGCTGAACTGAAAACCACC; α-Sma-F, CCCAGACATCAGGGAGTAATGG; α-Sma-R, TCTATCGGATACTTCAGCGTCA; IL-6-F, TCTATACCACTTCACAAGTCGGA; Il-6-R, GAATTGCCATTGCACAACTCTTT; Cofilin-F, ATGACATGAAGGTTCGCAAGT; Cofilin-R, GACAAAAGTGGTGTAGGGGTC;Ctnnb1-F, ATGGAGCCGGACAGAAAAGC; Ctnnb1-R, TGGGAGGTGTCAACATCTTCTT; Cdh2-F, AGGCTTCTGGTGAAATTGCAT; Cdh2-R: GTCCACCTTGAAATCTGCTGG; Cdh1-F, CAGTTCCGAGGTCTACACCTT; and Cdh1-R, TGAATCGGGAGTCTTCCGAAAA.
Western blotting
Protein lysates were prepared in Laemmli sample buffer, and the protein amount was estimated using a BCA protein assay kit (Pierce, catalog no. 23225). For DDR1 Western blotting, samples were run on SDS-PAGE and transferred to an H-bond nitrocellulose membrane using standard procedures. The membrane was blocked using 5% BSA and immunoblotted with anti-DDR1 (D1G6) XP rabbit mAb (Cell Signaling Technology, catalog no. 5583), GAPDH (14C10) rabbit mAb (Cell Signaling Technology, catalog no. 2118), Cofilin mouse mAb (Santa Cruz Biotechnology, catalog no. SC-53934), and α-tubulin mouse mAb (Calbiochem, catalog no. CP06). Protein detection was done using ECL Plus Western blotting substrate (Pierce, catalog no. 32132).
Cytokine array/ELISA
A membrane-based antibody array was used to detect the secretome of Ddr1 WT- and KO-SVF. To reduce the baseline levels of growth factors and cytokines in FBS, SVF was cultured in medium containing 1% FBS (heat-inactivated) for 2 days. Cell-free supernatant was collected and used in the Proteome Profiler Mouse XL cytokine array kit (R&D Systems, catalog no. ARY028) following the instructions of the manufacturer. For the ELISA, SVF was incubated in 1% FBS (heat-inactivated) medium for 2 days. Concentrations of IL-6 in the WT- and KO-SVF cell supernatant or AT-3/M-Wnt tumor cell supernatant were detected utilizing the mouse IL-6 ELISA kit (Invitrogen, catalog no. KMC0061) according to the kit instructions.
MTT proliferation assay
1 × 103 cells were seeded into a 96-well plate and incubated for 1–4 days. 50 μl of MTT solution (3 mg/ml) was added to the culture medium at the indicated time point and incubated at 37 °C for 1 h until the purple precipitate became visible. The medium was removed without disturbing the purple precipitate. 100 μl of DMSO was added to each well and mixed homogeneously, and the absorbance was recorded at 570 nm. For the experiment shown in Fig. S2, c and d, tumor cells were trypsinized from the top chamber of the inserts (0.4-μm pore; Millipore, catalog no. MCHT12H48) after 3 days of co-culture with either medium alone or SVF seeded in the bottom chamber. Tumor cells were seeded into a 96-well plate and incubated for 2 days to monitor the proliferation rate as described above.
Immunohistochemistry/TUNEL assay
Tumors were harvested from specified host mice and fixed with 10% neutral-buffered formalin (Fisher Scientific, catalog no. 23245685) overnight at 4 °C. Paraffin-embedded tumors were cut into 3-μm sections for staining. Slides were deparaffinized and rehydrated by 100% xylene and graded ethanol (100%, 95%, 70%, and 50%). The slides were then boiled with antigen-unmasking solution and washed with PBS. After pretreatment with 3% hydrogen peroxide for 10 min, the slides were subjected to immunostaining with anti-Ki67 (Thermo Fisher Scientific, catalog no. MA5-14520, 1:100) and anti-phospho-histone H3 (Cell Signaling Technology, catalog no. 9701, 1:200). After 2 h of incubation with primary antibody, the ABC peroxidase detection system (Vector Laboratories, catalog no. PK-6105) was used with 3,3′-diaminobenzidine as substrate (Vector Laboratories, catalog no. SK-4105) to detect the primary antibody. Enumeration of Ki67 and p-H3–positive cells was done using image analysis software (ImmunoRatio, http://153.1.200.58:8080/immunoratio/,7 Institute of Biomedical Technology, University of Tampere, Tampere, Finland) (49).
For the TUNEL assay, slides were deparaffinized by graded ethanol (100%, 90%, 70%, and 60%) and PBS wash and subsequently permeabilized by 0.2% Triton X-100 wash for 5 min. Nick-end DNA fragmentations were labeled with the DeadEnd Fluorometic TUNEL system kit (Promega, catalog no. G3250) according to the instructions of the manufacturer. Slides were washed with PBS and mounted with Vectashield mounting medium with DAPI (Vector Laboratories, catalog no. H-1200). TUNEL-positive cells were visualized by fluorescence microscopy and quantified by ImageJ software.
Statistical methods
Mean comparison of two groups was assessed by two-tailed Student's t test. Mean differences of multiple groups were examined by one-way analysis of variance followed by multiple comparison tests. Two-way analysis of variance was used for tumor growth curve analysis. In all assays, p < 0.05 was considered statistically significant.
Author contributions
X. S., K. G., B. W., D. Z., B. Y., X. Z., H.-C. C., and C. Z. data curation; X. S. and R. L. formal analysis; X. S., B. W., and Y. H. investigation; X. S. and K. G. methodology; X. S. and R. L. writing-original draft; B. W., T. J. C., M. P. B., S. H., Y. H., and R. L. writing-review and editing; T. J. C., M. P. B., S. H., Y. H., and R. L. supervision; Y. H. and R. L. funding acquisition; R. L. conceptualization; R. L. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Yi Li for the generous gift of the AT-3 cell line and Sabrina Smith for technical assistance with mouse genotyping.
This work was supported by NCI, National Institutes of Health Grants CA206529 and CA161349, Department of Defense Grant WSlXWH-14-1-0129), and the Tom C. and H. Frost Endowment (to R. L.); NCI, National Institutes of Health Grant CA212674, Department of Defense Grant W81XWH-17-1-0007, and Cancer Prevention and Research Institute of Texas Grant RP170126 (to Y.-F. H.); National Institutes of Health Postdoctoral Training Grant T32CA148724 (to H.-C. C.); and the Owens Foundation and the Skinner Endowment (to T. J. C.). This work was also supported by University of Texas Health San Antonio Cancer Center Grant CA054174. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- ECM
- extracellular matrix
- ASC
- adipose stem/stromal cell
- KO
- knockout
- SVF
- stromal–vascular fraction
- IL
- interleukin
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- PSR
- picrosirius red
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- RT-qPCR
- quantitative RT-PCR.
References
- 1. Moley K. H., and Colditz G. A. (2016) Effects of obesity on hormonally driven cancer in women. Sci. Transl. Med. 8, 323ps3 10.1126/scitranslmed.aad8842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park J., Morley T. S., Kim M., Clegg D. J., and Scherer P. E. (2014) Obesity and cancer: mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 10, 455–465 10.1038/nrendo.2014.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan D. S., Vieira A. R., Aune D., Bandera E. V., Greenwood D. C., McTiernan A., Navarro Rosenblatt D., Thune I., Vieira R., and Norat T. (2014) Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 25, 1901–1914 10.1093/annonc/mdu042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gilbert C. A., and Slingerland J. M. (2013) Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu. Rev. Med. 64, 45–57 10.1146/annurev-med-121211-091527 [DOI] [PubMed] [Google Scholar]
- 5. Olson O. C., Quail D. F., and Joyce J. A. (2017) Obesity and the tumor microenvironment. Science 358, 1130–1131 10.1126/science.aao5801 [DOI] [PubMed] [Google Scholar]
- 6. Iyengar N. M., Hudis C. A., and Dannenberg A. J. (2015) Obesity and cancer: local and systemic mechanisms. Annu. Rev. Med. 66, 297–309 10.1146/annurev-med-050913-022228 [DOI] [PubMed] [Google Scholar]
- 7. Iyengar N. M., Zhou X. K., Gucalp A., Morris P. G., Howe L. R., Giri D. D., Morrow M., Wang H., Pollak M., Jones L. W., Hudis C. A., and Dannenberg A. J. (2016) Systemic correlates of white adipose tissue inflammation in early-stage breast cancer. Clin. Cancer Res. 22, 2283–2289 10.1158/1078-0432.CCR-15-2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Picon-Ruiz M., Pan C., Drews-Elger K., Jang K., Besser A. H., Zhao D., Morata-Tarifa C., Kim M., Ince T. A., Azzam D. J., Wander S. A., Wang B., Ergonul B., Datar R. H., Cote R. J., et al. (2016) Interactions between adipocytes and breast cancer cells stimulate cytokine production and drive Src/Sox2/miR-302b–mediated malignant progression. Cancer Res. 76, 491–504 10.1158/0008-5472.CAN-15-0927 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y. Y., Attané C., Milhas D., Dirat B., Dauvillier S., Guerard A., Gilhodes J., Lazar I., Alet N., Laurent V., Le Gonidec S., Biard D., Hervé C., Bost F., Ren G. S., et al. (2017) Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2, e87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nieman K. M., Romero I. L., Van Houten B., and Lengyel E. (2013) Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 1831, 1533–1541 10.1016/j.bbalip.2013.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Howe L. R., Subbaramaiah K., Hudis C. A., and Dannenberg A. J. (2013) Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 19, 6074–6083 10.1158/1078-0432.CCR-12-2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang T., Tseng C., Zhang Y., Sirin O., Corn P. G., Li-Ning-Tapia E. M., Troncoso P., Davis J., Pettaway C., Ward J., Frazier M. L., Logothetis C., and Kolonin M. G. (2016) CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 7, 11674 10.1038/ncomms11674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bulun S. E., Lin Z., Zhao H., Lu M., Amin S., Reierstad S., and Chen D. (2009) Regulation of aromatase expression in breast cancer tissue. Ann. N.Y. Acad. Sci. 1155, 121–131 10.1111/j.1749-6632.2009.03705.x [DOI] [PubMed] [Google Scholar]
- 14. Clyne C. D., Speed C. J., Zhou J., and Simpson E. R. (2002) Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J. Biol. Chem. 277, 20591–20597 10.1074/jbc.M201117200 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh S., Ashcraft K., Jahid M. J., April C., Ghajar C. M., Ruan J., Wang H., Foster M., Hughes D. C., Ramirez A. G., Huang T., Fan J. B., Hu Y., and Li R. (2013) Regulation of adipose oestrogen output by mechanical stress. Nat. Commun. 4, 1821 10.1038/ncomms2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vogel W., Gish G. D., Alves F., and Pawson T. (1997) The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 10.1016/S1097-2765(00)80003-9 [DOI] [PubMed] [Google Scholar]
- 17. Fu H. L., Valiathan R. R., Arkwright R., Sohail A., Mihai C., Kumarasiri M., Mahasenan K. V., Mobashery S., Huang P., Agarwal G., and Fridman R. (2013) Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J. Biol. Chem. 288, 7430–7437 10.1074/jbc.R112.444158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leitinger B. (2014) Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 310, 39–87 10.1016/B978-0-12-800180-6.00002-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dorison A., Dussaule J. C., and Chatziantoniou C. (2017) The role of discoidin domain receptor 1 in inflammation, fibrosis and renal disease. Nephron 137, 212–220 10.1159/000479119 [DOI] [PubMed] [Google Scholar]
- 20. Valiathan R. R., Marco M., Leitinger B., Kleer C. G., and Fridman R. (2012) Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31, 295–321 10.1007/s10555-012-9346-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rammal H., Saby C., Magnien K., Van-Gulick L., Garnotel R., Buache E., El Btaouri H., Jeannesson P., and Morjani H. (2016) Discoidin domain receptors: potential actors and targets in cancer. Front. Pharmacol. 7, 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gao H., Chakraborty G., Zhang Z., Akalay I., Gadiya M., Gao Y., Sinha S., Hu J., Jiang C., Akram M., Brogi E., Leitinger B., and Giancotti F. G. (2016) Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 166, 47–62 10.1016/j.cell.2016.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finak G., Bertos N., Pepin F., Sadekova S., Souleimanova M., Zhao H., Chen H., Omeroglu G., Meterissian S., Omeroglu A., Hallett M., and Park M. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 14, 518–527 10.1038/nm1764 [DOI] [PubMed] [Google Scholar]
- 24. Gimble J. M., Bunnell B. A., Frazier T., Rowan B., Shah F., Thomas-Porch C., and Wu X. (2013) Adipose-derived stromal/stem cells: a primer. Organogenesis 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hou G., Vogel W., and Bendeck M. P. (2001) The discoidin domain receptor tyrosine kinase DDR1 in arterial wound repair. J. Clin. Invest. 107, 727–735 10.1172/JCI10720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart T. J., and Abrams S. I. (2007) Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J. Immunol. 179, 2851–2859 10.4049/jimmunol.179.5.2851 [DOI] [PubMed] [Google Scholar]
- 27. Dunlap S. M., Chiao L. J., Nogueira L., Usary J., Perou C. M., Varticovski L., and Hursting S. D. (2012) Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev. Res. (Phila.) 5, 930–942 10.1158/1940-6207.CAPR-12-0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verbrugge I., Hagekyriakou J., Sharp L. L., Galli M., West A., McLaughlin N. M., Duret H., Yagita H., Johnstone R. W., Smyth M. J., and Haynes N. M. (2012) Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 72, 3163–3174 10.1158/0008-5472.CAN-12-0210 [DOI] [PubMed] [Google Scholar]
- 29. Ford N. A., Rossi E. L., Barnett K., Yang P., Bowers L. W., Hidaka B. H., Kimler B. F., Carlson S. E., Shureiqi I., deGraffenried L. A., Fabian C. J., and Hursting S. D. (2015) Omega-3-acid ethyl esters block the protumorigenic effects of obesity in mouse models of postmenopausal basal-like and claudin-low breast cancer. Cancer Prev. Res. (Phila) 8, 796–806 10.1158/1940-6207.CAPR-15-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niu M., Valdes S., Naguib Y. W., Hursting S. D., and Cui Z. (2016) Tumor-associated macrophage-mediated targeted therapy of triple-negative breast cancer. Mol. Pharm. 13, 1833–1842 10.1021/acs.molpharmaceut.5b00987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Flanagan C. H., Rossi E. L., McDonell S. B., Chen X., Tsai Y. H., Parker J. S., Usary J., Perou C. M., and Hursting S. D. (2017) Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 3, 26 10.1038/s41523-017-0027-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bussard K. M., Mutkus L., Stumpf K., Gomez-Manzano C., and Marini F. C. (2016) Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 18, 84 10.1186/s13058-016-0740-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walter M., Liang S., Ghosh S., Hornsby P. J., and Li R. (2009) Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene 28, 2745–2755 10.1038/onc.2009.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ancrile B., Lim K. H., and Counter C. M. (2007) Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 10.1101/gad.1549407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kapur S. K., and Katz A. J. (2013) Review of the adipose derived stem cell secretome. Biochimie 95, 2222–2228 10.1016/j.biochi.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 36. Zahid H., Simpson E. R., and Brown K. A. (2016) Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharmacol. 31, 90–96 10.1016/j.coph.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 37. Crewe C., An Y. A., and Scherer P. E. (2017) The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest. 127, 74–82 10.1172/JCI88883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tamm I., Cardinale I., Krueger J., Murphy J. S., May L. T., and Sehgal P. B. (1989) Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. J. Exp. Med. 170, 1649–1669 10.1084/jem.170.5.1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sehgal P. B. (1990) Interleukin 6 in infection and cancer. Proc. Soc. Exp. Biol. Med. 195, 183–191 10.3181/00379727-195-43129D [DOI] [PubMed] [Google Scholar]
- 40. Bromberg J., and Wang T. C. (2009) Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 15, 79–80 10.1016/j.ccr.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sullivan N. J., Sasser A. K., Axel A. E., Vesuna F., Raman V., Ramirez N., Oberyszyn T. M., and Hall B. M. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947 10.1038/onc.2009.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ara T., Song L., Shimada H., Keshelava N., Russell H. V., Metelitsa L. S., Groshen S. G., Seeger R. C., and DeClerck Y. A. (2009) Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 69, 329–337 10.1158/0008-5472.CAN-08-0613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dethlefsen C., Højfeldt G., and Hojman P. (2013) The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat 138, 657–664 10.1007/s10549-013-2488-z [DOI] [PubMed] [Google Scholar]
- 44. Sasser A. K., Sullivan N. J., Studebaker A. W., Hendey L. F., Axel A. E., and Hall B. M. (2007) Interleukin-6 is a potent growth factor for ER-α-positive human breast cancer. FASEB J. 21, 3763–3770 10.1096/fj.07-8832com [DOI] [PubMed] [Google Scholar]
- 45. Knüpfer H, Preiss R. (2007). Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 102, 129–135 [DOI] [PubMed] [Google Scholar]
- 46. Hong D. S., Angelo L. S., and Kurzrock R. (2007) Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer 110, 1911–1928 10.1002/cncr.22999 [DOI] [PubMed] [Google Scholar]
- 47. Harkins J. M., Moustaid-Moussa N., Chung Y. J., Penner K. M., Pestka J. J., North C. M., and Claycombe K. J. (2004) Expression of interleukin-6 is greater in preadipocytes than in adipocytes of 3T3-L1 cells and C57BL/6J and ob/ob mice. J. Nutr. 134, 2673–2677 [DOI] [PubMed] [Google Scholar]
- 48. Nair S. J., Zhang X., Chiang H.-C., Jahid M. J., Wang Y., Garza P., April C., Salathia N., Banerjee T., Alenazi F. S., Ruan J., Fan J. B., Parvin J. D., Jin V. X., Hu Y., and Li R. (2016) Genetic suppression reveals DNA repair-independent antagonism between BRCA1 and COBRA1 in mammary gland development. Nat. Commun. 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tuominen V. J., Ruotoistenmäki S., Viitanen A., Jumppanen M., and Isola J. (2010) ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 12, R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.