

Abstract
Periodontal disease is initiated by microorganisms in dental plaque, and host immunoinflammatory response to the microbial challenge helps in disease progression. Conventional periodontal therapy was mainly targeted on the elimination of microbial component. However, a better understanding of molecular aspects in host response will enable the clinicians to formulate effective host modulation therapy (HMT) for the periodontal management. Inflammatory mediators were the main targets for HMT in the past. Transcription factors can regulate the production of multiple mediators simultaneously, and inhibition of these factors will be more beneficial than blocking individual molecule. Two important transcription factors implicated in chronic inflammatory diseases are nuclear factor kappa B (NF-κB) and signal transducers and activators of transcription 3. The role of these factors in periodontal disease is a less explored area. This comprehensive review is aimed at unveiling the critical role of NF-κB and signal transducers and activators of transcription 3 in periodontal pathogenesis. An online search was performed using MEDLINE/PubMed database. All publications till 2016 related to NF-κB, signal transducer and activator of transcription 3 (STAT3), and inflammation were included in writing this review. A total of 27,390 references were published based on the search terms used. Out of these, 507 were related to the periodontal research published in English till 2016. Relevant papers were chosen after carefully reading the abstract. This review has attempted to comprehend the existing knowledge regarding the role of transcription factors NF-κB and STAT3 in periodontal disease. Moreover, it also provides a connecting molecular link for the periodontal medicine concept.
Key words: Host response, inflammation, nuclear factor kappa B, periodontal disease, Signal Transducer and Activator of Transcription 3, transcription factors
INTRODUCTION
Periodontal disease is a common, chronic inflammatory disease of multifactorial etiology. Microbes in the dental plaque initiate periodontal inflammation, but host response to these microbial insults is mainly responsible for the progression of the disease.[1] Periodontal inflammation is a defense mechanism of the host against infection. Inflammatory mediators are produced from the host tissues to limit the activity of periodontopathogens, but paradoxically, it will create destruction of the periodontium. Genetic and environmental risk factors deregulate this host response further aggravating the disease process.
Microbial factors such as microbe-associated molecular patterns (MAMP) and host proteins like cell damage-associated molecular patterns (DAMP) communicate with periodontal cell surface receptors such as toll-like receptors (TLR), nod-like receptors, retinoic acid-inducible gene I-like receptors, and C-type lectin receptors.[2] When MAMPs and DAMPs are recognized by these receptors, a series of cellular signaling events take place which ultimately results in the production of various inflammatory mediators. The most important inflammatory mediators in periodontal disease are cytokines, prostaglandins, and matrix metalloproteinases.[1] Receptor activator nuclear factor kappa ligand (RANKL) is a critical molecular mediator of bone resorption. Along with these factors, oxidative stress also participates in both hard- and soft-tissue destruction of periodontium. During the cellular signaling event, transcription factors act as intermediate players to bring about activation of target genes. Epigenetic alterations in the host produced by polymicrobial synergy and dysbiosis also lead to activation of transcription factors as described in a recent study.[2]
Transcription factors are the factors present in cytoplasm of many cells and on activation, they are transported to nucleus to regulate the production of multiple inflammatory mediators. Even though lot of information is available on the inflammatory mediators of periodontal tissue destruction, little is known about the role of transcription factors in periodontal pathogenesis. This comprehensive review is aimed at unveiling the importance of two pivotal transcription factors, nuclear factor kappa B (NF-κB), and signal transducer and activator of transcription 3 (STAT3) in periodontal pathogenesis.
NUCLEAR FACTOR KAPPA B
NF-κB is a family of ubiquitous transcription factors first described by Sen and Baltimore in 1986 as the regulator of kappa light chain gene in murine B lymphocytes.[3] The family members are NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and cRel.[4] Any homo or heterodimer combination of these members is considered as NF-κB, but the classic form of NF-κB is the combination of p50 and p65.[4] Inside the cytoplasm, NF-κB exists as a complex with a protein known as inhibitor κB (IκB) and different types of IκB are IκBa, IκBβ, IκBέ, IκB-r, p100, p102, and BCL3.[4] When cells are stimulated with various activators such as tumor necrosis factor α (TNF-α), interleukin-1 (IL-1), lipopolysaccharide (LPS), oxidants and viruses phosphorylation, ubiquitination, and subsequent degradation of IκB occurs. Thus, NF-κB is made free and is transported to the nucleus where it activates target genes [Figure 1].[4]
Figure 1.
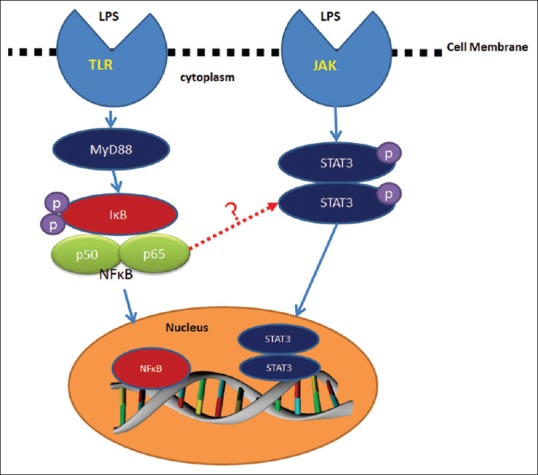
Activation of nuclear factor kappa B and signal transducer and activator of transcription 3 and possible cross talk (indicated by dotted red arrow) in their signaling in periodontal disease. TLR – Toll like receptors, LPS – Lipopolysaccharide, JAK – Janus activated kinase, STAT3 – Signal signal transducer and activator of transcription 3, MYD88 – Myeloid differentiation primary response 88, IXB – its not X but kappa (κ)- the expansion is inhibitor kappa B, P50 – PROTEIN 50, P65 – PROTEIN 65, NFB – nuclear factor-kappa B
SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION 3
The Janus kinase-signal transducers and activators of transcription (JAK-STAT) signaling is mainly activated by cytokines and control expression of genes related to inflammation. STATs are cytoplasmic proteins which are characterized by SH2 (Src Homology-2) domain. When the receptors associated with STAT signaling are stimulated by appropriate cytokines, initially dimerization of the receptors occur. Then, tyrosine kinases JAK1, JAK2, JAK3, and Tyk2 which are associated with these receptors are activated.[5] This receptor-kinase complex interacts with and activates members of the STAT family through tyrosine phosphorylation resulting in the formation of homo or heterodimers of STAT. These dimeric molecules are transported to the nucleus by exposure of nuclear localization signal to stimulate transcription of the responsive genes [Figure 1].[5] At present, seven STAT family members have been identified, that is, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. They have similarity in their molecular structure and function but play diverse physiological roles in a wide variety of biological processes.[5]
STAT3 was known earlier as acute-phase response factor (APRF), and Zhongin (1994) included it as a member of STAT family.[6] Gp130 cytokines such as IL-6, oncostatin M (OSM), leukemia inhibitory factor (LIF), and ciliary neurotrophic factor mediates STAT3 signaling.[6] STAT3 phosphorylation takes place at tyrosine (Tyr)-705, but maximal transcriptional activation requires additional phosphorylation at serine (Ser)-727.[7] SH2-containing phosphatases (SHPs), protein inhibitors of activated STAT3 (PIAS3), and suppressors of cytokine signaling (SOCS) are the endogenous regulators of STAT3 which prevent its activation in normal cells.[8]
CROSS TALK BETWEEN NUCLEAR FACTOR KAPPA B AND SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION 3
NF-κB and STAT3 synergistically control a common set of genes encoding for cytokines and chemokines.[9] NF-κB family members physically interact with STAT3 resulting in either transcriptional synergy or repression of NF-κB/STAT3 regulated genes, and many mechanisms for this have been suggested.[9] Unphosphorylated STAT3 bind to NF-κB/IκB complex and facilitate NF-κB activation. STAT3 may interact with p65 in the nucleus and increase its nuclear retention through acetylation and ensure constitutive NF-κB activation. NF-κB/STAT3 complex can activate target genes which cannot be induced by either factor alone.[9] STAT3 is an upstream signaling factor for NF-κB activation.[10] NF-κB activation will result in the production of IL-6 which in turn will activate STAT3.[11] Simultaneous activation may be beneficial to mediate effective inflammatory response in many diseases. NF-κB and STAT3 signaling cross talk was observed in many inflammatory conditions and cancers.[9,10] Even though functional studies have not yet identified such a cross talk in periodontal disease, there are every possibility for the same [Figure 1].
THE IMPORTANCE OF NUCLEAR FACTOR KAPPA B AND SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION 3 IN PERIODONTAL PATHOGENESIS
Among the 150 activators of NF-κB listed, majority are seen in abundance in periodontal disease, for example, bacterial LPS, prostaglandin E2, IL-1β, TNF alpha, stress, viruses, etc.[12] The keystone pathogen Porphyromonas gingivalis is used as an activator of NF-κB in many in vitro experiments.[13,14] NF-κB activation by P. gingivalis, Aggregatibacter actinomycetemcomitans, Treponema denticola, Fusobacterium nucleatum, and P. intermedia induced apoptosis of monocytes and neutrophils.[15] Target genes of NF-κB including cytokines, matrix metalloproteinases (MMP), cyclooxygenase 2 (COX 2), inducible nitric oxide synthase (iNOS), RANKL, etc., are beneficial to the progression of periodontal disease.[12] NF-κB activation by various activators such as IL-1β, TNF α has been observed in cultured periodontal cells which lead to production of inflammatory mediators such as prostaglandins and MMP.[16,17] Ninety-one genes which represented biological processes associated with periodontitis were upregulated by NF-κB activation induced by periodontopathogens P. gingivalis and F. nucleatum in H400 oral epithelial cell lines.[15]
The hallmarks of periodontal destruction are inflammation of the soft tissues, collagen degradation, and bone resorption. The major tissue destructive enzyme in periodontal disease is MMP and its production is controlled by NF-κB activation as evidenced by in vitro experiments.[17] Periodontal tissue remodeling is also mediated by the activation of NF-κB-dependent genes encoding inducible forms of COX-2 and iNOS enzymes participating in production of prostaglandins, nitric oxide, and nitric oxide metabolites.[12] Vascular endothelial growth factor activated by NF-κB in fibroblasts promotes formation of new blood vessels in inflamed periodontal tissues.[18]
NF-κB plays an important role in bone destruction as it can activate osteoclasts according to scientific evidence.[19,20] NF-κB-dependent cytokines such as IL-1α and IL-1 β, TNF-α, IL-6, and IL-17 induce osteoclast differentiation and activation mediated by RANK.[19] Osteoclastogenesis is controlled by NF-κB activation followed by c-Fos and NFATc1 (nuclear factor for activated T-cells) activation.[20] NF-κB activation impair Fos-related antigen-1 which is a transcription factor involved in bone matrix formation, thereby impairing bone formation.[21] Impairment of osteogenic differentiation of inflamed periodontal ligament stem cells is mediated through NF-κB signaling and inhibition of NF-κB activation can reverse this phenomenon.[22]
Activation of NF-κB in diseased human periodontal tissues has been documented in a few studies.[23,24] Increased activation of p50 and p65 subunits of NF-κB and decreased expression of IκB were expressed in diseased periodontal tissues compared to healthy ones. Increased formation of nuclear p50 homodimers were observed in the human gingiva from chronic periodontitis patients and activation of dendritic cells by P. gingivalis LPS resulted in increase in p50/p65 ratios.[25] Cytoplasmic expression of NF-κB was strong in all cell types in inflamed periodontal ligament, and intense nuclear expression was noted in immune and endothelial cells.[14]
STAT3 pathway is activated mainly by IL-6 and IL-6 family of cytokines which signal through gp130 receptors.[6] The importance of IL-6 in periodontal disease is well documented.[1] Other activators of STAT3 such as IL-22, INF-γ, TNF-α, IL-1, IL-4, IL-10, IL-17/23, and LPS, etc., are also seen associated with periodontal disease.[1] Signaling target of STAT3 include INF-γ, TNF-α, IL-1, IL-4, IL-6, and IL-10, MMP-2 and MMP-9, iNOS, VEGF, COX-2, etc., and many of these molecules have biologically significant role in the periodontal pathogenesis.[1]
Even though STAT3 activation has not been evaluated in human periodontal tissues; activation of STAT3 in human periodontal ligament cells was observed when they were stimulated with IL-1 β and IL-6.[26] The activated STAT3 lead to the expression of CC chemokine ligand 20 which recruit Th17 cells that play a central role in periodontal bone destruction.[26]
A few experimental studies in periodontal disease models also demonstrated STAT3 activation. Activation of STAT3 was observed in a ligature-induced periodontitis model in winstar rats.[27] Rapid and transient activation of extracellular-regulated kinases and p38 mitogen-activated protein kinase as well as NF-κB was also noted in the same specimens. This experiment indicates a cross talk and involvement of many signaling pathways in cytokine-mediated periodontal destruction. Upregulated expression of activated STAT3 was also induced by LPS in experimental animals.[28]
Risk factors of periodontal disease such as diabetes, smoking, and obesity activate STAT3 and NF-κB leading to persistent inflammation.[29,30,31,32] Nicotine and LPS-treated periodontal ligament cells increased the number of osteoclasts and inhibition of hypoxia inducible factor-2α reversed the effects through multiple signaling pathways including STAT3 and NF-κB.[33] STAT3 and NF-κB activation has also been reported in many chronic inflammatory diseases such as rheumatoid arthritis which share similarities with periodontal disease in pathogenesis.[34]
Microbial etiology is insufficient to explain the periodontal disease progression in a subgroup of patients where molecular mechanisms such as STAT3 and NF-κB activation can be considered as logical explanations for the exaggerated host response. It is evident that activation of these transcription factors is possible in periodontal disease and their cross talk and target gene upregulation can favor the rapid progression of the disease.
NUCLEAR FACTOR KAPPA B AND SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION 3: A MOLECULAR LINK IN PERIODONTAL MEDICINE CONCEPT
Recent studies have identified association between periodontal disease and cardiovascular disease, diabetes mellitus, preterm low birth weight, and many other systemic diseases.[35] Many mechanisms to explain this association has been proposed, including systemic dissemination of pathogens and stimulation of host response. However, none of them were able to give satisfactory explanation for this interrelation. NF-κB and STAT3 activation can be considered as an important molecular mechanism to explain the link between periodontal disease and systemic diseases. NF-κB as a connecting link between atherogenesis and periodontitis has been already proposed.[36] The role of activated STAT3 in atheroma formation also has been clearly understood from in vivo as well as in vitro studies.[37] STAT3 mediates inhibition of Pseudomonas aeruginosa and P. gingivalis-induced apoptosis in respiratory epithelial cells, thus contributing to the pathogenesis of respiratory disease.[38] In a recent study, placental tissues of preeclampsic women exhibited NF-κB upregulation giving supportive evidence for the role of NF-κB in the association between preeclampsia and periodontal disease.[39] NF-κB activation produce IL-6 which is an important activator of STAT3, and this molecular mechanism operates in association between obesity, aging, chronic inflammation, and cancer.[40]
Chronic infections and inflammation are found to be associated with about 20% of human cancers.[41] Chronic periodontal disease is the most abundant source of low-grade inflammation in the body. Studies have found out an association between chronic periodontal inflammation and cancer of oral cavity, pancreas, lungs, kidney, blood, and gastric cancers.[42] However, molecular mechanism pertaining to this association is still under investigation. NF-κB and STAT3 pathways may be the pivotal signaling mediators in inflammation associated tumor development.[43] NF-κB and STAT3 together activate FAT10 which inhibit tumor suppressor p53 facilitating tumor progression, and it can also upregulate oncogenes such as cyclin D1.[44,45] A recent study has demonstrated that P. gingivalis and F. nucleatum interact with oral epithelial cells through toll-like receptors and signal along IL-6/STAT3 resulting in oral squamous cell carcinoma in murine periodontitis model.[46]
CLINICAL IMPLICATIONS
Traditional periodontal therapy was focused on eliminating or reducing microorganisms in plaque biofilm using mechanical and chemical methods. Extended knowledge of the role of host response in periodontal pathogenesis has opened up a new treatment approach called host modulation therapy (HMT). HMT has proved as a beneficial adjunct to conventional periodontal therapy individuals who are susceptible to periodontal disease.[1] Drugs mainly utilized for host modulation were subantimicrobial dose doxycycline, NSAIDs, and bone-sparing drugs like bisphosphonates. Anticytokine therapy targeting IL-1, IL-6, and RANKL when administered either systemically or locally was also found to be beneficial for periodontal therapy. However, most of these drugs had unwanted side effects and inhibition of cytokines can affect its protective functions in the immune system.
Chronic diseases are connected with approximately 500 gene products. Thus, prevention and treatment of such diseases require inhibition of multiple molecular mediators. A more recent approach for HMT is by inhibiting major signaling pathways of inflammation.[47] Here, the advantage is that many molecules can be simultaneously targeted. Signaling blockade is more beneficial because when individual molecule is inhibited there can be compensating mechanism with another molecule. Rapid and transient activation of signaling can result in long-lasting modulation of cytokine gene expression. Hence, a short-term blockade without affecting normal physiological processes may be beneficial for periodontal disease.
Signaling blockade has been attempted and found to be useful in experimental periodontitis and also in other similar chronic inflammatory diseases.[48,49] NF-κB inhibitors include a wide variety of molecules such as antioxidants, proteasome inhibitors, IκB phosphorylation/degradation blockers, upregulators of IκB, inhibitors of Rel/NF-κB nuclear transport, inhibitors of Rel/NF-κB DNA binding, and transactivation. Genetically engineered proteins that block specific steps in NF-κB activation were also developed including I-TRAF (TNF receptor-associated factor interacting protein), IKK complex mutants, and IκBα superrepressor.[50] NF-κB is inhibited by drugs such as aspirin, NSAID's, glucocorticoids, cyclosporine, and tacrolimus different mechanisms. Recent research has shown promising results in preventing the progression of bone loss with topical application of NF-κB decoy oligodeoxynucleotides in experimental periodontitis in beagle dogs.[51] Clinical efficacy of these agents for periodontal therapy can be further established with human clinical trials.
STAT3 inhibition was attempted by targeting tyrosine kinases, and several inhibitors were developed. JAK2 inhibitors are already in the market.[52] JAK inhibitors such as tofacitinib and ruxolitinib are found to be useful in patients with rheumatoid arthritis.[53] Inhibitors of JAK3 and Tyk2 were mainly tried for the drug development because deficiency of JAK1 or JAK2 can be lethal. No Tyk2 inhibitor is developed so far, but JAK3 antagonist CP-690550 (Pfizer) showed potential benefits in the treatment of rheumatoid arthritis.[54]
STAT3 inhibition can also be achieved using peptidomimetics, STAT3 dimerization inhibiting molecules, competitive binding molecules for STAT3, antisense oligonucleotides, and small inhibitory RNA.[55] Cell-specific modulation of STAT3/SOCS3 decreases pro-inflammatory response in many inflammatory conditions like arthritis.[56] Photodynamic therapy which is an emerging treatment concept in periodontal disease produce cross-linking of STAT3, and cross-linked STAT3 is unable to enter the nucleus. The cells will be unresponsive to cytokines such as IL-6, oncostatin M at least for 24 h.[57]
Inhibition of one signaling pathway allows cells to survive using some other mechanism. Dual inhibition of STAT3–NF-κB may overcome problems associated with inhibition of either pathway. Dual inhibition has been reported to be beneficial in many studies. Blocking STAT3 activity preferentially inhibits LPS-mediated IL-1 β and IL-6 production, but not TNF-α, in RAW264.7 cells which require inhibition of NF-κB pathway.[58] Hence, agents that simultaneously block NF-κB and STAT3 may be more beneficial for inflammatory bone diseases like periodontal disease. Inhibitors of STAT3, NF-κB, or both were successfully used in treating many immune inflammatory conditions like arthritis.[59]
Inhibition of toll-like receptor 4 (TLR4) and STAT3 by 25-hydroxyvitamin D3 was utilized in experimental periodontitis.[60] Fasting blood glucose, glycosylated hemoglobin, and TNF-α levels were reduced in diabetic rats by 25-OHD3 intraperitoneal injection and correspondingly reduction in alveolar bone loss was also noted. In the same animals, increased expression of vitamin D receptor and reduction in TLR4, JAK1, STAT3, and their phosphorylation were also noted in gingival epithelia.
Basal activity of both NF-κB and STAT3 are required for many normal physiological functions. Moreover, drugs with NF-κB and STAT3 inhibiting property possess many side effects. Specific and potent inhibitors that provide an effective treatment while sparing the host from the side effects are actually required. The route of drug delivery might also affect the outcome of therapy. Local delivery of the agents in periodontal therapy reduces the side effects associated with systemic exposure and minimize the risk of general immunosuppression. Gene delivery also may appear promising for periodontal therapy.
Extracts of many medicinal plants can inhibit NF-κB and STAT3 signaling.[59,61] Curcumin which has proven to be effective in managing periodontal disease interfere with NF-κB STAT3 cross talk.[16] Nutritional modulation is a significant and emerging concept in periodontal management. Omega 3 fatty acid, one of the important nutritional supplements recommended for periodontal disease also reduce the activation of inflammatory transcription factors NF-κB and STAT3.[62] Specific inhibitors of these transcription factors for systemic administration, local drug delivery as well as HMT have to be identified for periodontal disease. Future controlled clinical trials are required to identify safe and effective NF-κB and STAT3 inhibitors that can revolutionize the existing periodontal treatment strategies and thereby help to improve patient's oral as well as systemic health.
CONCLUSION
Periodontal disease is a multifactorial disease, and the major role in its pathogenesis is played by immune inflammatory response of the host. So along with controlling microbial factors, host modulation is an integral part in periodontal therapy. NF-κB and STAT3 are two crucial transcription factors in chronic inflammatory diseases. The role of NF-κB and STAT3 in periodontal disease is clearly evident from the available literature. Activation and cross talk between STAT3 and NF-κB is possible in periodontal disease environment, and it can favor the disease progression if allowed to continue. Inhibition of these factors has been utilized successfully in the treatment of similar chronic inflammatory diseases. Simultaneous inhibition of NF-κB and STAT3 may constitute the next generation therapeutic modality for periodontal disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000. 2014;64:57–80. doi: 10.1111/prd.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappa B by a posttranslational mechanism. Cell. 1986;47:921–8. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin AS Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Akira S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells. 1999;17:138–46. doi: 10.1002/stem.170138. [DOI] [PubMed] [Google Scholar]
- 6.Zhong Z, Wen Z, Darnell JE Jr. Stat3 and stat4: Members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A. 1994;91:4806–10. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Z, Zhong Z, Darnell JE Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 8.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: Clinical implications. Clin Cancer Res. 2007;13:1362–6. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- 9.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–9. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cha B, Lim JW, Kim H. Jak1/Stat3 is an upstream signaling of NF-κB activation in Helicobacter pylori-induced IL-8 production in gastric epithelial AGS cells. Yonsei Med J. 2015;56:862–6. doi: 10.3349/ymj.2015.56.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, et al. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol. 2011;186:1199–208. doi: 10.4049/jimmunol.1002971. [DOI] [PubMed] [Google Scholar]
- 12.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe A, Takeshita A, Kitano S, Hanazawa S. CD14-mediated signal pathway of Porphyromonas gingivalis lipopolysaccharide in human gingival fibroblasts. Infect Immun. 1996;64:4488–94. doi: 10.1128/iai.64.11.4488-4494.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gölz L, Memmert S, Rath-Deschner B, Jäger A, Appel T, Baumgarten G, et al. Hypoxia and P. gingivalis synergistically induce HIF-1 and NF-κB activation in PDL cells and periodontal diseases. Mediators Inflamm. 2015;2015:438085. doi: 10.1155/2015/438085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milward MR, Chapple IL, Wright HJ, Millard JL, Matthews JB, Cooper PR, et al. Differential activation of NF-kappaB and gene expression in oral epithelial cells by periodontal pathogens. Clin Exp Immunol. 2007;148:307–24. doi: 10.1111/j.1365-2249.2007.03342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu P, Huang P, Chen MW. Curcumin attenuates cyclooxygenase-2 expression via inhibition of the NF-κB pathway in lipopolysaccharide-stimulated human gingival fibroblasts. Cell Biol Int. 2013;37:443–8. doi: 10.1002/cbin.10050. [DOI] [PubMed] [Google Scholar]
- 17.Kida Y, Kobayashi M, Suzuki T, Takeshita A, Okamatsu Y, Hanazawa S, et al. Interleukin-1 stimulates cytokines, prostaglandin E2 and matrix metalloproteinase-1 production via activation of MAPK/AP-1 and NF-kappaB in human gingival fibroblasts. Cytokine. 2005;29:159–68. doi: 10.1016/j.cyto.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol. 1997;17:4015–23. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 20.Edwards JR, Mundy GR. Advances in osteoclast biology: Old findings and new insights from mouse models. Nat Rev Rheumatol. 2011;7:235–43. doi: 10.1038/nrrheum.2011.23. [DOI] [PubMed] [Google Scholar]
- 21.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15:682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Hu C, Wang G, Li L, Kong X, Ding Y, et al. Nuclear factor-κB modulates osteogenesis of periodontal ligament stem cells through competition with β-catenin signaling in inflammatory microenvironments. Cell Death Dis. 2013;4:e510. doi: 10.1038/cddis.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambili R, Santhi WS, Janam P, Nandakumar K, Pillai MR. Expression of activated transcription factor nuclear factor-kappaB in periodontally diseased tissues. J Periodontol. 2005;76:1148–53. doi: 10.1902/jop.2005.76.7.1148. [DOI] [PubMed] [Google Scholar]
- 24.Arabaci T, Cicek Y, Canakci V, Canakci CF, Ozgoz M, Albayrak M, et al. Immunohistochemical and stereologic analysis of NF-κB activation in chronic periodontitis. Eur J Dent. 2010;4:454–61. [PMC free article] [PubMed] [Google Scholar]
- 25.Jotwani R, Moonga BS, Gupta S, Cutler CW. Nuclear factor-kappaB p50 subunits in chronic periodontitis and Porphyromonas gingivalis lipopolysaccharide-pulsed dendritic cells. Ann N Y Acad Sci. 2010;1192:278–85. doi: 10.1111/j.1749-6632.2009.05247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hosokawa Y, Shindo S, Hosokawa I, Ozaki K, Matsuo T. IL-6 trans-signaling enhances CCL20 production from IL-1β-stimulated human periodontal ligament cells. Inflammation. 2014;37:381–6. doi: 10.1007/s10753-013-9750-8. [DOI] [PubMed] [Google Scholar]
- 27.Garcia de Aquino S, Manzolli Leite FR, Stach-Machado DR, Francisco da Silva JA, Spolidorio LC, Rossa C Jr, et al. Signaling pathways associated with the expression of inflammatory mediators activated during the course of two models of experimental periodontitis. Life Sci. 2009;84:745–54. doi: 10.1016/j.lfs.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Chaves de Souza JA, Nogueira AV, Chaves de Souza PP, Kim YJ, Silva Lobo C, Pimentel Lopes de Oliveira GJ, et al. SOCS3 expression correlates with severity of inflammation, expression of proinflammatory cytokines, and activation of STAT3 and p38 MAPK in LPS-induced inflammation in vivo. Mediators Inflamm. 2013;2013:650812. doi: 10.1155/2013/650812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–8. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 30.Romeo GR, Kazlauskas A. Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. J Biol Chem. 2008;283:9595–605. doi: 10.1074/jbc.M710092200. [DOI] [PubMed] [Google Scholar]
- 31.Macha MA, Matta A, Chauhan SS, Siu KW, Ralhan R. Guggulsterone (GS) inhibits smokeless tobacco and nicotine-induced NF-κB and STAT3 pathways in head and neck cancer cells. Carcinogenesis. 2011;32:368–80. doi: 10.1093/carcin/bgq278. [DOI] [PubMed] [Google Scholar]
- 32.Shankar E, Vykhovanets EV, Vykhovanets OV, Maclennan GT, Singh R, Bhaskaran N, et al. High-fat diet activates pro-inflammatory response in the prostate through association of Stat-3 and NF-κB. Prostate. 2012;72:233–43. doi: 10.1002/pros.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae WJ, Shin MR, Kang SK, Zhang-Jun, Kim JY, Lee SC, et al. HIF-2 inhibition supresses inflammatory responses and osteoclastic differentiation in human periodontal ligament cells. J Cell Biochem. 2015;116:1241–55. doi: 10.1002/jcb.25078. [DOI] [PubMed] [Google Scholar]
- 34.Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176:5652–61. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 35.Cullinan MP, Ford PJ, Seymour GJ. Periodontal disease and systemic health: Current status. Aust Dent J. 2009;54(Suppl 1):S62–9. doi: 10.1111/j.1834-7819.2009.01144.x. [DOI] [PubMed] [Google Scholar]
- 36.Nichols TC, Fischer TH, Deliargyris EN, Baldwin AS Jr. Role of nuclear factor-kappa B (NF-kappa B) in inflammation, periodontitis, and atherogenesis. Ann Periodontol. 2001;6:20–9. doi: 10.1902/annals.2001.6.1.20. [DOI] [PubMed] [Google Scholar]
- 37.Gharavi NM, Alva JA, Mouillesseaux KP, Lai C, Yeh M, Yeung W, et al. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem. 2007;282:31460–8. doi: 10.1074/jbc.M704267200. [DOI] [PubMed] [Google Scholar]
- 38.Li Q, Pan C, Teng D, Lin L, Kou Y, Haase EM, et al. Porphyromonas gingivalis modulates Pseudomonas aeruginosa-induced apoptosis of respiratory epithelial cells through the STAT3 signaling pathway. Microbes Infect. 2014;16:17–27. doi: 10.1016/j.micinf.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Mahendra J, Parthiban PS, Mahendra L, Balakrishnan A, Shanmugam S, Junaid M, et al. Evidence linking the role of placental expressions of peroxisome proliferator-activated receptor-γ and nuclear factor-kappa B in the pathogenesis of preeclampsia associated with periodontitis. J Periodontol. 2016;87:962–70. doi: 10.1902/jop.2016.150677. [DOI] [PubMed] [Google Scholar]
- 40.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science. 2013;339:286–91. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaud DS, Liu Y, Meyer M, Giovannucci E, Joshipura K. Periodontal disease, tooth loss, and cancer risk in male health professionals: A prospective cohort study. Lancet Oncol. 2008;9:550–8. doi: 10.1016/S1470-2045(08)70106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–9. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi Y, Kim JK, Yoo JY. NFκB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Mol Oncol. 2014;8:642–55. doi: 10.1016/j.molonc.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li N, Grivennikov SI, Karin M. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 2011;19:429–31. doi: 10.1016/j.ccr.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 2015;6:22613–23. doi: 10.18632/oncotarget.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mbalaviele G, Anderson G, Jones A, De Ciechi P, Settle S, Mnich S, et al. Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. J Pharmacol Exp Ther. 2006;317:1044–53. doi: 10.1124/jpet.105.100362. [DOI] [PubMed] [Google Scholar]
- 48.Rogers JE, Li F, Coatney DD, Otremba J, Kriegl JM, Protter TA, et al. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. J Periodontol. 2007;78:1992–8. doi: 10.1902/jop.2007.070101. [DOI] [PubMed] [Google Scholar]
- 49.Souza JA, Rossa C, Jr, Garlet GP, Nogueira AV, Cirelli JA. Modulation of host cell signaling pathways as a therapeutic approach in periodontal disease. J Appl Oral Sci. 2012;20:128–38. doi: 10.1590/S1678-77572012000200002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: Balancing life and death – A new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimizu H, Nakagami H, Morita S, Tsukamoto I, Osako MK, Nakagami F, et al. New treatment of periodontal diseases by using NF-kappaB decoy oligodeoxynucleotides via prevention of bone resorption and promotion of wound healing. Antioxid Redox Signal. 2009;11:2065–75. doi: 10.1089/ars.2008.2355. [DOI] [PubMed] [Google Scholar]
- 52.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 54.West K. CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Curr Opin Investig Drugs. 2009;10:491–504. [PubMed] [Google Scholar]
- 55.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–32. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–22. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 57.Liu W, Oseroff AR, Baumann H. Photodynamic therapy causes cross-linking of signal transducer and activator of transcription proteins and attenuation of interleukin-6 cytokine responsiveness in epithelial cells. Cancer Res. 2004;64:6579–87. doi: 10.1158/0008-5472.CAN-04-1580. [DOI] [PubMed] [Google Scholar]
- 58.Samavati L, Rastogi R, Du W, Hüttemann M, Fite A, Franchi L, et al. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol. 2009;46:1867–77. doi: 10.1016/j.molimm.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 59.Saravanan S, Islam VI, Babu NP, Pandikumar P, Thirugnanasambantham K, Chellappandian M, et al. Swertiamarin attenuates inflammation mediators via modulating NF-κB/I κB and JAK2/STAT3 transcription factors in adjuvant induced arthritis. Eur J Pharm Sci. 2014;56:70–86. doi: 10.1016/j.ejps.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Wang Q, Li H, Xie H, Fu M, Guo B, Ding Y, et al. 25-hydroxyvitamin D3 attenuates experimental periodontitis through downregulation of TLR4 and JAK1/STAT3 signaling in diabetic mice. J Steroid Biochem Mol Biol. 2013;135:43–50. doi: 10.1016/j.jsbmb.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 61.Yang DJ, Chang YY, Lin HW, Chen YC, Hsu SH, Lin JT, et al. Inhibitory effect of litchi (Litchi chinensis sonn.) flower on lipopolysaccharide-induced expression of proinflammatory mediators in RAW2647 cells through NF-κB, ERK, and JAK2/STAT3 inactivation. J Agric Food Chem. 2014;62:3458–65. doi: 10.1021/jf5003705. [DOI] [PubMed] [Google Scholar]
- 62.Monk JM, Liddle DM, De Boer AA, Brown MJ, Power KA, Ma DW, et al. Fish-oil-derived n-3 PUFAs reduce inflammatory and chemotactic adipokine-mediated cross-talk between co-cultured murine splenic CD8+T cells and adipocytes. J Nutr. 2015;145:829–38. doi: 10.3945/jn.114.205443. [DOI] [PubMed] [Google Scholar]