

Abstract
Coding variants in the APOL1 gene are associated with kidney diseases in African ancestral populations; yet, the underlying biologic mechanisms remain uncertain. Variant-dependent autophagic and cytotoxic cell death have been proposed as pathogenic pathways mediating kidney injury. To examine this possibility, we conditionally expressed APOL1-G0 (reference), -G1, and -G2 (variants) using a tetracycline-regulated system in HEK293 cells. Autophagy was monitored biochemically and cell death was measured using multiple assays. We measured intracellular Na+ and K+ content with atomic absorption spectroscopy and APOL1-dependent currents with whole-cell patch clamping. Neither reference nor variant APOL1s induced autophagy. At high expression levels, APOL1-G0, -G1, and -G2 inserted into the plasma membrane and formed pH-sensitive cation channels, causing collapse of cellular Na+ and K+ gradients, phosphorylation of p38 mitogen-activated protein kinase, and cell death, without variant-dependent differences. APOL1-G0 and -G2 exhibited similar channel properties in whole-cell patch clamp experiments. At low expression levels, neither reference nor variant APOL1s localized on the plasma membrane, Na+ and K+ gradients were maintained, and cells remained viable. Our results indicate that APOL1-mediated pore formation is critical for the trypanolytic activity of APOL1 and drives APOL1-mediated cytotoxicity in overexpression systems. The absence of cytotoxicity at physiologic expression levels suggests variant-dependent intracellular K+ loss and cytotoxicity does not drive kidney disease progression.
Keywords: genetic renal disease, autophagy, cell death, kidney
Common genetic variations in the APOL1 gene have been linked to progressive kidney diseases in populations with African ancestry.1–9 APOL1-G0 is the reference sequence and the two kidney disease–associated haplotypes are APOL1-G1, consisting of two nonsynonymous sequence variants in high linkage disequilibrium (rs73885319, p.S342G and rs60910145, p.I384M), and APOL1-G2, a 6-bp in-frame deletion resulting in loss of two amino acids (rs71785313, p.N388_Y389del). The APOL1-G1 and -G2 variants are rare in non-African populations, but are common in populations with African ancestry, such as blacks, where the allele frequencies have been reported as 18%–21% and 13%–15%, respectively.1
The APOL1 gene is restricted to humans and a few nonhuman primate species and has a role in innate immunity. Trypanosoma brucei is a blood-borne parasite endemic to sub-Saharan Africa, which infects livestock, leading to nagana, but humans resist infection caused by this parasite. Circulating APOL1, contained in a subclass of HDL particles, mediates this resistance by lysing the trypanosome (trypanolysis).10 Its ability to form channels in lipid bilayers is central to its trypanolytic activity.11–14 The APOL1-G2 variant confers enhanced protection from infection with the trypanosomal subspecies, Trypanosoma brucei rhodesiense, and the APOL1-G1 variant is associated with less severe clinical disease after infection with another trypanosomal subspecies, Trypanosoma brucei gambiense.15 These trypanosomal subspecies cause African sleeping sickness in humans and a single APOL1-G1 or -G2 allele confers these selective advantages, which have likely driven their high frequencies in black populations.
APOL1 has also been localized in arteriolar endothelial cells and podocytes within the normal human kidney as well as in kidney biopsy samples of individuals with APOL1-associated diseases.16,17 The lack of an association between circulating levels of normal and variant APOL1 with kidney phenotypes18,19 and the positive association of kidney allograft failure in kidney transplant recipients that received organs with the risk genotypes20–22 point to APOL1 within the kidney as the driver of APOL1-associated kidney diseases. However, the functional role of APOL1 within the cells of the kidney is currently unknown.
Overexpression of APOL1 in mammalian cell lines or model organisms has been used to model the mechanisms mediating APOL1-associated kidney diseases. Overexpression of APOL1-G0 and/or the APOL1-G1 and -G2 variants causes cell death.23–30 Autophagic cell death was the first mechanism proposed for APOL1 toxicity in mammalian cells,23 but not all subsequent reports replicated this result.26,28,30 Lysosomal damage, a proposed mechanism of trypanolysis,12 has also been implicated in APOL1-mediated mammalian cytotoxicity.26 Finally, similar to other trypanolytic mechanisms,11–14 APOL1-mediated cell death can result from its channel activity when it inserts into the plasma membrane, which allows loss of cellular ion gradients, osmotic swelling, and ultimately cell death.28,30 A key question that remains outstanding is whether the APOL1-G1 and -G2 cause differential effects on cytotoxicity compared with APOL1-G0, and whether cytotoxicity is the mechanism driving the pathogenesis of APOL1-associated kidney diseases. Therefore, we have designed the following experiments in order to address the relative toxicity of APOL1-G0 compared with APOL1-G1 or -G2, the mechanism of APOL1-mediated cell death, and the effect of APOL1 expression on autophagic processes.
Results
We first generated cell lines that conditionally expressed with tetracycline treatment the reference sequence of APOL1 (APOL1-G0) and the APOL1 variants that have been associated with progressive kidney diseases, APOL1-G1 and -G2. APOL1 levels were measured after the addition of tetracycline (1 μg/ml) in three clones for each APOL1 genotype (Figure 1). Tetracycline-inducible APOL1 expression was variable across clones (Figure 1, A and B), detectable after 4 hours of tetracycline treatment, and continued to increase at the 8-hour time point (Figure 1B). Using tetracycline (at 1 μg/ml) minimized differences in the APOL1 induction rate and steady state abundance compared with lower tetracycline concentrations. One clone from each genotype with matched levels of APOL1 expression was examined for APOL1 expression using confocal immunofluorescent microscopy (Figure 1C). The APOL1 expression level across a population of cells and its subcellular distribution appeared qualitatively similar across all three genotypes.
Figure 1.

Comparable expression of APOL1-G0, -G1, and -G2 can be achieved in stable cell lines. APOL1 expression was assayed by immunoblotting 293 cell lines that conditionally express APOL1-G0, -G1, and -G2 (G0, G1, and G2) with tetracycline induction. Three clones of each genotype were studied (labeled A, B, C for each genotype). (A) Band density was quantitated using ImageJ software, and the expression of APOL1 normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is shown for each clone tested before tetracycline treatment (0 hour) and 4 and 8 hours after the addition of tetracycline (1 µg/ml). (B) APOL1 expression, determined by immunoblotting, after 0, 4, and 8 hours of tetracycline induction for three G0, G1, and G2 clones each. Blots were stripped and reprobed for GAPDH expression as loading controls. (C) One clone for each genotype (G0 C, G1 C, G2 B) judged to have comparable levels of APOL1 expression by immunoblotting was examined with confocal immunofluorescent microscopy for APOL1 expression before and 5 hours after the addition of tetracycline (1 μg/ml). Scale bars=25 µm.
Because APOL1 expression has been associated with autophagic cell death,23,26 we evaluated the effect of APOL1 expression on autophagy (Figure 2). We examined three clones for each genotype (Figure 1) to account for clone-dependent differences. The normalized expression of LC3II in each of the nine clones examined is shown (Figure 2, A and B) after 8 and 16 hours of induction, respectively, graphically illustrating the degree of variability between clones. The remaining graphs (Figure 2, C–F) depict average normalized LC3II or p62 abundance of three independent clones for each genotype in a single bar. APOL1 did not affect autophagy at 8 hours (Figure 2C, Supplemental Figure 1A) or 16 hours (Figure 2D, Supplemental Figure 1A). Consistent with these data, APOL1 did not significantly change the normalized p62 expression at 8 hours (Figure 2E) and 16 hours (Figure 2F). Autophagic flux was increased with serum starvation, but was not altered by the expression of APOL1-G0, -G1, or -G2 (Supplemental Figure 1B). Although p62 levels tend to be lower at 16 hours, the abundance of p62 did not increase with bafilomycin A treatment consistent with the conclusion that neither reference nor variant APOL1s activate autophagic flux. Tetracycline treatment alone did not alter autophagic flux in parental 293 cell lines or affect the expression of GAPDH used for normalization (Supplemental Figure 1C).
Figure 2.

Stable expression of APOL1 does not induce autophagy. The protein abundance of LC3II, p62, and GAPDH was assessed by immunoblotting in three clones of each genotype (G0, G1, and G2) after tetracycline induction (1 μg/ml). The bar graphs show the normalized expression of (A–D) LC3II or (E and F) p62, two biochemical markers of autophagy, 8 or 16 hours after tetracycline addition. (A and B) The results for three clones of each genotype, demonstrating genotype-independent, clonal variability in LC3II abundance. (C–F) Bar graphs of normalized LC3II and p62 abundance stratified by APOL1 genotype (mean±SD). Conditions include no treatment (Cont), treatment with bafilomycin A (Baf, 100 nM) alone to induce an autophagic block, treatment with tetracycline alone (Tet, 1 μg/ml), and treatment with tetracycline and bafilomycin A (Tet+Baf). Cells were treated with tetracycline for 8 or 16 hours, as indicated. Bafilomycin was added for the last 3 hours. Within-genotype treatment differences were not statistically significant (single-factor ANOVA).
Increased cytotoxicity of the APOL1-G1 and -G2 variants has been proposed as a mechanism of APOL1-associated kidney diseases; therefore, we examined our stable cell lines for variant-dependent death. In contrast to published data, cytotoxicity/viability ratios determined by a fluorogenic assay were similar in all genotypes compared with uninduced control cells (Figure 3A). The average cytotoxicity/viability ratio of the clones expressing APOL1-G0, the reference sequence, was higher than those expressing either the APOL1-G1 (P value 0.14) or APOL1-G2 (P value <0.001) kidney disease risk variants. An alternative assay of cell viability uses methyl-thiazolyl-tetrazolium (MTT), which is reduced in metabolically active cells to produce a blue precipitate, formazan.31 Eight hours after APOL1-G0, -G1, or -G2 induction, cell viability was equivalent to uninduced control cells (Figure 3B). However, after 16 hours of APOL1 expression cytotoxicity increased and was indistinguishable between genotypes (Figure 3C).
Figure 3.

APOL1-induced cytotoxicity is variant-independent. (A) Fluorescent assay demonstrating no difference in the average cytotoxicity-to-viability ratio, normalized to control untreated cells, in 293 cells stimulated with tetracycline to express APOL1-G0, -G1, or G2 (G0, G1, G2). Each bar represents mean±SD of three independent experiments with one to two independent clones, with two to four technical replicates (n=12–14). (B) MTT assays after 8 hours of tetracycline (1 μg/ml) induction demonstrated no loss of viability in G0, G1, or G2 cells compared with uninduced cells (mean±SD, n=2–3 independent experiments, each with three technical replicates, for each APOL1 genotype). (C) In contrast, at 16 hours of tetracycline (1 μg/ml) induction, cytotoxicity is marked in G0, G1, and G2 cells compared with uninduced control cells (mean±SD, n=2 independent experiments, each with two to three technical replicates, for each APOL1 genotype). MTT assays in control cells of each genotype were normalized to 100% viability in each independent experiment.
Many cell death assays use biochemical, morphologic, or physiologic surrogate measures for cell death, which quantify early kinetic processes that regulate but do not always result in cell death.32 To address this possibility we performed a clonogenic survival assay, which is generally regarded as the gold standard for the determination of cellular cytotoxicity/survival after a cytotoxic exposure such as APOL1. Two hours of induction (tetracycline, 1 μg/ml) of reference or variant APOL1s resulted in almost complete loss of cell viability (Figure 4). Treatment with glycine to nonspecifically block ion-fluxes in pyroptosis,33 digoxin to inhibit autosis,34 or wortmanin to inhibit autophagy35 failed to rescue the reference or variant APOL1 cell lines from cell death in the clonogenic assay (Figure 4).
Figure 4.

Clonogenic survival assays demonstrate variant-independent cell death after APOL1 expression. Bar graph shows the fraction of surviving cells in the test condition normalized to untreated control cells (mean+SD, n=3). The vertical axis indicates the APOL1 genotype and treatments. The normalized surviving fraction of 1.0 is highlighted in red. Respective APOL1 genotypes are abbreviated G0, G1, or G2. C-Del338+Tet, 16 hours after plating the cells tetracycline 1 µg/ml was added to media to induce expression in a stable 293 cell line of an APOL1 transgene with a deletion of carboxy terminal amino acids, 339–398; Dig, digoxin (1 nM, 6 hours); Gly, glycine (5 mM glycine added to culture media throughout assay); 2HrTet, tetracycline (1 μg/ml, 2 hours); LowTet, 16 hours after plating the cells tetracycline was added at 5 ng/ml for APOL1-G0 and APOL1-G1 and 10 ng/ml for APOL1-G2; NoTx, no additions; Tet, tetracycline (1 μg/ml, 16 hours); v., versus; Wort, wortmanin (50 nM, 16 hours).
APOL1 has been reported to have channel properties when expressed in oocytes or when purified and reconstituted into lipid bilayers.11–13,28 APOL1 appears to form monovalent cation channels permeable to both Na+ and K+. In order to determine the relationship between the appearance of channel activity and the onset of cell death markers, we used atomic absorption spectroscopy to measure cell-associated Na+ and K+ after 0, 4, 5, 6, 7, 8, 9, and 10 hours of tetracycline (1 μg/ml)-induced expression of APOL1-G0, -G1, and -G2. Cell-associated K+ and Na+ were similar in cell lines expressing the three APOL1 genotypes in the absence (time 0) and presence of tetracycline for up to 4 hours (Figure 5, A and C). However, continued treatment with tetracycline for 5–10 hours resulted in a progressive loss of cellular K+ and gain of cellular Na+ in clones from each APOL1 genotype, reaching maximal effect by 10 hours (Figure 5, B and D). Expression of APOL1 with a deletion of the carboxy-terminal amino acids 339–398, for up to 24 hours, failed to induce any change in cellular Na+ or K+, nor was there evidence of cell death using the clonogenic assay (Figure 4, Supplemental Figure 2B). Cytosolic free Ca2+ concentration ([Ca2+]i) measured using fura-2, was similar with or without 8 hours of induction of any of the APOL1 genotypes. Furthermore, expression of the APOL1 genotypes had no effect on the transient increase in [Ca2+]i observed after stimulation of endogenous purinergic receptors (Supplemental Figure 3). Together, these results demonstrate that APOL1 expression correlates with a variant-independent loss of K+ and a gain of Na+ by the cell, without a change in Ca2+ homeostasis or signaling, which precedes loss of cell viability.
Figure 5.

APOL1-G0, -G1, -G2 cause a time-dependent loss of cellular K+ and a gain of cellular Na+. Cell-associated (A) K+ and (C) Na+ were measured as described in the Concise Methods in G0, G1, or G2 cells without or with tetracycline (1 µg/ml) for 4 hours (mean±SD, n=5–6). Cellular (B) K+ and (D) Na+ content as a function of time after tetracycline addition is shown, normalized to time 0 (mean±SD, n=3). (B) Hatches between the time 0 and the 5-hour time measurement added for clarity, illustrating this line connects these two time points and does not represent additional interval measurements. Tet, tetracycline.
Previous reconstitution studies showed that the channels formed by APOL1 are cation selective and inhibited by a reduction in bath pH.13 If in fact APOL1 channels are responsible for the loss of K+ and gain of Na+, we reasoned that APOL1 channel activity should be measurable at induction times >4 hours. In order to test this hypothesis, whole-cell patch-clamp experiments were performed on APOL1-G2 cells. Preliminary studies showed the channels induced by APOL1-G2 expression were permeable to Na+, K+, and Cs+. In subsequent experiments, we used a normal bath solution, and a Cs+ pipette solution to eliminate endogenous K+ currents. Under these recording conditions, whole-cell currents were small in APOL1 cells in the absence (not shown) or presence of tetracycline for 4–5 hours (Figure 6B). When measured 6–7 hours after induction, large currents were observed immediately upon establishment of whole-cell recording mode (Figure 6, A and B). The currents had a reversal potential near 0 mV and showed a slight outwardly rectifying I-V relationship (Figure 6C).
Figure 6.

APOL1 expression is associated with the appearance of cation-selective and pH-sensitive whole-cell membrane currents. (A) Whole-cell membrane currents were recorded in an APOL1-G2–expressing 293 cell 6 hours after addition of tetracycline (1 µg/ml). Voltage ramps were applied every 6 seconds and the outward current at +60 mV (red) and inward current at −80 mV (black) during each ramp are plotted as a function of time after rupture of the patch for whole-cell recording. At the time indicated by the horizontal bar (top), the bath solution was changed from pH 7.2 to pH 5.1 and subsequently returned to pH 7.2. (B) Summary of inward (−80) and outward (+60) current amplitudes recorded at pH 7.2 at 4–5 or 6–7 hours after tetracycline induction. Values are mean±SD, n=3–5. (C) Representative current-voltage relationships (I-V) of the cell shown in (A) obtained in bath solution of the indicated pH. Note that the blue and black traces are virtually superimposable. (D) Summary of inward (−80) and outward (+60) current amplitudes at the different pH values; mean±SD, n=3. (E) Representative I-Vs showing the inhibition by reduced pH and the shift in reversal potential upon replacement of extracellular Na+ with NMDG (n=3). (F) Experiments were performed as in (A). The I-Vs show inhibition by reduced pH in APOL1-G0–expressing 293 cells (representative of n=3). Note that the blue and black traces are superimposed. G0, APOL1-G0; G2, APOL1-G2; TET, tetracycline.
To determine if these currents in cells have characteristics previously reported for APOL1 channels reconstituted in lipid bilayers, the extracellular bath solution was rapidly changed from pH 7.2 to pH 5.1. Within 12 seconds, both inward and outward currents were reduced to near background levels (Figure 6, A, C, and D). The inhibition was rapidly reversed upon return of the extracellular pH to 7.2. Inhibition by reduced pH and the subsequent recovery could be repeated multiple times in the same cell. Another characteristic of reconstituted APOL1 channels is their cation selectivity. Under our recording conditions, a reversal potential near 0 mV could be explained by activity of either a cation channel or a Cl− channel. To evaluate selectivity, we first confirmed that the recorded cell expressed a large reversible pH-sensitive current with a reversal potential near zero (Figure 6E). Subsequent superfusion of the cell with bath solution in which the Na+ was replaced with the impermeable cation, N-methyl-D-glucamine (NMDG), resulted in a shift of the reversal potential to approximately −50 mV and a pronounced outward rectification of the I-V relationship (Figure 6E), consistent with a channel permeable to both Na+ and Cs+, but not Cl−. A similar negative shift in reversal potential with NMDG was observed during recording of APOL1 currents using pipette solutions in which the Cl− was replaced by aspartate (not shown), again consistent with a lack of anion permeability of the APOL1 channels. Lastly, similar pH-sensitive currents were observed in cells expressing the G0 variant of APOL1 (Figure 6F). Taken together, our results are consistent with the conclusion that both overexpressed APOL1-G0 and -G2 form constitutively active cation channels in the plasma membrane, which leads to the dissipation of Na+ and K+ concentration gradients without a change in [Ca2+]i and before cell death.
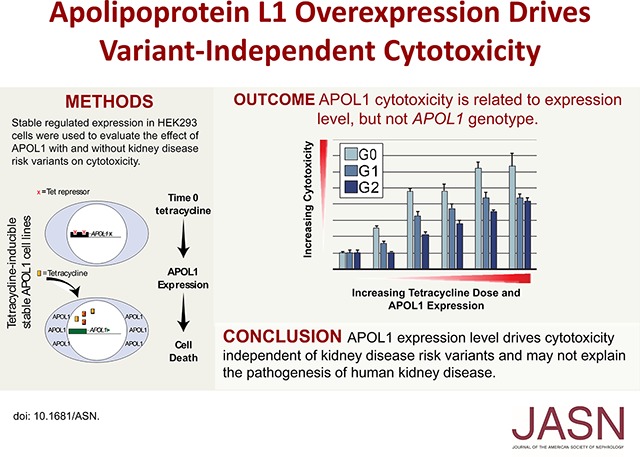
Using a similar stable expression system, others have reported variant-dependent cytotoxicity after insertion of APOL1 into the plasma membrane.30 We also examined lower APOL1 expression for variant-dependent differences in cytotoxicity (Figure 7). We first established the dose-response relationship between low tetracycline concentrations and APOL1 expression (Supplemental Figure 4). As expected, lower tetracycline concentrations reduced APOL1 abundance in all stable lines (Figure 7A); in addition, the rate and steady state levels (Supplemental Figure 4) of APOL1 were more variable than at high-tetracycline concentrations. Within this lower dose range, we chose tetracycline concentrations to induce comparable APOL1 levels in G0, G1, and G2 clones (Figure 7A, Supplemental Figure 4). Because cytotoxicity is preceded by plasma membrane channel activity, we assessed the presence of APOL1 at the plasma membrane using biotinylation and precipitation of surface proteins in cells in the absence of tetracycline and after 8 and 24 hours of incubation in the presence of high and low concentrations of tetracycline, respectively (Figure 7A). Biotinylated reference and variant APOL1s were only precipitated from plasma membranes isolated from cells exposed to a high concentration of tetracycline. Consistent with the absence of plasma membrane APOL1 in cells exposed to no or matched low dose tetracycline concentrations, cytotoxicity was markedly attenuated but increased as the tetracycline concentration was increased from the matched low-dose concentrations across genotypes (Figure 7B). There was little evidence of cell death using clonogenic survival assays in APOL1-G0, -G1, or -G2 cells induced with matched low-dose tetracycline concentrations and APOL1 expression levels (Figure 4). MTT assays of APOL1-G0, -G1, and -G2 cells treated for 24 and 48 hours with the matched low-dose tetracycline concentration also demonstrated no cytotoxicity (Figure 7C). We were unable to find a level of APOL1 expression that resulted in variant-dependent cell death. Phosphorylation of p38 MAPK has been reported to be a downstream effector of APOL1-mediated loss of intracellular potassium.30 Therefore, we assessed phosphorylation of p38 MAPK in cells expressing APOL1 at low and high levels. p38 MAPK was phosphorylated in the presence of APOL1-G0, -G1, and -G2 and phosphorylated p38 MAPK amounts were similar across genotypes and increased with higher APOL1 expression independent of genotype (Figure 7D).
Figure 7.

APOL1-G0, -G1, and -G2 proteins, induced with low doses of tetracycline, do not localize to plasma membrane, cause cell death, or stimulate p38 MAPK phosphorylation. (A) APOL1-G0, -G1, or -G2 clones were cultured without tetracycline (0); with low (L) concentrations of tetracycline adjusted to induce equivalent amounts of G0, G1, and G2 protein (G0, 5 ng/ml; G1, 5 ng/ml; G2, 10 ng/ml); or with high (H) tetracycline concentrations (1 μg/ml, all genotypes). Cell surface proteins were biotinylated and cells were collected in lysis buffer. APOL1 was detectable in the input lysates of tetracycline-stimulated but not untreated cells (left panel). Biotinylated APOL1 was only identified in membranes from cells treated with high concentrations of tetracycline (right panel, labeled IP). (B) Fluorogenic cytotoxicity/viability assay demonstrates dose-dependent cytotoxicity with increasing tetracycline doses across APOL1 genotypes. An increased cytotoxicity/viability ratio was observed in APOL1-G0 and APOL1-G1 clones at the “1×” tetracycline dose compared with the no tetracycline control, despite using doses chosen to provide matched APOL1 expression. Cells were treated for 24 hours with no tetracycline (0), or increasing doses of tetracycline (1×–5×); these doses correspond to 5, 10, 15, 20, and 25 ng/ml for APOL1-G0 and -G1; and, 10, 20, 30, 40, and 50 ng/ml for APOL1-G2, respectively. (C) MTT assays also show no significant cytotoxicity after treatment with low concentrations of tetracycline. Bars represent mean±SD (n=2). (D) Immunoblots of APOL1, phosphorylated p38 MAPK, total p38 MAPK, and GAPDH from one of three experiments yielding similar results. Lanes 1–3, lysates from 293 cells used to generate the stable cell lines (293 No. Tet); lanes 4–6, lysates from G0 (0), G1 (1), and G2 (2) cells treated for 24 hours with low concentrations of tetracycline, as above; lanes 7–9, lysates from G0 (0), G1 (1), and G2 (2) cells treated for 8 hours with tetracycline, 1 μg/ml. IB, immunoblot; IP, immunoprecipitate; M.W., molecular weight; Phospho-p38, phosphorylated-p38 mitogen-activated protein kinase; Tet, tetracycline.
Discussion
We generated and characterized 293 cells that conditionally expressed APOL1-G0, -G1, or -G2 to study biologic mechanisms underpinning the genetic association of APOL1 variants with kidney disease. Building on APOL1’s known trypanolytic activity, published in vitro evidence has suggested that variant-dependent, autophagic, or cytotoxic cell death is responsible for progressive kidney disease. We did not detect variant-dependent, differential cytotoxicity, findings which differ from several prior reports using HEK293 cells,25,30,36,37 HEK293T cells,38 or human podocytes.26 The reasons that our data differ from earlier publications are unclear. Established cell lines such as 293 cells can exhibit heterogeneous phenotypes between laboratories.39 We also found significant variability in the tetracycline induction kinetics of APOL1 expression across independent clones expressing all three genotypes. These results suggest that APOL1 induction kinetics are as important as APOL1 expression level in determining the phenotypic response to a particular tetracycline dose. Variations in APOL1 induction kinetics could confound interpretation of short-term, surrogate assays for cell death used in prior studies. We confirmed variant-independent cytotoxicity by measuring cell survival after 7–10 days.32 We found that APOL1-G0, -G1, and -G2 can all be cytotoxic, an outcome that depends on expression levels and not upon nonsynonymous changes in the amino acid sequences of the variant APOL1s.
The notion that cytotoxicity drives variant APOL1-associated kidney disease is on the basis of data generated using model systems ectopically expressing APOL1 from heterologous promoters. However, no studies have reported APOL1-mediated cytotoxicity when APOL1 is expressed under its endogenous promoter. Importantly, we were able to induce reference and variant APOL1 expression without causing cytotoxicity, better recapitulating human kidney phenotypes. APOL1 is normally expressed in healthy human kidney cells16,17,40 and most blacks that carry the high risk APOL1 genotype do not develop kidney diseases. Therefore a “second hit” seems to be required for kidney disease initiation and progression in individuals with high-risk APOL1 genotypes.
Reference and variant APOL1s target to the plasma membrane when sufficiently overexpressed in cell culture. However, APOL1 does not localize to podocyte plasma membranes nor colocalize with the podocyte plasma membrane receptor phosphatase, GLEPP1, in human kidneys.16 When associated with plasma membranes in cells, some report variant APOL1s permit greater intracellular potassium depletion than reference APOL1.30,38 However, we found no variant-dependent differences in the intracellular potassium loss and found comparable channel activity in cells expressing APOL1-G0 compared with those expressing APOL1-G2. We characterized the mechanism of K+ efflux for the first time in mammalian cells and found APOL1-G0 and APOL1-G2 both had pH-sensitive, nonselective, cation channel activity, consistent with previous reports.11,13 Channel characteristics of cells expressing APOL1-G0 or -G2 were identical. In contrast to these data with full length APOL1 proteins, chloride channel activity has been recorded when the pore-forming domain of APOL1 (amino acids 60–325) was reconstituted in lipid bilayers.12 A recent report has provided evidence that may reconcile these observations, reporting that APOL1 channel conducts chloride at low pH and potassium when the pH is neutralized.41 Taken together, our data would suggest a model where increasing the overexpression of APOL1 leads to variant-independent APOL1 localization to the plasma membrane, ion channel opening, loss of transcellular Na+ and K+ gradients, and cell death.
We have also previously generated transgenic mice expressing APOL1-G0 or APOL1-G2 under control of the Nephrin promoter for podocyte-specific expression.42 Similar to the APOL1 expression levels in cells that were not cytotoxic, these mice did not develop kidney damage as assessed by serum creatinine levels, proteinuria, and histology even when aged to 300 days, and we could not demonstrate necrotic, apoptotic, or autophagic podocyte cell death. Interestingly, the APOL1-G2 mice did have a postnatal reduction of glomerular podocyte density by 200 days when compared with wild-type or APOL1-G0 transgenic mice, suggesting transgenic expression of APOL1-G2 in podocytes may promote accelerated but clinically silent podocyte detachment. Another murine model of inducible, podocyte-specific APOL1-G0, -G1, or -G2 expression also reported variant-dependent podocyte depletion.37 Unlike mice expressing APOL1 under the Nephrin promoter, these animals consistently demonstrated albuminuria, azotemia, and histologic kidney damage with the expression of variant but not reference APOL1s, which were attributed to cytotoxicity. These data are not entirely consistent with the epidemiology of common human kidney disease, where the majority of individuals with APOL1 risk genotypes do not develop overt kidney disease in the absence of an additional stressor. A third mouse model utilizing hydrodynamic gene delivery of APOL1-G0, -G1, or -G2 also found variant-dependent cytotoxicity and proteinuria, which was dependent upon the soluble urokinase plasminogen activator receptor, suPAR.43 In vitro APOL1-G1 and -G2 augmented suPAR-dependent αvβ3 activation, permitting podocyte detachment. Transgenic expression of human APOL-G1 and -G2 in zebrafish glomeruli caused histologic abnormalities but not proteinuria or edema.44 Two reports of ectopic expression of variant but not reference APOL1s in the Drosophila nephrocyte, a cell type with some similarities to the podocyte, increased endocytosis and loss of nephrocytes with aging.45,46 Experiments in Saccharomyces cerevisiae also found a genetic interaction between APOL1 expression and deletion of components in the endocytic pathway.46 Together, these results suggest that processes such as podocyte adhesion and endocytic trafficking, in addition to cytotoxicity, may also contribute to renal pathology associated with APOL1 risk variants.
In summary, our data fail to demonstrate variant-dependent cytotoxic or autophagic cell death, suggesting neither of these pathways mediate progressive APOL1-associated kidney disease. APOL1-G0, -G1, and -G2 can all localize to the plasma membrane at high expression levels, which leads to channel formation, and may cause cell death. The channel activity characteristics of APOL1-G0 were similar to those of APOL1-G2, suggesting that kidney diseases do not result from variant-dependent changes in APOL1 channel activity. Importantly, this report demonstrates that APOL1-G0, -G1, and -G2 can all be expressed without causing cytotoxicity, and this system can be used to identify alternative cell processes that mediate APOL1-associated kidney diseases.
Concise Methods
Cell Line Creation and Culture
Tetracycline-regulated expression (T-Rex)–293 cells (Life Technologies) with the tetracycline repressor were used to generate stable transfectants for the expression of APOL1-G0, -G1, or -G2. Clones for each genotype were derived from independent single-cell clones. Cells were maintained in culture according to manufacturer instructions in DMEM+Glutamax (Life Technologies), 10% tetracycline-free, FBS (GE Healthcare Hyclone), 2 mM L-glutamine, 1% Pen-Strep, 5 µg/ml blasticidin, and 400 μg/ml zeocin. APOL1 expression was induced by the addition of a stock solution of tetracycline in sterile water to achieve the indicated final concentrations for the specified time periods.
Antibodies and Reagents
Antibodies for immunoblotting include a rabbit polyclonal anti-APOL1 (HPA01885; Sigma) at 1:4000 or rabbit monoclonal anti-APOL1 (ab108315, lot#GR8593–2; AbCam) at 1:4000, rabbit monoclonal anti-LC3II (3868; Cell Signaling) at 1:1000, rabbit polyclonal anti-p62 (P0067, lot#108K4764; Sigma) at 1:2000, rabbit monoclonal anti-GAPDH (2118; Cell Signaling) at 1:2000, mouse monoclonal anti–Na/K-ATPase (Cat#sc-21712, Lot#G1615; Santa Cruz Biotech) at 1:200, and rabbit monoclonal anti-tubulin (2128; Cell Signaling) at 1:1000. For secondary detection of primary antibodies in immunoblotting HRP-conjugated protein A (P8651, lot#110M6029; Sigma) was used at 1:15,000.
Assays of Cell Death and Cytotoxicity
The MTT assay was performed using the thiazolyl blue tetrazolium bromide reagent (M2128; Sigma). The fluorogenic MultiTox-Fluor Multiplex Cytotoxicity Assay (Promega) was used according to manufacturer instructions. Clonogenic survival assays were done as described with the following minor differences.47 Briefly, cells from each APOL1 genotype were grown in culture without APOL1 induction, trypsinized, and harvested in a single-cell suspension, and counted using a hemocytometer. Between 50 and 200 cells were plated in each well of a six-well culture dish; cells were allowed to attach for 8–16 hours, before addition of tetracycline for APOL1 induction or the experimental drug treatment. Cells were maintained in culture for 7–10 days, changing media every 48 hours, until single clonogenic colonies were visible with light microscopy; wells were then washed with PBS, and cells fixed and stained with 6% gluteraldehyde and 0.5% crystal violet in water for 20 minutes. Fixative was removed and wells thoroughly rinsed and colonies counted by two independent observers. Three wells on each plate were used as control to calculate plating efficiency and three wells were treated as the experimental wells and used to calculate surviving fraction. Cells were then exposed to specified treatment conditions and maintained in culture for 10 days. Media with treatment supplements were changed every 48 hours and at least three replicates were done for each condition.
Biotinylation of Cell Surface Proteins
After tetracycline induction of cultured cells, they were washed with ice-cold PBS, and incubated with EZ-Link Sulfo-NHS-LC-biotin (Thermo Fisher Scientific) in PBS (1 mg/ml), 4 ml total volume per 10-cm dish for 5 minutes on ice. Plates were washed once with PBS and residual NHS quenched with 4 ml of 0.1 M glycine in PBS, then washed with ice-cold PBS and lysed with the addition of 300 μl of lysis buffer (10 mM Tris, pH=7.5, 1 mM EDTA, 175 mM NaCl, 1% Triton X-100, PMSF 0.7 mM, 1× protease inhibitor cocktail [P8340; Sigma]), incubated on ice for 10 minutes, and centrifuged at 13,000 × g for 10 minutes. Next, each sample was quantitated with DC protein assay kit (Biorad) and 500–1000 μg of total protein was transferred to a fresh microfuge, transferred to a fresh microcentrifuge tube containing 60 µl of streptavidin resin (Thermo Fisher Scientific), and incubated overnight at 4°C. Beads were collected with centrifugation at 13,000 rpm for 1 minute at 4°C, washed four times with 500 μl of lysis buffer, resuspended in Laemmli sample buffer for separation of proteins with SDS-PAGE, and transferred to PVDF membranes for immunoblotting.
Immunoblotting
Immunoblots were done as previously described,16 images were captured as TIFF files, and band densities were determined using ImageJ software available from the National Institutes of Health (http://rsb.info.nih.gov/ij/).
Confocal Microscopy
Cells were grown on glass coverslips, fixed in PBS containing 4% paraformaldehyde for 20 minutes at room temperature, permeabilized with 0.2% Triton X-100 for 10 minutes at room temperature, blocked with 10% normal goat serum (NGS) for 1 hour at room temperature, then incubated with anti-APOL1 antibody diluted 1:50 in PBS with 1% NGS overnight at 4°C, then washed and incubated with goat anti-rabbit-FITC secondary antibody diluted 1:400 in PBS with 1% NGS for 1 hour in the dark. Coverslips were washed and mounted using Vectashield with DAPI. Images were captured on a Leica SPE RGB+405 one Spectral Channel System with ACS Objectives.
Cellular Na+ and K+ Content
The cellular content of Na+ and K+ was measured as previously described with minor modifications.48 Briefly, cells were grown in 30-mm culture dishes in the absence or presence of tetracycline to induce the expression of APOL1-G0, -G1, or -G2 variants. At selected time points, the culture medium was aspirated from the dish and the cells were rapidly washed three times with 2 ml of ice-cold solution containing 110 mM MgCl2 and 5 mM Tris-Cl, pH 7.4 with HCl. After aspiration of the final wash, the cells were solubilized in 2 ml of 10% nitric acid. Cellular particulate matter was removed by centrifugation and the resultant supernatants were diluted (generally 1:5 or 1:10) in 10% nitric acid for the subsequent measurement of Na+ and K+ by atomic absorption spectroscopy (Agilent 55B AA Spectrometer). Concentrations of Na+ and K+ in the diluted samples were compared with standard curves for Na+ and K+, and are expressed as micrograms per liter.
Whole-Cell Patch Clamping
The giga-seal technique for current recording was utilized in the whole-cell mode as previously described.49 Briefly, cells, grown on 35-mm plastic cell culture dishes, were placed on the stage of a Nikon inverted microscope immediately before use. The normal extracellular solution contained (in millimolar) 137 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.2). The normal pipette solution contained (in millimolar) 124 CsCl2, 2 MgCl2, 1 CaCl2, 11 EGTA, and 10 HEPES (pH 7.2 and pCa 8). In some experiments, the Na+ in the bath solution was isosmotically replaced with NMDG. To change extracellular pH, the cells were rapidly superfused (via a perfusion pipette placed in close proximity to the cell) with normal bath solution or with bath solution in which the Hepes buffer was replaced with MES buffer, pH 5.2. Data were obtained using an Axopatch 1C amplifier (Axon Instruments) and sampled on-line using pCLAMP 9.0 software. The ground electrode was an Ag-AgCl pellet connected to the bath via an agar bridge containing 150 mM KCl. All recordings were made at room temperature (approximately 22°C). To generate current-voltage (I-V) relations, voltage ramps from −80 to +60 mV over 1 second were repetitively applied at 6-second intervals. Unless otherwise indicated, the holding potential between ramps was −60 mV. All figures show representative current traces normalized to cell capacitance (pA/pF).
Statistical Analyses
To correct for multiple comparisons, the single-factor ANOVA test was used to assess for significant differences across multiple groups. Where significance was detected, the Tukey honestly significant difference test was used to assess for significance between specific groups. A significance threshold of <0.05 was used for all experiments.
Disclosures
None.
Supplementary Material
Acknowledgments
J.F.O., L.A.B., and J.R.S. are supported by grants from the National Institutes of Health, R01 DK108329, R01 DK097836, and UL1 TR000439; S.M.M. was supported by training grant DK007470.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016121322/-/DCSupplemental.
References
- 1.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR: Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K: Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128: 345–350, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA: APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, Limou S, Sezgin E, Nelson GW, Fogo AB, Goetsch S, Kopp JB, Winkler CA, Naicker S: APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol 26: 2882–2890, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsen CP, Beggs ML, Saeed M, Walker PD: Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol 24: 722–725, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, Birmingham DJ, Hebert LA, Hicks PJ, Segal MS, Edberg JC, Brown EE, Alarcón GS, Costenbader KH, Comeau ME, Criswell LA, Harley JB, James JA, Kamen DL, Lim SS, Merrill JT, Sivils KL, Niewold TB, Patel NM, Petri M, Ramsey-Goldman R, Reveille JD, Salmon JE, Tsao BP, Gibson KL, Byers JR, Vinnikova AK, Lea JP, Julian BA, Kimberly RP; Lupus Nephritis–End‐Stage Renal Disease Consortium : End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol 66: 390–396, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR: Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol 22: 2098–2105, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulasi II, Tzur S, Wasser WG, Shemer R, Kruzel E, Feigin E, Ijoma CK, Onodugo OD, Okoye JU, Arodiwe EB, Ifebunandu NA, Chukwuka CJ, Onyedum CC, Ijoma UN, Nna E, Onuigbo M, Rosset S, Skorecki K: High population frequencies of APOL1 risk variants are associated with increased prevalence of non-diabetic chronic kidney disease in the Igbo people from south-eastern Nigeria. Nephron Clin Pract 123: 123–128, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, Boerwinkle E, Parekh RS, Kao WH: APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 24: 1484–1491, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Van Den Abbeele J, Pays A, Tebabi P, Van Xong H, Jacquet A, Moguilevsky N, Dieu M, Kane JP, De Baetselier P, Brasseur R, Pays E: Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 422: 83–87, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Molina-Portela MP, Lugli EB, Recio-Pinto E, Raper J: Trypanosome lytic factor, a subclass of high-density lipoprotein, forms cation-selective pores in membranes. Mol Biochem Parasitol 144: 218–226, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homblé F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E: Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309: 469–472, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Thomson R, Finkelstein A: Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: Relevance to trypanosome lysis. Proc Natl Acad Sci U S A 112: 2894–2899, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene AS, Hajduk SL: Trypanosome lytic factor-1 initiates oxidation-stimulated osmotic lysis of Trypanosoma brucei brucei. J Biol Chem 291: 3063–3075, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper A, Ilboudo H, Alibu VP, Ravel S, Enyaru J, Weir W, Noyes H, Capewell P, Camara M, Milet J, Jamonneau V, Camara O, Matovu E, Bucheton B, MacLeod A: APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. eLife 6:e25461, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR: APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol 22: 2119–2128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, Kretzler M, Hemal AK, Rudel LL, Petrovic S, Weckerle A, Pollak MR, Ross MD, Parks JS, Freedman BI: Localization of APOL1 protein and mRNA in the human kidney: Nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol 26: 339–348, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruggeman LA, O’Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, Bosch RJ, Gupta S, Pollak MR, Sedor JR, Kalayjian RC: Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol 25: 634–644, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlitina J, Zhou H, Brown PN, Rohm RJ, Pan Y, Ayanoglu G, Du X, Rimmer E, Reilly DF, Roddy TP, Cully DF, Vogt TF, Blom D, Hoek M: Plasma levels of risk-variant APOL1 do not associate with renal disease in a population-based cohort. J Am Soc Nephrol 27: 3204–3219, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeves-Daniel AM, DePalma JA, Bleyer AJ, Rocco MV, Murea M, Adams PL, Langefeld CD, Bowden DW, Hicks PJ, Stratta RJ, Lin JJ, Kiger DF, Gautreaux MD, Divers J, Freedman BI: The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 11: 1025–1030, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee BT, Kumar V, Williams TA, Abdi R, Bernhardy A, Dyer C, Conte S, Genovese G, Ross MD, Friedman DJ, Gaston R, Milford E, Pollak MR, Chandraker A: The APOL1 genotype of African American kidney transplant recipients does not impact 5-year allograft survival. Am J Transplant 12: 1924–1928, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freedman BI, Pastan SO, Israni AK, Schladt D, Julian BA, Gautreaux MD, Hauptfeld V, Bray RA, Gebel HM, Kirk AD, Gaston RS, Rogers J, Farney AC, Orlando G, Stratta RJ, Mohan S, Ma L, Langefeld CD, Bowden DW, Hicks PJ, Palmer ND, Palanisamy A, Reeves-Daniel AM, Brown WM, Divers J: APOL1 genotype and kidney transplantation outcomes from deceased African American donors. Transplantation 100: 194–202, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA: Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem 283: 21540–21549, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, Winkler CA, Kopp J, Rotimi C, Adeyemo A, Doumatey A, Ayodo G, Alper SL, Pollak MR, Friedman DJ, Raper J: Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A 111: E2130–E2139, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ: Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 87: 332–342, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC: APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol 307: F326–F336, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khatua AK, Cheatham AM, Kruzel ED, Singhal PC, Skorecki K, Popik W: Exon 4-encoded sequence is a major determinant of cytotoxicity of apolipoprotein L1. Am J Physiol Cell Physiol 309: C22–C37, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heneghan JF, Vandorpe DH, Shmukler BE, Giovinazzo JA, Raper J, Friedman DJ, Pollak MR, Alper SL: BH3 domain-independent apolipoprotein L1 toxicity rescued by BCL2 prosurvival proteins. Am J Physiol Cell Physiol 309: C332–C347, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng D, Weckerle A, Yu Y, Ma L, Zhu X, Murea M, Freedman BI, Parks JS, Shelness GS: Biogenesis and cytotoxicity of APOL1 renal risk variant proteins in hepatocytes and hepatoma cells. J Lipid Res 56: 1583–1593, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olabisi OA, Zhang JY, VerPlank L, Zahler N, DiBartolo S 3rd, Heneghan JF, Schlöndorff JS, Suh JH, Yan P, Alper SL, Friedman DJ, Pollak MR: APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A 113: 830–837, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosmann T: Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63, 1983 [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, Candi E, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, Di Daniele N, Dixit VM, Dynlacht BD, El-Deiry WS, Fimia GM, Flavell RA, Fulda S, Garrido C, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Joseph B, Jost PJ, Kaufmann T, Kepp O, Klionsky DJ, Knight RA, Kumar S, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lugli E, Madeo F, Malorni W, Marine JC, Martin SJ, Martinou JC, Medema JP, Meier P, Melino S, Mizushima N, Moll U, Muñoz-Pinedo C, Nuñez G, Oberst A, Panaretakis T, Penninger JM, Peter ME, Piacentini M, Pinton P, Prehn JH, Puthalakath H, Rabinovich GA, Ravichandran KS, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Shi Y, Simon HU, Stockwell BR, Szabadkai G, Tait SW, Tang HL, Tavernarakis N, Tsujimoto Y, Vanden Berghe T, Vandenabeele P, Villunger A, Wagner EF, Walczak H, White E, Wood WG, Yuan J, Zakeri Z, Zhivotovsky B, Melino G, Kroemer G: Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ 22: 58–73, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergsbaken T, Fink SL, Cookson BT: Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B: Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 110: 20364–20371, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. : Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma L, Chou JW, Snipes JA, Bharadwaj MS, Craddock AL, Cheng D, Weckerle A, Petrovic S, Hicks PJ, Hemal AK, Hawkins GA, Miller LD, Molina AJ, Langefeld CD, Murea M, Parks JS, Freedman BI: APOL1 renal-risk variants induce mitochondrial dysfunction. J Am Soc Nephrol 28: 1093–1105, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K: Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23: 429–438, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Granado D, Müller D, Krausel V, Kruzel-Davila E, Schuberth C, Eschborn M, Wedlich-Söldner R, Skorecki K, Pavenstädt H, Michgehl U, Weide T: Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J Am Soc Nephrol 28: 3227–3238, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stepanenko AA, Dmitrenko VV: HEK293 in cell biology and cancer research: Phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 569: 182–190, 2015 [DOI] [PubMed] [Google Scholar]
- 40.Kotb AM, Simon O, Blumenthal A, Vogelgesang S, Dombrowski F, Amann K, Zimmermann U, Endlich K, Endlich N: Knockdown of ApoL1 in zebrafish larvae affects the glomerular filtration barrier and the expression of nephrin. PLoS One 11: e0153768, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruno J, Pozzi N, Oliva J, Edwards JC: Apolipoprotein L1 confers pH-switchable ion permeability to phospholipid vesicles. J Biol Chem 292: 18344–18353, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruggeman LA, Wu Z, Luo L, Madhavan SM, Konieczkowski M, Drawz PE, Thomas DB, Barisoni L, Sedor JR, O’Toole JF: APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol 27: 3600–3610, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayek SS, Koh KH, Grams ME, Wei C, Ko YA, Li J, Samelko B, Lee H, Dande RR, Lee HW, Hahm E, Peev V, Tracy M, Tardi NJ, Gupta V, Altintas MM, Garborcauskas G, Stojanovic N, Winkler CA, Lipkowitz MS, Tin A, Inker LA, Levey AS, Zeier M, Freedman BI, Kopp JB, Skorecki K, Coresh J, Quyyumi AA, Sever S, Reiser J: A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat Med 23: 945–953, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olabisi O, Al-Romaih K, Henderson J, Tomar R, Drummond I, MacRae C, Pollak M: From man to fish: What can Zebrafish tell us about ApoL1 nephropathy? Clin Nephrol 86: 114–118, 2016 [DOI] [PubMed] [Google Scholar]
- 45.Fu Y, Zhu JY, Richman A, Zhang Y, Xie X, Das JR, Li J, Ray PE, Han Z: APOL1-G1 in nephrocytes induces hypertrophy and accelerates cell death. J Am Soc Nephrol 28: 1106–1116, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, Wasser WG, Magen D, Zaknoun E, Schuldiner M, Salzberg A, Kornitzer D, Marelja Z, Simons M, Skorecki K: APOL1-mediated cell injury involves disruption of conserved trafficking processes. J Am Soc Nephrol 28: 1117–1130, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C: Clonogenic assay of cells in vitro. Nat Protoc 1: 2315–2319, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Katsnelson MA, Rucker LG, Russo HM, Dubyak GR: K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194: 3937–3952, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ: Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391: 85–100, 1981 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.