

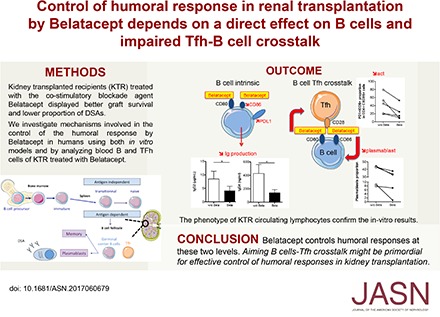
Abstract
Generation of de novo donor-specific antibodies (dnDSAs) after renal transplant is recognized as the leading cause of late transplant failure. Hence, the optimal immunosuppressive strategies to limit dnDSA development need to be defined. Recent clinical trials using the novel costimulatory blockade agent CTLA4-Ig (Belatacept) have shown that kidney transplant recipients (KTRs) treated with Belatacept have better graft survival and function and a lower proportion of dnDSAs than control-treated KTRs. Mechanisms involved in the control of humoral responses by Belatacept remain to be investigated. Here, we analyzed the effect of Belatacept on different steps of the B cell–mediated response in humans. In vitro, Belatacept reduced plasmablast differentiation, Ig production, and the expression of the major transcription factor involved in plasma cell function, Blimp-1, in a T cell–independent manner. Moreover, Belatacept induced activation of the STAT3 transcription factor in stimulated B cells and reduced the expression of CD86. Additionally, Belatacept blocked CD28-mediated activation of T follicular helper cells (Tfhs) in an autologous Tfh-memory B cells model. We then validated these observations in KTRs treated with Belatacept, who had a reduced proportion of blood effector B cells and activated Tfh (PD1+ICOS+) compared with control-treated KTRs. Our in vitro and in vivo results suggest that Belatacept modulates B cell function directly and at the level of B cell-Tfh interaction. These mechanisms likely account for the optimal control of humoral responses observed in KTRs treated with Belatacept.
Keywords: kidney transplantation, Belatacept (CTLA4-Ig), B cells, T follicular helper cells
Over the last decade, anti-HLA donor-specific antibodies (DSAs) arising after kidney transplantation, also called de novo donor-specific antibodies (dnDSAs), have increasingly been recognized as the leading cause of late transplant failure in kidney transplanted recipients (KTRs).1,2 Approximately 15%–30% of KTRs develop dnDSAs.3,4 Despite the numerous treatment strategies directed to boost conventional immunosuppression or suppress B cells activity by targeting plasma cells, antibodies, and/or complement, there has been no satisfactory therapy that reliably reverses the effects of DSAs after they are established.5 Therefore, clinicians have to focus on the primary prevention of dnDSAs generation. One of the risk factors associated with the development of dnDSAs is the therapeutic immunosuppression, which obviously represents the parameter with the widest range of potential improvement in terms of patient’s compliance, the degree of immunosuppression, and maybe the nature of the drugs itself.
Belatacept (cytotoxic T lymphocyte–associated antigen 4 [CTLA4]-Ig; LEA29Y; Bristol Myers Squibb) is a human fusion protein combining the extracellular portion of CTLA4 that has been mutated to confer greater binding avidity to CD80 and CD86 (B7 molecules) and the constant region fragment of human IgG1. CTLA4 binds to surface costimulatory ligands (CD80 and CD86) of antigen-presenting cells (APCs) and thus, prevents their interaction with CD28, thereby blocking T cell activation.6 Belatacept was granted approval by the US Food and Drug Administration and the European Medicines Agency in 2011 on the basis of the 3-year data from two phase 3 studies: the Belatacept Evaluation of Nephroprotection and Efficacy as First-Line Immunosuppression Trial (BENEFIT) and the BENEFIT Extended Criteria Donors.7,8 In their follow-up analysis of the BENEFIT, Vincenti et al.9 found that recipient mortality and the graft failure rate at 7 years after transplantation were significantly lower in the group of recipients treated with Belatacept compared with the control recipients treated with calcineurin inhibitors (CNIs). Interestingly, incidence of dnDSAs at year 7 was significantly lower in Belatacept-treated patients than in CNI-treated patients.9 This latter result can be considered as an active determinant of long-term graft outcome in patients treated with Belatacept. In those trials, it was suggested that improved adherence to the intravenous regimen could explain why Belatacept was associated with less dnDSAs formation.10 However, in our cohort of KTRs included in the BENEFIT, we previously showed that the proportion and the absolute number of CD19+CD24hiCD38hi B cells, a subset known to be associated with operational tolerance,11,12 are increased compared with controls. This suggests that Belatacept may have regulatory effect on B cells.13
The mechanisms of action of Belatacept begin to be elucidated. In animal models, Belatacept inhibits T-dependent antibody production. Larsen et al.14 showed enhanced inhibition of anti-sheep red blood cell antibodies by Belatacept compared with its parent Abatacept. The combined blockade of both CD28:B7 and CD40:40L pathways suppressed DSAs formation in kidney-transplanted macaques.15 Moreover, Kim et al.16 showed in a nonhuman primate model that Belatacept or anti-CD40 mAb combined with lymphodepletion suppressed the humoral response, which led to the prevention of antibody-mediated rejection. This observation was associated with a reduced frequency and size of germinal centers and a reduced proportion of T follicular helper cells (Tfhs).16 Together, these results led us to consider whether Belatacept in patients with transplants could act (1) directly on Ig production by B cells and/or (2) on their APC property.
The results presented herein show that Belatacept works directly by decreasing the plasmablasts differentiation and IgG secretion. Furthermore, Belatacept induces direct signaling in stimulated B cells that may be triggered by CD86. We also showed using both an in vitro model and in vivo data collected on KTRs that Belatacept, by modifying the expression of costimulatory molecules on the surface of activated B cells, decreases the proportion and activation status of circulating Tfhs and impairs Tfh/B cells crosstalk.
Results
Belatacept Modulates Plasmablasts Differentiation and Function
Because patients treated with Belatacept show reduced DSAs production, we first assessed whether Belatacept could have a direct effect on B cells. To assess plasmablast differentiation (defined as CD19loCD38hiCD27hi) (Figure 1A), untouched B cells were stimulated with CD40L and IL-21 to mimic Tfh help. After 5 days, such stimulation significantly increased plasmablasts by 29-fold compared with unstimulated B cells (Figure 1B). In the presence of Belatacept, the plasmablast proportion was slightly but significantly decreased compared with untreated cells (Figure 1C). The mortality of B cells and the proportion of memory B cells (defined as CD19+CD38−CD27+) were not affected by Belatacept (Supplemental Figure 1, A and B). Additionally, the proliferative capacities of plasmablasts (Supplemental Figure 1, C and D) and more generally, B cells (data not shown) were not altered by Belatacept.
Figure 1.

Belatacept alters differentiation of plasmablasts and function of stimulated B cells in vitro. B cells were cultured with CD40L and IL-21 stimulation for 10 days. (A) Analysis of plasmablast (CD19loCD27hiCD38hi) differentiation on day 5 using the indicated gating strategy. (B and C) Proportion of plasmablasts (B) before and after stimulation (n=18) and (C) with Belatacept (Bela) or without Belatacept (w/o Bela; n=25). (D–F) Quantification of (D) IgG2 (n=14), (E) IgG4 (n=8), and (F) IgM (n=6) after 10 days of culture. (G) Expression of Blimp-1 in B cells lysate by Western blot on day 5. Tubulin was used as a loading control. One representative of eight immunoblots is shown. (H) Ratio of Blimp-1 signal on tubulin signal (n=8). (I) Proportion of IFNγ-producing plasmablasts on day 5 with or without Belatacept. Data are given as mean±SEM for each group. *P<0.05 versus Belatacept+ (Wilcoxon matched pairs signed rank test).
Next, we assessed the functionality of activated B cells in terms of IgG and IgM secretions. Given that Belatacept is composed of type 1 human γ-chain, total IgG and IgG1 production could not be quantified. We also observed that Belatacept interfered with IgG3 detection (data not shown). However, the secretions of both IgG2 and IgG4 were significantly decreased in the presence of Belatacept (Figure 1, D and E). The production of IgM was not statistically different between treated and untreated cultures, although it showed a trend to decrease in Belatacept-treated cultures (Figure 1F).
Blimp-1 is the major transcription factor involved in plasmablasts differentiation and antibodies secretion. Importantly, using Western blot, we showed a decrease in Blimp-1 expression in Belatacept-stimulated culture compared with control on day 5 (Figure 1, G and H).
We next assessed the cytokine profile of stimulated B cells and plasmablasts cultured with or without Belatacept. Although IL-6, TNFα, IL-21, and IL-10 secretion did not differ between groups (data not shown), we did observe a significant decrease in IFNγ-producing B cells, most notably in the plasmablasts compartment in the presence of Belatacept (Figure 1I).
Thus, our model reveals that the reduced amount of DSAs observed in treated patients could at least in part rely on a direct effect on B cells independent of T cell participation.
Belatacept Induces a Direct Signaling in Stimulated B Cells and Modifies the Expression of Costimulatory Molecules on B Cell Surface
It was previously shown in dendritic cells (DCs) that CTLA4-Ig could induce a reverse signaling through the STAT3 pathway, whereas CD28-Ig works through the activation of the Akt/NFkB pathway.17,18 Thus, we assessed both STAT3 and Akt/NFkB signaling in stimulated B cells at 15 and 30 minutes in the presence and absence of Belatacept (Figure 2). Although Belatacept did not alter Akt signaling (Figure 2, A and B), there was a significant increase in STAT signaling determined by increased STAT3 phosphorylation in stimulated B cells in the presence of Belatacept (Figure 2, A and C).
Figure 2.

Belatacept induces STAT3 phosphorylation in stimulated B cells. B cells were cultured with or without CD40L and IL-21 stimulation for 15 and 30 minutes in the presence or absence of Belatacept. (A) Expression of phospho-Akt (Ser473), total Akt, phospho-STAT3 (Tyr705), and STAT3. Tubulin was used as a loading control. One representative of five immunoblots is shown. (B) Ratio of p-Akt (Ser473) signal on Akt total signal (n=5). (C) Ratio of p-STAT3 (Tyr705) signal on STAT3 total signal (n=5). Data are given as mean±SEM for each group. *P<0.05 without Belatacept (w/o Bela) versus Belatacept (Bela); #P<0.05 unstimulated versus stimulated (two-way ANOVA test).
We next aimed to determine if this signaling was receptor mediated and more specifically, if Belatacept directly acts on CD80 and/or CD86, both of which are known CTLA4 ligands. In vitro stimulation with CD40L and IL-21 induced high expression of CD80 and CD86 on cultured B cells on day 5 (i.e., the proportions of CD80+ and CD86+ B cells increased by 128% and 413%, respectively, after stimulation) (Supplemental Figure 2, A and B). Interestingly, different patterns of CD80 and CD86 expression were observed on memory B cells and on plasmablasts, the two types of fully differentiated B cells. Indeed, CD80 mean proportion was similar on memory B cells and plasmablasts, but CD80 mean fluorescence index (MFI) was threefold higher in memory B cells compared with plasmablasts. CD86 proportion was significantly lower in memory B cells compared with plasmablasts, and the same trend was seen for MFI (Supplemental Figure 2, C and D).
Belatacept treatment significantly decreased both the proportion and MFI of CD80 and CD86 in activated B cells (Supplemental Figure 2E), memory B cells, and plasmablasts compared with untreated cultures (Figure 3, A–D). To assess if this decreased expression was related to the ligation of Belatacept on these receptors (hence inducing a steric hindrance and/or competition for the same epitope with flow cytometry antibodies), protein expression of CD80 and CD86 by immunoblot was studied (Figure 3, E–H). Interestingly, CD86 expression was decreased on Belatacept-treated B cells lysates compared with untreated cells (Figure 3, E and F), whereas CD80 was not. This result strongly suggests that Belatacept triggers a direct signal in stimulated B cells via CD86. We also observed that about 6% of B cells expressed CD28 and that CD28 was not modulated by Belatacept (Figure 3I). Additionally, inducible T cell costimulator ligand expression was not modified by Belatacept (data not shown). In contrast, programmed cell death ligand 1 (PDL1) expression on total activated B cells and plasmablasts was significantly increased in the presence of Belatacept (Figure 3J).
Figure 3.

Belatacept modifies the pattern of expression of costimulatory molecules on the surface of B cells were cultured with CD40L and IL-21 stimulation in the presence or absence of Belatacept for 5 days (n=10). (A) Proportion and (B) MFI of CD86 in total B cells, memory B cells, and plasmablasts. (C) Proportion and (D) MFI of CD80 in B cells subsets described in Figure 1A. (E) Expression of CD86 in B cells lysates on day 5 by Western blot. Tubulin was used as a loading control. One representative of eight immunoblots is displayed. (F) Ratio of CD86 signal on Tubulin signal (n=8). (G) Expression of CD80 in B cells lysates on day 5 by Western blot. Tubulin was used as a loading control. One representative of five immunoblots is displayed. (H) Ratio of CD80 signal on Tubulin signal (n=8). (I) Proportion of CD28+ B cells after Belatacept treatment analyzed by flow cytometry (n=5). (J) Proportion of PDL1+ B cells after Belatacept treatment analyzed by flow cytometry (n=7). Data are given as mean±SEM for each group. Bela, Belatacept; w/o Bela, without Belatacept. *P<0.05 versus Belatacept (Wilcoxon matched pairs signed rank test).
Belatacept Disrupts Tfh-B Cells Interaction Both In Vitro and In Vivo
Given that Belatacept directly induced modification of costimulatory molecules expression on B cells, we wondered whether Belatacept could also affect Tfh-B cells crosstalk. As such, the ability of B cells generated with or without Belatacept to stimulate CD4 and Tfh proliferation was evaluated (Figure 4). B cells previously generated in the presence of Belatacept had a reduced capacity to induce T cells proliferation (Figure 4, A and B), Tfh proliferation (Figure 4, C–E) and Tfh activation (proportion of inducible T cell costimulatory-positive [ICOS+] Tfh) (Figure 4F).
Figure 4.

Stimulated B cells generated with Belatacept decrease proliferation of CD4+ cells and Tfhs. B cells were cultured with CD40L and IL-21 stimulation in the presence or absence of Belatacept for 5 days (n=6). After 5 days, stimulated B cells generated with or without Belatacept were washed and cocultured with autologous CD4 cells labeled with cell proliferation die (C.P.D.) at a 1:1 ratio (5×104) in the presence of soluble anti-CD3 (2 μg/ml) for 4 days. Proliferation and ICOS proportion were analyzed by flow cytometry. (A) Dot plots of a representative donor showing gating strategies for proliferative CD4 cells. (B) Proportion of proliferative CD4 cells cocultured with stimulated B cells generated with or without Belatacept for each donor. (C) Dot plots showing the gating strategy for Tfhs, proliferative Tfhs, and ICOS+ Tfhs. (D) Proportion of Tfhs in CD4+ cells. (E) Proportion of proliferative Tfhs in Tfhs. (F) Proportion of ICOS+ Tfhs in Tfhs. Bela, Belatacept; w/o Bela, without Belatacept. *P<0.05 versus Belatacept (Wilcoxon matched pairs signed rank test).
To better characterize the specific interaction between Tfhs and B cells in the presence of Belatacept, sorted autologous Tfhs (CD4+CD45RA−CXCR5+PD1+) and memory B cells (CD19+IgD−CD38dimCD27±) from healthy blood donors (HBDs) were cocultured for 5 days with or without Belatacept (Supplemental Figure 3). We did not observe any difference in Tfh proportion and survival in the presence of Belatacept (data not shown). However, the proportion and MFI of activated ICOS+ and programmed cell death-1 (PD1) ICOS+ Tfhs were reduced in the presence of Belatacept (Figure 5, A–D). Notably, we observed that plasmablasts proportion was lower in the Belatacept-treated cultures (Figure 5E).
Figure 5.

Tfhs-memory B cells crosstalk is altered in the presence of Belatacept. Purified blood memory B cells (CD19+IgD−CD38dimCD27±) were cocultured with Tfhs (CD4+CD45RA−CXCR5+PD1+) at a 1:1 ratio (1–3×104) in the presence of the superantigen staphylococcal enterotoxin B (SEB) for 5 days with or without Belatacept. (A) Dot plot of ICOS in CD4+CXCR5+ cells for a representative donor with or without Belatacept or stimulation by SEB. Notably, the expressions of ICOS (left panel) and PD1 (data not shown) were negative without SEB stimulation. (B and C) Proportions of (B) ICOS+ and (C) ICOS+PD1+ Tfhs were analyzed by flow cytometry. (D) MFI of ICOS+ Tfhs. (E) Plasmablasts (CD19+CD38hi) proportion. Data are given as mean±SEM for each group. Bela, Belatacept; w/o Bela, without Belatacept.
Then, to test the clinical relevance of our data in the peripheral blood of patients with grafts, we analyzed blood samples of KTRs who had stable graft functions treated with either Belatacept (Belatacept group; n=10) or CNI (CNI group; n=12) and an additional cohort of HBDs (n=12). Basic clinical characteristics of KTRs treated with Belatacept or CNI are displayed in Table 1.
Table 1.
Clinical and demographic characteristic of KTRs
| Demographic and Transplant Characteristics | KTR | P Value | |
|---|---|---|---|
| CNI | Belatacept | ||
| Patients, n | 12 | 10 | |
| Demography | |||
| Recipient age at the time of follow-up, yr | 64±3 | 65±4 | 0.90 |
| Sex, men/women | 5/7 | 6/4 | 0.70 |
| Donor age, yr | 50±5 | 54±4 | >0.99 |
| Living/cadaveric donor | 1/11 | 0/10 | |
| HLA A-B-DR incompatibilities | 3±0.4 | 3±0.4 | >0.99 |
| Time after transplantation, yr | 11.2±1 | 10.6±0.7 | 0.85 |
| Primary renal disease, n (%) | 0.20 | ||
| Immunologica | 2 (17) | 5 (50) | |
| Nonimmunologicb | 10 (83) | 5 (50) | |
| Immunosuppressive treatment, n (%) | |||
| Belatacept | 0 | 10 (100) | <0.001 |
| CNI | 12 (100) | 0 | <0.001 |
| Tacrolimus | 9 (75) | 0 | |
| Cyclosporin A | 3 (25) | 0 | |
| Steroid | 10 (83) | 9 (90) | >0.99 |
| MPA | 12 (100) | 10 (100) | >0.99 |
| Episode of biopsy proven acute rejection, n (%) | 1 (8) | 1 (10) | >0.99 |
| Biologic data | |||
| GFR, ml/min per 1.73 m2 | 60±5 | 65±4 | 0.40 |
| Proteinuria, g/24 h, median (minimum to maximum) | 0.1 (0.05–0.2) | 0.025 (0–0.6) | 0.06 |
| DSA score 4 (MFI<1100), n (%) | 4 (33) | 3 (30) | >0.99 |
| MFI, mean±SEM | 803±80 | 935±127 | 0.40 |
Data are presented as mean±SEM or n (percentage) unless otherwise indicated. GFR was estimated according to the Modification of Diet in Renal Disease formula. P value was calculated by two-tailed Mann–Whitney U test or Fisher exact test. MPA, Mycophenolic Acid.
Immunologic renal diseases include IgA nephropathy and Wegener granulomatosis.
Nonimmunologic diseases include uropathy, nephroangiosclerosis, autosomal dominant polycystic kidney disease, diabetic nephropathy, and tubulointerstitial disease.
Although lymphocytes counts and proportion of CD19+ cell did not differ between both groups of KTRs, recipients treated with Belatacept displayed significantly reduced absolute numbers of CD19+ (Table 2). Moreover, the proportions and absolute numbers of memory B cells (CD19+CD27+), switched memory B cells (CD19+CD27+IgD−), and unswitched memory B cells (CD19+CD27+IgD+) were significantly reduced in KTRs treated with Belatacept compared with patients treated with CNI (Figure 6, A–G). Finally, the Belatacept group also displayed lower proportions of blood plasmablasts (CD19+CD38hiCD24−) in CD19+ cells compared with the CNI group (Figure 6H), consistent with our in vitro cultures (Figure 5E).
Table 2.
Summary of immunologic characteristics of KTRs
| Lymphocytes Substets | KTR | P Value | |
|---|---|---|---|
| CNI | Belatacept | ||
| No. of lymphocytes, no. per 1 mm3 | 1858±178 | 1448±282 | 0.15 |
| CD4+ proportion, % in lymphocytes | 46.5±2.6 | 42.6±3.8 | 0.80 |
| No. of CD4+, no. per 1 mm3 | 849±107 | 624±147 | 0.07 |
| CD4+CD45RA− proportion | 52±7 | 40±5 | 0.35 |
| No. of CD4+CD45RA− | 425±62 | 231±49 | 0.01 |
| Frequency of CD19+ | 6.6±1 | 5.2±1.2 | 0.35 |
| No. of CD19+ | 123±19 | 100±52 | 0.03 |
Data are presented as mean±SEM.
Figure 6.

KTRs treated with Belatacept display lower proportion of circulating effector B cells. Blood B cells immunophenotyping of HBDs (n=12) or KTRs treated with CNI (n=12) or Belatacept (n=10). (A) Representative dots plots showing gating strategies for switched and unswitched memory B cells (upper panel) and memory cells (CD24+CD38−), plasmablasts (CD24−CD38hi), and transitional (CD24hiCD38hi) B cells. (B) Proportion and (C) number of memory (CD27+) B cells among CD19 cells. (D) Proportion and (E) number of switched memory (CD19+CD27+IgD−) B cells in CD19+ cells. (F) Proportion and (G) number of unswitched memory (CD19+CD27+IgD+) B cells in CD19+ cells. (H) Proportion of plasmablasts (CD19+CD24−CD38hi) in CD19+ cells. Data are given as mean±SEM for each group. Bela, Belatacept. *P<0.05 versus CNI (Kruskal–Wallis test [B, D, F, and H] or Mann–Whitney U test [C, E, and G]); #P<0.05 versus HBD (Kruskal–Wallis test [B, F, and H]).
To assess B cell capacities to interact with Tfh, patterns of expression of Belatacept ligands (i.e., CD80 and CD86) were analyzed on circulating B cells. CD80 and CD86 were not expressed on the same circulating B cell subpopulations of HBDs, and their expression varied according to the maturation stages of B cells (Figure 7, A–C). For instance, CD80 was mainly expressed on memory B cells (CD19+CD24+CD38−), whereas CD86 expression was predominant on immature transitional B cells (CD19+CD24hiCD38hi) (Figure 7, A–C). In KTRs treated with Belatacept, the proportion and MFI of CD80+ B cells were significantly reduced in B cells compared with those in CNI-treated patients (Figure 7, D and E). A similar significant reduction of MFI of CD80+ B cells was observed in memory B cells of the Belatacept group (Figure 7G), whereas the proportion of CD80+ B cells tended to be lower in the Belatacept group (P=0.07; n=6) (Figure 7F). No difference was detected in the proportion of CD86+ B cells between Belatacept and CNI groups (data not shown). The analysis of CD86 MFI could not be interpreted because of the scarce number of events.
Figure 7.

CD80 and CD86 expression varies according to the maturation stages of circulating B cells and KTRs treated with Belatacept display decreased CD80 expression on B cells, particularly on memory B cells. (A) Staining of CD19+ B cells with CD80 and CD86 antibodies (upper panel) and isotype control (lower panel). (B and C) Proportions of (B) CD80+ and (C) CD86+ B cells of HBDs were evaluated according to B cell subsets described in Figure 6A (n=10). Data are given as mean±SEM for each group. *P<0.05 memory versus mature B cells (Kruskal–Wallis test); #P<0.05 memory versus transitional B cells (Kruskal–Wallis test); &P<0.05 transitional versus mature B cells (Kruskal–Wallis test). (D) CD80 proportion and (E) MFI in B cells of HBDs (n=10) or KTRs treated with CNI (n=12) or Belatacept (n=10) described in Table 1. (F) CD80 proportion and (G) MFI in memory B cells of patients described above. Data are given as mean±SEM for each group. Bela, Belatacept. *P<0.05 versus CNI (Kruskal–Wallis test); #P<0.05 versus HBD (Kruskal–Wallis test).
We next aimed to compare the proportion and activation status of circulating Tfhs between the three groups of individuals (KTRs, those treated with CNI, and HBDs). The proportions of total CD4+ and memory CD4+CD45RA− were similar in both groups of KTRs (Table 2). However, KTRs treated with Belatacept had lower absolute numbers of memory CD4+CD45RA− T cells than those treated with CNI (Figure 8, A–C, Table 2). The proportion and absolute numbers of circulating Tfhs (CD4+CD45RA−CXCR5+) were significantly lower in KTRs treated with Belatacept than in CNI-treated patients (Figure 8, D and E). The quantitative Tfh defect observed in KTRs treated with Belatacept affected the three subgroups of functional Tfhs (Tfh1, -2, and -17) (Figure 8, F–H). Interestingly, 99% of Tfhs expressed CD28 in three subgroups (Figure 9A). To evaluate the activation status of blood Tfhs, we quantified highly activated Tfhs (ICOS+ and ICOS+PD1+) in CD4+ cells and Tfhs. The proportion in CD4+ cells and absolute numbers of activated Tfhs ICOS+ and ICOS+PD1+ were significantly reduced in KTRs treated with Belatacept compared with patients treated with CNI (Figure 9, B–F). Moreover, in Tfhs, the proportion of ICOS+PD1+ Tfhs was significantly lower in the Belatacept group than in the CNI group (Figure 9, G and H). Altogether, our results highlight both quantitative and functional defects of circulating Tfhs in KTRs treated with Belatacept.
Figure 8.

KTRs treated with Belatacept display decreased proportion of circulating Tfh. (A) Representative dot plot showing gating strategy of memory (CD4+CD45RA−) T cells and Tfhs (CD4+CD45RA−CXCR5+) (upper panel) as well as Tfh subsets: Tfh17 (CCR6+CXCR3), Tfh2 (CCR6−CXCR3−), and Tfh1 (CCR6−CXCR3+; lower panel). (B) Proportion and (C) number of memory CD4 cells (CD45RA− cells) in CD4+ cells. (D) Proportion and (E) number of Tfhs (CD45RA−CXCR5+ cells) in CD4+ cells. (F–H) Proportions of (F) Tfh1 (CCR6−CXCR3+ cells), (G) Tfh2 (CCR6−CXCR3−), and (H) Tfh17 (CCR6+CXCR3−) in CD4+ cells. Data are given as mean±SEM for each group (Belatacept, n=10; CNI, n=8; HBD, n=10). Bela, Belatacept. *P<0.05 versus CNI (Kruskal–Wallis test [D and F–H] or Mann–Whitney U test [C and E]); #P<0.05 versus HBD (Kruskal–Wallis test [B, D, and F–H]).
Figure 9.

The circulating Tfh of KTRs treated with Belatacept display lower levels of markers of activation. PD1, ICOS, and CD28 expressions were studied on gated Tfhs (CD4+CD45RA−CXCR5+) by flow cytometry. (A and B) Dot plots are shown for Fluorescence Minus One, one representative of KTRs treated with CNI, and another with Belatacept. (C) Proportion and (D) number of activated ICOS+ Tfhs in CD4+ cells. (E) Proportion and (F) number of activated PD1+ICOS+ Tfhs in CD4+ cells. (G and H) Proportions of (G) ICOS+ Tfhs or (H) PD1+ICOS+ Tfhs in Tfhs. Data are given as mean±SEM for each group (Belatacept, n=6; CNI, n=6; HBD, n=6). Bela, Belatacept. *P<0.05 versus CNI (Kruskal–Wallis test [D, F, and G] or Mann–Whitney U test [C, E, and H]); #P<0.05 versus HBD (Mann–Whitney U test [C, E, and H]).
Discussion
Prevention of dnDSAs formation is a major step in the prevention of the subsequent humoral rejection, a leading cause of graft loss in kidney transplantation. Our study reveals that the lowest level of dnDSAs observed in Belatacept-treated KTRs is related to a direct control of B cells responses by Belatacept.
Whereas Belatacept was initially developed to target T cells, these findings show that this therapeutic molecule also directly modulates effector B cells responses and humoral function, independent of T cell modulation. When we used highly purified B cells that we stimulated in vitro to reproduce Tfh help, we saw that Belatacept reduced plasmablasts proportion and more significantly, IgG2 and IgG4 secretion. Thus, we were able to show for the first time that Belatacept directly modulates plasma cells function. Additionally, our data show that Belatacept reduces the expression of the transcription factor, Blimp-1, majorly involved in plasma cells generation and antibodies secretion.19 Tellier et al.20 have already shown that the inactivation of Blimp-1 in mature plasma cells impaired their ability to secrete antibodies but not their survival. In accordance, we did not observe any increase in B cell mortality in vitro or any modification in B cell and plasmablast proliferation in the presence of Belatacept. We thus hypothesize that the reduction of Blimp-1 by Belatacept does not primarily affect plasma cells differentiation but rather “blocks” their Ig production.
To assess if Belatacept could also influence antibody-independent functions of treated B cells,21–23 we evaluated whether it modified the cytokine profile secreted by B cells and specifically, plasma cells. We found a significant decrease in IFNγ-secreting plasmablasts and B cells as a whole. Interestingly, IFNγ has been implicated in Ig class switching.24–27 Whether this particular subset of IFN-secreting plasma cells, which seems to be specifically targeted by Belatacept, plays a role in the decreased Ig secretion observed in our model needs to be further evaluated.
The rational for the development and use of Belatacept molecule is mainly on the basis of its ability to provide an optimal blockade of naïve T cell activation to prevent allograft rejection.28 By binding on B7 receptors (CD80/CD86) on APCs, Belatacept prevents APCs delivery of costimulatory signal to T cell by inhibiting CD28 (expressed on T cells) interactions with its APC ligands. Kowalczyk et al.17 were the first to suggest that CTLA4-Ig may also have direct effect on APCs. Indeed, CTLA4-Ig could induce a reverse signaling by B7 molecules in human DCs that led to the generation of tolerogenic DCs. In contrast, triggering CD86 on human or murine splenic B cells with an agonist antibody or CD28-Ig could activate B cells, leading to increased IgG production.29–31 However, before this work, a direct effect of Belatacept on B cells had not been assessed. Here, we showed that Belatacept activates the STAT3 signaling pathway in stimulated B cells, a mechanism previously described in DCs by Kowalczyk et al.32
We next aimed to assess which receptor could account for the direct signaling triggered by Belatacept in stimulated B cells. Unlike CD80, CD86 molecule has a longer cytoplasmic tail and potential phosphorylation sites that could mediate intracellular signaling. Immunoblot experiments strongly suggest that ligation of Belatacept on CD86 (but not on CD80) specifically triggers a direct signaling in stimulated B cells and decreases its expression. These results are coherent with the described structure of these two molecules33–35 and suggest a novel mechanism by which CD80 and CD86 play differential roles in B cells. In addition, we show that CD86 was mainly expressed on plasmablasts after activation and that expression was significantly repressed in the presence of Belatacept. This reinforces the possibility that Belatacept could directly target CD86 on plasmablasts and may inhibit their secretion of Ig by downregulating Blimp-1 expression and/or IFNγ secretion. Other mechanisms may also be implicated in the modulation of humoral responses observed in our in vitro study. After 5 days of stimulation, B cells expressed low levels of CD28 and PDL1. CD28 expression on long-lived plasma cells was shown to be necessary for their function and maintenance in bone marrow.36,37 Moreover, PDL1 can bind to CD80, and PDL1 expression was upregulated in the presence of Belatacept. Still, admitting that the blockade of CD28 or PDL1 signaling by Belatacept in B cells participates in the direct modulation of humoral responses in vitro remains to be further investigated.
Our results suggest that Belatacept modifies the pattern of expression of costimulatory molecules on the surface of B cells by several mechanisms, including (1) occupancy by Belatacept on both CD80 and CD86 ligands, (2) decrease in free expression of CD86, and (3) increased expression of PDL1. Importantly, we revealed that, after B cells were generated in the presence of Belatacept, they had impaired abilities to stimulate T cell proliferation in vitro without needing a continuous exposure to the drug. This result supports that the modified pattern of costimulatory receptors induced by Belatacept is functionally relevant and durably impairs B cell APCs capacities.
It has been previously suggested that Belatacept alters germinal center responses caused by Tfhs priming blockade by DC.16 We hypothesize that Belatacept could also impair germinal center reactions by targeting B-Tfh interaction in human patients with transplants.
We showed that Belatacept decreased the expression of Tfh activation markers in an in vitro model of coculture using purified Tfhs and memory B cells. This suggests that blocking access to costimulatory molecules, such as CD80/86, on B cells reduces Tfh activation, given that all circulating Tfhs highly express CD28. Our results are in accordance with the work of Ville et al.,38 which reported, in a model of Tfhs-B cells coculture isolated from pediatric tonsil, a reduced proportion of Tfhs in the presence of Belatacept or a selective antagonist of CD28, intimating that both molecules control proliferative capacities of human Tfhs. Previous studies identified ICOS-inducible T cell costimulator ligand as the main driver for Tfhs differentiation and maintenance.39,40 Our results bring further evidence that CD28 expression on Tfh could also be a determinant pathway for Tfh activation.
Importantly, we assessed whether the above-described in vitro effects of Belatacept could also be observed in KTRs treated with Belatacept.
The decreased B cell expression of CD80 that we observed by flow cytometry in these patients and particularly, on memory B cells highly suggests an efficient binding of Belatacept to its receptor in physiologic condition. CD86 expression, however, was very low and did not differ between both groups of patients. We believe that this very low level of CD86 expression at basal state on circulating B cells prevents us from assessing any effect of Belatacept on CD86 ligands in vivo.
Notably, we showed that these patients displayed lower proportions of circulating effector B cells (i.e., memory B cells, switched memory B cells, and plasmablasts) associated with diminished proportion of circulating Tfhs compared with control patients treated with CNI. Furthermore, the circulating Tfhs of KTRs treated with Belatacept displayed lower levels of markers of activation as assessed by the proportion of ICOS+PD1+ Tfhs in Tfh populations. Interestingly, the use of Tfhs/B cells defect in the control of DSAs secretion was suggested in operational tolerant KTRs who also displayed the lowest levels of dnDSAs. These patients also had Tfh defect and impaired Tfhs-B cells crosstalk in vitro. Moreover, an increase in the proportion of circulating Tfhs preceded the appearance of dnDSAs in a cohort of stable KTRs.41 Altogether, these results suggest that therapeutic intervention on Tfhs-B cells crosstalk may be a crucial step to control HLA sensitization.
In conclusion, our study strongly suggests that immunosuppressive therapy using Belatacept plays a direct and active role at several steps of the humoral response. Belatacept may uniquely control B cell responses by modulating both their antigen-presenting capacities, leading to the impairment of Tfhs-B cells crosstalk, and the production of antibodies by effector B cells. These results allow us, at least in part, to account for the lesser extent of HLA sensitization observed in KTRs. They also raise new clinical perspectives in the development of immunosuppressive strategies aiming to target costimulatory molecules in the fields of both transplantation and autoimmune disease.
Concise Methods
A detailed description, including information on materials and reagents, flow cytometry, and Western blotting, is in Supplemental Material.
Isolation and Culture of B Cells
Peripheral blood of HBDs was obtained from Etablissement Français du Sang (Creteil, France). PBMCs were isolated by density gradient centrifugation with lymphocyte separation medium (Laboratoires Eurobio, Les Ulis, France). Total B cells from freshly isolated PBMCs were purified by negative selection using the B cell isolation kit II for magnetic cell separation (Miltenyi Biotec SAS, Paris, France). Belatacept was added to the cell cultures at the concentration of 40 μg/ml during 5 or 10 days.17 The controls consisted of cultures without drug.
Supplemental Material has culture details.
Flow Cytometry, CFSE Labeling, and Ig Production in B Cell Assays
After 5 and 10 days of culture, cell culture supernatants were collected and stored at −80°C. Cultured B cells were analyzed for expression of surface markers by multicolor flow cytometry. Cell aliquots were stained for 20 minutes with selected combinations of fluorochrome-conjugated antibodies at 4°C. Supplemental Material has details.
For the proliferation assay, 1–10×106 cells of the concerned cell populations were labeled with 1 μM cell proliferation dye (eBioscience, Paris, France) before culture initiation. After 5 days of culture, cells were harvested and incubated with antibodies, and the dilution of the cell proliferation dye was assessed by flow cytometry.
Ig concentrations were measured in the culture supernatants by Luminex following the manufacturer’s protocol (Affymetrix E-bioscience; Human Isotyping procartaplex).
Western Blot Analyses of Purified B Cells
On day 5, activated B cells were lysed in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, and 0.1% SDS) containing a cocktail of protease and phosphatase inhibitors (Roche). After concentration assessment, proteins were separated on SDS gels and transferred on nitrocellulose membranes (Amersham; GE Healthcare Life Sciences). The blots were incubated with different primary antibodies. Supplemental Material has details.
Isolation and Culture of CD4 Cells
CD4 cells from frozen PBMCs were purified by negative selection using the CD4 cell isolation kit II for magnetic cell separation (Miltenyi Biotec SAS, Paris, France). Isolated CD4 cells were then labeled with cell proliferation dye (eBioscience) and cultured with anti-CD3 (Miltenyi Biotec SAS) at the concentration of 2 μg/ml with or without B cells at a 1:1 ratio during 4 days.
In Vitro Tfh-B Cell Coculture Assays
B cells and Tfhs were isolated from HBD whole blood using the MACSprep HLA B/T Cell Isolation Kit (Miltenyi Biotec SAS). Sorting conditions were detailed in Supplemental Material. Memory B cells (1×104) were cocultured with blood Tfhs at a 1:1 ratio with endotoxin-reduced staphylococcal enterotoxin B (100 ng/ml; Sigma-Aldrich). Belatacept was added to the cell cultures at different concentrations (10 and 40 μg/ml).
Supplemental Material has details.
Patients
Two groups of first KTRs were included in the study. (1) The first group included KTRs treated with Belatacept (LEA29Y), mycophenolate mofetil, and steroids (n=10). We picked out patients from phase 3 clinical studies cohorts (BMS) from Henri Mondor and Bicêtre Hospitals. These patients had had stable renal function (Modification of Diet in Renal Disease >35 ml/min and proteinuria <0.5 g/g). Time between transplantation and sample analysis ranged from 8 to 14 years. (2) The second group included patients with stable graft function treated with CNI, mycophenolate mofetil, and/or steroids (n=12). Patients were selected from our database, were not immunized at the time of transplantation, and were matched to patients on Belatacept for the date of transplantation.
In both groups, we excluded patients with history of humoral rejection or HLA sensitization (defined by DSAs with MFI>1100) and those with a recent event of acute T cell–mediated rejection, infection, or tumor (<6 months).
Detailed clinical data are presented in Table 1. All participating patients gave written informed consent.
A control group of HBDs was included (n=12).
Immunophenotyping of Blood B Cells and Tfhs
B and Tfh subsets were characterized from freshly isolated PBMCs by flow cytometry (BD Canto II) with the following markers: CD19, CD24, CD38, CD80, CD3, CD4, CD45RA, CXCR5, CD274, CD278, CD27, IgD, IgM, CD86, CD197, CXCR5, CCR6, CXCR3, and CD279. Supplemental Material has further details.
Markers of activation and costimulation were analyzed according to the percentage of positive cells and the intensity of expression as evaluated by MFI with the FlowJo v.10 software (Treestar).
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software. Statistical analyses were adapted to data distribution (Kruskal–Wallis test, Mann–Whitney U test, and nonparametric paired Wilcoxon test). Continuous variables from patients and HBD groups were expressed as mean±SEM and compared using the Kruskal–Wallis test. Paired biologic observations from cells cultures were analyzed using Wilcoxon paired nonparametric tests (flow cytometry and Western blot data). Significance was defined as a P value of <0.05.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank A. Guguin and A. Henry from Institut Mondor de Recherche Biomédicale cells sorting facility, M. Surenaud from IMRB luminex plateform, and K. Bailly from Cochin Hospital cells sorting facility.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017060679/-/DCSupplemental.
References
- 1.Loupy A, Hill GS, Jordan SC: The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol 8: 348–357, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Terasaki PI: A personal perspective: 100-Year history of the humoral theory of transplantation. Transplantation 93: 751–756, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Hidalgo LG, Campbell PM, Sis B, Einecke G, Mengel M, Chang J, Sellares J, Reeve J, Halloran PF: De novo donor-specific antibody at the time of kidney transplant biopsy associates with microvascular pathology and late graft failure. Am J Transplant 9: 2532–2541, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, Nickerson PW: Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant 12: 1157–1167, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Everly MJ, Terasaki PI: The state of therapy for removal of alloantibody producing plasma cells in transplantation. Semin Immunol 24: 143–147, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, Lang P, Grinyo J, Halloran PF, Solez K, Hagerty D, Levy E, Zhou W, Natarajan K, Charpentier B; Belatacept Study Group : Costimulation blockade with belatacept in renal transplantation. N Engl J Med 353: 770–781, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Rostaing L, Vincenti F, Grinyó J, Rice KM, Bresnahan B, Steinberg S, Gang S, Gaite LE, Moal M-C, Mondragón-Ramirez GA, Kothari J, Pupim L, Larsen CP: Long-term belatacept exposure maintains efficacy and safety at 5 years: Results from the long-term extension of the BENEFIT study. Am J Transplant 13: 2875–2883, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Vincenti F, Tedesco Silva H, Busque S, O’Connell P, Friedewald J, Cibrik D, Budde K, Yoshida A, Cohney S, Weimar W, Kim YS, Lawendy N, Lan S-P, Kudlacz E, Krishnaswami S, Chan G: Randomized phase 2b trial of tofacitinib (CP-690,550) in de novo kidney transplant patients: Efficacy, renal function and safety at 1 year. Am J Transplant 12: 2446–2456, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, Moal M-C, Mondragon-Ramirez GA, Kothari J, Polinsky MS, Meier-Kriesche H-U, Munier S, Larsen CP: Belatacept and long-term outcomes in kidney transplantation. N Engl J Med 374: 333–343, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Heher E, Markmann JF: The clearer BENEFITS of belatacept. N Engl J Med 374: 388–389, 2016 [DOI] [PubMed] [Google Scholar]
- 11.Pallier A, Hillion S, Danger R, Giral M, Racapé M, Degauque N, Dugast E, Ashton-Chess J, Pettré S, Lozano JJ, Bataille R, Devys A, Cesbron-Gautier A, Braudeau C, Larrose C, Soulillou JP, Brouard S: Patients with drug-free long-term graft function display increased numbers of peripheral B cells with a memory and inhibitory phenotype. Kidney Int 78: 503–513, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, Chapman S, Craciun L, Sergeant R, Brouard S, Rovis F, Jimenez E, Ballow A, Giral M, Rebollo-Mesa I, Le Moine A, Braudeau C, Hilton R, Gerstmayer B, Bourcier K, Sharif A, Krajewska M, Lord GM, Roberts I, Goldman M, Wood KJ, Newell K, Seyfert-Margolis V, Warrens AN, Janssen U, Volk H-D, Soulillou J-P, Hernandez-Fuentes MP, Lechler RI: Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest 120: 1848–1861, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leibler C, Matignon M, Pilon C, Montespan F, Bigot J, Lang P, Carosella ED, Cohen J, Rouas-Freiss N, Grimbert P, Menier C: Kidney transplant recipients treated with belatacept exhibit increased naïve and transitional B cells. Am J Transplant 14: 1173–1182, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, Anderson D, Cowan S, Price K, Naemura J, Emswiler J, Greene J, Turk LA, Bajorath J, Townsend R, Hagerty D, Linsley PS, Peach RJ: Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant 5: 443–453, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Pearson TC, Trambley J, Odom K, Anderson DC, Cowan S, Bray R, Lin A, Hollenbaugh D, Aruffo A, Siadak AW, Strobert E, Hennigar R, Larsen CP: Anti-CD40 therapy extends renal allograft survival in rhesus macaques. Transplantation 74: 933–940, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Kim EJ, Kwun J, Gibby AC, Hong JJ, Farris AB 3rd, Iwakoshi NN, Villinger F, Kirk AD, Knechtle SJ: Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection. Am J Transplant 14: 59–69, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowalczyk A, D’Souza CA, Zhang L: Cell-extrinsic CTLA4-mediated regulation of dendritic cell maturation depends on STAT3. Eur J Immunol 44: 1143–1155, 2014 [DOI] [PubMed] [Google Scholar]
- 18.Koorella C, Nair JR, Murray ME, Carlson LM, Watkins SK, Lee KP: Novel regulation of CD80/CD86-induced phosphatidylinositol 3-kinase signaling by NOTCH1 protein in interleukin-6 and indoleamine 2,3-dioxygenase production by dendritic cells. J Biol Chem 289: 7747–7762, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M: Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17: 331–343, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, Nutt SL: Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 17: 323–330, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen P, Fillatreau S: Antibody-independent functions of B cells: A focus on cytokines. Nat Rev Immunol 15: 441–451, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Lund FE, Randall TD: Effector and regulatory B cells: Modulators of CD4+ T cell immunity. Nat Rev Immunol 10: 236–247, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fillatreau S: Regulatory plasma cells. Curr Opin Pharmacol 23: 1–5, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, Lund FE: Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 1: 475–482, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Bossie A, Vitetta ES: IFN-gamma enhances secretion of IgG2a from IgG2a-committed LPS-stimulated murine B cells: Implications for the role of IFN-gamma in class switching. Cell Immunol 135: 95–104, 1991 [DOI] [PubMed] [Google Scholar]
- 26.Kawano Y, Noma T, Yata J: Regulation of human IgG subclass production by cytokines. IFN-gamma and IL-6 act antagonistically in the induction of human IgG1 but additively in the induction of IgG2. J Immunol 153: 4948–4958, 1994 [PubMed] [Google Scholar]
- 27.Lund FE: Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 20: 332–338, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ford ML: T cell cosignaling molecules in transplantation. Immunity 44: 1020–1033, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeannin P, Delneste Y, Lecoanet-Henchoz S, Gauchat J-F, Ellis J, Bonnefoy J-Y: CD86 (B7-2) on human B cells. A functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells. J Biol Chem 272: 15613–15619, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe A, Sanders VM: Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J Immunol 165: 680–690, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Rau FC, Dieter J, Luo Z, Priest SO, Baumgarth N: B7-1/2 (CD80/CD86) direct signaling to B cells enhances IgG secretion. J Immunol 183: 7661–7671, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowalczyk A, D’Souza CA, Zhang L: Cell-extrinsic CTLA4-mediated regulation of dendritic cell maturation depends on STAT3. Eur J Immunol, 44: 1143–1155, 2014 [DOI] [PubMed] [Google Scholar]
- 33.Hathcock KS, Hodes RJ: Role of the CD28-B7 costimulatory pathways in T cell-dependent B cell responses. Adv Immunol 62: 131–166, 1996 [DOI] [PubMed] [Google Scholar]
- 34.Marín LA, Moya-Quiles MR, Miras M, Muro M, Minguela A, Bermejo J, Ramírez P, García-Alonso AM, Parrilla P, Alvarez-López MR: Evaluation of CD86 gene polymorphism at +1057 position in liver transplant recipients. Transpl Immunol 15: 69–74, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Kin NW, Sanders VM: CD86 regulates IgG1 production via a CD19-dependent mechanism. J Immunol 179: 1516–1523, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, Schoenberger SP, Lee KP: Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med 208: 1435–1446, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozanski CH, Utley A, Carlson LM, Farren MR, Murray M, Russell LM, Nair JR, Yang Z, Brady W, Garrett-Sinha LA, Schoenberger SP, Green JM, Boise LH, Lee KP: CD28 promotes plasma cell survival, sustained antibody responses, and BLIMP-1 upregulation through its distal PYAP proline motif. J Immunol 194: 4717–4728, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ville S, Poirier N, Branchereau J, Charpy V, Pengam S, Nerriere-Daguin V, Le Bas-Bernardet S, Coulon F, Mary C, Chenouard A, Hervouet J, Minault D, Nedellec S, Renaudin K, Vanhove B, Blancho G: Anti-CD28 antibody and belatacept exert differential effects on mechanisms of renal allograft rejection. J Am Soc Nephrol 27: 3577–3588, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crotty S: T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529–542, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S: ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34: 932–946, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chenouard A, Chesneau M, Bui Nguyen L, Le Bot S, Cadoux M, Dugast E, Paul C, Malard-Castagnet S, Ville S, Guérif P, Soulillou J-P, Degauque N, Danger R, Giral M, Brouard S: Renal operational tolerance is associated with a defect of blood Tfh cells that exhibit impaired B cell help. Am J Transplant 17: 1490–1501, 2017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.