

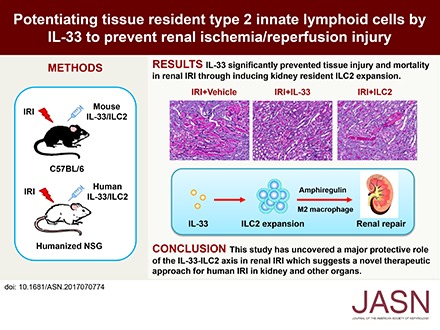
Abstract
The IL-33-type 2 innate lymphoid cell (ILC2) axis has an important role in tissue homeostasis, inflammation, and wound healing. However, the relative importance of this innate immune pathway for immunotherapy against inflammation and tissue damage remains unclear. Here, we show that treatment with recombinant mouse IL-33 prevented renal structural and functional injury and reduced mortality in mice subjected to ischemia-reperfusion injury (IRI). Compared with control-treated IRI mice, IL-33–treated IRI mice had increased levels of IL-4 and IL-13 in serum and kidney and more ILC2, regulatory T cells (Tregs), and anti-inflammatory (M2) macrophages. Depletion of ILC2, but not Tregs, substantially abolished the protective effect of IL-33 on renal IRI. Adoptive transfer of ex vivo–expanded ILC2 prevented renal injury in mice subjected to IRI. This protective effect associated with induction of M2 macrophages in kidney and required ILC2 production of amphiregulin. Treatment of mice with IL-33 or ILC2 after IRI was also renoprotective. Furthermore, in a humanized mouse model of renal IRI, treatment with human IL-33 or transfer of ex vivo–expanded human ILC2 ameliorated renal IRI. This study has uncovered a major protective role of the IL-33–ILC2 axis in renal IRI that could be potentiated as a therapeutic strategy.
Keywords: ischemia/reperfusion injury, IL-33, innate lymphoid cells, macrophages, immunotherapy
Innate lymphoid cells (ILCs) are a newly identified heterogeneous family of innate immune cells with critical roles in immunity, tissue homeostasis, and pathologic inflammation.1,2 They are categorized into three groups on the basis of their cytokine production and transcription factors: ILC1, ILC2, and ILC3, which represent the innate counterparts of CD4+ THelper1 (Th1), Th2, and Th17 cells.3 Type 2 innate lymphoid cells (ILC2s) require the transcription factor GATA-3; produce type 2 cytokines IL-4, IL-5 and IL-13; and play important roles in immunity against pathogens, type 2 inflammation, and tissue homeostasis and repair.1–3 For example, in influenza virus infection of mice, ILC2s were activated by lung epithelial cell–derived IL-25 and IL-33 and produced amphiregulin (Areg), which promotes repair of the airway epithelium.4 IL-33–dependent ILC2 responses were also shown to contribute to cutaneous wound healing.5 Thus, modulation of ILC2 activity may provide a basis for improved immunotherapy.
IL-33 is a nuclear cytokine from the IL-1 family that is mainly produced by stromal cells after proinflammatory stimulation.6–8 IL-33 has pleiotropic effects in immune and inflammatory responses.9 Previous studies have shown that IL-33 exacerbates allergic inflammation and tissue fibrosis10–12 but attenuates atherosclerosis and protects against parasite infection.13,14 Recent findings showed that the IL-33–driven ILC2 axis plays a critical role in cutaneous wound healing, limitation of intestinal and adipose tissue inflammation, and protection against experimental cerebral malaria.5,15–17 IL-33 has been shown to induce expansion of ILC2 in kidney and protect against renal injury in Adriamycin nephropathy.18
Renal ischemia-reperfusion injury (IRI) is a major cause of AKI, and it causes significant morbidity and mortality. Ischemia induces a complex series of events that lead to altered hemodynamics, tubular injury, and inflammation.19,20 Current treatments for human IRI are nonspecific and have limited efficacy. It is well established that both innate and adaptive immune responses are important contributors to the pathology of ischemic injury.20 In this study, we sought to determine the role of the IL-33–ILC2 pathway in renal IRI. Here, we found that IL-33 significantly prevented renal structural and functional injury and reduced mortality in mice with IRI through inducing kidney resident ILC2 expansion. Importantly, we showed that human IL-33 is a critical cytokine in both promoting ILC2 responses and preventing renal injury in humanized mice with IRI and that adoptive transfer of ex vivo–expanded human ILC2 attenuated renal injury in humanized mice with IRI.
Results
IL-33 Protected Against Renal Injury in IRI Mice
To determine whether IL-33 could modulate AKI, we treated C57BL/6 mice with mouse recombinant IL-33 (0.3 μg per mouse per day intraperitoneally) for 5 consecutive days before bilateral IRI (Figure 1A). Serum creatinine was increased in IRI mice compared with that of control mice, and it was significantly improved in IRI mice treated with IL-33 (Figure 1B). We evaluated overall tissue injury using a semiquantitative tubular injury score and found that IL-33–treated mice showed significantly reduced tissue injury (Figure 1, C and D). Gr-1+ neutrophil infiltration in the outer medulla of postischemic kidney was significantly reduced in IRI mice treated with IL-33 (Figure 1E). Moreover, IL-33 treatment markedly improved the survival rate of moderate IRI mice (30 minutes of bilateral renal pedicle clamping at 37°C)—none of the IL-33–injected mice died, whereas 44% of PBS-injected mice died by day 7 (Figure 1F). Notably, IL-33 treatment significantly reduced mortality in severe IRI mice (38 minutes of bilateral renal pedicle clamping at 37°C) compared with PBS-treated controls—only 16% of the IL-33–injected mice died, whereas 100% of PBS-injected mice died by day 7 (Figure 1G). Together, these results show that IL-33 markedly attenuated renal IRI and postischemic renal failure. The effect of IL-33 on renal fibrosis was also assessed on day 28 after unilateral IRI by histologic examination. Immunostaining of markers associated with fibrosis, including collagen I and α-smooth muscle actin, revealed that IL-33 treatment did not induce renal interstitial fibrosis in sham-operated kidney. More importantly, we also found that IL-33 treatment significantly reduced renal fibrosis in postischemic kidney of IRI mice (Supplemental Figure 1).
Figure 1.

IL-33 protected against renal injury in IRI mice. (A) C57BL/6 mice were treated with mouse recombinant IL-33 daily for 5 consecutive days before bilateral IRI. Mice were euthanized 1 day after IRI. (B) Serum creatinine levels were assessed in control, IRI + Vehicle (PBS), and IRI + IL-33 at 1 day after IRI. ***P<0.001 versus IRI + vehicle. (C) Representative periodic acid–Schiff-stained sections of kidney outer medulla from IRI mice treated with PBS or IL-33. Original magnification, ×200. (D) Semiquantitative assessment of tubular injury in control, IRI + Vehicle, and IRI + IL-33 1 day after IRI. ***P<0.001 versus IRI + vehicle. (E) Number of Gr-1+ neutrophils was assessed by immunofluorescence staining in outer medulla of kidney. Data shown are the mean±SEM (n=12 per group) from three independent experiments. hpf, High-power field. ***P<0.001 versus IRI + vehicle. (F and G) Survival rate of C57BL/6 mice (male, 8–10 weeks of age) subjected to IRI for (F) 30 minutes or (G) 38 minutes and injected with IL-33 (n=18) or PBS (n=18). Data are from three independent experiments. **P<0.01 between IRI + Vehicle and IRI + IL-33 mice as assessed by log rank test; ***P<0.001 between IRI + Vehicle and IRI + IL-33 mice as assessed by log rank test.
IL-33 Induced Th2 Cytokine, ILC2, Regulatory T Cells, and Anti-Inflammatory Macrophages In Vivo
We then investigated the mechanism by which IL-33 protects mice against IRI. In IRI mice treated with IL-33, serum levels of the Th2 cytokines IL-4 and IL-13 were significantly increased compared with those of control and IRI mice (Figure 2A). IL-33 treatment also enhanced the expression of IL-4 and IL-13 in kidney and kidney-draining lymph nodes (Figure 2B). Meanwhile, IL-33 treatment reduced the expression of various inflammatory genes TNF-α, IL-1β, IL-6, CXCL1, and CXCL2 in kidney (Figure 2C).
Figure 2.

IL-33 induced ILC2s, M2 macrophages, and Tregs in IRI mice. (A) IL-4 and IL-13 levels in serum were assessed in control, IRI + Vehicle, and IRI + IL-33 groups day 1 after bilateral IRI. (B and C) The mRNA expression of IL-4, IL-13, and various inflammatory genes in kidney was examined by quantitative PCR and expressed relative to the control of each experiment. (D) Representative FACS analysis showing the gating strategy to identify CD45+Lin−CD127+ST2+GATA3+ ILC2 in mouse kidney. Lin mixture includes CD3, CD5, TCRβ, TCRγδ, CD19, B220, CD49b, CD11b, CD11c, FcεRIα, Gr-1, and Ter-119. (E) Histogram showing expression of CD90, KLRG1, CD25, and IL-17RB on kidney ILC2s. Specific markers (red lines) and isotype controls (gray-filled areas) are shown. (F and G) Percentage and absolute number of ST2+GATA3+ ILC2s in the kidneys of control, IRI + Vehicle, or IRI + IL-33 mice. (H) Number of F4/80+ macrophages was assessed by immunofluorescence staining in kidney. (I and J) F4/80+ kidney macrophages were sorted by flow cytometry in control, IRI + Vehicle, and IRI + IL-33 mice on day 1 after IRI. The mRNA expression of MR, arginase, HO-1, YM1, IL-10, iNOS, TNF-α, IL-1β, IL-6, and CCL2 was quantified by quantitative PCR in F4/80+ kidney macrophages. (K and L) Percentage of CD4+Foxp3+ Tregs in the CD4+ T cell compartment from the (K) spleens and (L) kidneys of control, IRI + Vehicle, or IRI + IL-33 mice. Data shown are the mean±SEM (n=8 per group). **P<0.01 versus IRI + vehicle; ***P<0.001 versus IRI + vehicle.
Recently, IL-33 has been found to directly induce ILC2 expansion in multiple anatomic sites, where they contribute to Th2 immune responses and regulate inflammation.5,6,16,17 We, therefore, analyzed the effect of IL-33 on ILC2 in kidney of IRI mice by multicolor flow cytometry. After pregating on CD45+ cells to exclude any contaminating kidney epithelial cells, lineage-negative (Lin−) CD127+ cells in kidney were analyzed for expression of key markers that define Lin−CD127+ST2+GATA3+ ILC2 (Figure 2D). In addition, Lin−CD127+ST2+GATA3+ cells in kidney expressed CD90, CD25, KLRG1, and IL-17RB (Figure 2E), a phenotype consistent with ILC2.13,21 In comparison with control and IRI mice, IL-33–treated IRI mice exhibited significant increases in the number of ILC2 in kidney (70-fold) and other anatomic sites (Figure 2, F and G, Supplemental Figure 2). ILC2, but not CD4+ T cells, CD8+ T cells, and NK cells, were shown to be tissue-resident cells in parabiosis experiments (Figure 3, A–D). ILC2 proliferated strongly in IL-33–treated mice (Figure 3, E and F). Therefore, these data indicate that IL-33 elicits an ILC2 response in kidney, which may be associated with the induction of Th2 responses and renoprotection by IL-33.
Figure 3.

Kidney resident ILC2s proliferated on IL-33 treatment. (A) Congenically marked CD45.1+ and CD45.2+ C57BL/6 mice underwent parabiosis surgery. After 30 days, parabiotic mice were administered 0.5 μg mouse recombinant IL-33 or PBS daily for 5 consecutive days. (B) Representative FACS analysis showing the gating strategy to determine the percentage of host-derived ILC2s in the kidney on day 35 after parabiosis surgery. Lin mixture includes CD3, CD5, TCRβ, TCRγδ, CD19, B220, CD49b, CD11b, CD11c, FcεRIα, Gr-1, and Ter-119. (C and D) The percentages of host-derived ILC2s, CD4+ and CD8+ T cells, and NK cells in kidney on day 35 after parabiosis surgery. Data shown are the mean±SEM (n=6 per group). *P<0.01 versus Parabiont. (E and F) C57BL/6 mice were treated with 0.3 μg mouse recombinant IL-33 or PBS daily for 5 consecutive days. The percentages of (E) BrdU+ ILC2s and (F) Ki-67+ ILC2s in total kidney ILC2s were analyzed in C57BL/6 mice with PBS or IL-33 treatment. Data shown are the mean±SEM (n=6 per group). ***P<0.001 versus C57BL/6.
IL-33 also acts on other immune cells, such as macrophages and regulatory T cells (Tregs).16,22 Macrophages are highly heterogeneous cells that can be divided into two major subsets: classically activated (M1) macrophages produce proinflammatory cytokines and cause tissue injury and inflammation, whereas alternatively activated macrophages (anti-inflammatory [M2]) are involved in immune regulation and wound healing.23,24 The number of F4/80+ macrophages in the kidney of mice treated with IL-33 did not change compared with those treated with PBS (Figure 2H). Kidney macrophages from IRI mice treated with IL-33 had significantly increased expression of M2 macrophage markers, including mannose receptor (MR), arginase, heme oxygenase-1 (HO-1), FIZZ1, and IL-10 (Figure 2I), but reduced expression of M1 macrophage markers, including iNOS, TNF-α, IL-1β, IL-6, and CCL2 (Figure 2J). Therefore, our data indicate that IL-33 did not reduce macrophage number in the kidney but promoted their polarization toward an M2 phenotype. IL-33 has been reported to expand a subset of ST2+Foxp3+ Tregs in vivo.22 We found that IL-33 treatment enhanced the number of Foxp3+ Tregs in spleen and kidney of IRI mice (Figure 2, K and L), which may be involved in IL-33–mediated renoprotection.
M2 Macrophages but Not Tregs Contributed to IL-33–Mediated Renoprotection in IRI Mice
Previous studies have shown that Tregs can limit tissue injury and contribute to recovery in IRI.25 IL-33–mediated cardiac allograft survival and acute graft-versus-host disease protection are dependent on Tregs.26,27 To test the role of Tregs in protection against AKI through delivery of IL-33, transgenic depletion of regulatory T cells (DEREG) mice were administered diphtheria toxin (DT) to selectively deplete Tregs during IL-33 administration (Figure 4A). Treg depletion in DEREG mice was confirmed in the spleen and kidney by flow cytometry (Figure 4, B and C). Treg depletion worsened renal histologic and functional injury in IRI mice, but Treg depletion did not affect IL-33–mediated renoprotection in IRI, indicating that Tregs are unlikely to be an important mechanism in IL-33–mediated protection against IRI (Figure 4, D–F). Macrophages have been shown to play a reparative role during the recovery phase of experimental IRI.28 Kidney F4/80+ macrophages were depleted by administration of a c-fms inhibitor (GW2580) in IRI mice (Figure 5, A–C). Kidney macrophage depletion reduced tubular injury and improved renal function in IRI mice without IL-33 treatment. However, the protective effect of IL-33 was only partially reversed in macrophage-depleted IRI mice, suggesting that M2 macrophage is not the key player in IL-33–mediated renoprotection in IRI mice (Figure 5, D–F).
Figure 4.

Tregs did not contribute to IL-33–mediated renoprotection in IRI mice. (A) DEREG C57BL/6 mice were treated with mouse recombinant IL-33 daily for 5 consecutive days as well as DT on days −5, −3, and −1 before bilateral IRI. Mice were euthanized 1 day after IRI. (B and C) Frequency of CD4+Foxp3+ Tregs in the CD4+ T cell compartment from the spleens and kidneys of control, IRI + Vehicle, IRI + DT, IRI + IL-33, or IRI + IL-33 + DT mice. (D) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice 1 day after IRI. Original magnification, ×200. (E and F) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, IRI + DT, IRI + IL-33, or IRI + IL-33 + DT mice. Data shown are the mean±SEM (n=6–8 per group). *P<0.05, ***P<0.001.
Figure 5.

M2 macrophages contributed to IL-33–mediated renoprotection in IRI mice. (A) C57BL/6 mice were treated with GW2580 and/or mouse recombinant IL-33 daily for 5 consecutive days before bilateral IRI. Mice were euthanized 1 day after IRI. (B and C) Frequency of F4/80+ macrophages in the total kidney cells and number of F4/80+ macrophages per kidney section (number per high-power field [hpf]) were assessed in control, IRI + Vehicle, IRI + GW2580, IRI + IL-33, or IRI + IL-33 + GW2580 mice. (D) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice 1 day after IRI. Original magnification, ×200. (E and F) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, IRI + GW2580, IRI + IL-33, or IRI + IL-33 + GW2580 mice. Data shown are the mean±SEM (n=6–8 per group). *P<0.05; **P<0.01, ***P<0.001.
ILC2s Played Critical Roles in IL-33–Mediated Renoprotection in IRI Mice
ILC2s play important roles in tissue homeostasis and repair.1 Recent studies have shown that ILC2s were critical for lung and intestine tissue protection after influenza virus or dextran sodium sulfate–induced damage.4,17 The potential contribution of ILC2 to IL-33–mediated protection of IRI was assessed in Rag1−/− mice, in which administration of anti-CD90 antibodies leads to depletion of ILC2 (Figure 6A). Flow cytometric analysis of CD45+Lin−GATA3+ ILC2 in kidney confirmed significant depletion of ILC2 in mice with anti-CD90 antibody treatment compared with IgG-treated control mice (Figure 6, B and C). Anti-CD90 antibody treatment also partially depleted NK cells in Rag1−/− mice. However, depletion of CD90+ cells in IRI Rag1−/− mice without IL-33 administration did not affect renal histologic and functional injury. Notably, the protective effect of IL-33 on tubular injury and renal function was significantly abolished in ILC2-depleted IRI mice, indicating a critical role for ILC2 in IL-33–mediated protection of kidney injury after IRI (Figure 6, D–F). Furthermore, ILC2s separated from C57BL/6 mice treated with IL-33 were expanded in culture with IL-2/IL-7/IL-33 for 12 days (Figure 6G). The cultured ILC2s maintained the expression of key markers, including ST2, GATA3, CD90, and CD25, and produced large amounts of Areg and IL-13 but low amounts of IL-4 (Figure 6, H and I). To directly test whether ILC2s were responsible for attenuation of tubular injury in IRI model, we adoptively transferred ex vivo–expanded ILC2d into IRI mice 1 day before IRI. Renal function and tubular cell damage were significantly improved in IRI mice treated with ILC2s compared with those of control IRI mice (Figure 6, J–L). These results further show that IL-33–expanded ILC2s play a major role in protecting against acute tubular injury in IRI mice.
Figure 6.

ILC2s played a key role in IL-33–mediated renoprotection in IRI mice. (A) Rag−/− mice were treated with mouse recombinant IL-33 daily for 5 consecutive days as well as anti-CD90 antibody or control rat IgG twice before bilateral IRI surgery. Mice were euthanized 1 day after IRI. (B and C) Percentage of GATA3+ ILC2s in the CD45+ leukocyte compartment from the kidneys of control, IRI + Vehicle, IRI + anti-CD90, IRI + IL-33 + rat IgG, or IRI + IL-33 + anti-CD90 mice. (D) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice 1 day after IRI. Original magnification, ×200. (E and F) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, IRI + anti-CD90, IRI + IL-33 + rat IgG, or IRI + IL-33 + anti-CD90 mice. Data shown are the mean±SEM (n=8 per group) from two independent experiments. ***P<0.001. (G–I) ILC2s were isolated from C57BL/6 mice treated with IL33 by flow sorting, and sort purified ILC2 cultured with IL-2, IL-7, and IL-33 for 12 days. The number of ILC2 was calculated on days 3, 6, and 12. Areg, IL-4, and IL-13 were measured in culture supernatant via ELISA. Data are representative of at least three independent experiments. (J–M) Rag−/− mice were treated with ex vivo–expanded ILC2s 1 day before IRI and GW2580 daily for 5 consecutive days before IRI. Mice were euthanized 1 day after IRI. (J) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice 1 day after IRI. Original magnification, ×200. (K and L) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, IRI + ILC2, or IRI + ILC2 + GW2580 mice. (M) The mRNA expression of MR, arginase, HO-1, FIZZ1, and IL-10 was quantified by quantitative PCR in F4/80+ kidney macrophages sorted from control, IRI + Vehicle, IRI + ILC2, or IRI + ILC2 + GW2580 mice. Data shown are the mean±SEM (n=8 per group) from two independent experiments. **P<0.01; ***P<0.001.
We then investigated the mechanism by which ILC2 protects mice against IRI. Fluorescently labeled ILC2s were observed in both sham and IRI kidneys, whereas the number of ILC2s in IRI kidney was significantly higher than that in sham kidney (Supplemental Figure 3). Adoptive transfer of ILC2s elevated the expression of M2 macrophage markers (MR, arginase, HO-1, FIZZ1, and IL-10) in endogenous kidney macrophages (Figure 6M). In vitro, bone marrow macrophages, when cocultured with ILC2, had increased expression of M2 macrophage markers, and the expression of these markers was further enhanced by the addition of IL-33 (Supplemental Figure 4). However, macrophage depletion by administration of GW2580 had no discernible effect on ILC2-mediated protection of IRI (Figure 6, K and L), indicating that ILC2-mediated protection of IRI is independent of M2 macrophages.
ILC2 Protects Against Renal IRI in an Areg-Dependent Manner
The expanded ILC2 in response to IL-2/7/33 stimulation produced greater amounts of Areg (Figure 6H), a ligand of the EGF receptor, which has been shown to ameliorate intestinal inflammation and tissue damage.17 To confirm the importance of Areg in ILC2-mediated protection of IRI, we deleted Areg in ILC2 using CRISPR-Cas9. ILC2s transfected with control empty vector produced a large amount of Areg in the supernatant, whereas ILC2s transfected with Areg CRISPR-Cas9 did not produce Areg (Figure 7A). The protective effect of ILC2 on renal function and tubular injury was significantly abolished in IRI mice treated with Areg-deleted ILC2 (Figure 7, B and C). Moreover, simulated ischemic injury of renal tubular epithelial cells (TECs) was induced by immersing the cellular monolayer in mineral oil; these cells were then cocultured with ILC2 transfected with Areg CRISPR-Cas9 or control vector. Apoptosis of ischemic TECs was significantly increased compared with that of control TECs, and it was reduced by coculture with ILC2, whereas coculture with Areg-deleted ILC2 resulted in an increase of apoptosis in ischemic TECs (Figure 7, D and E). These findings confirmed that Areg primarily from ILC2 played a significant role in ILC2-mediated renoprotection in IRI mice.
Figure 7.

ILC2s protected against renal IRI in an Areg-dependent manner. (A) Ex vivo–expanded ILC2s were transfected with control (type 2 innate lymphoid cell-control [ILC2-C]) or Areg CRISPR-Cas9 (type 2 innate lymphoid cell-amphiregulin [ILC2-Areg]). Areg was measured in culture supernatant of ILC2-C and ILC2-Areg via ELISA. (B and C) Transfected ILC2s were adoptively transferred into C57BL/6 1 day before IRI. Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, IRI + ILC2-C, or IRI + ILC2-Areg mice 1 day after IRI. Data shown are the mean±SEM (n=4–5 per group). **P<0.01; ***P<0.001. (D and E) Renal TEC ischemia was induced in vitro by immersing the cellular monolayer in mineral oil for 60 minutes at 37°C. Transfected ILC2s (ILC2-C or ILC2-Areg) were cocultured with ischemic renal tubular epithelial cells (IRI-TECs) for 1 day. TECs were exposed to serum-free K1 medium alone as the nonischemic control (control tubular epithelial cell [Ctrl-TEC]). (D) Representative FACS analysis of apoptosis in TECs after a 1-day coculture. (E) Frequency of early apoptosis (Annexin V+7AAD− cells) and late apoptosis (Annexin V+7AAD+ cells) in TECs after a 1-day coculture. Data shown are the mean±SEM (n=4 per group). **P<0.01; ***P<0.001.
IL-33 and ILC2 Were Protective Post-IRI
We investigated whether IL-33 and ILC2 can be used therapeutically. IL-33 and ILC2 were administrated beginning 24 hours after bilateral IRI surgery (Figure 8A). IL-33 or ILC2 treatment markedly improved the survival rate of mice with moderate IRI (30 minutes of bilateral renal pedicle clamping at 37°C); all of the IL-33– or ILC2-treated mice survived until 4 weeks after IRI when the experiment was terminated, whereas 50% of PBS-injected mice died by day 4 (Figure 8B). IL-33– or ILC2-treated mice were protected from IRI-induced renal dysfunction (Figure 8C) and tubular injury (Figure 8, D and E).
Figure 8.

IL-33 and ILC2 protected against IRI when administered post-IRI surgery. (A) C57BL/6 mice were treated with mouse recombinant IL-33 daily for 5 consecutive days or ILC2 twice beginning 24 hours after bilateral IRI surgery. Mice were euthanized on day 28 after IRI. (B) Survival rate of C57BL/6 mice subjected to IRI and injected with PBS, IL-33, or ILC2 (n=8 per group). **P<0.01 between IRI + Vehicle and IRI + IL-33/IRI + ILC2 mice as assessed by log rank test. (C) Serum creatinine levels were assessed in IRI + Vehicle (PBS), IRI + IL-33, and IRI + IL-33 at different time points after IRI. Data shown are the mean±SEM. *P<0.05 versus IRI + IL-33 and IRI + ILC2. (D) Representative periodic acid–Schiff-stained sections of kidney outer medulla from IRI mice treated with PBS, IL-33, or ILC2. Original magnification, ×200. (E) Tubular injury score was assessed in necropsy of moribund IRI mice or control, IRI + Vehicle, IRI + IL-33, and IRI + ILC2 mice on day 28 after IRI. Data shown are the mean±SEM. ***P<0.001.
Human IL-33 Reduced Renal IRI in Humanized Mice
Previous studies have described ILC2s in human lung, intestine, and skin.5,17 A recent study also identified ILC2s in healthy human kidney.18 We identified a Lin− population of cells in healthy human kidney that lacked expression of markers for T cells (CD3 and TCRαβ), B cells (CD19 and CD20), monocytes/macrophages (CD14/16 and CD11b), dendritic cells (CD11c and CD123), NK cells (CD56), or mast cells (FcεRIα) but that expressed CD127 and CRTH2. The Lin−CD127+CD161+CRTH2+ ILC population in the kidney parenchyma expressed ST2, KRLG1, and CD25 (Supplemental Figure 5), consistent with the cell surface phenotype of the ILC2 population found in other human organs. The demonstration of over 3000 ILC2s per gram of healthy human kidney suggests that ILC2 could have an important role in tissue homeostasis.
To investigate the possibility of IL-33–mediated protection of IRI in humans, we established immune system humanized NOD scid-γ (NSG) mice using human hematopoietic progenitor cells (HPCs). In this model, human immune cells were shown to be essential contributors to IRI (Supplemental Figure 6). After administration of human recombinant IL-33, tubular cell injury and serum creatinine were reduced significantly compared with those of humanized IRI mice treated with PBS (Figure 9, A–D). In humanized mice, CD45+Lin–CD127+CD161+CRTH2+ human ILC2s were found in the kidney of all mice at 8–12 weeks after reconstitution of HPCs (Figure 9E). The human ILC2s showed expression of key surface markers ST2, KRLG1, and CD25 (Figure 9F). Notably, human IL-33 treatment significantly increased the number of human ILC2s in kidney of humanized mice (Figure 9, G and H). In addition, CD14+ human monocytes/macrophages isolated from kidney of humanized IRI mice with IL-33 treatment had enhanced expression of M2 macrophage markers, including MR, CCL18, and IL-10 (Figure 9I), and reduced expression of M1 macrophage markers, including TNF-α, IL-1β, and CCL2 (Figure 9J). Furthermore, a method for human ILC2 expansion in vitro was established. CD45+Lin–CD127+CRTH2+ human ILC2s sorted from PBMCs proliferated markedly in culture with IL-2/IL-7/IL-33 for 12 days (Figure 9K). The cultured human ILC2s maintained the expression of key markers, including CD161, CRTH2, ST2, KRLG1, and CD25 (data not shown), and produced large amounts of Areg and IL-13 (Figure 9, L and M). The in vivo function of these expanded human ILC2s was examined by their adoptive transfer into humanized mice with IRI. The transfused human ILC2s were observed in both sham and IRI kidneys, but there were greater numbers of ILC2s in IRI kidney than sham kidney (Supplemental Figure 7). ILC2 treatment significantly improved renal function and reduced tubular cell injury in humanized IRI mice (Figure 9, N–P), indicating that ex vivo–expanded human ILC2 can effectively prevent IRI.
Figure 9.

Human IL-33 reduced renal IRI in humanized NOD scid-γ (h-NSG) mice. (A) h-NSG mice were treated with human recombinant IL-33 daily for 5 consecutive days before bilateral IRI surgery. Mice were euthanized 1 day after IRI. (B) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice day 1 after IRI. Original magnification, ×200. (C and D) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, or IRI + IL-33 mice. (E) Representative FACS analysis showing the gating strategy to identify human CD45+Lin−CD127+CD161+CRTH2+ ILC2s in the kidneys of h-NSG mice. Lin mixture includes CD3, TCRαβ, CD19, CD20, CD14, CD16, CD11b, CD11c, CD123, CD56, and FcεRIα. (F) Histogram showing expression of ST2, KLRG1, and CD25 on human kidney ILC2s. Specific markers (red lines) and isotype controls (gray-filled areas) are shown. (G and H) Percentage of CD161+CRTH2+ ILC2s in the CD45+LIN−CD127+ cell compartment from the kidneys of control, IRI + Vehicle, or IRI + IL-33 mice. (I and J) CD14+ human monocytes/macrophages were sorted from kidneys of control, IRI + Vehicle, and IRI + IL-33 mice on day 1 after IRI. The mRNA expression of MR, CCL18, IL-10, TNF-α, IL-1β, and CCL2 was quantified by quantitative PCR in kidney CD14+ human monocytes/macrophages. Data shown are the mean±SEM (n=6 per group) from two independent experiments. **P<0.01 versus IRI + vehicle; ***P<0.001 versus IRI + vehicle. (K–M) Human ILC2s were isolated from PBMCs and cultured with IL-2, IL-7, and IL-33 for 12 days. The number of ILC2s was calculated on days 3, 6, and 12. Areg, IL-4, and IL-13 were measured in culture supernatant via ELISA. Data are representative of at least three independent experiments. (N–P) h-NSG mice were treated with ex vivo–expanded human ILC2s 1 day before IRI. Mice were euthanized 1 day after IRI. (N) Representative periodic acid–Schiff-stained sections of kidney outer medulla from mice 1 day after IRI. Original magnification, ×200. (O and P) Serum creatinine levels and tubular injury score were assessed in control, IRI + Vehicle, or IRI + ILC2 mice. Data shown are the mean±SEM (n=6 per group) from two independent experiments. #P<0.01 versus IRI + vehicle.
Discussion
We found that IL-33 significantly prevented renal structural and functional injury and reduced mortality in mice with IRI. The mechanisms underlying the protective effect of IL-33 are associated with its initiation of Th2 immune responses through expansion of ILC2s, M2 macrophages, and Tregs. On the basis of our data, ILC2s seem to be an important pathway in IL-33–mediated renoprotection in this model. Furthermore, we have shown a homologous population of ILCs in human kidney and IL-33–mediated renoprotection in humanized mice, suggesting that the beneficial effect of IL-33 in murine IRI can be extended to human IRI.
An important finding in this study is that IL-33–elicited ILC2s play a critical role in protection against IRI, which is shown here for the first time. We have previously reported that IL-25 induced type 2 immune responses, including expansion of Th2 cells, M2 macrophages, and ILC2s, which may underlie IL-25’s protective role in IRI and Adriamycin nephropathy.29,30 However, whether expansion of ILC2 is responsible for IL-25–mediated renoprotection was not addressed clearly. In this study, we showed that depletion of ILC2 significantly eliminated the therapeutic effect of IL-33 in Rag1−/− mice, and transfused ILC2s significantly attenuated IRI, clearly showing that ILC2 is an important pathway in IL-33–mediated renoprotection in this model. Depletion of ILC2 using anti-CD90 antibody did not completely reverse IL-33–mediated protection, indicating that other cells and mechanisms may contribute to IL-33–mediated protection in IRI. Recently, Riedel et al.18 also showed that ILC2s are responsible for IL-33–mediated renoprotection in the Adriamycin nephropathy model by using Rag2−/−Il2rg−/− mice that lack ILCs. However, in another earlier report, high-dose IL-33 (1 mg twice a day for 3 days) exacerbated renal injury in cisplatin-induced AKI.31 These contradictory results could be explained by difference in the kidney disease models or the dose and duration of IL-33 treatment. More recently, IL-33 treatment post-IRI procedure showed both a protective and a harmful role in two studies, contrasting results that could be explained by the timing of treatment. Short-term application of IL-33 at early stage of disease was protective, whereas long-term application of IL-33 worsened renal fibrosis.32,33 We also found that IL-33 and ILC2 are protective post-IRI. The possible mechanisms underlying the protective role of ILC2 include at least three possibilities. First, IL-33–responsive ILC2 produced greater amounts of Th2 cytokines, which suppress the proinflammatory activity of type 1 immune responses. This is supported by our data showing that proinflammatory factors TNF-α, IL-1β, IL-6, CXCL1, and CXCL2 were reduced in kidney of IL-33–treated mice. Second, ILC2s directly modulate macrophage phenotype via generating IL-13, thereby contributing to protection of renal injury in IRI mice. Here, we observed that IL-33 did not promote induction of M2 macrophage directly but enhanced M2 macrophage polarization in the presence of ILC2 in vitro. Third, ILC2s directly promote tubule repair through producing Areg in IRI, which is consistent with an earlier report showing that transfer of ILC2 ameliorated intestinal disease severity in an Areg-dependent manner.17 However, other mechanisms may be used by ILC2s to mediate renoprotection. A recent study showed that ILCs are tissue-resident cells that are maintained locally by self-renewal under physiologic conditions in lung, liver, and intestine,34 but whether ILC2 can be expanded locally in response to IL-33 stimulation is unknown. Here, we provide data supporting that the majority of kidney ILCs are tissue-resident cells that are maintained in steady state and strongly expanded locally with partial replenishment from circulating ILCs in IL-33–treated mice.
IL-33–induced Th2 immune response in both the periphery and within the kidneys of IRI mice is characterized by the overproduction of IL-4 and IL-13. Previous studies support the hypothesis that enhancing Th2 immunity with exogenous IL-4 or IL-13 reduces renal inflammation and injury in experimental immune-mediated GN.35–38 The mechanisms underlying the renoprotective effects of IL-33 could involve its initiation of Th2 immune responses. Moreover, M2 macrophages were significantly increased in kidney of IL-33–treated IRI mice. M2 macrophages exhibit potent anti-inflammatory properties and play important roles in immune regulation, tissue repair, and remodeling.24,29,39–41 Macrophages modulated ex vivo to an anti-inflammatory phenotype have been used successfully as a cell-based therapy in IRI.42 M2 macrophages are able to produce anti-inflammatory cytokines, which suppress inflammation and tissue injury. M2 macrophages also express HO-1, an anti-inflammatory enzyme that has been shown to be beneficial in renal and liver IRI.42–44 Here, we found that kidney macrophage expressed higher levels of HO-1 in IL-33–treated mice. Depletion of kidney macrophages by administration of c-fms inhibitor partially abolished the IL-33–mediated protection of IRI, indicating the M2 macrophages contribute to the renoprotective effects of IL-33 in IRI. IL-33 can directly stimulate Treg proliferation and activation.22 Recent reports have highlighted that Tregs contribute to the protective effect of IL-33 in models of cardiac allograft rejection and acute graft-versus-host disease.26,27 However, we found that depletion of Tregs using DT did not eliminate protection of IRI by IL-33, suggesting that Tregs are not responsible for IL-33’s protective effect in IRI. This is also supported by our data showing that IL-33 markedly attenuated renal injury in lymphocyte-deficient mice (Rag1−/−) with IRI.
To investigate the relevance of these findings in experimental models to human disease, we identified ILC2s in human kidney and found that their phenotypes are consistent with the ILC2 population found in human organs, suggesting that ILC2 may be a potential therapeutic target for kidney disease in humans. Next, we revealed that human ILC2s can develop normally in humanized mice, providing a valuable platform for studying the interaction of IL-33 with the human immune system in vivo. Then, we showed that human IL-33 effectively expanded human ILC2 in kidney of humanized mice and significantly reduced renal IRI. A previous study has shown that IL-33 protected against cardiac allograft rejection through Treg induction.26 Combining this with our observations, we propose that IL-33 could be an attractive future therapeutic approach for organ transplantation via both suppression of allograft rejection and protection against IRI. Moreover, we reported a set of culture conditions that allows the expansion of ILC2 isolated from human PBMC, providing feasibility for clinical application of ILC2. Ex vivo–expanded human ILC2 significantly reduced tubular injury in humanized IRI mice, indicating that ILC2 could be an effective therapeutic approach for IRI.
Over several decades, immunotherapy has focused predominantly on the adaptive immune system, especially dendritic cell mediation of T cell immunity.45,46 Targeting innate immune cells for immunotherapy has emerged in recent years,47 because the innate immune system is a key player in initiating and regulating adaptive immune responses, such as those elicited against pathogens and cancer.48 The concept of maneuvering innate immunity to control adaptive immune responses has been considered as a novel immunotherapeutic strategy.2,3 In this study, we explore the strategies of promoting ILC2 expansion or transferring ILC2 to reduce immune-mediated renal injury in IRI. The results show that promoting the innate immune axis of IL-33–ILC2 has strong efficacy in limiting kidney IRI, showing a novel strategy of manipulating ILC2 to treat disease.
As a cytokine, IL-33 has side effects, especially in patients with history of chronic allergic inflammatory disorders, which may impede its application in clinical settings. IL-33 induces Th2 immune responses, which are characterized by high levels of IL-4, IL-5, IL-9, and IL-13. Th2 cytokines along with activated eosinophils and mast cells are key players in the development of chronic allergic inflammatory disorders, such as asthma.49,50 However, use of IL-33 selectively in patients with renal disease who have no history of allergy may avoid this side effect. The other concern regarding IL-33 is that its long-term use could cause organ fibrosis.11,12 To prevent development of fibrosis, IL-33 should be applied selectively and for short periods in acute diseases, such as IRI, rather than as a long-term treatment in chronic diseases. Our data showed that short-term IL-33 treatment actually reduced renal fibrosis in IRI. Although we have shown that human IL-33 mediated ILC2 expansion and reduced kidney injury in a humanized mouse model, studies in primates are more likely to indicate efficacy and identify possible side effects of IL-33 in humans.
In conclusion, this study has uncovered a strong protective effect of IL-33 in renal IRI. The renoprotective effect of IL-33 depends on its ability to drive ILC2 expansion in kidney. Furthermore, it seems that the beneficial effect of enhancing the IL-33–ILC2 pathway can be extended to human IRI, because a homologous population of cells was identified in human kidney and human IL-33 effectively reduced kidney injury in a humanized mouse model of IRI. Transfer of ex vivo–expanded human ILC2 significantly reduced kidney injury in humanized mice with IRI. This study suggests a novel therapeutic approach of promoting the innate immune axis of IL-33–ILC2 for human IRI in kidney and other organs.
Concise Methods
Mice
C57BL/6 (CD45.2+), congenic C57BL/6 (CD45.1+), Rag1−/−, and NSG mice were purchased from the Animal Resources Centre (Perth, Australia) or Australian BioResources (Sydney, Australia). NSG mice and DEREG mice were bred at the Department of Animal Care at Xinxiang Medical University (XMU). For all studies, adult (8–10 weeks of age) male mice were used in accordance with the animal care and use protocol approved by the Animal Ethics Committee of Western Sydney Local Health District (WSLHD) or XMU.
IRI Murine Model and IL-33 Administration
IRI induction was performed as previously described. Briefly, bilateral renal ischemia was induced via a midline abdominal excision; both renal pedicles were clamped for 30 minutes (C57BL/6) or 35 minutes (Rag1−/−) at 37°C using a nontraumatic sterile clamp (Roboz). After the ischemic period, clamps were released to induce reperfusion. After the procedure, mice were given 1 ml of warm normal saline intraperitoneally to prevent dehydration. Animals subjected to sham operation were used as controls.
For IL-33 treatment, C57BL/6 (male, 8–10 weeks of age) mice were administered 0.3 μg mouse recombinant IL-33 (Biolegend) intraperitoneally daily for 5 consecutive days before or after IRI surgery. The dose and duration were selected according to previously published studies.6,16,17,22 Control animals received PBS only. To deplete Tregs, DEREG mice (male, 8–10 weeks of age) were injected intraperitoneally with DT (30 mg/kg; Sigma-Aldrich) on days −5, −3, and −1 before IRI surgery. Tregs were examined in spleen and kidney by flow cytometry. Monocyte/macrophage depletion was induced by administration of GW2580 (an inhibitor of c-fms; Biovision) once daily at a dose of 160 mg/kg by oral gavage for 5 consecutive days before IRI surgery.51 For ILC2 depletion, Rag1−/− mice (male, 8–10 weeks of age) were given anti-CD90.2 antibody (30-H12; 250 μg per mouse; Biolegend) or control rat IgG2b on days −5 and −2 before IRI surgery. Mice were euthanized on day 1 or day 28 after IRI surgery. Blood, spleen, kidney-draining lymph nodes, and kidneys were harvested for analysis. Blood creatinine levels were measured using a Hitachi 747 automatic analyzer.
Murine ILC2 Cells Expansion and Adoptive Transfer to IRI Mice
ILC2s were isolated from kidneys of C57BL/6 mice treated with IL-33 daily for 5 consecutive days. ILC2s were cultured in RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) plus IL-2 (10 ng/ml), IL-7 (10 ng/ml), and IL-33 (50 ng/ml) for 12 days. Cellfree supernatants were assessed for Areg, IL-4, and IL-13 cytokine production by ELISA.
Rag1−/− mice (male, 8–10 weeks of age) were divided into four groups—control, IRI with vehicle, IRI with ILC2 treatment, and IRI with ILC2 and GW2580 treatment. Monocyte/macrophage depletion was induced by administration of GW2580 (160 mg/kg, oral gavage, daily) for 5 consecutive days before bilateral IRI surgery. For ILC2 treatment, 1×106 ILC2s were transferred into Rag1−/− mice by a single tail vein injection 1 day before bilateral IRI surgery. In parallel, CFSE-labeled ILC2s (1×106) were transfused into Rag1−/− mice 1 day before unilateral IRI surgery. All mice were euthanized on day 1 after IRI surgery. The distribution of CFSE-labeled ILC2s was analyzed in kidney sections by fluorescence microscopy. The number of transfused ILC2s was quantitated in eight to ten nonoverlapping high-power fields (×400).
Human Tissue
Fresh kidney tissue was taken from ten patients undergoing nephrectomy because of trauma or renal tumor. All tissues were obtained with informed patient consent and in accordance with local ethics approval from the Human Research Ethics Committee of XMU or the WSLHD. Kidney tissue was enzymatically digested with 1 mg/ml collagenase IV (Sigma-Aldrich) and 100 μg/ml DNase I (Roche) for 40 minutes at 37°C with intermittent agitation. The digested cell suspension was then passed through 40-μm cell strainer for subsequent assays.
IRI Model in Humanized Mice and IL-33 Administration
Umbilical cord blood was collected from mothers attending the Third Affiliated Hospital of XMU after informed consent through a protocol approved by the Human Research Ethics Committee of XMU. Mononuclear cells were obtained by Ficoll density centrifugation and ammonium chloride red cell lysis. The Lin−CD34+ HPCs were isolated using a CD34+ isolation kit (Miltenyi Biotec). NSG mice were housed in microisolators and fed sterile food and water. Mice aged 8–10 weeks of age were sublethally irradiated (3.75 Gy) up to 24 hours before intravenous injection of 1×105 Lin–CD34+ human HPCs. Mice were analyzed 8–12 weeks after reconstitution. More than 95% of the humanized NSG mice were stably reconstituted with human leukocytes in the blood (60%–90% at 10–12 weeks). Each cohort had similar levels of engraftment.
IRI induction was performed as described above. Bilateral renal ischemia was induced via a midline abdominal excision; both renal pedicles were clamped for 35 minutes at 37°C. Animals subjected to sham operation were used as controls. For IL-33 treatment, humanized NSG mice were administered 0.3 μg human recombinant IL-33 (Biolegend) intraperitoneally daily for 5 consecutive days before IRI surgery. Control animals received PBS only. Humanized NSG mice were euthanized on day 1 after IRI surgery. Blood, spleen, and kidneys were harvested for analysis. Blood creatinine levels were measured using a Hitachi 747 automatic analyzer.
Human ILC2s Expansion and Adoptive Transfer to Humanized IRI Mice
Human ILC2s were isolated from PBMCs of mothers attending the Third Affiliated Hospital of XMU. Human ILC2s were cultured in RPMI 1640 medium containing 100 U/ml penicillin-streptomycin supplemented with 10% human AB serum plus IL-2 (20 ng/ml), IL-7 (20 ng/ml), and IL-33 (50 ng/ml) for 12 days. Cellfree supernatants were assessed for IL-4 and IL-13 cytokine production by ELISA (eBioscience). For ILC2 treatment, 1×106 human ILC2s were transferred into humanized NSG mice by a single tail vein injection 1 day before bilateral IRI surgery. In parallel, CFSE-labeled human ILC2s (1×106) were transfused into humanized NSG mice 1 day before unilateral IRI surgery. All mice were euthanized on day 1 after IRI surgery. The distribution of CFSE-labeled ILC2 was analyzed in kidney sections by fluorescence microscope. The number of transfused ILC2 was quantitated in eight to ten nonoverlapping high-power fields (×400).
Cell Suspensions Preparation
Spleen and KLDN were isolated, minced, and digested for 30 minutes at 37°C in RPMI 1640 containing 1 mg/ml collagenase D (Roche) and 100 μg/ml DNase I (Roche). The digested cell suspension was then passed through a 40-μm cell strainer. Kidney, liver, and lung were perfused with saline before removal and digested with collagenase and DNase as previously described.30 Kidney, liver, and lung were cut into 1- to 2-mm3 pieces and placed in DMEM containing 1 mg/ml collagenase IV (Sigma-Aldrich) and 100 μg/ml DNase I (Roche) for 40 minutes at 37°C with intermittent agitation. The digested cell suspension was then passed through a 40-μm cell strainer. The cell suspensions were allowed to settle for 10 minutes, after which the upper three fourths (lower-density cells) were removed for subsequent assays. F4/80+ macrophages were sorted from kidney by FACS. Sorted macrophages were used for real-time PCR analyses to detect macrophage phenotypes.
Flow Cytometry and Cell Sorting
For FACS analysis of mouse cells, single-cell suspensions were stained with Fc block/anti-CD16/32 (2.4G2) and antibodies to CD45.2 (104), ST2 (RMST2–2), CD127 (A7R34), GATA3 (TWAJ), KLRG1 (2F1), IL-17RB (9B10), CD90.2 (30-H12), and CD25 (PC61) as well as antibodies to T cell, B cell, NK cell, monocyte/macrophage, dendritic cell, eosinophil, neutrophil, and erythroid cell lineages: CD3 (145–2C11), CD5 (53–7.3), TCRβ (H57–597), TCRγδ (eBioGL3), CD19 (1D3), B220 (RA3–6B2), CD49b (DX5), CD11b (M1/70), CD11c (N418), FcεRIα (MAR-1), Gr-1 (RB6–8C5), and Ter-119 (all purchased from eBioscience or Biolegend). Cells were analyzed on an LSR Fortessa flow cytometer (BD Biosciences). For FACS sorting, single-cell suspensions were pregated on hematopoietic cells using anti-CD45.2 antibody; then, lineage markers were used to exclude immune cells, and 4′,6-diamidino-2-phenylindole was used to exclude dead cells. Mouse ILC2s (CD45+Lin−CD127+ST2+CD90+) were sorted using a FACSAria II (BD). After sorting, cells were used for phenotypic and functional assays.
For FACS analysis of human cells, single-cell suspensions were stained with antibodies to CD45 (HI30), CD127 (A019D5), CRTH2 (BM16), GATA3 (TWAJ), CD161 (HP-3G10), KLRG1 (13F12F2), ST2 (HB12), CD117 (104D2), and CD25 (M-A251) as well as antibodies to T cell, B cell, NK cell, monocyte/macrophage, dendritic cell, eosinophil, neutrophil, and erythroid cell lineages: CD3 (UCHT1), TCRαβ (IP26), CD19 (HIB19), CD20 (2H7), CD14 (M5E2), CD16 (3G8), CD11b (ICRF44), CD11c (BU15), CD123 (6H6), CD56 (HCD56), and FcεRIα (AER-37; all purchased from Biolegend or BD Biosciences). Cells were analyzed on an LSR Fortessa flow cytometer (BD Biosciences). For FACS sorting, single-cell suspensions were pregated on hematopoietic cells using anti-CD45 antibody; then, lineage markers were used to exclude immune cells, and 4′,6-diamidino-2-phenylindole was used to exclude dead cells. Human ILC2s (CD45+Lin−CD127+CRTH2+CD161+) were sorted using a FACSAria II (BD). After sorting, cells were used for phenotypic and functional assays.
5-Bromo-2′-Deoxyuridine Incorporation
For 5-bromo-2′-deoxyuridine (BrdU) incorporation, mice were given 50 mg/kg BrdU (Sigma-Aldrich) intraperitoneally after administration of IL-33 for 5 consecutive days. The kidney tissues were harvested at 4 hours after injection and enzymatically digested with collagenase. Cell suspensions after ILC2 staining were incubated with Foxp3 Fixation/Permeabilization solution (eBioscience) and then incubated with anti-BrdU or anti-mouse Ki67 (BioLegend). Cells were analyzed on an LSR Fortessa flow cytometer (BD Biosciences).
Parabiosis
Parabiotic mice were generated as reported34,52 using 6- to 8-week-old weight-matched CD45.1 and CD45.2 female mice and maintained for 30 days postsurgery. Briefly, matching skin incisions were made from elbow to knee in each mouse, and the subcutaneous fascia was bluntly dissected to create about 0.5 cm of free skin. Forelimb and hind limb were tied together using 2–0 silk suture, and skin incisions were closed using wound clips. Postoperatively, mice received analgesia and antibiotics for 7 days and then as needed for the duration of the experiment. Parabiotic mice were administered 0.5 μg mouse recombinant IL-33 (Biolegend) intraperitoneally daily for 5 consecutive days 30 days after parabiotic surgery. The procedure was approved by the Animal Ethics Committee at XMU.
ILC2 Coculture with Macrophages
ILC2s were isolated from C57BL/6 mice treated with IL-33. ILC2s were expanded as described above. Primary cultures of murine macrophages were obtained from bone marrow of C57BL/6 mice by a technique previously described. Macrophages derived from bone marrow were cultured in RPMI 1640 medium and supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) plus 10 ng/ml M-CSF for 6 days, and F4/80+ macrophages were sorted by flow cytometry. Cell viability was >95%. Bone marrow macrophages were seeded onto 12-well culture plates (2×105 cells per well), preincubated with or without mouse recombinant IL-33 (20 ng/ml) for 1 hour, and then, cocultured with ILC2 (2×104 cells per well) for 6 hours. Macrophage phenotype was examined by quantitative PCR.
CRISPR-Cas9 Transfection
Ex vivo–expanded ILC2s were transfected with AREG CRISPR-Cas9 plasmid or its control (sc-419185; Santa Cruz) in accordance with the manufacturer’s instructions. Transfected cells were cultured for 7 days before transfer to recipient mice as indicated in Figure 7.
Primary Culture of Mouse Renal TECs
Primary mouse TECs were generated following established methods adapted from Doctor et al.53 In brief, kidneys were harvested after cardiac perfusion with saline to remove blood cells. The tissue from the outer cortex was cut into pieces of approximately 1 mm3 and then digested in DMEM containing 1 mg/ml collagenase IV (Sigma-Aldrich) and 100 μg/ml DNase I (Roche) for 40 minutes at 37°C with intermittent agitation. Renal TECs were separated by centrifugation using Percoll solution and cultured in defined K1 medium: DMEM/F12 medium supplemented with 10 ng/ml EGF, 1 ng/ml PGE1, 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 5×10−11 M tri-iodothyronine, 5×10−8 M hydrocortisone, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES, and 5% FBS.
Simulated Ischemic Renal TECs Coculture with ILC2
Ischemia in renal TECs was simulated by immersing the cellular monolayer in mineral oil according to the protocol of Meldrum et al.54 This immersion simulated ischemia by restricting cellular exposure to oxygen and nutrients as well as by limiting metabolite washout. Briefly, renal TECs (5×104) were placed in 12-well tissue culture plates in serum-free K1 medium for 24 hours, washed twice with PBS, and immersed in mineral oil for 60 minutes at 37°C. After washing with PBS, TECs were incubated in K1 medium. Transfected ILC2s (2×105) were cocultured with ischemic renal TECs for 1 day. TECs were exposed to serum-free K1 medium alone as the nonischemic control. Apoptosis of TECs at 1 day after the coculture was measured by staining with 7-AAD and Annexin V following the manufacturer’s protocol (BD Biosciences).
ELISA of Cytokines
IL-4 and IL-13 levels in sera and culture supernatants were assayed using an ELISA kit (eBioscience). Areg levels in culture supernatants were analyzed using an ELISA kit (R&D Systems). ELISA was performed according to the manufacturer’s protocol.
Quantitative PCR
Total RNA was isolated from kidney or endogenous renal macrophages by the RNeasy Mini Kit (Qiagen) and then reverse transcribed with First Strand cDNA Synthesis Kit (Fermantas). Real-time PCR was performed on the Rotogene-6000 Real-Time Thermo cycler (Corbett Research) using the SYBR mastermix (Invitrogen). The analysis method was as described before, and the PCR primer sequences are presented in Supplemental Table 1.
Histology and Immunofluorescence
Coronal sections of kidney tissue were stained with periodic acid–Schiff. Tubular injury score in the corticomedullary junction and outer medulla were evaluated semiquantitatively.28,55 Briefly, tubular damage was estimated in 8–12 high-power fields per section by using a scoring system on the basis of the percentage of damaged tubules per field (0: normal; 1: <10%; 2: 11%–25%; 3: 26%–50%; 4: 51%–75%; and 5: >75%). The mean score of each animal was compared. To avoid selection bias, the areas to be viewed for morphometric analysis were anatomically identical for each section and positioned before microscopic visualization.
For immunofluorescence staining, rat anti-mouse Gr-1 (Ly-6G, 1/100; Biolegend) or F4/80 (1/200; eBiosciences) or mouse anti-mouse collagen I (1/1000; Abcam) or α-smooth muscle actin (1/500; Sigma-Aldrich) was used as the primary antibody, and AF488 goat anti-rat IgG (1/1000; Invitrogen) and AF546 goat anti-mouse IgG (1/1000; Invitrogen) were used as the secondary antibodies. Control rat or mouse IgG to primary antibodies was included in staining. The number of interstitial Gr-1– and F4/80-positive cells was quantitated in eight to ten nonoverlapping fields in the corticomedullary junction and outer medulla (×400).
Statistical Analyses
Renal functional data were log transformed before analysis to stabilize the variance. Statistical tests included unpaired, two-tailed t test using Welch correction for unequal variances and one-way ANOVA with Tukey multiple comparison test. Analysis of Kaplan–Meier survival curves was performed using a log rank test with a Gehan–Breslow–Wilcoxon post-test. Statistical analyses were performed using Prism (Version 6; GraphPad). Results are expressed as the mean±SEM. A P<0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank the Western Sydney Local Health District and Xinxiang Medical University animal facilities for their conscientious care of mice.
This work was supported by National Health and Medical Research Council of Australia grants 1061785 (to Y.W. and D.C.H.H.) and 1061848 (to Y.W. and D.C.H.H.) and National Natural Science Foundation of China grants 81200506 and 81570624.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017070774/-/DCSupplemental.
References
- 1.Sonnenberg GF, Artis D: Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21: 698–708, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eberl G, Colonna M, Di Santo JP, McKenzie AN: Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 348: aaa6566, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eberl G, Di Santo JP, Vivier E: The brave new world of innate lymphoid cells. Nat Immunol 16: 1–5, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D: Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12: 1045–1054, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, Artis D, Volk SW: IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Invest Dermatol 136: 487–496, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA: IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23: 479–490, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Liew FY, Pitman NI, McInnes IB: Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat Rev Immunol 10: 103–110, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Mirchandani AS, Salmond RJ, Liew FY: Interleukin-33 and the function of innate lymphoid cells. Trends Immunol 33: 389–396, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Molofsky AB, Savage AK, Locksley RM: Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 42: 1005–1019, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, Sudo K, Okumura K, Saito H, Nakae S: IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 107: 18581–18586, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, Voehringer D, McKenzie AN, Neurath MF, Pflanz S, Wirtz S: Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39: 357–371, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, Graham GJ, Kurowska-Stolarska M, Liew FY, McSharry C, Xu D: IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 134: 1422–1432.e11, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN: Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464: 1367–1370, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, Baker AH, McInnes IB, Liew FY: IL-33 reduces the development of atherosclerosis. J Exp Med 205: 339–346, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P, Artis D: Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519: 242–246, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, Couper KN, Ryffel B, Liew FY: IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog 11: e1004607, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D: IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 112: 10762–10767, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riedel JH, Becker M, Kopp K, Düster M, Brix SR, Meyer-Schwesinger C, Kluth LA, Gnirck AC, Attar M, Krohn S, Fehse B, Stahl RAK, Panzer U, Turner JE: IL-33-mediated expansion of type 2 innate lymphoid cells protects from progressive glomerulosclerosis. J Am Soc Nephrol 28: 2068–2080, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eltzschig HK, Eckle T: Ischemia and reperfusion--from mechanism to translation. Nat Med 17: 1391–1401, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonventre JV, Yang L: Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S: Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463: 540–544, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BMJ, Löhning M, Belkaid Y, Fallon PG, Powrie F: The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513: 564–568, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon S: Alternative activation of macrophages. Nat Rev Immunol 3: 23–35, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Murray PJ, Wynn TA: Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, Crow MT, King LS, Rabb H: Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int 76: 717–729, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, Wang Z, Lang M, Stolz DB, Zheng XX, Demetris AJ, Liew FY, Wood KJ, Thomson AW: IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol 187: 4598–4610, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matta BM, Reichenbach DK, Zhang X, Mathews L, Koehn BH, Dwyer GK, Lott JM, Uhl FM, Pfeifer D, Feser CJ, Smith MJ, Liu Q, Zeiser R, Blazar BR, Turnquist HR: Peri-alloHCT IL-33 administration expands recipient T-regulatory cells that protect mice against acute GVHD. Blood 128: 427–439, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Q, Wang C, Zheng D, Wang Y, Lee VW, Wang YM, Zheng G, Tan TK, Yu D, Alexander SI, Harris DC, Wang Y: IL-25 induces M2 macrophages and reduces renal injury in proteinuric kidney disease. J Am Soc Nephrol 22: 1229–1239, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Q, Niu Z, Tan J, Yang J, Liu Y, Ma H, Lee VW, Sun S, Song X, Guo M, Wang Y, Cao Q: IL-25 elicits innate lymphoid cells and multipotent progenitor type 2 cells that reduce renal ischemic/reperfusion injury. J Am Soc Nephrol 26: 2199–2211, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akcay A, Nguyen Q, He Z, Turkmen K, Won Lee D, Hernando AA, Altmann C, Toker A, Pacic A, Ljubanovic DG, Jani A, Faubel S, Edelstein CL: IL-33 exacerbates acute kidney injury. J Am Soc Nephrol 22: 2057–2067, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stremska ME, Jose S, Sabapathy V, Huang L, Bajwa A, Kinsey GR, Sharma PR, Mohammad S, Rosin DL, Okusa MD, Sharma R: IL233, A novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J Am Soc Nephrol 28: 2681–2693, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang H, Xu F, Wen XJ, Liu HZ, Wang HB, Zhong JY, Yang CX, Zhang B: Interleukin-33 signaling contributes to renal fibrosis following ischemia reperfusion. Eur J Pharmacol 812: 18–27, 2017 [DOI] [PubMed] [Google Scholar]
- 34.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY: Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350: 981–985, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tam FW, Smith J, Karkar AM, Pusey CD, Rees AJ: Interleukin-4 ameliorates experimental glomerulonephritis and up-regulates glomerular gene expression of IL-1 decoy receptor. Kidney Int 52: 1224–1231, 1997 [DOI] [PubMed] [Google Scholar]
- 36.Tipping PG, Kitching AR, Huang XR, Mutch DA, Holdsworth SR: Immune modulation with interleukin-4 and interleukin-10 prevents crescent formation and glomerular injury in experimental glomerulonephritis. Eur J Immunol 27: 530–537, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Kitching AR, Turner AL, Wilson GR, Edgtton KL, Tipping PG, Holdsworth SR: Endogenous IL-13 limits humoral responses and injury in experimental glomerulonephritis but does not regulate Th1 cell-mediated crescentic glomerulonephritis. J Am Soc Nephrol 15: 2373–2382, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Sandovici M, Henning RH, van Goor H, Helfrich W, de Zeeuw D, Deelman LE: Systemic gene therapy with interleukin-13 attenuates renal ischemia-reperfusion injury. Kidney Int 73: 1364–1373, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Yang Z, Grinchuk V, Urban JF Jr., Bohl J, Sun R, Notari L, Yan S, Ramalingam T, Keegan AD, Wynn TA, Shea-Donohue T, Zhao A: Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of type 2 immunity. PLoS One 8: e59441, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wynn TA, Vannella KM: Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44: 450–462, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VW, Zheng G, Tan TK, Ince J, Alexander SI, Harris DC: IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol 21: 933–942, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J, Kluth DC: Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol Ther 18: 1706–1713, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, Wigmore SJ: Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther 17: 65–72, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferenbach DA, Nkejabega NC, McKay J, Choudhary AK, Vernon MA, Beesley MF, Clay S, Conway BC, Marson LP, Kluth DC, Hughes J: The induction of macrophage hemeoxygenase-1 is protective during acute kidney injury in aging mice. Kidney Int 79: 966–976, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Sabado RL, Balan S, Bhardwaj N: Dendritic cell-based immunotherapy. Cell Res 27: 74–95, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newick K, O’Brien S, Moon E, Albelda SM: CAR T cell therapy for solid tumors. Annu Rev Med 68: 139–152, 2017 [DOI] [PubMed] [Google Scholar]
- 47.Curran E, Corrales L, Kline J: Targeting the innate immune system as immunotherapy for acute myeloid leukemia. Front Oncol 5: 83, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT: Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12: 631–638, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY: IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol 185: 3472–3480, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, Lewis L, Finkelman FD, Smith DE, Bryce PJ, Kurt-Jones EA, Wang TC, Sivaprasad U, Hershey GK, Herbert DR: Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med 209: 607–622, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J, Chang I, Singh A, Harris RC: CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest 122: 4519–4532, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO: The cellular and molecular origin of tumor-associated macrophages. Science 344: 921–925, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doctor RB, Chen J, Peters LL, Lux SE, Mandel LJ: Distribution of epithelial ankyrin (Ank3) spliceoforms in renal proximal and distal tubules. Am J Physiol 274: F129–F138, 1998 [DOI] [PubMed] [Google Scholar]
- 54.Meldrum KK, Meldrum DR, Hile KL, Burnett AL, Harken AH: A novel model of ischemia in renal tubular cells which closely parallels in vivo injury. J Surg Res 99: 288–293, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Alikhan MA, Jones CV, Williams TM, Beckhouse AG, Fletcher AL, Kett MM, Sakkal S, Samuel CS, Ramsay RG, Deane JA, Wells CA, Little MH, Hume DA, Ricardo SD: Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am J Pathol 179: 1243–1256, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.