

Abstract
The majority of environmental bacteria are not readily cultured in the lab, leaving the natural products they make inaccessible using culture-dependent discovery methods. Cloning and heterologous expression of DNA extracted from environmental samples (environmental DNA, eDNA) provides a means of circumventing this discovery bottleneck. To facilitate the identification of clones containing biosynthetic gene clusters, we developed a model heterologous expression reporter strain Streptomyces albus::bpsA ΔPPTase. This strain carries a 4΄-phosphopantetheinyl transferase (PPTase)-dependent blue pigment synthase A gene, bpsA, in a PPTase deletion background. eDNA clones that express a functional PPTase restore production of the blue pigment, indigoidine. As PPTase genes often occur in biosynthetic gene clusters (BGCs), indigoidine production can be used to identify eDNA clones containing BGCs. We screened a soil eDNA library hosted in S. albus::bpsA ΔPPTase and identified clones containing non-ribosomal peptide synthetase (NRPS), polyketide synthase (PKS) and mixed NRPS/PKS biosynthetic gene clusters. One NRPS gene cluster was shown to confer the production of myxochelin A to S. albus::bpsA ΔPPTase.
Keywords: functional metagenomics, environmental DNA, PPTase, Streptomyces albus, eDNA, myxochelin
We have created a model heterologous expression strain to help with identification of biosynthetic gene clusters from environmental DNA libraries.
INTRODUCTION
Bacterial natural products have been a prolific source of structurally and functionally diverse small molecules used in numerous therapeutic areas including infectious disease, oncology and immunosuppression (Newman and Cragg 2016). Traditional methods used to identify bacterial natural products require that the producing organism be cultured in the laboratory. Unfortunately, only a small fraction of environmental bacteria are amenable to culture-based approaches. Metagenomics, which involves the characterization of DNA extracted directly from environmental samples (environmental DNA, eDNA), provides an alternative, culture-independent means of studying environmental microbiomes (Craig et al.2010). In one application of this approach known as functional metagenomics, libraries of clones containing eDNA are directly screened for phenotypes commonly associated with the production of small molecules (e.g. antibiosis, toxicity, iron chelation, pigmentation). Phenotypically distinct clones are then screened for the production of clone-specific metabolites. Although small molecules have been identified using functional metagenomics, these methods have not yet been developed to the extent that they can be used to rapidly harvest molecules from the environment. For functional metagenomics to be a truly useful natural product discovery tool, we must (i) improve methods for cloning eDNA fragments large enough to regularly capture complete biosynthetic gene clusters (BGCs), (ii) identify model bacterial hosts with improved ability to promiscuously express exogenous gene clusters and (iii) develop methods capable of identifying eDNA clones containing functional BGCs (Katz, Hover and Brady 2016). Here, we report on the improvement of methods to address the last of these three issues by engineering Streptomyces albus to report on the expression of phosphopantetheine transferase (PPTase) genes (Owen et al.2012; Charlop-Powers et al.2013; Iqbal et al.2016).
Most bacteria devote only a small fraction of their genome to secondary metabolism and therefore only a small fraction of the clones found in any metagenomic library is expected to encode for a natural product (Garcia, Fernández-Guerra and Casamayor 2011). The development of methods capable of readily identifying clones containing BGCs would undoubtedly increase the utility of these libraries as a source of small molecules (Owen et al.2012; Charlop-Powers et al.2013). Natural product biosynthesis employs a diverse collection of biosynthetic enzymes. Each of these enzymes tends to interact with a narrow set of substrates. In addition to the large collection of substrate-specific biosynthetic enzymes, there is a smaller group of biosynthetic enzymes that are functionally redundant across BGCs that encode diverse natural products. These functionally redundant enzymes are potentially useful for identifying BGC-containing metagenomic clones using complementation-based screening methods (Fig. 1A).
Figure 1.

Identification of eDNA clones containing biosynthetic genes by using complementation of functionally redundant biosynthetic genes. (A) Complementation of functionally redundant genes found in natural product biosynthesis can be used to identify eDNA clones containing BGCs (Owen et al.2012; Charlop-Powers et al.2013). (B) One example of a family of functionally redundant BGC-associated enzymes is the PPTase family that is responsible for the transfer of 4-phosphopantetheine from coenzyme A to PCP and ACP domains in non-ribosomal peptide and polyketide synth(et)ases, respectively. (C) Production of the blue pigment, indigoidine, by the NRPS encoded by bpsA (the blue pigment synthase A gene) is dependent on the post-translational modification by a PPTase enzyme (top panel). Deletion of the Sfp-like PPTase gene in S. albus abrogates heterologous production of indigoidine by BpsA in this host (middle panel). Complementation of PPTase function in a PPTase knockout background expressing bpsA (e.g. S. albus::bpsA ΔPPTase) will restore indigoidine production (bottom panel). Color production by eDNA clones hosted in S. albus::bpsA ΔPPTase can be used to identify PPTase expressing clones and in turn clones containing biosynthetic genes.
One example of a functionally redundant family of natural product biosynthetic genes is PPTases. PPTases are required for activating peptidyl-carrier-protein (PCP) and acyl-carrier-protein (ACP) domains of non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) gene clusters, respectively (Beld et al.2014). NRPS and PKS gene clusters encode the majority of pharmacologically relevant microbial secondary metabolites (Sieber and Marahiel 2005). In these common, assembly line-like systems, PPTases post-translationally modify NRPS/PKS megasynth(et)ases with the addition of a phosphopantetheine prosthetic group to a conserved serine residue of ACP or PCP domains. This post-translational modification is used to shuttle biosynthetic intermediates from one assembly line module to the next (Fig. 1B) (Lambalot et al.1996). PPTase genes frequently appear in bacterial NRPS/PKS gene clusters. They have also been shown to be promiscuous, with PPTase enzymes encoded by one gene cluster capable of complementing the PPTase function in another gene cluster (Nakano, Marahiel and Zuber 1988; Huang, Wendt-Pienkowski and Shen 2006; Owen et al.2012; Charlop-Powers et al.2013; Martin et al.2013). The dependence of NRPS/PKS biosynthesis on PPTase function together with the frequent occurrence of PPTases in BGCs has formed the basis of two different methods for identifying biosynthetic gene sequences in Escherichia coli-based metagenomic libraries. One approach uses PPTase-dependent pigment production by BpsA to screen for BGC containing clones. The authors of this study proposed that this approach could be applied to other bacterial hosts and this formed the basis for our screening strategy (Owen et al.2012). The other approach uses PPTase-dependent siderophore biosynthesis to select for BGC containing clones under low iron conditions (Charlop-Powers et al.2013). While E. coli is a great genetic workhorse, it has historically not been particularly useful as a heterologous host for secondary metabolite BGCs. We recently described an optimized method for constructing metagenomic libraries in S. albus, a cultured bacterium that displays promiscuous heterologous expression properties (Iqbal et al.2016). This method has now allowed us to extend bpsA gene expression-type BGC screening method (Owen et al.2012) to metagenomic libraries hosted in S. albus (Fig. 1C).
MATERIALS AND METHODS
Preparation of Streptomyces albus::bpsA
The indigoidine biosynthesis gene, blue pigment synthase A (bpsA), was amplified using pCDF-duet::bpsA as template (Owen et al.2012) (Primers: bpsA-NdeI-f 5΄-AAACATATGACTCTTCAGGAGACCAG and bpsA-PacI-r 5΄-AAATTAATTAACTACTCTCCGAGCAGGTACC), digested with NdeI/PacI (sites underlined) and inserted into the NdeI/PacI sites of the Streptomyces:Escherichia coli shuttle expression vector pIJ10257 containing a hygromycin selection marker (Hong et al.2005). The resultant construct was transformed into E. coli S17.1 and transferred by conjugation into S. albus J1074 (Zaburannyi et al.2014) to give S. albus::bpsA.
Deletion of the Sfp-type PPTase in Streptomyces albus::bpsA
Based on the sequence of Sfp-type PPTase gene (xnr_5716) in S. albus J1074 (Zaburannyi et al.2014), a 20-nucleotide protospacer was selected, and this sequence was appended with BbsI sites (underlined) to give the final protospacer (pptase-spacer-f-BbsI 5΄-ACGCCCAGGCCGACCACCGCGGCC and pptase-spacer-r-BbsI: 5΄-AAACGGCCGCGGTGGTCGGCCTGG). The protospacer was inserted to the BbsI sites of the pCRISPomyces2 vector by Golden Gate assembly. The vector was transformed into E. coli and selected using apramycin (50 μg/ml) (Cobb, Wang and Zhao 2015). The repair template was designed to consist of ∼800 bp immediately upstream and downstream of the Sfp-type PPTase in S. albus J1074. To create the final template, two amplicons (xnr_5715 and xnr_5717) were created using the following: xnr_5717 primers: 5717-pCRISP-HA-f 5΄-CCGGGCGTTTTTTATCTAGGATGTCCGGGAGTCGCCGG & 5717–5715-HA-r: 5΄-GGGAGGCAGGGCTGCTGATCAGCGTCCTCGGTTCGCG and xnr_5715 primers: 5715–5717-HA-f CGCGAACCGAGGACGCTGATCAGCAGCCCTGCCTCCC & 5715-pCRISP-HA-r TACGGTTCCTGGCCTCTAGGAGGACCGATGACCCCGCC. Gibson assembly was used to insert these amplicons into XbaI-digested pCRISPomyces2 vector containing the protospacer sequence (Cobb, Wang and Zhao 2015). The resulting vector was electroporated into E. coli S17.1 and transferred by conjugation to S. albus::bpsA. After 16 h, the conjugation plates were overlaid with apramycin, hygromycin and nalidixic acid at final concentrations of 50, 100 and 30 μg/ml, respectively. Exconjugants were picked and grown at 39°C to clear the CRISPR/Cas9 plasmid. Gene disruption was confirmed by PCR screening genomic DNA (Primers: PPTase-KO-scrn-f 5΄-AGGAAGGGACGGGTGAGGAA and PPTase-KO-scrn-r 5΄-TCGACGAGGAACTGGCCG) (Fig. S1, Supporting Information). The final strain was named S. albus::bpsA ΔPPTase.
Streptomyces albus library construction
The original construction of the cosmid library from Texas soil eDNA was described previously (Brady 2007; Iqbal et al.2016). This library was conjugated from E. coli into S. albus::bpsA ΔPPTase. Briefly, 1 L of Luria-Bertani (LB) broth was inoculated with 10 ml of an overnight culture and grown with shaking at 37°C to an OD600 = 0.5–0.6. Aliquots (80 ml) of cells were washed with ice cold LB, mixed with 25 × 108S. albus::bpsA ΔPPTase spores (heat-shocked at 50°C) and plated on ISP4 (Kieser et al.2000) media supplemented with 10 mM MgCl2. These plates were incubated at 30°C for 12–16 h and then overlaid with antibiotics to give final concentrations of 50 μg/ml apramycin and 25 μg/ml nalidixic acid. After 10–14 days of incubation at 30°C, blue colonies were picked and analyzed.
Recovery of cosmid DNA from Streptomyces albus and analysis of the eDNA inserts
A single S. albus colony containing each hit was used to inoculate 5 ml of tryptone soya broth (TSB) media and grown at 30°C until the culture was turbid. The cell pellet was resuspended in 750 μl of Qiagen's buffer P1 containing 3 mg/ml lysozyme and incubated with shaking for 2 h at 37°C. This mixture was divided into three equal volumes. Buffer P2 (250 μl) was added to each tube. Tubes were mixed by inversion and after 15–20 min they were neutralized with the addition of 350 μl of buffer N3. Centrifugation (15 000 rpm, 30 min) was used to pellet the cell debris. The supernatant from all three tubes was combined and applied to a single Qiagen miniprep column. The column was washed three times with 500 μl PB buffer, three times with 750 μl PE buffer and centrifuged (10 000 rpm, 2 min) to remove residual wash buffers. The DNA was eluted using 50 μl of warm (50°C) Qiagen EB, desalted (Peng et al.2009) and transformed into E. coli EC100. Single colonies were grown in LB media, miniprepped and sequenced using the Ion Personal Genome Machine (PGM) system. Open reading frames (ORFs) were predicted using MetaGeneMark (Zhu, Lomsadze and Borodovsky 2010). ORFs were scanned for the presence of PPTase (PF01648), NRPS (PF00668) and PKS (PF00109, PF00698 and PF16197) domains using HMM scan (Finn, Clements and Eddy 2011) and annotated accordingly. The cosmid DNA was also electroporated in to E. coli S17.1 and transformed back to the S. albus::bpsA ΔPPTase strain to re-confirm the color production.
GenBank accession numbers
The clones with BGCs are deposited with the following GenBank accession codes KY560349 – KY560368.
Streptomyces liquid cultures and UPLC-MS analysis
Each clone was inoculated into 5 ml TSB media containing the following antibiotics: 50 μg/ml apramycin, 25 μg/ml nalidixic acid and 50 μg/ml nystatin. After 2 days of incubation (30°C, 250 rpm), 50 μl of the TSB culture was used to inoculate 50 ml R5A (Kieser et al.2000) media without antibiotics. The cultures were incubated at 30°C with shaking (250 rpm) for 10–14 days. R5A culture (25 ml) was extracted using an equal volume of ethyl acetate, and the solvent was removed by evaporation. Each ethyl acetate extract was dissolved in 300 μl methanol, and 3 μl was analyzed by high-resolution mass spectrometry (HRMS) and ultraperformance liquid chromatography mass spectrometry (UPLC-MS) [2.1 × 50 mm 1.7μ C18 column, 0.6 ml/min, mobile phase gradient used solvent A = dH2O + 0.1% formic acid and solvent B = MeOH + 0.1% formic acid: 0 to 0.9 min (10% B), 0.9 min to 9.2 min (linear gradient from 10 to 100% B), 9.2 min to 10.8 min (hold at 100% B), 10.80 min to 10.90 min (step down to 10% B), 10.90 min to 12.0 min (equilibrate to 10% B)].
Comparison of clone m9-20 extracts to myxochelin A standard
Analysis was done by UPLC-MS using the following extended mobile phase conditions: [2.1 × 50 mm 1.7μ C18 column, 0.6 ml/min, mobile phase gradient used solvent A = dH2O + 0.1% formic acid and solvent B = MeOH + 0.1% formic acid: 0 to 3.6 min (10% B), 3.6 min to 36.8 min (linear gradient from 10 to 100% B), 36.8 min to 43.2 min (hold at 100% B), 43.2 min to 43.6 min (step down to 10% B), 43.6 min to 48 min (equilibrate to 10% B)]. For the co-injection experiment, 0.5 μl of a 10 mM solution of myxochelin A in methanol was added to 20 μl of the m9–20 ethyl acetate extract that was previously dissolved in 300 μl methanol as described above. This mixture (3 μl) was analyzed using the UPLC extended method. For the myxochelin A trace, 3 μl of a 1 mM solution in methanol was analyzed.
RESULTS AND DISCUSSION
In Escherichia coli, NRPS encoded production of the colored metabolite indigoidine and the requirement for an NRPS-encoded siderophore biosynthesis to support growth on low iron have been used to identify eDNA clones containing PPTases (Owen et al.2012; Charlop-Powers et al.2013). Compared to E. coli, siderophore biosynthesis in Streptomyces is more complex as it uses both NRPS-dependent and NRPS-independent pathways to produce siderophores for iron sequestration (Barona-Gomez et al.2006). We elected to explore PPTase-dependent indigoidine color production as a tool for identifying PPTase-expressing clones in Streptomyces albus-hosted metagenomic libraries. Indigoidine production is biosynthetically encoded by a single, multiple-domain NRPS gene, bpsA, from Streptomyces lavendulae (Takahashi et al.2007). BpsA was first integrated into the S. albus genome under the control of the strong constitutive promoter ermE* using the phiBT1 integration locus (Gregory, Till and Smith 2003). As expected, the resulting strain, S. albus::bpsA, produces the dark blue indigoidine pigment (Fig. 1C). Streptomyces albus contains two PPTases: one that is used in fatty acid biosynthesis and another, an Sfp family PPTase, that is specific to natural product biosynthesis (Bunet et al.2014). We performed a knockout of the Sfp-type PPTase in S. albus::bpsA using CRISPR/Cas9 (Fig. S1, Supporting Information) (Cobb, Wang and Zhao 2015). This strain, S. albus::bpsA ΔPPTase, grows normally but no longer produces indigoidine. Production of indigoidine was restored when the deleted PPTase was reintroduced in trans at the phiC31 integration site under the control of the ermE* constitutive promoter, confirming that this S. albus::bpsA ΔPPTase strain could be used as a reporter of PPTase expression (Gregory, Till and Smith 2003). A previously constructed 500 000-membered cosmid library (Iqbal et al.2016) was used to screen for eDNA-derived secondary metabolite gene clusters using this S. albus::bpsA ΔPPTase reporter strain. The resulting exconjugants were allowed to mature at room temperature for 10–14 days.
Blue colonies, expected to result from the complementation of indigoidine biosynthesis by an eDNA-derived PPTase gene, were picked directly from the conjugation plates (Fig. 1C). Cosmid DNA isolated from small-scale cultures of each blue clone was transformed into E. coli, and aliquots of DNA isolated from these cultures were restriction mapped to identify clones with unique eDNA inserts. Clones containing unique restriction maps were sequenced and conjugated back into S. albus::bpsA ΔPPTase to confirm that each clone was in fact sufficient to confer a blue phenotype to the host and that these clones encoded PPTases. We identified 40 environmental clones that were capable of restoring indigoidine biosynthesis to the S. albus::bpsA ΔPPTase reporter strain, all of which were predicted to encode PPTases.
Secondary metabolism-associated Sfp-type PPTases are easily distinguished from primary metabolism-associated AcpS-like PPTase by the difference in size (Beld et al.2014). Thirty six of the 40 identified PPTases resemble the larger (212–275 amino acids) Sfp-type PPTases, while three resemble the smaller (100–158 amino acids) AcpS-like PPTase. One clone (m1-11) encodes a two-domain, 451-amino acid PPTase. The second domain is predicted to be a cystathionine beta synthase regulatory domain (PF00571). Phylogenetic analysis shows that eDNA-derived PPTases cluster with PPTases originating from diverse bacterial taxa, including actinomycetes, proteobacteria and cyanobacteria. No eDNA-derived PPTases fall into the clade associated with PPTases from the firmicute or the eukaryotic branches of the PPTase phylogenetic tree (Figs 2 and S2, and Table S1, Supporting Information).
Figure 2.
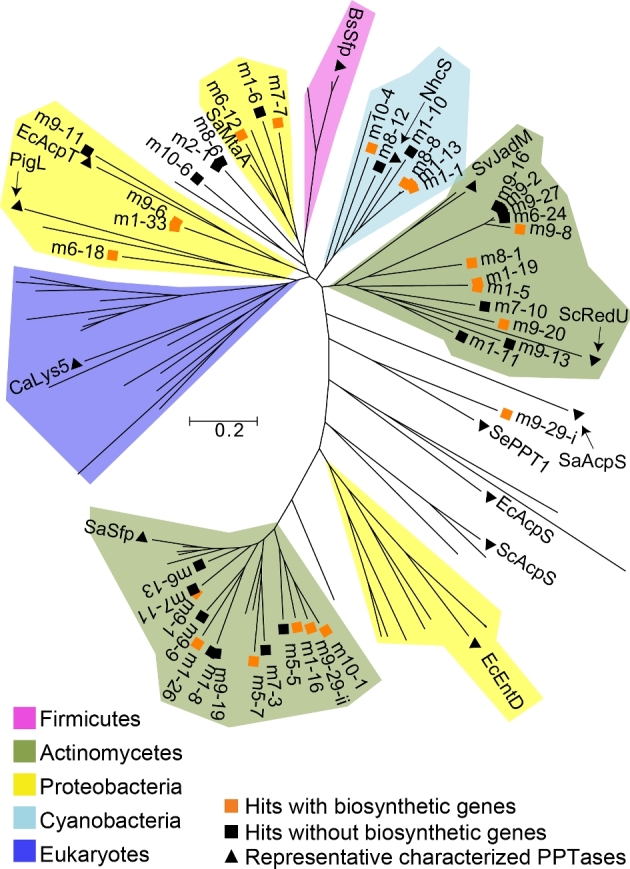
PPTase phylogenetic tree. eDNA-derived PPTases along with ∼60 functionally characterized PPTases of diverse phylogenetic origin (Table S1) were used to construct a phylogenetic tree (Beld et al.2014). Characterized PPTases largely group by phylogenetic origin and therefore clades have been colored using these genes as a guide. Representative functionally characterized PPTases of diverse phylogenetic origin are labeled: S. albus Sfp (SaSfp), E. coli EntD (EcEntD), S. albus AcpS (SaAcpS), S. coelicolor AcpS (ScAcpS), E. coli AcpS (EcAcpS), Saccharopolyspora erythraea PPT1 (SePPT1), S. coelicolor RedU (ScRedU), S. venezuelae JadM (SvJadM), Nodularia spumigena NSOR10 (NhcS), Bacillus subtilis Sfp (BsSfp), S. aurantiaca Sg. a15 MtaA (SaMtaA), E. coli AcpT (EcAcpT), Serratia marcescens PigL (PigL) and Candida albicans Lys5 (CaLys5). The tree was constructed using MEGA (Kumar et al.2016) and the neighbor joining method. A phylogenetic tree where all branches are labeled is provided in Fig. S2.
Half of the PPTase-containing clones were predicted to contain natural product biosynthesis genes. Eleven encode NRPS genes, two encode PKS genes and seven encode mixed NRPS/PKS biosynthesis genes (Fig. 3, Table S2). Each of these eDNA clones was grown in R5A liquid medium to test for secondary metabolite production (Kieser et al.2000). After 10 days, cultures were extracted with ethyl acetate and the extracts were examined by UPLC-MS for the production of clone-specific metabolites (Fig. S3, Supporting Information). This analysis identified a single, clone-specific peak produced by clone m9–20. This peak was not observed in extracts from the empty vector control cultures or in extracts from any other colored clone (Fig. 4A and B). In silico analysis of the m9–20 clone suggested the presence of a myxochelin-like gene cluster (Fig. 4C, Table S3) (Silakowski et al.2000). HRMS analysis of the clone-specific peak found in the m9–20 ethyl acetate extract indicated a molecular formula of C20H24N2O7 [HRMS (ESI/Orbitrap) m/z: (M + H)+ Calcd for C20H24N2O7 405.1656; Found 405.1657]. Upon further analysis, the major peak in clone m9–20 was found to co-elute with a purified standard of myxochelin A (Fig. S4, Supporting Information), a dihydroxybenzoate-containing siderophore that was first characterized from myxobacteria (Kunze et al.1989).
Figure 3.

Annotation of genes found on PPTase-containing clones. (A) ORFs identified on each PPTase clone are shown. Predicted PPTases and biosynthetic genes associated with PPTase post-translational modifications (PKS, NRPS and mixed PKS/NRPS) are color coded according to the key provided in the figure. Clones have an average insert size of 31 kb. (B) A BLAST analysis was performed to identify the phylogenetic origin of the closest sequenced relative of all predicted genes on each clone. The origin of the gene with highest sequence identify to each PPTase in shown in the first column. The most common phylum of the closest relative for each individual gene in a clone is shown in column 2. In the absence of other means of identifying the source microbe, these analyses provide some insight into the potential phylogenetic origin of the BGC fragment captured on each eDNA clone.
Figure 4.

Clone m9–20 produces myxochelin A. (A) UPLC traces show the presence of a major, clone-specific metabolite in culture broth extracts from clone m9–20. HRMS analysis of this peak indicates that the predicted molecular formula corresponds to that of myxochelin A. (B) Clone m9–20 restored PPTase function and indigoidine production leading to a blue clone that produces myxochelin A. (C) Comparison of the genes found in the myxochelin (mxc) biosynthetic gene cluster from S. aurantiaca Sg. a15 (Silakowski et al.2000) with genes present on clone m9–20. The % identity between each related gene is shown. (D) Biosynthesis of the DHBA pre-cursor used in the biosynthesis of myxochelin A. (E) Proposed biosynthesis of myxochelin A through condensation of lysine and two molecules of DHBA (Silakowski et al.2000).
A comparison of the genes present in the biosynthetic cluster captured on the m9–20 clone to that of the myxochelin A BGC (Mxc cluster) from Stigmatella aurantiaca Sg a15 reveals homologs to all genes predicted to be required for the biosynthesis of myxochelin A (Fig. 4C, Table S3, Supporting Information) (Silakowski et al.2000). A number of predicted Mxc BGCs appear in sequenced bacterial genomes. Based on sequence identity and gene organization, the closest relative of the Mxc-like gene cluster present in m9–20 is found in Streptomyces sp. FxanaC1 (Fig. S5, Supporting Information). Myxochelins arise from the condensation of dihidroxybenzoic acid (DHBA) with lysine. The DHBA substrate is produced from isochorismate by MxcC, MxcE and MxcF (Fig. 4D). The condensation of two molecules of DHBA with lysine occurs on the NRPS megasynthase MxcG. This trimer is released by the terminal reductive domain in MxcG to yield myxochelin A (Fig. 4E) (Silakowski et al.2000). The m9–20 clone lacks the aldehyde amino transferase (MxcL) required for the production of myxochelin B by the S. aurantiaca Sg. a15 Mxc cluster. As expected, we did not detect any myxochelin B in cultures of m9–20. Interestingly, the isochorismatase gene (mxcF) from the m9–20 clone appears be truncated, suggesting it may no longer encode a functional enzyme. We assume that this function is complemented by the native S. albus metabolism. One candidate for this is a PhzD-like enzyme encoded by the S. albus genome. Although the PhzD enzyme from phenazine biosynthesis uses 2-amino-2-deoxychorismic acid as a substrate, other PhzD-like enzymes have also been reported to have isochorismatase activity (Parsons et al.2003; Hubrich, Muller and Andexer 2014).
Even though m9–20 was the only clone that conferred the production of a detectable clone-specific small molecule to S. albus, 19 other PPTase-containing clones had secondary metabolite biosynthetic genes. We did not see any significant relationships between the collections of biosynthetic genes found on these clones and sequenced gene clusters present in publicly available databases by BLAST and antiSMASH analysis (Altschul et al.1990; Medema et al.2011). In the case of the two type II PKS-containing clones, m1–26 and m6–12, phylogenetic analysis of the ketosynthase β gene, which serves as a robust indicator of the compound class encoded by the BGC, suggests that both clusters encode pentangular polyphenols (Tang, Tsai and Khosla 2003). Pentangular polyphenol BGCs encode production of the largest known aromatic polyketides. Apart from their interesting complex structures, the pentangular polyphenol group of the aromatic polyketides, which includes xantholipin, (Terui et al.2003) benastatin A, (Aoyagi et al.1992) and arixanthomycins (Kang and Brady 2014) among others, displays a wide range of biological activity.
In conclusion, we created a Streptomyces-based PPTase complementation reporter strain, S. albus::bpsA ΔPPTase, enabling us to identify metagenomic clones containing BGCs. The screening of a soil eDNA cosmid library using this strain led to the identification of 20 clones containing biosynthetic genes. Clone m9–20 was found to confer the production of myxochelin A to S. albus::bpsA ΔPPTase. Detailed in silico analysis of this clone indicated that it contains all of the genes predicted to be required for myxochelin A biosynthesis in S. albus. Our bioinformatics analysis suggests that clone m9–20 was in fact the only PPTase containing clone that contained a complete BGC. In the remaining clones containing large modular NRPS/PKS genes, biosynthetic genes are present at the edges of the eDNA inserts, suggesting that the BGCs likely extend beyond the end of the cosmid clones. The upper limit of eDNA inserts captured using cosmid-based methodologies is 40 kb, making the capture of a complete NRPS or PKS gene cluster on a single clone unlikely. As outlined above, we have proposed that three issues must be addressed for functional metagenomics to provide regular access to secondary metabolites: (i) development of methods for larger eDNA inserts, (ii) identification of promiscuous heterologous expression host strains and (iii) development of methods for identifying clones containing BGCs. We believe that the development of the S. albus::bpsA ΔPPTase strain will prove useful for addressing the third of these three bottlenecks. The true potential of this strain will only be fully realized once methods are developed for generating eDNA clones that are large enough to regularly capture entire NRPS/PKS gene clusters.
Supplementary Material
Supplementary data are available at FEMSLE online.
Acknowledgments
We thank Dr David F. Ackerley for the pCDF-duet::bpsA vector. HRMS was done at The Rockefeller University Proteomics Resource Center.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSLE online.
FUNDING
This research was funded by NIH grant R01-GM115331-02 to SFB. JKB was supported in part by Merck postdoctoral fellowship.
Conflict of interest. None declared.
REFERENCES
- Altschul SF, Gish W, Miller W et al. . Basic local alignment search tool. J Mol Biol 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- Aoyagi T, Aoyama T, Kojima F et al. . Benastatin-A and benastatin-B, new inhibitors of glutathione-S-transferase, produced by Streptomyces Sp MI384-DF12 .1. Taxonomy, production, isolation, physicochemical properties and biological activities. J Antibiot 1992;45:1385–90. [DOI] [PubMed] [Google Scholar]
- Barona-Gomez F, Lautru S, Francou FX et al. . Multiple biosynthetic and uptake systems mediate siderophore-dependent iron acquisition in Streptomyces coelicolor A3(2) and Streptomyces ambofaciens ATCC 23877. Microbiology-(UK) 2006;152:3355–66. [DOI] [PubMed] [Google Scholar]
- Beld J, Sonnenschein EC, Vickery CR et al. . The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Natu Prod Reps 2014;31:61–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SF. Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat Protoc 2007;2:1297–305. [DOI] [PubMed] [Google Scholar]
- Bunet R, Riclea R, Laureti L et al. . A single Sfp-Type phosphopantetheinyl transferase plays a major role in the biosynthesis of PKS and NRPS derived metabolites in streptomyces ambofaciens ATCC23877. PLoS One 2014;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlop-Powers Z, Banik JJ, Owen JG et al. . Selective enrichment of environmental DNA libraries for genes encoding nonribosomal peptides and polyketides by phosphopantetheine transferase-dependent complementation of siderophore biosynthesis. ACS Chem Biol 2013;8:138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb RE, Wang YJ, Zhao HM. High-efficiency multiplex genome editing of streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 2015;4:723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig JW, Chang FY, Kim JH et al. . Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microb 2010;76:1633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 2011;39:W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JAL, Fernández-Guerra A, Casamayor EO. A close relationship between primary nucleotides sequence structure and the composition of functional genes in the genome of prokaryotes. Mol Phylogenet Evol 2011;61:650–8. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Till R, Smith MCM. Integration site for streptomyces phage phi BT1 and development of site-specific integrating vectors. J Bacteriol 2003;185:5320–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong HJ, Hutchings MI, Hill LM et al. . The role of the novel fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J Biol Chem 2005;280:13055–61. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wendt-Pienkowski E, Shen B. A dedicated phosphopantetheinyl transferase for the fredericamycin polyketide synthase from Streptomyces griseus. J Biol Chem 2006;281:29660–8. [DOI] [PubMed] [Google Scholar]
- Hubrich F, Muller M, Andexer JN. In vitro production and purification of isochorismate using a two-enzyme cascade. J Biotechnol 2014;191:93–8. [DOI] [PubMed] [Google Scholar]
- Iqbal HA, Low-Beinart L, Obiajulu JU et al. . Natural product discovery through improved functional metagenomics in streptomyces. J Am Chem Soc 2016;138:9341–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, Brady SF. Arixanthomycins A-C: phylogeny-guided discovery of biologically active eDNA-Derived pentangular polyphenols. ACS Chem Biol 2014;9:1267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M, Hover BM, Brady SF. Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biot 2016;43:129–41. [DOI] [PubMed] [Google Scholar]
- Kieser T, Bibb MJ, Buttner MJ et al. . Practical Streptomyces Genetics. Norwich, UK: The John Innes Foundation, 2000. [Google Scholar]
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze B, Bedorf N, Kohl W et al. . Myxochelin A, a new iron-chelating compound from Angiococcus disciformis (Myxobacterales). Production, isolation, physico-chemical and biological properties. J Antibiot 1989;42:14–7. [DOI] [PubMed] [Google Scholar]
- Lambalot RH, Gehring AM, Flugel RS et al. . A new enzyme superfamily - the phosphopantetheinyl transferases. Chem Biol 1996;3:923–36. [DOI] [PubMed] [Google Scholar]
- Martin P, Marcq I, Magistro G et al. . Interplay between siderophores and colibactin genotoxin biosynthetic pathways in escherichia coli. PLoS Pathog 2013;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P et al. . antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 2011;39:W339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano MM, Marahiel MA, Zuber P. Identification of a genetic-locus required for biosynthesis of the lipopeptide antibiotic surfactin in Bacillus subtilis. J Bacteriol 1988;170:5662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 2016;79:629–61. [DOI] [PubMed] [Google Scholar]
- Owen JG, Robins KJ, Parachin NS et al. . A functional screen for recovery of 4΄-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. Environ Microbiol 2012;14:1198–209. [DOI] [PubMed] [Google Scholar]
- Parsons JF, Calabrese K, Eisenstein E et al. . Structure and mechanism of Pseudomonas aeruginosa PhzD, an isochorismatase from the phenazine biosynthetic pathway. Biochemistry 2003;42:5684–93. [DOI] [PubMed] [Google Scholar]
- Peng D, Luo Y, Guo S et al. . Elaboration of an electroporation protocol for large plasmids and wild-type strains of Bacillus thuringiensis. J Appl Microbiol 2009;106:1849–58. [DOI] [PubMed] [Google Scholar]
- Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem Rev 2005;105:715–38. [DOI] [PubMed] [Google Scholar]
- Silakowski B, Kunze B, Nordsiek G et al. . The myxochelin iron transport regulon of the myxobacterium Stigmatella aurantiaca Sg a15. Eur J Biochem 2000;267:6476–85. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kumagai T, Kitani K et al. . Cloning and characterization of a Streptomyces single module type non-ribosomal peptide synthetase catalyzing a blue pigment synthesis. J Biol Chem 2007;282:9073–81. [DOI] [PubMed] [Google Scholar]
- Tang Y, Tsai SC, Khosla C. Polyketide chain length control by chain length factor. J Am Chem Soc 2003;125:12708–9. [DOI] [PubMed] [Google Scholar]
- Terui Y, Chu YW, Li JY et al. . Xantholipin, a novel inhibitor of HSP47 gene expression produced by Streptomyces sp. Tetrahedron Lett 2003;44:5427–30. [Google Scholar]
- Zaburannyi N, Rabyk M, Ostash B et al. . Insights into naturally minimised Streptomyces albus J1074 genome. BMC Genomics 2014;15:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 2010;38:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSLE online.