

Abstract
Takayasu’s arteritis is a chronic, non-specific primary and inflammatory disease of large vessels, especially the aorta and its branches. Although pulmonary artery involvement is common, clinical signs of pulmonary artery involvement as the initial presentation is rare. Considering Takayasu’s arteritis in the early diagnosis is important. In this article, we report a 23-year-old female patient of Takayasu’s arteritis who was treated with successful surgery combined with medical therapy.
Keywords: Pulmonary endarterectomy, pulmonary hypertension, Takayasu’s arteritis
Introduction
Takayasu’s arteritis is a chronic, non-specific primary and granulomatous vasculitis of large vessels such as the aorta and other major branches.(1) Clinical signs of pulmonary artery (PA) involvement as the initial presentation are unusual and there are only few case reports of Takayasu’s arteritis patients complaining of symptoms of pulmonary hypertension (PH).(2-4) Treating this disease with pulmonary endarterectomy (PEA) is also rare.
Case Report
A 23-year-old female patient was referred to our hospital due to three-week history of progressive dyspnea with nonproductive cough and poor exercise tolerance which deteriorated within two days. Her right brachial and radial pulses were diminished. There was a right ventricular heave, loud S2 and a grade 2/6 systolic murmur heard throughout the pericardium. Forced expiratory volume in 1 second/forced vital capacity ratio was 89%, forced vital capacity was 71%, and forced expiratory volume in 1 second was 74% in lung function test. Initial laboratory analysis revealed C-reactive protein of 3.76 mg/dL (0-0.5 mg/dL) and erythrocyte sedimentation rate of 33 mm/h (0-20 mm/h). On admission, the patient was in the New York Heart Association functional class III with a six minutes walk distance of 270 meters.
The echocardiography showed an ejection fraction of 70%, right ventricular enlargement with D septum, second degree tricuspid regurgitation, and 89 mmHg systolic PA pressure. Pulmonary computed tomographic angiography showed contrast filling defect of right middle lobe PA, left lower lobe PA, right and left upper lobe PA, and distal left main PA which was compatible with pulmonary embolism. PH due to acute/subacute thromboembolism was considered in diagnosis. So, tissue plasminogen activator (100 mg) was administered as a continuous intravenous infusion over a two hour period. The patient was coumadinized and discharged after clinical stabilization.
Despite adequate anticoagulation for three months, control transthoracic echocardiography revealed no significant decrease in PA pressure. Evidence of chronic thromboembolic pulmonary hypertension (CTEPH) was then confirmed by pulmonary angiography and right heart catheterization. Pulmonary angiography showed a filling defect in left upper lobe PA and right upper lobe PA.
Systolic PA pressure was 95 mmHg and mean PA pressure was 45 mmHg. Vasoreactivity testing was performed with inhaler iloprost which showed that patient had negative vasodilator response.
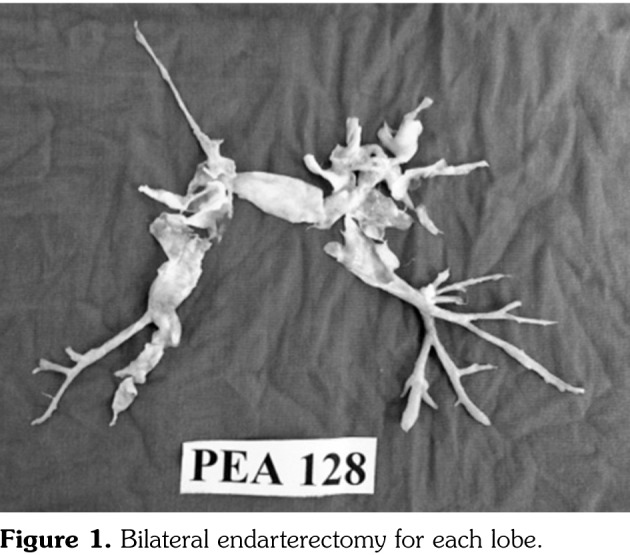
We decided to perform PEA, since our patient was in New York Heart Association functional class III, had PA hypertension, had no significant comorbidities, and showed computed tomography angiographic evidence of PA tree obstruction despite three months of anticoagulation. Other causes of PA hypertension were excluded. During surgery, the pulmonary arteries were thick walled and fibrotic, making the operation technically challenging. However, successful bilateral endarterectomy was performed for each lobe (Figure 1). Postoperative period was unremarkable since the mean PA pressure decreased from 45 mmHg to 25 mmHg and the patient was extubated on postoperative day one and discharged on postoperative day 10.
Figure 1. Bilateral endarterectomy for each lobe.
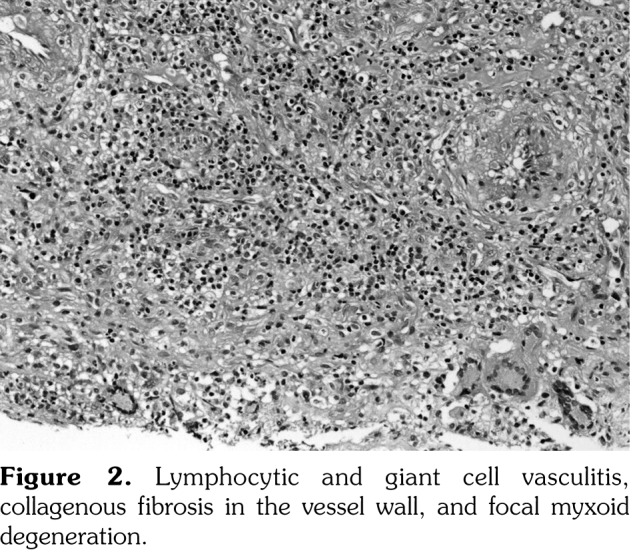
The histological work up showed lymphocytic and giant cell vasculitis, collagenous fibrosis in the vessel wall, and focal myxoid degeneration (Figure 2). Takayasu’s arteritis was considered in differential diagnosis and a magnetic resonance angiography scan of thorax and abdomen showed significantly decreased right subclavian artery calibration whereas abdominal aorta, renal arteries, and celiac trunk calibration was normal. Rheumatoid factor, antinuclear antibodies, anti deoxyribonucleic acid antibody, viral serology, and antineutrophil cytoplasmic antibody were all negative.
Figure 2. Lymphocytic and giant cell vasculitis, collagenous fibrosis in the vessel wall, and focal myxoid degeneration.
The patient fulfilled three of the six American College of Rheumatology 1990 classification criteria: development of symptoms or findings at age<40 years, decreased brachial artery pulse, and arteriogram abnormality.(5) Immunosuppresive therapy (methotrexate and oral corticosteroid) was started. Additionally, the patient was treated with iloprost. At postoperative three months, iloprost treatment was discontinued. Patient’s clinical symptoms improved significantly: she improved to New York Heart Association class II, her six minutes walk distance increased to 460 meters, and echocardiography revealed reduced systolic PA pressure.
Surgery combined with medical treatment improved patient’s symptoms significantly and her condition has been stable for the last two years with normal erythrocyte sedimentation rate and C-reactive protein.
Discussion
The arteritic process in patients with Takayasu’s arteritis is not only limited to the aortic arch. PA involvement was found in an average of 50% of cases studied.(6, 7) In a study with 60 cases of Takayasu’s arteritis, Kerr et al.( 8) demonstrated a 10-month delay between the onset of the first symptoms and diagnosis. It may have been challenging to distinguish these cases from primary lung disease and chronic pulmonary embolism.
The angiographic pattern of arteritic lesions seems to be the same as that observed in pulmonary thromboembolism. Also, it must be emphasized that pulmonary embolism due to flow limit of PA has been documented in these patients.(9, 10) In our case, patient was diagnosed with pulmonary embolism initially and thrombolytic therapy was administered. Improved angiographic appearance detected after thrombolysis suggested that concomitant pulmonary embolism was present in our patient.
Moreover, our patient had severe PA hypertension accompanied by progressive dyspnea and poor exercise tolerance. After three months of clinical follow-up, surgical treatment was recommended to relieve the PA obstruction and PH.
Pulmonary endarterectomy is the procedure of choice and potentially a curative option for patients with CTEPH. The technical aspects of the surgery have been refined over the past 20 years and the procedure is currently performed with limited risk in experienced centers. Surgical candidates undergo PEA according to the standardized technique previously described.(11)
We treated the patient with immunosupressive therapy and inhaler iloprost -a stable analogue of prostacyclin- postoperatively. Following extubation, inhaled iloprost was started at doses of 2.5 μg, six times per day since iloprost is known to reduce residual postoperative PH, decrease right ventricular afterload, and may facilitate the early postoperative management after PEA.(11) It has been shown that inhaler iloprost was effective in PAH associated with connective tissue diseases and in operable CTEPH.( 12)
In conclusion, Takayasu’s arteritis may mimic CTEPH. Entertaining the diagnosis preoperatively is of benefit so that the patient can be informed about the differential diagnosis and the surgeon can plan to undertake a reconstructive procedure of a PA as well as PEA. The surgery combined with medical therapy may be effective in PH associated with Takayasu’s arteritis.
Footnotes
Conflict of Interest: The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Financial Disclosure: The authors received no financial support for the research and/or authorship of this article.
References
- 1.Gravanis MB. Giant cell arteritis and Takayasu aortitis: morphologic, pathogenetic and etiologic factors . Int J Cardiol. 2000;75:21–33. doi: 10.1016/s0167-5273(00)00184-4. [DOI] [PubMed] [Google Scholar]
- 2.Brugiere O, Mal H, Sleiman C, Groussard O, Mellot F, Fournier M. Isolated pulmonary arteries involvement in a patient with Takayasu's arteritis . Eur Respir J. 1998;11:767–770. [PubMed] [Google Scholar]
- 3.Hayashi K, Nagasaki M, Matsunaga N, Hombo Z, Imamura T. Initial pulmonary artery involvement in Takayasu arteritis. Radiology. 1986;159:401–403. doi: 10.1148/radiology.159.2.3961173. [DOI] [PubMed] [Google Scholar]
- 4.Şentürk T, Kaderli Aydın A, Karabacak S, Yeşilbursa D, Serdar OA. Pulmonary artery hypertension as an initial manifestation of Takayasu’s arteritis: A case report . Respir Med. 2010;3:211–213. [Google Scholar]
- 5.Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis . Arthritis Rheum. 1990;33:1129–1134. doi: 10.1002/art.1780330811. [DOI] [PubMed] [Google Scholar]
- 6.Sharma S, Kamalakar T, Rajani M, Talwar KK, Shrivastava S. The incidence and patterns of pulmonary artery involvement in Takayasu's arteritis . Clin Radiol. 1990;42:177–181. doi: 10.1016/s0009-9260(05)81929-4. [DOI] [PubMed] [Google Scholar]
- 7.Liang P, Hoffman GS. Advances in the medical and surgical treatment of Takayasu arteritis. Curr Opin Rheumatol. 2005;17:16–24. doi: 10.1097/01.bor.0000146607.65808.37. [DOI] [PubMed] [Google Scholar]
- 8.Kerr GS, Hallahan CW, Giordano J, Leavitt RY, Fauci AS, Rottem M, et al. Takayasu arteritis. Ann Intern Med. 1994;120:919–929. doi: 10.7326/0003-4819-120-11-199406010-00004. [DOI] [PubMed] [Google Scholar]
- 9.Nasu T. Pathology of pulseless disease. A systematic study and critical review of twenty-one autopsy cases reported in Japan. Angiology. 1963;14:225–242. doi: 10.1177/000331976301400502. [DOI] [PubMed] [Google Scholar]
- 10.Lupi Herrera E, Contreras R, Espino Vela J, Sánchez Torres G, Horwitz S. Nonspecific arteritis in childhood. Clinical and anatomo-pathological observations. Arch Inst Cardiol Mex. 1972;42:477–493. [PubMed] [Google Scholar]
- 11.Yıldızeli B, Taş S, Yanartaş M, Kaymaz C, Mutlu B, Karakurt S, et al. Pulmonary endarterectomy for chronic thrombo-embolic pulmonary hypertension: an institutional experience . Eur J Cardiothorac Surg. 2013;44:219–227. doi: 10.1093/ejcts/ezt293. [DOI] [PubMed] [Google Scholar]
- 12.Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–329. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]