

Abstract
Protein tyrosine phosphatases (PTP) are exciting and novel targets for cancer drug discovery that work in concert with protein tyrosine kinases (PTK) in controlling cellular homeostasis. Given the activating role that some PTKs play in initiating growth factor-mediated cellular processes, PTPs are usually perceived as the negative regulators of these events and therefore tumor suppressive in nature. However, mounting evidence indicate that PTPs do not always antagonize the activity of PTKs in regulating tyrosine phosphorylation, but can also play dominant roles in the initiation and progression of signaling cascades that regulate cell functions. It follows, therefore, that PTP malfunction can actively contribute to a host of human disorders, in particular, cancer, metabolic syndromes, and autoimmune diseases. The Src homology domain containing phosphatase 2 (SHP2) and the three-membered family of phosphatases of regenerating liver (PRL) are infamously oncogenic members of the PTP superfamily. Both are established regulators of major cancer pathways such as Ras/ ERK1/2, Src, JAK/STAT, JNK, NF-κB, and PTEN/PI3K/AKT. Furthermore, upregulation, mutation, or other dysregulation of these PTPs has been positively correlated with cancer initiation and progression. This review will provide topical coverage of target validation and drug discovery efforts made in targeting these oncogenic PTPs as compelling candidates for cancer therapy.
Introduction
Protein tyrosine phosphorylation is essential for regulating a myriad of cellular processes, including cell growth and survival. Dysregulation of tyrosine phosphorylation mediated cell signaling is a well-recognized cause for diseases. Numerous medicinal agents acting on protein tyrosine kinases (PTK) have reached the clinic in recent years. Because protein tyrosine phosphorylation is a dynamic and reversible posttranslational modification that is orchestrated by a regulatory partnership between PTKs and protein tyrosine phosphatases (PTP), there is the potential to modulate disease progression by targeting the PTPs. Indeed, malfunction of PTP activity contributes to the development and progression of aberrations such as cancer, metabolic and autoimmune disorders, infectious disease and neurodegeneration. Given the involvement of PTPs to human malady, a more comprehensive investigation of them is paramount to the development of more effective therapeutic interventions. In this review, we will focus on the PTPs and their eligibility as targets of drug discovery in cancer. Although several PTPs have been implicated as potential tumor suppressors, growing evidence establish that a large number of PTPs function as powerful tumor promoters in many types of cancers (1). Over the years, our understanding of how PTPs contribute to signaling and disease has expanded to include a better coverage of PTP function and target validation. In turn, this has generated heightened interest over their candidacy for therapeutic development. Previously insurmountable obstacles such as inhibitor potency, specificity and bioavailability are being addressed by methods of drug design that exploit unique structural features proximal to the catalytic and regulatory sites. This review aims to shine the spotlight on two oncogenic PTPs: the Src homology 2 domain containing phosphatase 2 (SHP2) and the phosphatases of regenerating liver (PRL). SHP2 and PRLs play critical roles in cancer progression and are emerging as compelling therapeutic targets for cancer drug discovery.
SHP2 Is a Bona Fide Oncoprotein
SHP2, encoded by the PTPN11 gene, is an allosteric phosphatase containing two SH2 domains (i.e., N-SH2 and C-SH2), a PTP catalytic domain and a C-terminal tail (Fig. 1A). In the basal state, SHP2 is kept in an autoinhibited conformation by intramolecular interactions between the N-SH2 domain and the catalytic cleft of the PTP domain (2). However, upon growth factor or cytokine stimulation, binding of specific pTyr motifs from growth factor receptors or adaptor proteins to the N-SH2 domain releases autoinhibition and activates SHP2 (Fig. 1A). This elegant allosteric mechanism ensures that SHP2 exerts its phosphatase activity only when recruited to appropriate cellular locales (3). SHP2 is required for full activation of the Ras/ERK1/2 pathway, a major signaling cascade in cancer biology. Although there is general agreement that SHP2 acts downstream of growth factor receptors and upstream of Ras, the mechanistic underpinnings of how SHP2 mediates Ras/ERK1/2 activation remains an active area of research. Current models suggest that SHP2 fulfills its positive role in signaling by dephosphorylating pTyr residues, such as the RasGAP-binding site on receptor tyrosine kinases, the CSK-binding sites on paxillin and PAG/CBP, or the Grb2/SOS binding site on Sprouty1 (Fig. 1B), which negatively regulate Ras/ERK1/2 pathway activation (4). Most recent data indicate that dephosphorylation of Ras/pTyr32 by SHP2 can also lead to Ras activation (5). In addition to the Ras/ERK1/2 pathway, SHP2 has also been shown to promote PI3K/AKT, JAK/STAT, JNK, and NF-κB signaling (Fig. 1B), which are strongly associated with various human cancers (3, 4, 6).
Figure 1.
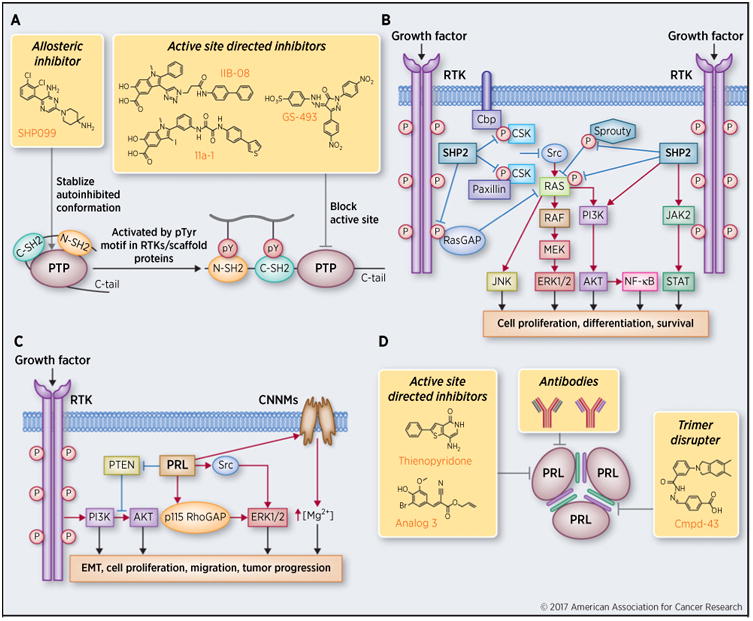
Signaling mechanisms and therapeutic targeting of SHP2 and PRLs. A, The schematic structure of SHP2, strategies of targeting SHP2 for cancer therapy, and the latest small-molecule SHP2 inhibitors. B, SHP2 promotes multiple oncogenic signaling pathways. C, PRL-mediated signaling in cancer. D, Strategies of targeting PRLs for cancer therapy and representative PRL inhibitors. EMT, epithelial–mesenchymal transition.
Genetic and clinical studies have linked SHP2 with many human diseases including cancers. Germline gain-of-function PTPN11 mutations cause approximately 40% to 50% of Noonan syndrome, an autosomal dominant developmental disorder with increased risk of malignancy. Somatic gain-of-function PTPN11 mutations are broadly associated with leukemia (including juvenile myelomonocytic leukemia) and various solid tumors including lung adenocarcinoma, colon cancer, neuroblastoma, melanoma and hepatocellular carcinoma (4, 6, 7). Recent evidence also show that the leukemia associated PTPN11 E76K mutation in the bone marrow microenvironment promotes the development and progression of myeloproliferative neoplasm (8). Although PTPN11 mutations are less frequently observed in solid tumors than in leukemia, SHP2 overexpression is common in many types of carcinoma. For example, SHP2 is overexpressed in approximately 80% of HER2+ ERα/PR+ infiltrating ductal carcinoma, and elevated SHP2 level in breast cancer tissues is positively correlated with lymph node metastasis and higher tumor grade (9). Furthermore, SHP2 signature genes are overexpressed in approximately 55% of human primary breast tumors and correlated with invasive ductal carcinoma and poor prognosis (10). In addition, SHP2 is positively correlated with EGFR expression in breast cancers (11), consistent with the obligatory requirement of SHP2 in receptor tyrosine kinase signaling. SHP2 is also upregulated in human prostate cancer and positively correlates with advanced disease stage, tumor metastasis and shortened patient survival (12). In fact, increased SHP2 expression is also found in gastric, oral, lung, head and neck, thyroid, liver, pancreatic cancers and melanoma (13). Taken together, there is substantial evidence to support SHP2 as a bona fide oncoprotein in the PTP family.
Proof of Concept for Targeting SHP2
Given the obligatory requirement of SHP2 in multiple growth factor–mediated oncogenic pathways and the involvement of SHP2 in leukemia and solid tumors, inhibition of SHP2 is expected to have broad therapeutic applications in oncology. Moreover, since SHP2 is directly downstream of growth factor receptors, targeting oncogenic receptor tyrosine kinase pathways at the level of SHP2 may offer unique advantages in receptor tyrosine kinase drug resistance settings. Indeed, a recent study indicates that SHP2 is a central node in intrinsic and acquired resistance to tyrosine kinase–targeted cancer drugs (14). Herein, we will focus on the latest pharmacological studies furnishing proof-of-concept validation of targeting SHP2 for novel anti-cancer agents. Compound 11a-1 (Fig. 1A) is an indole salicylic acid inhibitor (IC50 = 200 nmol/L) that binds the SHP2 active site and a nearby unique peripheral pocket (15). It displays >5-fold preference for SHP2 over 20 other PTPs. Notably, 11a-1 efficaciously attenuates cell viability and blocks EGF-induced ERK1/2 activation in H1975 lung cancer cells; abrogates ERK1/2 and AKT activation in SKBR3 breast cancer cells and suppresses cell growth in a 3D Matrigel environment; and inhibits oncogenic KITD814V-induced constitutive cell growth. More recent data show that 11a-1 can also effectively block SHP2-mediated ERK1/2 and AKT activation and attenuates melanoma cell viability, migration and colony formation, and significantly suppress both MeWo and B16F10 melanoma xenograft growth (13). Strong in vivo efficacy of IIB-08 (16), a precursor of 11a-1, was also demonstrated in mast cell leukemia (17) and an orthotopic intracranial GBM xenograft mouse model (5), indicating that this class of compounds can cross the blood–brain barrier. GS-493 (Fig. 1A) is an aryl sulfonic acid–based active site directed inhibitor with an IC50 value of 71 nmol/L for SHP2, and a 29- and 45-fold selectivity over SHP1 and PTP1B, respectively (18). GS-493 blocks HGF/SF induced scattering of human HPAF II pancreatic cancer cells, and attenuates growth of lung cancer cell line LXFA 526L in soft agar and in a nude mouse xenograft model (18). Most recently, an allosteric SHP2 inhibitor SHP099 (Fig. 1A) was developed by targeting the unique SHP2 molecular switch mechanism (19). The X-ray crystal structure of SHP2·SHP099 complex revealed that SHP099 occupies a tunnel at the junction of the N-SH2/C-SH2/PTP domains, serving as molecular glue that holds SHP2 in the autoinhibited closed conformation. SHP099 inhibits SHP2 activation with an IC50 value of 70 nmol/L and exhibits no detectable activity against 21 PTPs and 66 kinases. SHP099 suppresses pERK1/2 in MDA-MB-468 and KYSE520 cells with high EGFR amplification, but not in A2058 cells bearing a K-Ras mutation. When administered by oral gavage at 100 mg/kg daily to nude mice harboring KYSE520 xenografts, SHP099 exhibited significant antitumor activity over a 24-day period. In an orthotopic human-primary-tumor–derived FLT3-ITD acute myeloid leukemia model, SHP099 treatment by 75 mg/kg daily oral administration led to a near-complete eradication of circulating CD45+ leukemic cells and significantly reduced splenomegaly in the mice. Collectively, these promising pharmacological results obtained with SHP2 inhibitors of diverse structural scaffolds and distinct mode of action strongly support the validity of therapeutic targeting of SHP2 for various cancers.
Roles of PRLs in Cancer
PRL1 was first discovered as an immediate early gene during liver regeneration after partial hepatectomy (20). PRL2 and PRL3 were identified based on their high sequence homology with PRL1. The PRLs have a PTP catalytic domain and a characteristic C-terminal prenylation CAAX motif that enables their plasma membrane association (20). Among the PRLs, PRL3 was first found to be involved in colorectal cancer metastasis, and was subsequently reported to be involved in the progression and metastasis of several other types of human cancer (20). For example, it was determined that PRL3 is strikingly overexpressed in human metastatic colorectal cancer samples but not in normal, benign or primary cancer tissue and is thought to contribute to the establishment of liver and lung metastases in particular (21, 22). Overexpression of PRLs is associated with increased cell proliferation and cell migration and also correlated with late-stage metastasis and poor clinical outcomes, whereas knock-down of PRLs reduces cell proliferation and cell migration (20). Indeed, the PRLs have been proposed as novel prognostic markers and drivers for breast, lung, liver, skin, gastric, and ovarian cancers (23–25). Recent studies also demonstrated that PRL2 is important for the leukemogenic potential of oncogenic NOTCH1 in vivo (26). In cell culture studies, upregulation of the PRLs promotes activation of both ERK1/2 and PI3K/AKT pathway (Fig. 1C; ref. 20). In addition, PRL3 can activate the Src kinase and regulate Rho family GTPases to promote invasion, proliferation and motility (Fig. 1C; ref. 20).Furthermore, PRL3 has also been shown to stimulate epithelial–mesenchymal transition through downregulation of the tumor-suppressor PTEN (Fig. 1C; ref. 27). Interestingly, recent studies show that PRLs can also regulate intracellular magnesium homeostasis through direct binding interaction with the magnesium transporters of the cyclin M (CNNM) family of proteins (Fig. 1C; refs. 28–31). To uncover the in vivo function of PRLs, gene knockout was employed to study the effect of PRL deletion at the organismal level. Mice deficient in either PRL1 or PRL3 are grossly normal (32, 33). In contrast, mice lacking PRL2, the most ubiquitously and abundantly expressed PRL family member, display placenta insufficiency (34), impaired spermatogenesis (35), and defects in hematopoietic stem cell self-renewal (36). Mechanistically, these phenotypes are caused by decreased AKT activity as a result of increased PTEN level (33–36). Consequently, PRLs may serve as oncogenes by promoting AKT activation through downregulation of PTEN. PTEN is one of the most frequently inactivated tumor suppressors that antagonizes the PI3K/AKT pathway by dephosphorylating phosphatidylinositol (3,4,5)-triphosphate (37). Importantly, PTEN levels are frequently found downregulated in cancer patients in the absence of genetic loss or mutation, and PTEN deficiency can increase cancer risk in a dose-dependent manner (37). Therefore, therapeutic targeting of the PRLs may constitute a novel strategy to combat cancer by restoring PTEN, especially in tumors induced by partial PTEN loss. Further study of the role of PRL in promoting PTEN down-regulation has both merit in cancer biology and relevance to therapeutic advancement. Therefore, PRLs are compelling diagnostic markers and promising therapeutic targets in cancer.
Proof of Concept for Targeting the PRL Phosphatases
PRLs have been suggested as potential therapeutic targets for cancer treatment, and pharmacological inhibition of PRLs has attracted attention in the PTP field. To date, several PRL inhibitors have been reported. Thienopyridone (Fig. 1D) potently and selectively inhibited all three PRLs in vitro with IC50 values of 173, 277, and 128 nmol/L for PRL1, PRL2, and PRL3, respectively (38). Thienopyridone can induce cancer cell anoikis, which is a type of programmed cell death initiated by p130Cas cleavage. Thienopyridone also prevents the association of PRLs with CNNMs (39). Most recently, a more potent derivative of thienopyridone, Analog 13, has been developed, which showed IC50 values of 50, 52 and 18 nmol/L against PRL1, PRL2, and PRL3 respectively (40). Analog 3 is an easily accessible, low micromolar PRL inhibitor identified through virtual screening and structural activity relationship efforts and has proven useful for florescence microscopy and cellular based studies (Fig. 1D; ref. 41). Given the functional requirement of PRL trimerization, pharmacologic disruption of PRL trimer formation represents an innovative approach for the treatment of human cancers with elevated PRL expression (42). To overcome the specificity issue associated with the active site inhibitors, Cmpd-43 was identified as a specific binder of the PRL trimer interface, which is a unique feature of the PRL phosphatases (Fig. 1D; ref. 42). Cmpd-43 blocks PRL trimerization and inhibits the PRL-mediated cell proliferation and migration through attenuation of both ERK1/2 and AKT activity. Importantly, Cmpd-43 exhibits excellent anticancer activity both in vitro and in a xenograft mouse model of melanoma. These results provide pharmacological support that trimerization is important for PRL function and targeting PRL trimerization represents another example of exploiting the regulatory mechanisms of the PTPs for therapeutic development. Monoclonal antibodies are an attractive cancer therapy owing to better specificity and efficacy compared to standard small-molecule inhibitors. Most recently, a novel PRL3 antibody that showed strong antitumor activity in vitro and in vivo has been developed, although the mechanism by which this antibody targets intracellular PRL3 is not completely understood (Fig. 1D; ref. 43). Together, studies with small-molecule inhibitors and biologics further validate PRLs as viable targets for cancer drug discovery and provide a solid foundation upon which novel PRL-based–targeted anticancer agents can be developed.
Outlook
The evidence summarized herein expose oncogenic roles of SHP2 and PRLs in many cancers, raising the possibility that inhibition of these phosphatases may have broader therapeutic applications in oncology. Available pharmacological data with small-molecule inhibitors and biologics also highlight the promise of targeting SHP2 and PRLs for drug discovery in cancer. In addition to the development of SHP2-targeted monotherapy using more specific and potent inhibitors, the combination of SHP2 inhibitor with existing kinase-targeted drugs to improve the curative effect and/or combat drug resistance should be another focus of attention. The rapid emergence of drug resistance has largely limited the therapeutic benefit in targeted cancer therapy. Usually, mutation of the target itself or activation of alternate oncogenic signaling pathway confer drug resistance. Given that SHP2 acts downstream of most receptor tyrosine kinases to activate Ras, a key node of multiple oncogenic signaling pathways, inhibition of SHP2 is anticipated to be effective in suppressing the reactivated signals. Moreover, immunotherapy is an emerging strategy to fight cancers. SHP2 is also implicated in the suppression of the TCR signaling pathway after the PD-1 receptor is engaged. It will be important to further define the precise role of SHP2 in PD-1 signaling pathway and evaluate whether a SHP2 inhibitor can boost the immune responses against cancer. As for the PRLs, the foundation for more innovative approaches to target the PRLs has been laid. Exciting advances in gaining specificity have been made by exploiting humanized antibodies or a unique structural feature of this PTP family, trimer formation. Furthermore, given the role of PRL in downregulating the PTEN tumor suppressor, tumors initiated by a loss of PTEN may provide an ideal opportunity to target PRL to abrogate cancer progression. We anticipate that the future could invite the development of more potent and isoform specific compounds to individually target each PRL protein. Identification of PRL substrates will be essential for a better understanding of their roles in tumorigenesis and for evaluation of PRL target engagement in drug discovery.
Acknowledgments
We gratefully acknowledge the financial support of NIH grants RO1 CA69202 and RO1 CA207288.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Julien SG, Dubé N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 2.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 3.Chan G, Neel BG. Role of PTPN11 (SHP2) in cancer. In: Neel BGand-Tonks NK, editor. Protein tyrosine phosphatases in cancer. New York: Springer Science+Business Media; 2016. pp. 115–43. [Google Scholar]
- 4.Huang WQ, Lin Q, Zhuang X, Cai LL, Ruan RS, Lu ZX, et al. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr Cancer Drug Targets. 2014;14:567–88. doi: 10.2174/1568009614666140717105001. [DOI] [PubMed] [Google Scholar]
- 5.Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun. 2015;6:8859. doi: 10.1038/ncomms9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;2:179–92. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 7.Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007;3:862–7. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–8. doi: 10.1038/nature20131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopath. 2008;53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x. [DOI] [PubMed] [Google Scholar]
- 10.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. 2012;18:529–37. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- 11.Matalkah F, Martin E, Zhao H, Agazie YM. SHP2 acts both upstream and downstream of multiple receptor tyrosine kinases to promote basal-like and triple-negative breast cancer. Breast Cancer Res. 2016;18:2. doi: 10.1186/s13058-015-0659-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang K, Zhao H, Ji Z, Zhang C, Zhou P, Wang L, et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene. 2016;35:1271–82. doi: 10.1038/onc.2015.184. [DOI] [PubMed] [Google Scholar]
- 13.Zhang RY, Yu ZH, Zeng L, Zhang S, Bai Y, Miao J, et al. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget. 2016;7:73817–29. doi: 10.18632/oncotarget.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, et al. PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep. 2015;12:1978–85. doi: 10.1016/j.celrep.2015.08.037. [DOI] [PubMed] [Google Scholar]
- 15.Zeng LF, Zhang RY, Yu ZH, Li S, Wu L, Gunawan AM, et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. 2014;57:6594–609. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, et al. Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) J Med Chem. 2010;53:2482–93. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mali RS, Ma P, Zeng LF, Martin H, Ramdas B, He Y, et al. Role of SHP2 phosphatase in KIT induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood. 2012;120:2669–78. doi: 10.1182/blood-2011-08-375873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grosskopf S, Eckert C, Arkona C, Radetzki S, Böhm K, Heinemann U, et al. Selective inhibitors of the protein tyrosine phosphatase SHP2 block cellular motility and growth of cancer cells in vitro and in vivo. Chem-MedChem. 2015;10:815–26. doi: 10.1002/cmdc.201500015. [DOI] [PubMed] [Google Scholar]
- 19.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–52. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 20.Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–52. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 21.Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–6. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 22.Bardelli A, Saha S, Sager JA, Romans KE, Markowitz SD, Lengauer C, et al. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003;15:5607–15. [PubMed] [Google Scholar]
- 23.den Hollander P, Rawls K, Tsimelzon A, Shephard J, Mazumdar A, Fuqua SA, et al. Phosphatase PTP4A3 promotes triple-negative breast cancer growth and predicts poor patient survival. Cancer Res. 2016;76:1942–53. doi: 10.1158/0008-5472.CAN-14-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gari HH, Gearheart CM, Fosmire S, DeGala GD, Fan Z, Torkko KC, et al. Genome-wide functional genetic screen with the anticancer agent AMPI-109 identifies PRL-3 as an oncogenic driver in triple-negative breast cancers. Oncotarget. 2016;7:15757–71. doi: 10.18632/oncotarget.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Peng L, Dong B, Kong L, Meng L, Yan L, et al. Overexpression of phosphatase of regenerating liver-3 in breast cancer: association with a poor clinical outcome. Ann Oncol. 2006;17:1517–22. doi: 10.1093/annonc/mdl159. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi M, Bai Y, Chen S, Gao R, Yao C, Cai W, et al. Phosphatase PRL2 promotes oncogenic NOTCH1-Induced T-cell leukemia. Leukemia. 2017;31:751–4. doi: 10.1038/leu.2016.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–6. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 28.Hardy S, Uetani N, Wong N, Kostantin E, Labbe DP, Begin LR, et al. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene. 2015;34:986–95. doi: 10.1038/onc.2014.33. [DOI] [PubMed] [Google Scholar]
- 29.Funato Y, Yamazaki D, Mizukami S, Du L, Kikuchi K, Miki H. Membrane protein CNNM4-dependent Mg2+ efflux suppresses tumor progression. J Clin Invest. 2014;124:5398–410. doi: 10.1172/JCI76614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulerez I, Funato Y, Wu H, Yang M, Kozlov G, Miki H, et al. Phospho-cysteine in the PRL-CNNM pathway mediates magnesium homeostasis. EMBO Rep. 2016;17:1890–900. doi: 10.15252/embr.201643393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giménez-Mascarell P, Oyenarte I, Hardy S, Breiderhoff T, Stuiver M, Kostantin E, et al. Structural basis of the oncogenic interaction of phosphatase PRL-1 with the magnesium transporter CNNM2. J Biol Chem. 2017;292:786–801. doi: 10.1074/jbc.M116.759944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmerman MW, Homanics GE, Lazo JS. Targeted deletion of the metastasis-associated phosphatase Ptp4a3 (PRL-3) suppresses murine colon cancer. PLoS One. 2013;8:e58300. doi: 10.1371/journal.pone.0058300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai Y, Zhou HM, Zhang L, Dong Y, Zeng Q, Shou W, et al. Role of Phosphatase of Regenerating Liver 1 (PRL-1) in spermatogenesis. Sci Rep. 2016;6:34211. doi: 10.1038/srep34211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong Y, Zhang L, Zhang S, Bai Y, Chen H, Sun X, et al. Phosphatase of regenerating liver 2 (PRL2) is essential for placental development by down-regulating PTEN (Phosphatase and Tensin Homologue Deleted on Chromosome 10) and activating Akt protein. J Biol Chem. 2012;287:32172–9. doi: 10.1074/jbc.M112.393462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong Y, Zhang L, Bai Y, Zhou HM, Campbell AM, Chen H, et al. Phosphatase of regenerating liver 2 (PRL2) deficiency impairs Kit signaling and spermatogenesis. J Biol Chem. 2014;289:3799–810. doi: 10.1074/jbc.M113.512079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi M, Bai Y, Dong Y, Chen S, Gao R, Zhang L, et al. PRL2/PTP4A2 phosphatase is important for hematopoietic stem cell self-renewal. Stem Cells. 2014;32:1956–7. doi: 10.1002/stem.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daouti S, Li WH, Qian H, Huang KS, Holmgren J, Levin W, et al. A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130Cas cleavage. Cancer Res. 2008;68:1162–9. doi: 10.1158/0008-5472.CAN-07-2349. [DOI] [PubMed] [Google Scholar]
- 39.Kostantin E, Hardy S, Valinsky WC, Kompatscher A, de Baaij JH, Zolotarov Y, et al. Inhibition of PRL-2·CNNM3 protein complex formation decreases breast cancer proliferation and tumor growth. J Biol Chem. 2016;291:10716–25. doi: 10.1074/jbc.M115.705863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salamoun JM, McQueeney KE, Patil K, Geib SJ, Sharlow ER, Lazo JS, et al. Photooxygenation of an amino-thienopyridone yields a more potent PTP4A3 inhibitor. Org Biomol Chem. 2016;14:6398–402. doi: 10.1039/c6ob00946h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoeger B, Diether M, Ballester PJ, Köhn M. Biochemical evaluation of virtual screening methods reveals a cell-active inhibitor of the cancer-promoting phosphatases of regenerating liver. Eur J Med Chem. 2014;88:89–100. doi: 10.1016/j.ejmech.2014.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bai Y, Yu ZH, Liu S, Zhang L, Zhang RY, Zeng LF, et al. Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 2016;76:4805–15. doi: 10.1158/0008-5472.CAN-15-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thura M, Al-Aidaroos AQ, Yong WP, Kono K, Gupta A, Lin YB, et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight. 2016;1:e87607. doi: 10.1172/jci.insight.87607. [DOI] [PMC free article] [PubMed] [Google Scholar]