

Abstract
The gastrointestinal (GI) epithelium is the fastest renewing adult tissue and is maintained by tissue-specific stem cells. Treatment-induced GI side effects are a major dose-limiting factor for chemotherapy and abdominal radiotherapy and can decrease the quality of life in cancer patients and survivors. p53 is a key regulator of the DNA damage response, and its activation results in stimulus- and cell type–specific outcomes via distinct effectors. We demonstrate that p53-dependent PUMA induction mediates chemotherapy-induced intestinal injury in mice. Genetic ablation of Puma, but not of p53, protects against chemotherapy-induced lethal GI injury. Blocking chemotherapy- induced loss of LGR5+ stem cells by Puma KO or a small-molecule PUMA inhibitor (PUMAi) prevents perturbation of the stem cell niche, rapid activation of WNT and NOTCH signaling, and stem cell exhaustion during repeated exposures. PUMAi also protects human and mouse colonic organoids against chemotherapy-induced apoptosis and damage but does not protect cancer cells in vitro or in vivo. Therefore, targeting PUMA is a promising strategy for normal intestinal chemoprotection because it selectively blocks p53-dependent stem cell loss but leaves p53-dependent protective effects intact.
INTRODUCTION
Gastrointestinal (GI) side effects are a major dose-limiting factor in chemotherapy and abdominal radiotherapy and can cause long-term complications in cancer survivors (1, 2). Colorectal cancer patients are commonly treated with irinotecan hydrochloride (CPT-11) in combination with 5-fluorouracil (5-FU). Radiation and most chemotherapeutic agents damage multiple tissues and organ systems, whereas CPT-11 causes selective GI injury, with severe diarrhea and nausea in more than 50% of patients (3). This acute toxicity is caused by the accumulation of high concentrations of SN-38, the active metabolite of CPT-11, in the intestine (4), as well as the conversion of nontoxic SN-38 glucuronide back to SN-38 by gut bacteria, followed by intestinal reabsorption (5). SN-38 is an inhibitor of topoisomerase I, a key enzyme in both DNA replication and RNA transcription, and causes replication stress and DNA damage in proliferating cells (6). Anti-diarrhea medication can help alleviate CPT-11–induced diarrhea but has a limited effect on reducing long-term GI dysfunction (1). There is currently no U.S. Food and Drug Administration–approved agent that prevents or treats CPT-11–induced GI injury or complications.
Chemotherapy or radiation-induced acute enteropathy is characterized by loss of proliferating crypt cells, epithelial barrier breakdown, and inflammation during or shortly after the treatment. In many patients, delayed enteropathy can occur months or later after therapy and is characterized by intestinal dysfunction associated with pathological changes in the epithelial and stromal compartments, such as vascular sclerosis and progressive intestinal wall fibrosis. Emerging evidence suggests that chronic intestinal damage is likely a consequence of early toxicity (1, 7). Enterotoxicity caused by chemotherapeutics, such as CPT-11 and 5-FU, is associated with rapid crypt apoptosis in mice and humans (8, 9). The small intestinal epithelium is the fastest renewing adult tissue, and intestinal stem cells (ISCs) located in the bottom of crypts drive the renewal and regeneration after injury. ISCs include the columnar cells at the crypt bottom (CBCs) and some cells at position 4 relative to the crypt bottom (+4 cells) (10, 11). Genetic ablation of leucine-rich repeat-containing G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor 5–expressing (LGR5+) CBCs is well tolerated in healthy mice but strongly exacerbates radiation-induced intestinal injury, although underlying mechanisms remain unknown (12).
Activation of p53 after DNA damage results in tissue- and target- specific outcomes such as tumor suppression or acute toxicity (13–16). p53 activation is a double-edged sword in controlling intestinal regeneration after high-dose radiation. On one hand, p53-dependent PUMA induction accounts for most radiation-induced apoptosis and acute loss of intestinal and hematopoietic stem and progenitor cells, and PUMA deficiency or down-regulation protects against radiation-induced lethality (17–20) and lymphomagenesis (21, 22). However, p53-dependent induction of p21, and perhaps other targets, is essential for productive intestinal regeneration by preventing DNA damage accumulation, replication stress, and delayed nonapoptotic cell death, and p53/p21 deficiency exacerbates intestinal injury (20, 23). The role of p53 in chemotherapy-induced GI injury is not well understood. The p53 inhibitor pifithrin-α reduced CPT-11–induced intestinal apoptosis within the first several hours in rats but did not affect delayed cell death or the onset of mucositis (8).
Here, we sought to determine the role of p53-dependent apoptosis in chemotherapy-induced intestinal injury. We demonstrated that genetic or pharmacological inhibition of Puma by a small-molecule PUMA inhibitor (PUMAi), but not p53 deficiency, provides potent protection against GI injury induced by single and repeated exposures to CPT-11 but does not compromise its antitumor activity in vivo. This protection is associated with selective preservation of LGR5+ stem cells, stem cell niche, and genome integrity. Our findings provide a mechanism-based approach and a compound for intestinal chemoprotection through selective inhibition of p53-dependent apoptosis and enterotoxicity.
RESULTS
PUMA mediates chemotherapy-induced crypt apoptosis and lethality
To investigate a potential role of p53-dependent apoptosis in chemotherapy-induced acute intestinal injury, we treated wild-type (WT), p53 knockout (KO), and Puma KO mice with a single dose of irinotecan (200 mg/kg; CPT-11). CPT-11 induced robust crypt apoptosis in WT mice, as measured by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) and active caspase-3 staining, which peaked at 6 hours and decreased gradually by 48 hours. Apoptosis was observed in both the stem cells at the crypt base (CBCs at positions 1 to 3 relative to the crypt bottom) and +4 to 9 cells (relative to the crypt bottom), but it was abrogated in p53 KO and Puma KO mice (Fig. 1, A and B, and fig. S1, A to D). CPT-11 treatment induced p53 and its targets PUMA and p21 within 6 hours in the intestinal mucosa of WT but not p53 KO mice (Fig. 1C and fig. S1E). Puma KO did not significantly affect the expression of p53 or p21 (Fig. 1C). BH3-only proteins BIM and NOXA were not induced by CPT-11 or affected by Puma KO (fig. S1F). RNA in situ hybridization (ISH) confirmed that Puma mRNA was undetectable in the crypts of untreated WT mice and strongly induced in the CBCs and some +4 cells by CPT-11 treatment (Fig. 1D).
Fig. 1. PUMA mediates CPT-11–induced intestinal injury.
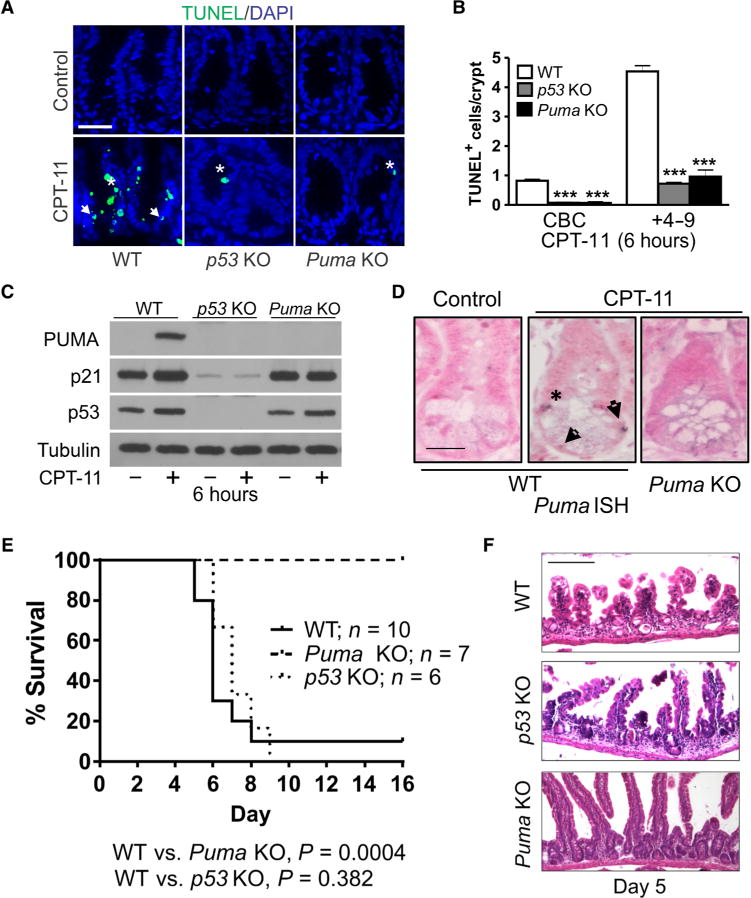
Mice with indicated genotypes were treated with CPT-11 (200 mg/kg) once or as specified and analyzed at indicated times. (A) Representative images of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining in intestinal crypts at 6 hours. DAPI (4′,6-diamidino-2-phenylindole) was used to stain nuclei. Scale bar, 50 μm. (B) Quantitation of TUNEL+ columnar cells at the crypt bottom (CBCs) and +4 to 9 cells in (A). (C) Expression of indicated proteins was analyzed by Western blotting at 6 hours. Lysates were pooled from the intestinal mucosa of three mice. Representative results are shown, and similar results were obtained in at least three independent experiments. (D) Puma RNA in situ hybridization (ISH) in the crypts at 6 hours. Scale bar, 20 μm. In (A) and (D), arrows indicate CBCs at positions 1 to 3 below Paneth cells, and asterisks indicate +4 to 9 cells at positions 4 to 9 above the CBCs. In (A) to (D), n = 3 mice per group. In (B), values are means + SEM. ***P < 0.001 (two-tailed Student’s t test), knockout (KO) versus wild type (WT). (E) Survival of mice treated with three consecutive daily doses of CPT-11 (215 mg/kg per day) on days 0, 1, and 2. WT versus Puma KO, P = 0.0004; WT versus p53 KO, P = 0.382 (log-rank test). (F) Hematoxylin and eosin (H&E) staining of the small intestine from mice on day 5 treated as in (E). Scale bar, 200 μm.
CPT-11 causes dose-dependent lethal GI injury in mice (24). To determine the sensitivity of C57BL/6J mice, we monitored survival after three consecutive daily doses of CPT-11. The 180, 215, and 250 mg/kg per day doses resulted in 30, 90, and 100% lethality, respectively, and all deaths occurred between days 5 and 8, characteristic of lethal GI injury (fig. S2). At the 90% lethal dose (LD90) of WT mice (215 mg/kg per day for 3 days), all Puma KO mice survived for 30 days, whereas all p53 KO mice died between days 6 and 9 (Fig. 1E). Histological analysis confirmed lethal GI injury in WT and p53 KO mice on day 5, showing severe loss of crypts and shortening of villi, which was abrogated in Puma KO mice (Fig. 1F). Consistent with a dual role of p53 in intestinal recovery from radiation (20), our data demonstrate that selective inhibition of p53-dependent induction of PUMA and apoptosis, but not p21 or other functions, results in intestinal chemoprotection.
A small-molecule PUMAi protects against chemotherapy-induced crypt apoptosis and lethality
We previously identified a number of potential PUMAis using a pharmacophore model of the key interactions of the PUMA BH3 domain with BCL-2 family proteins, in silico compound screening, and apoptosis assays (25, 26). One of these compounds (PUMAi) was confirmed to inhibit interaction of PUMA/BCL-xL but not of BIM/MCL-1 (fig. S3, A to C). PUMAi did not reduce camptothecin (CPT)–induced apoptosis in colon cancer cell lines with different p53 or PUMA genotypes, including p53 WT (HCT 116), null (p53 KO), mutant (HT 29, SW480, and DLD1), or PUMA null (PUMA KO) (fig. S3D). PUMA was only induced by CPT treatment in p53 WT HCT 116 cells, not in any p53-deficient line (fig. S3E) (27–29). The basal amount of PUMA was much higher in HCT 116 cells, compared to very low or undetectable expression in mouse intestinal mucosa (Fig. 1, C and D) (17) and in normal human small intestine and colon (30).
To examine the efficacy of PUMAi in vivo, we treated WT mice with vehicle or PUMAi (10 mg/kg, an empirical dose) 2 hours before a single dose of CPT-11 (200 mg/kg) and analyzed crypt apoptosis 6 and 24 hours after CPT-11 administration. PUMAi reduced TUNEL and active caspase-3 staining preferentially in the CBCs, compared to transit-amplifying (TA), +4 to 9, cells (Fig. 2, A and B, and fig. S4, A to D). PUMAi treatment did not further reduce crypt apoptosis in Puma KO mice (fig. S4E). We then developed a high-performance liquid chromatography (HPLC) assay and assessed PUMAi distribution in the intestinal mucosa and plasma at three time points, −1.5, +1, and +6 hours of CPT-11 administration (0 hour) (fig. S4F). PUMAi concentration remained at 67 and 30% in the intestinal mucosa at +1 and +6 hours, respectively, compared to −1.5 hours, but dropped more quickly in the plasma (Fig. 2C). CPT-11 is rapidly cleared from the plasma, with a half-life around 36 min (31). We found that PUMAi given 1 hour after CPT-11 treatment still reduced apoptosis effectively, suggesting that PUMAi does not affect CPT-11 conversion or intestinal absorption (Fig. 2D).
Fig. 2. PUMAi protects against CPT-11–induced intestinal injury.
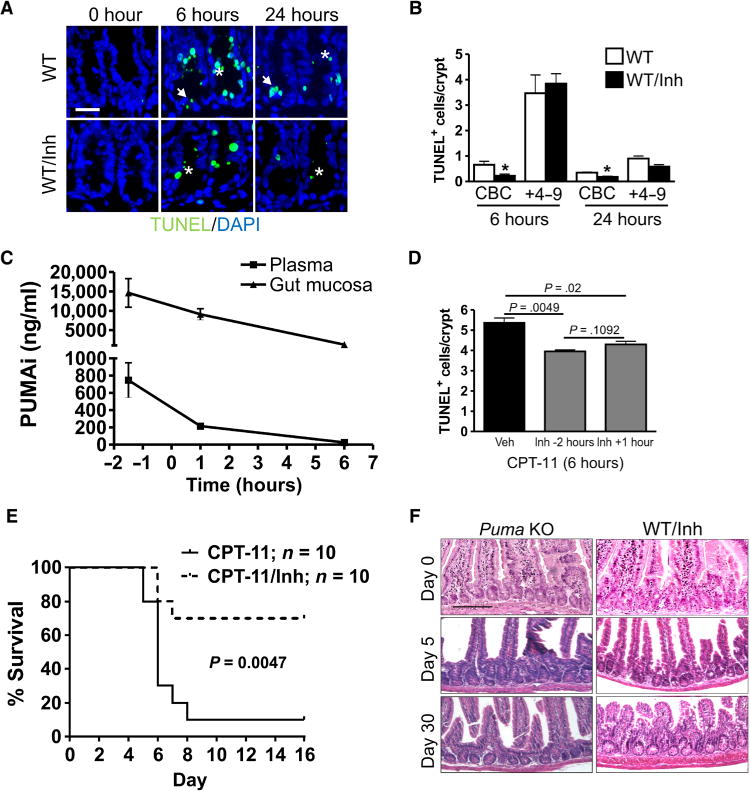
Mice were treated with CPT-11 with or without PUMA inhibitor (PUMAi). PUMAi (10 mg/kg) or vehicle was given intraperitoneally for 2 hours before CPT-11 or as specified. The small intestine or indicated tissue was analyzed at the indicated times. (A) Representative images of TUNEL staining in intestinal crypts of mice treated with CPT-11 (200 mg/kg). DAPI was used to stain nuclei. Scale bar, 50 μm. Arrows indicate CBCs, and asterisks indicate +4 to 9 cells. (B) Quantitation of TUNEL+ CBCs and transit-amplifying cells from (A). Inh, inhibitor. (C) Tissue distribution of PUMAi at different times after a single injection. (D) Quantitation of TUNEL+ crypt cells in mice 6 hours after CPT-11 (200 mg/kg) treatment. PUMAi (10 mg/kg) was given 2 hours before or 1 hour after CPT-11 treatment. n = 3 mice per group (B to D). In (B) and (D), values are means + SEM. *P < 0.05 (two-tailed Student’s t test). Veh, vehicle. (E) Survival of WT mice after three doses of CPT-11 (215 mg/kg per day) (done with the control in Fig. 1E). PUMAi (10 mg/kg) was given 2 hours before each dose of CPT-11. P = 0.0047 (log-rank test). (F) H&E staining of the small intestine from mice treated as in (E) on days 0, 5 and 30. Scale bar, 200 μm.
Further, PUMAi decreased CPT-11–induced lethality at three different doses (Fig. 2E and fig. S5, A and B). At the LD90, PUMAi improved survival from 10 to 70%. Histological analysis confirmed reduced GI injury on day 5 in the PUMAi and Puma KO groups, compared to WT mice (Figs. 1F and 2F). PUMAi also increased the survival of mice after 9.5- and 15-gray total body irradiation (fig. S5, C and D), further supporting the idea that targeting p53-dependent apoptosis, but not other functions, selectively reduces treatment-induced normal tissue toxicity.
PUMA inhibition does not reduce antitumor activity of CPT-11
To determine whether PUMA inhibition provides selective intestinal chemoprotection, we established subcutaneous Lewis lung carcinoma (LLC) tumors in immunocompetent WT and Puma KO mice. After tumors reached an average of 100 mm3 (day 11), mice received six doses of CPT-11 (200 mg/kg, intraperitoneally) over 2 weeks (fig. S6A). Tumor engraftment and CPT-11 response were indistinguishable in WT and Puma KO hosts (Fig. 3A and fig. S6B). In contrast, CPT-11–induced weight loss was reduced in Puma KO mice (Fig. 3B), correlated with preserved intestinal structure and barrier evident by higher villus height, crypt numbers, and lower neutrophil infiltration (Fig. 3, C to G).
Fig. 3. PUMA deficiency protects tumor-bearing mice from chemotherapy-induced GI injury.
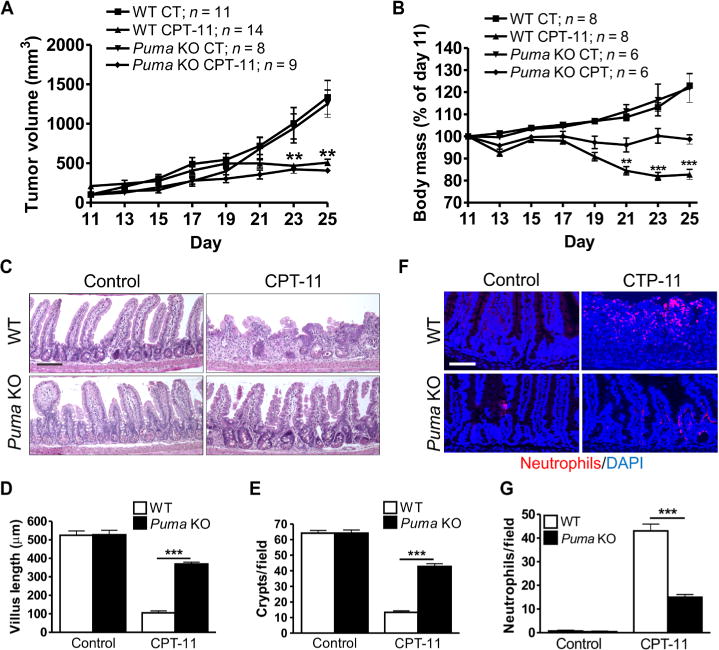
WT and Puma KO mice bearing Lewis lung carcinoma (LLC) tumors were treated with CPT-11 (200 mg/kg) six times over 15 days, on days 11, 14, 17, 19, 23, and 25. Mice or tumors were analyzed at the indicated times. (A) Tumor volumes were measured every other day from days 11 to 25. CT, vehicle control. (B) Body mass was expressed as a percentage of that on day 11 for each mouse from days 11 to 25. (C) H&E staining of the small intestine tissue on day 25. Scale bar, 100 μm. (D) Villus height from (C). Measurements were from a minimum of 40 villi per mouse. (E) Quantitation of intestinal crypts per field from (C). (F) Intestinal neutrophils detected by immunofluorescence on day 25. DAPI was used to stain nuclei. Scale bar, 100 μm. (G) Quantitation of neutrophils per 400× field from the same slides as (F). In (A), (B), (D), (E), and (G), values are means + SEM; n = indicated (A and B) or 3 to 4 mice per group (D, E, and G). **P < 0.01 and ***P < 0.001 (two-tailed Student’s t test). Puma KO, CPT versus control (A); CPT-11–treated, Puma KO versus WT (B).
The effects of PUMAi were then determined in tumor-bearing mice. PUMAi (10 mg/kg) or vehicle was given 2 hours before and 20 hours after each dose of CPT-11. The PUMAi group showed comparable tumor response compared with the vehicle group (Fig. 4A and fig. S6B) but was highly resistant to CPT-11–induced weight loss, intestinal damage (Fig. 4, B to G), and lethality (table S1). Furthermore, PUMAi or PUMA KO groups exhibited improved grooming and physical activity throughout the study based on daily inspections. LLC tumors in all three CPT-11–treated groups showed similar effects on proliferation or apoptosis (fig. S6, C and D) but lacked induction of p53, PUMA, or p21 (fig. S6E), consistent with mutant p53 (32). These studies demonstrate that PUMA inhibition attenuates CPT-11– induced enterotoxicity but not the establishment or therapeutic responses of p53 mutant tumors in immunocompetent hosts.
Fig. 4. PUMAi protects tumor-bearing mice from chemotherapy-induced GI injury.
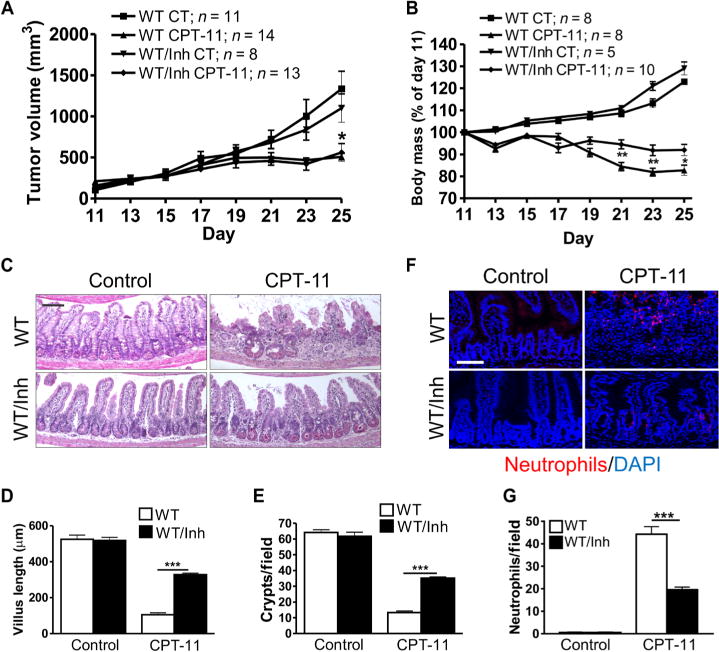
WT mice bearing LLC tumors were treated with CPT-11 (200 mg/kg) six times over 15 days on days 11, 14, 17, 19, 23, and 25. Vehicle or PUMAi was given 2 hours before and 20 hours after each dose of CPT-11. Mice or tumors were analyzed at the indicated times. (A) Tumor volumes were measured starting on day 11. (B) Body mass was expressed as a percentage of that on day 11 for each mouse from days 11 to 25. (C) H&E staining of the small intestine tissue on day 25. Scale bar, 100 μm. (D) Villus height from (C). Measurements were from a minimum of 40 villi per mouse. (E) Quantitation of intestinal crypts per field from (C). (F) Intestinal neutrophils detected by immunofluorescence on day 25. DAPI was used to stain the nuclei. Scale bar, 100 μm. (G) Quantitation of neutrophils per 400× field from the same slides as (F). In (A), (B), (D), (E), and (G), values are means + SEM; n = indicated (A and B) or 3 to 4 mice per group (D, E, and G). *P < 0.05, **P < 0.01, and ***P < 0.001 (two-tailed Student’s t test). PUMAi, CPT versus control (A); CPT-11–treated mice, PUMAi versus CT (B).
Targeting PUMA potently protects the LGR5+ stem cells and niche against chemotherapy
Little is known about the role of the ISC niche in injury-induced regeneration. We hypothesized that the loss of CBCs, not of more chemosensitive TA cells, sends “injury” signals via niche “emptying” to activate remaining stem cells. PUMAi provides a pharmacological way to test this because it selectively blocks CPT-11–induced CBC apoptosis (Fig. 2B). Using LGR5–EGFP (enhanced green fluorescent protein) reporter mice, we found that Puma KO or PUMAi decreases CPT-11–induced apoptosis of LGR5+ cells by more than 90 or 70%, respectively (Fig. 5, A and B). Puma KO also blocked LGR5+ cell apoptosis induced by another commonly used chemotherapy drug, 5-FU (fig. S7).
Fig. 5. Targeting PUMA protects the LGR5+ stem cells against CPT-11.
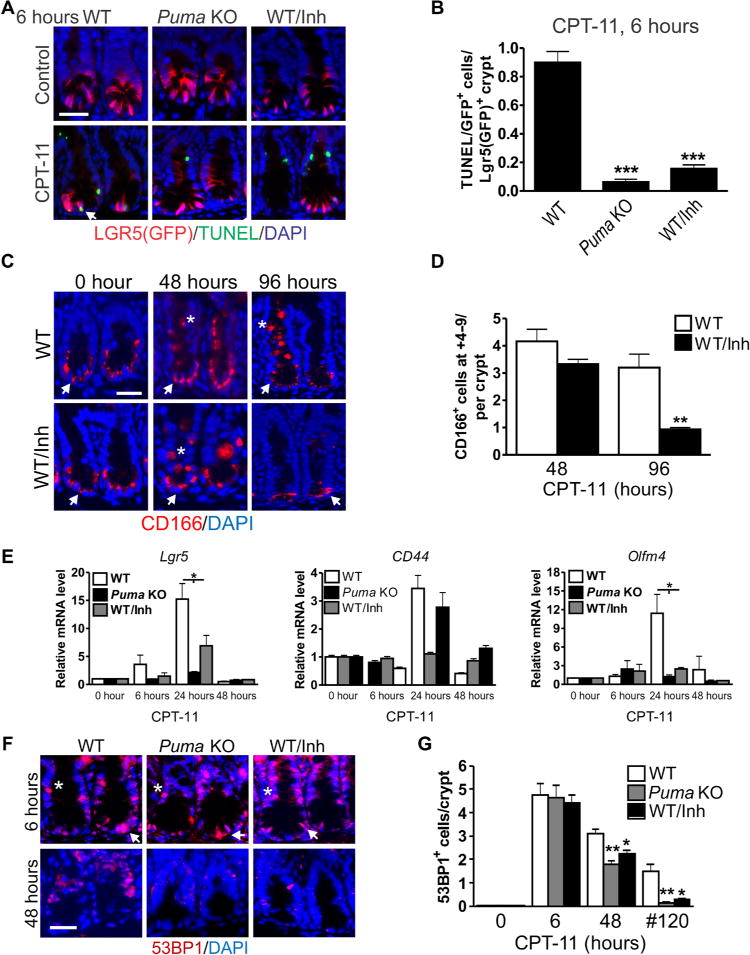
WT and Puma KO mice were treated with CPT-11 (200 mg/kg) and analyzed at the indicated times. PUMAi (10 mg/kg) was given once 2 hours before CPT-11 treatment. (A) Green fluorescent protein (GFP) (LGR5) and TUNEL immunofluorescence staining in intestinal crypts. Scale bar, 50 μm. Arrow indicates double positive cell. (B) Quantitation of GFP/TUNEL–double-positive cells per GFP-positive crypt from (A). (C) Immunofluorescence staining for CD166 in the intestinal crypts. Scale bar, 50 μm. (D) Quantitation of CD166 cells in the +4 to 9 region in crypts. (E) Indicated mRNAs were analyzed by quantitative reverse transcription polymerase chain reaction (qRT-PCR). Complementary DNAs (cDNAs) were synthesized from RNA pooled from three mice. Values were normalized to Gapdh expression and expressed relative to each gene’s own 0-hour control. (F) 53BP1 immunofluorescence staining in intestinal crypts. Scale bar, 50 μm. (G) Quantitation of 53BP1+ crypt cells at 6 and 48 hours after single CPT-11 treatment or #120 hours with three daily doses of CPT-11 (215 mg/kg per day) as in Fig. 2E. In (C) and (F), arrows indicate CBCs, and asterisks indicate +4 to 9 cells. In (A), (C), and (F), DAPI was used to stain the nuclei. In (B), (E), and (G), values are means + SEM; n = 3 mice per group. *P < 0.05, **P < 0.01, and ***P < 0.001 [one-way analysis of variance (ANOVA) with Tukey post hoc test performed separately for each time point]. In (D), values are means + SEM; n = 3 mice per group. **P < 0.01 (two-tailed Student’s t test).
CD166 is a WNT target that marks the ISC niche, including CBCs and Paneth cells at the crypt bottom, in homeostasis (33). CPT-11 treatment induced a marked expansion of CD166+ cells from 48 to 96 hours in WT mice, which was fully suppressed by PUMAi treatment by 96 hours (Fig. 5, C and D). This was preceded by transient but strong activation of WNT (Lgr5, CD44, and Wnt3A)– and NOTCH (Olfm4, Math1, Hes1, Hes5, and Dll1)–responsive genes as early as 6 hours, which was largely suppressed in Puma KO and PUMAi-treated mice (fig. S8). The only exception is the TA maker CD44, which was strongly suppressed by Puma KO but not by PUMAi (Fig. 5E). We then monitored crypt DNA damage by 53BP1 foci, which was comparable in Puma KO, PUMAi, and control groups at 6 hours. However, DNA damage in Puma KO and PUMAi groups was lower by 48 hours after a single dose of CPT-11 and, more so, by 120 hours after three doses of CPT-11 (Fig. 5, F and G). These data strongly suggest that the loss of LGR5+ cells disrupts the niche and triggers WNT and Notch signaling and intestinal regeneration after chemotherapy and that blocking LGR5+ cell loss likely delays compensatory proliferation and improves DNA repair.
LGR5+ cells are the critical target in intestinal chemoprotection by PUMAi
We further hypothesized that WNT and NOTCH activation triggered by LGR5+ cell loss will likely sensitize remaining stem cells to repeated exposures and stem cell exhaustion. After six doses of CPT-11 treatment, the LGR5+ crypt cells and the fraction of LGR5+ crypts (34) were reduced by more than 90% in WT mice, compared to only 40 to 60% in the Puma KO and PUMAi groups (Fig. 6, A and B). Olfm4 (10) RNA ISH indicated a 93% drop in Olfm4+ crypts in WT mice, compared to 8 and 49% in the Puma KO and PUMAi groups, respectively (Fig. 6C). Paneth cells are the niche for LGR5+ cells and marked by MMP7 expression. Repeated CPT-11 treatment, but not single treatment, diminished MMP7+ cells within the crypts, with prominent mislocalization scattered through the epithelium and nearly complete loss of the close contact of MMP7+ and LGR5+ cells, which was suppressed in the Puma KO and PUMAi groups (Fig. 6, D to F). Overall, these data demonstrate LGR5+ cell exhaustion as the key mechanism underlying chemotherapy-induced enterotoxicity, which can be effectively targeted by PUMAi.
Fig. 6. Targeting PUMA prevents LGR5+ stem cell exhaustion after repeated CPT-11 exposure.
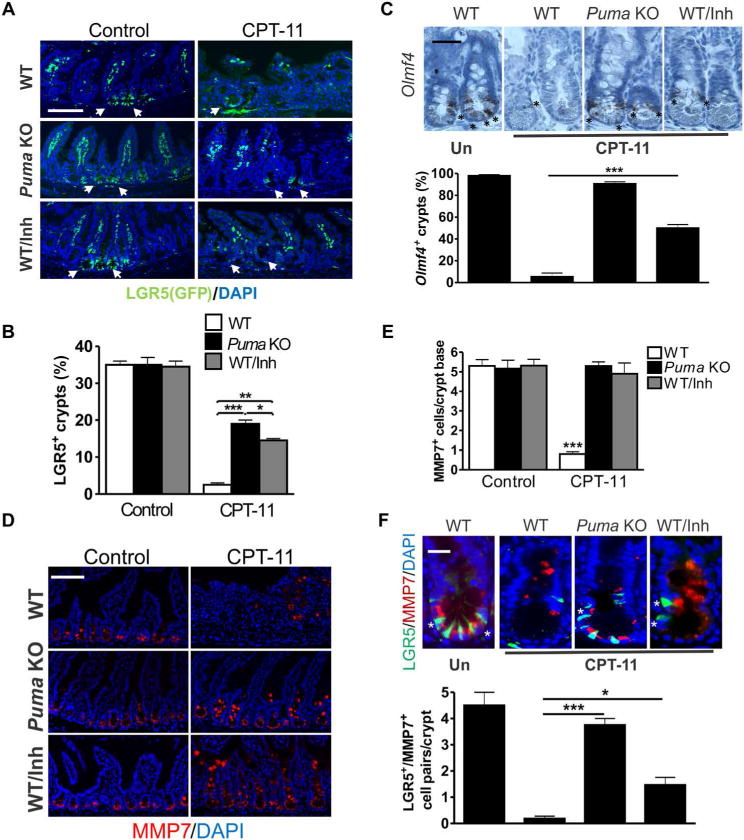
WT and Puma KO mice carrying the Lgr5-EGFP-IRES-creERT2 marking allele were treated with CPT-11 and PUMAi over 2 weeks as in Fig. 5 and analyzed for intestinal phenotypes. (A) GFP (LGR5) immunofluorescence in intestinal crypts. Arrows indicate LGR5+ (GFP) crypts. Scale bar, 100 μm. (B) Quantitation of intestinal crypts containing at least one GFP+ (LGR5+) cell. (C) Top: Olfm4 RNA ISH in intestinal crypts. Scale bar, 50 μm. Asterisks indicate Olfm4+ crypt cells. Bottom: Percentage of crypts containing at least one Olfm4+ cell. (D) MMP7 immunofluorescence in intestinal crypts. Scale bar, 100 μm. (E) Quantitation of MMP7+ crypt cells at the crypt base (CBC region). (F) Top: GFP (LGR5) and MMP7 immunofluorescence in intestinal crypts. Scale bar, 20 μm. Asterisks indicate LGR5+/MMP7+ cell pairs. Bottom: Quantitation of LGR5+/MMP7+ cell pairs per crypt. In (A), (D), and (F), DAPI was used to stain nuclei. In (B), (C), (E), and (F), values are means + SEM; n = 3 mice. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA with Tukey post hoc test).
PUMAi protects mouse and human colonic organoids against CPT-induced injury
To determine whether PUMAi acts directly on crypt cells, we used a three-dimensional epithelial organoid (35) or enteroid (36) system previously used to demonstrate radiation-induced, PUMA-dependent LGR5+ cell apoptosis (37). CPT induced growth suppression and activation of caspase-3 in mouse colonic organoids, which was strongly blocked by PUMAi (Fig. 7, A to C). PUMAi also suppressed CPT-induced activation of WNT and NOTCH targets (Lgr5, CD44, and Olfm4) (Fig. 7D). The suppression of TA marker CD44 was less pronounced compared to Lgr5 or Olfm4, similar to what we observed in vivo.
Fig. 7. PUMAi protects mouse and human colonic culture against CPT-induced damage.
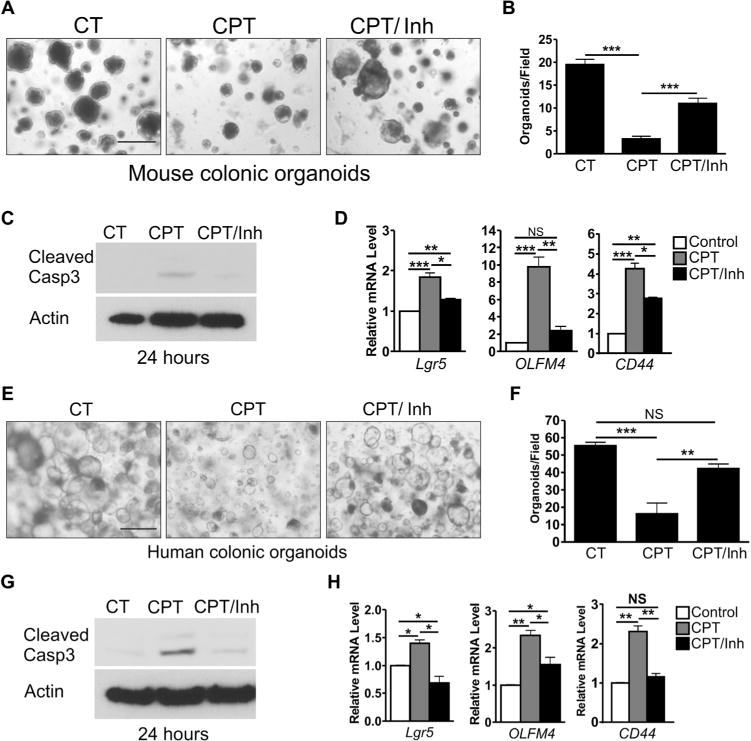
(A) Representative images of mouse colonic organoids 6 days after CPT treatment. Organoids were treated with vehicle control (CT) or 500 nM CPT (day 1) for 24 hours with or without 50 μM PUMAi and monitored for growth until day 7. Scale bar, 500 μm. (B) Quantitation of organoids 100 μm or greater in diameter per field from (A). (C) Western blots for active (cleaved) caspase-3 (Casp3) from organoids 24 hours after CPT treatment. (D) Indicated mouse mRNAs were analyzed by qRT-PCR 24 hours after CPT treatment. NS, not significant. (E) Representative images of human colonic organoids treated as in (A). Scale bar, 500 μm. (F) Quantitation of organoids 100 μm or greater in diameter per field from (E). (G) Western blots for active (cleaved) caspase-3 from human organoids 24 hours after CPT treatment. (H) Indicated human mRNAs analyzed 24 hours after CPT treatment. In (C) and (G), lysates were prepared from three wells. Actin was used as a control for protein loading. In (D) and (H), cDNAs were synthesized from RNA pooled from three cultured wells. Values were normalized to Gapdh and expressed relative to vehicle controls. In (B), (D), (F), and (H), values are means + SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA with Tukey post hoc test).
To demonstrate the translational potential of PUMAi, we used primary human colonic organoids. PUMAi enhanced organoid growth (Fig. 7, E and F) and inhibited CPT-induced caspase-3 activation (Fig. 7G). PUMAi suppressed CPT-induced expression of WNT and NOTCH targets (Fig. 7H), establishing that the p53- and PUMA-dependent loss of LGR5+ cells triggers a substantial activation of ISC-related pathways within the epithelial compartment.
DISCUSSION
GI complications are a major dose-limiting factor in patients receiving chemotherapy and abdominal radiation and can reduce the quality of life in cancer survivors. Identification of druggable targets is likely to have a major impact on cancer care by alleviating debilitating side effects, as well as improving outcomes by potentially increasing maximum tolerated doses. Our current study establishes p53- and PUMA-dependent LGR5+ stem cell loss as a major mediator of chemotherapy-induced enterotoxicity. Intestinal injury induced by repeated cytotoxic treatment is likely a consequence of the acute toxicity (1, 7), attributable to p53-dependent ISC loss, defective intestinal recovery, differentiation, and barrier, and persistent DNA damage and inflammation that further perpetuate ISC dysfunction and exhaustion (11). We speculate that compromised ISC functionality underlies “leaky gut syndrome” seen in some patients after pelvic or abdominal radiotherapy or chemotherapy, which is characterized by increased mucosal permeability, chronic fatigue, digestive problems, and systemic toxicities associated with persistent DNA damage (1).
p53 loss (38), but not Puma loss (26, 39, 40), strongly predisposes to spontaneous cancer development in mice. Our current work complements and extends several studies using single high-dose radiation models to uncouple the destructive and protective arms of p53 responses (17, 20, 23, 41). These findings strongly support the notion that targeting pathologic effects and apoptotic mediators of p53, not p53 itself, is a safer and more selective way to protect against cancer treatment–induced enterotoxicity (15, 16). This is consistent with stimulus and tissue-specific roles of BH3-only proteins in apoptosis and compromised apoptosis in cancer cells even with WT p53 due to oncogenic drivers such as WNT, phosphoinositide 3-kinase, and RAS/RAF (26, 42, 43). We discovered a promising lead compound, PUMAi, for intestinal chemoprotection, and its intestinal accumulation and normal tissue selectivity warrant further investigation.
Maintenance of intestinal epithelial integrity and function is vital, and it is driven by ISCs (10, 11, 44). Our data support position-sensitive loss of LGR5+ cells as the key pathology in chemotherapy- and radiation-induced enterotoxicity and the trigger for WNT and NOTCH activation and intestinal regeneration. This helps explain intestinal radiosensitization by genetic ablation of LGR5+ cells (12), and radio- or chemoprotection via LGR5+ cell preservation afforded by structurally and functionally diverse agents, including growth factors, Toll-like receptor 4 (TLR4) and TLR9 agonists, WNT agonists [reviewed in (11)], an hypoxia-inducible factor (HIF) activator (45), or inhibitors of glycogen synthase kinase β (37) and cyclin-dependent kinases 4 and 6 (46). Most, if not all, of these agents inhibit crypt apoptosis, and some also cause LGR5+ expansion (37, 47). These findings caution against the use of pro-proliferating agents, such as growth factors and WNT agonists, during repeated exposures to cytotoxic agents, which might result in ISC exhaustion via exacerbated DNA damage and prolonged proliferation. The sensitivity of WNT and NOTCH targets to CPT or PUMAi differed between human and mouse colonic organoids, as well as between organoids and mice, suggesting potentially species-specific differences in target regulation (37) and involvement of nonepithelial factors. Considering that PUMA mediates both p53-dependent and p53-independent apoptosis (26), it would be interesting to determine the effects of PUMAi in other injury models including DNA damage and inflammation (48–51).
Rapid renewal predisposes the GI epithelium to common side effects seen in cancer patients receiving radio- or chemotherapy, bone marrow transplant (1, 11), and even immunotherapy, with limited options for prevention or mitigation, representing a major unmet medical need. Here, we demonstrate PUMA as a promising target for selective intestinal chemoprotection and identify a small-molecule PUMAi active in vivo. Future studies will be needed to further define the mechanisms and pharmacokinetic and pharmacodynamic properties of PUMAi in normal and cancer tissues and to develop improved analogs.
MATERIALS AND METHODS
Study design
The goals of this study were to determine whether PUMA- and p53-dependent apoptosis mediates chemotherapy-induced intestinal stem cell loss and enteropathy and whether its genetic and pharmacological inhibition provides intestinal chemoprotection. Non–tumor-bearing and tumor-bearing mice, p53 and Puma KO mice, a small-molecule PUMAi, and in vitro mouse and human colonic organoids were used to measure the effects of PUMA inhibition on intestinal injury and regeneration, as well as tumor responses after chemotherapy. Sample size was determined using published work and power calculations. Experimenters were not blinded to treatment groups during the acquisition of data.
Mice and treatment
The procedures for all animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Puma+/+ (WT) and Puma−/− (Puma KO) mice (52) were generated in-house by heterozygote breeding. WT and Puma KO alleles were genotyped from genomic DNA isolated from tail snips, as described (52). p53−/− (p53 KO) mice were purchased from the Jackson Laboratory. The LGR5 marking mice Lgr5-EGFP (Lgr5-EGFP-IRES-creERT2) have been described (53). All strains are in or have been backcrossed with the C57BL/6 background for more than 10 generations (F10). Mice were housed in microisolator cages in a room illuminated from 07:00 to 19:00 hours (12:12-hour light/dark cycle), with access to water and chow ad libitum. CPT-11 (irinotecan hydrochloride; Camptosar, Pfizer) treatments were administered at 200 mg/kg in saline, unless otherwise indicated, by intraperitoneal injection (200 μl for a 20-g mouse). PUMAi was custom-synthesized by ChemBridge with a minimum purity of 95% by HPLC/mass spectrometry (MS), prepared as a stock (50 mg/ml) in dimethyl sulfoxide, then freshly diluted in phosphate-buffered saline (PBS) (2 mg/ml), and given at 10 mg/kg intraperitoneally (200 μl for a 20-g mouse).
For tumor experiments, 4 million LLC cells [American Type Culture Collection (ATCC)] were injected into the flanks of WT and Puma KO mice and allowed to grow for 11 days. Mice were then treated with CPT-11 (200 mg/kg) three times per week for 2 weeks. PUMAi (10 mg/kg) or vehicle control was administered by intraperitoneal injection 2 hours before and 20 hours after each dose of CPT-11. Mice were sacrificed 4 hours after the final CPT-11 treatment. Details on the dosing of CPT-11 and PUMAi, tumor measurements, and 5-FU and radiation models are found in Supplementary Materials and Methods.
Tissue processing and histological analysis
Immediately after sacrifice, about 10-cm portion of the jejunum was removed and carefully rinsed with ice-cold saline. The tissue was opened longitudinally and tacked to a foam board for fixation overnight in 10% formalin. Tissues were then rolled up into “swiss rolls” and embedded in paraffin. For tumor analysis, flank tumors were removed, measured, and cut into pieces for either formalin fixation or flash freezing in a dry ice/ethanol bath. Tissue sections (5 μm) were deparaffinized and rehydrated through graded ethanols. Histological analysis was performed by hematoxylin and eosin (H&E) staining.
Average villus height was determined by measuring 40 to 50 villi from different locations of the small intestine, from at least three different animals per group, and reported as means ± SEM. Measurements were made from the top of the crypt to the tip of the villus using 100× H&E images and SPOT 5.1 Advanced software (Diagnostic Instruments Inc.).
Immunohistochemistry and immunofluorescence
Rehydrated sections were treated with 3% hydrogen peroxide (immunochemistry only), followed by antigen retrieval for 10 min in boiling 0.1 M citrate buffer (pH 6.0) with 1 mM EDTA. Apoptosis was analyzed by TUNEL staining with the ApopTag Peroxidase In Situ Apoptosis Detection kit (Chemicon International) according to the manufacturer’s instructions. Details on other staining are found in Supplementary Materials and Methods.
In situ hybridization
ISH for Olfm4 was carried out with the RNAscope 2.0 BROWN kit (Advanced Cell Diagnostics) according to the manufacturer’s instructions. Briefly, tissue sections were baked for 60 min at 60°C, then deparaffinized in xylene, and rehydrated through graded ethanols. Sections then underwent three pretreatment steps, probe hybridization, six amplification steps, development, and counterstaining. ISH for Puma was carried out as previously described (17).
Western blotting
Fresh mucosal scrapings or minced tumor tissues were washed in 1 ml of ice-cold PBS and pelleted at 400g. Pellets were resuspended in 700 μl of homogenization buffer (0.25 M sucrose, 10 mM Hepes, and 1 mM EGTA) supplemented with protease inhibitors (complete EDTA-free mini, Roche) and homogenized in a Dounce homogenizer with 50 strokes of the pestle. After clearing by centrifugation at 16,000g, protein concentrations in the supernatant were determined by a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific).
Proteins (30 μg) were separated by SDS–polyacrylamide gel electrophoresis using the NuPAGE system (Invitrogen) and transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore). Representative results are shown, and similar results were obtained in at least three independent experiments. Antibodies used include PUMA (ab9643, Abcam), p53, p21, hemagglutinin (HA) (sc-6243, sc-397, and sc-805; Santa Cruz Biotechnology), V5 (R960-25, Invitrogen), tubulin (CP06, Oncogene Science), and actin (A5541, Sigma-Aldrich).
Quantitative real-time polymerase chain reaction
Fresh mucosal scrapings from 10 cm of jejunum were washed in cold PBS, resuspended in 700 μl of RNA lysis buffer, and homogenized in a Dounce homogenizer. RNA was isolated using the Quick-RNA MiniPrep kit (Zymo Research) according to the manufacturer’s instructions. Organoids were freed from Matrigel using Cell Recovery Solution (Corning), as described in (37), before RNA isolation. Complementary DNA was generated from 2 μg of total RNA from mice or ~100 ng of total RNA from colonic organoids and was pooled from three mice or three wells of organoid culture per treatment group using SuperScript III reverse transcriptase (Invitrogen) and random primers. Gene expression was normalized to Gapdh. Representative results are shown, and similar results were obtained in at least three independent experiments. Details on mouse and human primers are found in tables S2 and S3.
Cells and treatment
HCT 116, DLD1, SW480, and HT29 human colon cancer cells (ATCC), as well as HCT 116 p53 KO (54) and PUMA KO (27), were maintained at 37°C and 5% CO2 in McCoy’s 5A medium (Invitrogen) supplemented with 10% defined fetal bovine serum (HyClone), penicillin (100 U/ml), and streptomycin (100 μg/ml) (Invitrogen). Human embryonic kidney 293 cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) with the same supplements. For treatments, cells were plated in 12-well plates at ~30% density for 24 hours and then treated with CPT (Sigma-Aldrich). PUMAi was added to cells at 25 μM at the same time as CPT.
Measurement of apoptosis
After treatment, floating and adhering cells were collected and stained in a solution containing 3.7% formaldehyde, 0.5% NP-40, and Hoechst 33258 (10 μg/ml) (Molecular Probes) in PBS. Apoptosis was assessed through microscopic visualization of condensed and fragmented nuclei, as previously described (25, 27). A minimum of 300 cells per treatment were analyzed in triplicate. Representative results are shown, and similar results were obtained in at least three independent experiments.
Immunoprecipitation
HEK293 cells were transfected with previously described plasmids expressing HA-tagged PUMA (HA-PUMA), HA-tagged inactive PUMA (ΔBH3), or V5-tagged Bcl-xL (V5-Bcl-xL) (55) using Lipofectamine 2000 according to the manufacturer’s instructions. To test the PUMAi, 293 lysates containing HA-PUMA or HA-ΔBH3 were incubated for 15 min with 25 μM PUMAi, then mixed with V5-Bcl-xL lysates for 1 hour, and subjected to immunoprecipitation with an anti-HA antibody (sc-805, Santa Cruz Biotechnology) (25, 30). BIM/Mcl-1 interaction was tested in a similar fashion using lysates containing HA-BIM and V5-Mcl-1. Representative results are shown, and similar results were obtained in at least three independent experiments.
Measurement of PUMAi in tissues
PUMAi {1-[4-(2-hydroxyethyl)-1-piperazinyl]-3-(2-naphthyloxy)-2-propanol dihydrochloride} was synthesized by ChemBridge, with a purity of 95% by HPLC-MS, and an internal isotopic standard PUMAih7 (D5, five internal hydrogens replaced by deuterium) was synthesized by Alsachim, with purity approaching 100% by HPLC-MS. Nuclear magnetic resonance confirmed the isotope enrichment by 99.4% or more. Pooled plasma from untreated mice was used as control plasma. Detailed methods on LC-MS/MS (liquid chromatography–tandem MS) quantitation of PUMAi are found in Supplementary Materials and Methods.
Mouse and human colonic organoids
Mouse crypt isolation, organoid development, and passage were carried out as previously described (37, 56). Human normal colon crypt isolation and organoid development were performed as previously described (57). Modifications included the use of conditioned medium from L-WRN cells (ATCC CRL-3276) to supplement growth factors. Detailed protocols, reagents, and drug treatment are found in Supplementary Materials and Methods.
Statistical analysis
Statistical analyses were performed with GraphPad Prism IV software. Multiple comparisons of the responses were analyzed by one-way analysis of variance (ANOVA) and Tukey’s post hoc test, whereas those between two groups were made by two-tailed, unpaired t test. Survival data were analyzed by log-rank test. Differences were considered significant if the probability of the difference occurring by chance was less than 5 in 100 (P < 0.05). Sample size was determined using a combination of published work and power calculations. For ANOVA, we have computed the power for a test of interaction in a two-way factorial design applied by constructing mixed linear growth models to calculate the needed sample size. We estimated that usually 5 to 10 per group will provide 80% power to detect a standardized interaction of 1.5 SDs.
Supplementary Material
Fig. S1. PUMA KO inhibits CPT-11–induced crypt apoptosis.
Fig. S2. CPT-11 causes dose-dependent lethality in mice.
Fig. S3. A small-molecule PUMAi does not protect colon cancer cells against CPT-11–induced apoptosis.
Fig. S4. PUMAi inhibits CPT-11–induced CBC apoptosis.
Fig. S5. PUMAi protects against chemotherapy- and radiation-induced lethality.
Fig. S6. Targeting PUMA does not compromise tumor response to CPT-11.
Fig. S7. 5-FU–induced LGR5+ cell apoptosis is PUMA-dependent.
Fig. S8. Puma KO and PUMAi suppress CPT-11–induced expression of WNT and NOTCH targets.
Table S1. Treatment-associated lethality in LLC tumor experiments.
Table S2. Mouse-specific primers used for real-time reverse transcription polymerase chain reaction.
Table S3. Human-specific primers used for real-time reverse transcription polymerase chain reaction.
Acknowledgments
We would like to thank P. Buckhaults (University of South Carolina) for sharing the protocols on colonic cultures. Funding: This work is supported, in part, by American Cancer Society grant RGS-10-124-01-CCE, NIH grant U19-A1068021, Pennsylvania Tobacco funds and institutional funds (to J.Y.), CA172136 and CA203028 (to L.Z.), and U01 CA152753 (to R.E.S.). This project used the UPMC Hillman Cancer Center shared cancer pharmacokinetics and pharmacodynamics facility, glassware, animal, and cell and tissue imaging facilities that were supported, in part, by National Cancer Institute award P30CA047904.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/10/427/eaam7610/DC1 Materials and Methods
Author contributions: B.J.L., L.Y., L.W., J.H.B., J.L.E., L.Z., and J.Y. designed the experiments. B.J.L., L.Y., L.W., M.E.B., M.R., and R.A.P. performed the experiments. L.Z. and R.E.S. provided the key reagents including access to human tissues. B.J.L., L.Y., L.W., J.H.B., and J.Y. analyzed the data. B.J.L. and J.Y. wrote the manuscript. J.Y. conceived and supervised all experiments.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Hauer-Jensen M, Denham JW, Andreyev HJ. Radiation enteropathy—Pathogenesis, treatment, and prevention. Nat Rev Gastroenterol Hepatol. 2014;11:470–479. doi: 10.1038/nrgastro.2014.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee CS, Ryan EJ, Doherty GA. Gastro-intestinal toxicity of chemotherapeutics in colorectal cancer: The role of inflammation. World J Gastroenterol. 2014;20:3751–3761. doi: 10.3748/wjg.v20.i14.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein A, Voigt W, Jordan K. Chemotherapy-induced diarrhea: Pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol. 2010;2:51–63. doi: 10.1177/1758834009355164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araki E, Ishikawa M, Iigo M, Koide T, Itabashi M, Hoshi A. Relationship between development of diarrhea and the concentration of SN-38, an active metabolite of CPT-11, in the intestine and the blood plasma of athymic mice following intraperitoneal administration of CPT-11. Jpn J Cancer Res. 1993;84:697–702. doi: 10.1111/j.1349-7006.1993.tb02031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takasuna K, Hagiwara T, Hirohashi M, Kato M, Nomura M, Nagai E, Yokoi T, Kamataki T. Involvement of β-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 1996;56:3752–3757. [PubMed] [Google Scholar]
- 6.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51:4187–4191. [PubMed] [Google Scholar]
- 7.Berbée M, Hauer-Jensen M. Novel drugs to ameliorate gastrointestinal normal tissue radiation toxicity in clinical practice: What is emerging from the laboratory? Curr Opin Support Palliat Care. 2012;6:54–59. doi: 10.1097/SPC.0b013e32834e3bd7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowen JM, Gibson RJ, Stringer AM, Chan TW, Prabowo AS, Cummins AG, Keefe DM. Role of p53 in irinotecan-induced intestinal cell death and mucosal damage. Anticancer Drugs. 2007;18:197–210. doi: 10.1097/CAD.0b013e328010ef29. [DOI] [PubMed] [Google Scholar]
- 9.Keefe DM, Brealey J, Goland GJ, Cummins AG. Chemotherapy for cancer causes apoptosis that precedes hypoplasia in crypts of the small intestine in humans. Gut. 2000;47:632–637. doi: 10.1136/gut.47.5.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker N, van Oudenaarden A, Clevers H. Identifying the stem cell of the intestinal crypt: Strategies and pitfalls. Cell Stem Cell. 2012;11:452–460. doi: 10.1016/j.stem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Yu J. Intestinal stem cell injury and protection during cancer therapy. Transl Cancer Res. 2013;2:384–396. [PMC free article] [PubMed] [Google Scholar]
- 12.Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell. 2014;14:149–159. doi: 10.1016/j.stem.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 14.Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun. 2005;331:851–858. doi: 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 16.Gudkov AV, Komarova EA. Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol. 2010;2:a001180. doi: 10.1101/cshperspect.a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, Zhang L, Yu J. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell. 2008;2:576–583. doi: 10.1016/j.stem.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene. 2010;29:1622–1632. doi: 10.1038/onc.2009.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu H, Shen H, Yuan Y, XuFeng R, Hu X, Garrison SP, Zhang L, Yu J, Zambetti GP, Cheng T. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose γ-irradiation. Blood. 2010;115:3472–3480. doi: 10.1182/blood-2009-10-248278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leibowitz BJ, Qiu W, Liu H, Cheng T, Zhang L, Yu J. Uncoupling p53 functions in radiation-induced intestinal damage via PUMA and p21. Mol Cancer Res. 2011;9:616–625. doi: 10.1158/1541-7786.MCR-11-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010;24:1602–1607. doi: 10.1101/gad.1940210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, Strasser A. Apoptosis-promoted tumorigenesis: γ-Irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608–1613. doi: 10.1101/gad.1940110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23:3265–3271. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- 24.Kunimoto T, Nitta K, Tanaka T, Uehara N, Baba H, Takeuchi M, Yokokura T, Sawada S, Miyasaka T, Mutai M. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxycamptothecin, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987;47:5944–5947. [PubMed] [Google Scholar]
- 25.Mustata G, Li M, Zevola N, Bakan A, Zhang L, Epperly M, Greenberger JS, Yu J, Bahar I. Development of small-molecule PUMA inhibitors for mitigating radiation-induced cell death. Curr Top Med Chem. 2011;11:281–290. doi: 10.2174/156802611794072641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27(suppl. 1):S71–S83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U.S.A. 2003;100:1931–1936. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J, Yue W, Wu B, Zhang L. PUMA sensitizes lung cancer cells to chemotherapeutic agents and irradiation. Clin Cancer Res. 2006;12:2928–2936. doi: 10.1158/1078-0432.CCR-05-2429. [DOI] [PubMed] [Google Scholar]
- 29.Sun Q, Sakaida T, Yue W, Gollin SM, Yu J. Chemosensitization of head and neck cancer cells by PUMA. Mol Cancer Ther. 2007;6:3180–3188. doi: 10.1158/1535-7163.MCT-07-0265. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 31.Bissery MC, Vrignaud P, Lavelle F, Chabot GG. Experimental antitumor activity and pharmacokinetics of the camptothecin analog irinotecan (CPT-11) in mice. Anticancer Drugs. 1996;7:437–460. doi: 10.1097/00001813-199606000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Rizzo MG, Soddu S, Tibursi G, Calabretta B, Sacchi A. Wild-type p53 differentially affects tumorigenic and metastatic potential of murine metastatic cell variants. Clin Exp Metastasis. 1993;11:368–376. doi: 10.1007/BF00132980. [DOI] [PubMed] [Google Scholar]
- 33.Levin TG, Powell AE, Davies PS, Silk AD, Dismuke AD, Anderson EC, Swain JR, Wong MH. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology. 2010;139:2072–2082. doi: 10.1053/j.gastro.2010.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Stange DE, Ferrante M, Vries RG, van Es JH, van den Brink S, van Houdt WJ, Pronk A, van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 36.Stelzner M, Helmrath M, Dunn JC, Henning SJ, Houchen CW, Kuo C, Lynch J, Li L, Magness ST, Martin MG, Wong MH, Yu J. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1359–G1363. doi: 10.1152/ajpgi.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Wei L, Cramer JM, Leibowitz BJ, Judge C, Epperly M, Greenberger J, Wang F, Li L, Stelzner MG, Dunn JC, Martin MG, Lagasse E, Zhang L, Yu J. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci Rep. 2015;5:8566. doi: 10.1038/srep08566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 39.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 40.Villunger A, Michalak EM, Coultas L, Mullauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 41.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, Jeffords LB, Sodha P, Mercer KL, Cohen R, Takeuchi O, Korsmeyer SJ, Bronson RT, Kim CF, Haigis KM, Jain RK, Jacks T. p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science. 2010;327:593–596. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Yu J. Role of apoptosis in colon cancer biology, therapy, and prevention. Curr Colorectal Cancer Rep. 2013;9:331–340. doi: 10.1007/s11888-013-0188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud Å, Grosse AS, Gumucio DL, Ernst SA, Tsai YH, Dempsey PJ, Samuelson LC. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taniguchi CM, Miao YR, Diep AN, Wu C, Rankin EB, Atwood TF, Xing L, Giaccia AJ. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med. 2014;6:236ra64. doi: 10.1126/scitranslmed.3008523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei L, Leibowitz BJ, Wang X, Epperly M, Greenberger J, Zhang L, Yu J. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. J Clin Invest. 2016;126:4076–4087. doi: 10.1172/JCI88410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou WJ, Geng ZH, Spence JR, Geng JG. Induction of intestinal stem cells by R-spondin 1 and Slit2 augments chemoradioprotection. Nature. 2013;501:107–111. doi: 10.1038/nature12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu W, Wang X, Leibowitz B, Yang W, Zhang L, Yu J. PUMA–mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology. 2011;54:1249–1258. doi: 10.1002/hep.24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang P, Qiu W, Dudgeon C, Liu H, Huang C, Zambetti GP, Yu J, Zhang L. PUMA is directly activated by NF-κB and contributes to TNF-α-induced apoptosis. Cell Death Differ. 2009;16:1192–1202. doi: 10.1038/cdd.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu W, Wu B, Wang X, Buchanan ME, Regueiro MD, Hartman DJ, Schoen RE, Yu J, Zhang L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest. 2011;121:1722–1732. doi: 10.1172/JCI42917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dirisina R, Katzman RB, Goretsky T, Managlia E, Mittal N, Williams DB, Qiu W, Yu J, Chandel NS, Zhang L, Barrett TA. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology. 2011;141:1036–1045. doi: 10.1053/j.gastro.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu B, Qiu W, Wang P, Yu H, Cheng T, Zambetti GP, Zhang L, Yu J. p53 independent induction of PUMA mediates intestinal apoptosis in response to ischaemia–reperfusion. Gut. 2007;56:645–654. doi: 10.1136/gut.2006.101683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barker N, Clevers H. Tracking down the stem cells of the intestine: Strategies to identify adult stem cells. Gastroenterology. 2007;133:1755–1760. doi: 10.1053/j.gastro.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 54.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 55.Ming L, Wang P, Bank A, Yu J, Zhang L. PUMA dissociates Bax and Bcl-XL to induce apoptosis in colon cancer cells. J Biol Chem. 2006;281:16034–16042. doi: 10.1074/jbc.M513587200. [DOI] [PubMed] [Google Scholar]
- 56.Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, Wang Q, Yu J. ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013;4:e599. doi: 10.1038/cddis.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujii M, Matano M, Nanki K, Sato T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat Protoc. 2015;10:1474–1485. doi: 10.1038/nprot.2015.088. [DOI] [PubMed] [Google Scholar]
- 58.Qiu W, Wang X, Leibowitz B, Liu H, Barker N, Okada H, Oue N, Yasui W, Clevers H, Schoen RE, Yu J, Zhang L. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc Natl Acad Sci U.S.A. 2010;107:20027–20032. doi: 10.1073/pnas.1010430107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leibowitz BJ, Wei L, Zhang L, Ping X, Epperly M, Greenberger J, Cheng T, Yu J. Ionizing irradiation induces acute haematopoietic syndrome and gastrointestinal syndrome independently in mice. Nat Commun. 2014;5:3494. doi: 10.1038/ncomms4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. PUMA KO inhibits CPT-11–induced crypt apoptosis.
Fig. S2. CPT-11 causes dose-dependent lethality in mice.
Fig. S3. A small-molecule PUMAi does not protect colon cancer cells against CPT-11–induced apoptosis.
Fig. S4. PUMAi inhibits CPT-11–induced CBC apoptosis.
Fig. S5. PUMAi protects against chemotherapy- and radiation-induced lethality.
Fig. S6. Targeting PUMA does not compromise tumor response to CPT-11.
Fig. S7. 5-FU–induced LGR5+ cell apoptosis is PUMA-dependent.
Fig. S8. Puma KO and PUMAi suppress CPT-11–induced expression of WNT and NOTCH targets.
Table S1. Treatment-associated lethality in LLC tumor experiments.
Table S2. Mouse-specific primers used for real-time reverse transcription polymerase chain reaction.
Table S3. Human-specific primers used for real-time reverse transcription polymerase chain reaction.