

Abstract
We report the development of a new technology for monitoring multidimensional protein-protein interactions (PPIs) inside live mammalian cells using split RNA polymerase (RNAP) tags. In this new system, a protein-of-interest is tagged with an N-terminal split RNAP (RNAPN) and multiple potential binding partners are each fused to orthogonal C-terminal RNAPs (RNAPC). Assembly of RNAPN with each RNAPC is highly dependent on interactions between the tagged proteins. Each PPI-mediated RNAPN-RNAPC assembly transcribes from a separate promoter on a supplied DNA substrate, thereby generating a unique RNA output signal for each PPI. We develop and validate this new approach in the context of the Bcl-2 family of proteins. These key regulators of apoptosis are important cancer mediators, but are challenging to therapeutically target due to imperfect selectivity that leads to either off-target toxicity or tumor resistance. We demonstrated binary (1×1) and ternary (1×2) Bcl-2 PPI analyses by imaging fluorescent protein translation from mRNA outputs. Next, we performed a 1×4 PPI network analysis by direct measurement of four unique RNA signals via RT-qPCR. Finally, we used these new tools to monitor pharmacological engagement of Bcl-2 protein inhibitors, and uncover inhibitor-dependent competitive PPIs. The split RNAP tags improve upon other protein fragment complementation (PFC) approaches by offering both multidimensionality and sensitive detection using nucleic acid amplification and analysis techniques. Furthermore, this technology opens up new opportunities for synthetic biology applications due to the versatility of RNA outputs for cellular engineering applications.
Graphical abstract
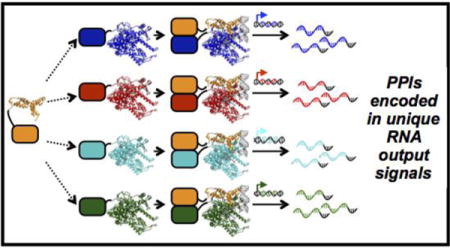
INTRODUCTION
Protein-protein interactions (PPIs) are fundamental to cellular organization, regulation, and cell signaling1-3. Although often considered “undruggable”, the centrality of dysregulated PPIs in many complex disease states makes these biomolecular interactions enticing targets for therapeutic development4-7. A key challenge to understanding the physiological and pathological roles of target PPIs are a lack of tools for measuring competitive interactions within complex PPI networks in native biological contexts. An exemplar of both the challenges and opportunities in PPI-targeted therapeutics is the B cell lymphoma 2 (Bcl-2) family of apoptotic regulatory proteins, which function through highly interconnected and competitive interaction networks to control cell death8.
A key hallmark of cancer is evasion of apoptosis, which is mediated by anti-apoptotic Bcl-2 family proteins such as Bcl-2, Bcl-XL and myeloid cell leukemia 1 (Mcl-1). These proteins maintain cell survival by binding and sequestering their pro-apoptotic multidomain counterparts, such as Bcl-2-associate X protein (BAX) and Bcl-2 antagonist killer 1 (BAK), and Bcl-2 homology 3 (BH3)-only proteins such as Bcl‑2 antagonist of cell death (BAD), truncated BH3‑interacting domain death agonist (tBID), and Bcl‑2‑interacting mediator of cell death (BIM)9,10, through competitive PPIs. Venetoclax, a recently approved Bcl-2 inhibitor and first FDA approved intracellular PPI inhibitor, is effective in patients with hematologic malignancies11. Mcl-1 dysregulation is a key mode of resistance to both Bcl-2 inhibitors such as Venetoclax12, and chemotherapy more broadly, driving myriad cancers. Mcl-1 inhibitors, therefore, are a current focal point of cancer drug discovery efforts13-16. The complexity and interconnectivity of Bcl-2-family apoptotic regulation illustrates the need for tools to monitor the interactions in a native biological context, but is also representative of the broader challenges and opportunities in both understanding and exploiting disease-relevant PPIs.
A multitude of live cell-deployable technologies for measuring PPIs have been developed17-26. The yeast 2-hybrid system (Y2H) is being utilized to catalog the potential binary PPIs across the entire human proteome27. However, a key limitation of such yeast-based studies is that biological relevance is not guaranteed for each identified interaction, and the interactions may be differentially regulated in mammalian systems. Förster/bioluminescent resonance energy transfer (FRET/BRET)-based sensors permit detection of subtle changes in distance with high spatiotemporal resolution28, and have even been adapted to rapidly and temporally measure changes in 40 cell signals under identical conditions29. Another widespread method for measuring PPIs in live mammalian cells is protein fragment complementation (PFC), which involves tagging two potential binding partners of interest with separate halves of a split protein reporter. PFC systems have been developed for a variety of imaging modalities, such as green fluorescent protein (GFP)21,30-32, luciferase33,34, and horseradish peroxidase (HRP)35, as well as other reporter systems such as tobacco etch virus (TEV) protease36,37 and ubiquitin38. The key advantage of most PFC technologies is that PPIs can be measured in real-time using simple imaging techniques. However, multiplexed and/or competitive detection of PPIs has only been reported in proof-of-concept demonstrations39-42, and at most for a trimolecular PPI system. Although the need for multidimensional PPI analysis techniques is widely acknowledged19, challenges with spectral overlap of reporters, incompatible PFC technologies, and differential sensitivities have largely precluded their development. Therefore, there is a need for generalizable PFC technologies to detect multidimensional PPIs in a competitive manner within mammalian systems.
Recently, we developed a proximity-dependent split RNA polymerase (RNAP) reporter system as a new method to measure target interactions in biological systems43. This involved evolving an N-terminal split RNAP tag, RNAPN, to assemble with a C-terminal split RNAP tag, RNAPC, in a PPI-dependent manner. The advantage of an RNAP-based reporter is that the RNA signal can be measured in a variety of sensitive ways, including fluorescence, sequencing, nanoparticle detection, etc.44-48, and can be amplified by polymerase chain reaction (PCR)49 or recombinase polymerase amplification (RPA)50 for even more sensitive detection. Moreover, unlike optical reporters, RNAP-based reporters are not limited by spectral overlap considerations, as unique “barcode” sequences can, in principle, be encoded in orthogonal RNA outputs45,51. Indeed, we recently demonstrated the possibility of performing multidimensional biochemical assays in mammalian cells using orthogonal protease-responsive RNAPs52. Finally, an RNA output offers control over genomic and transcriptomic processing through the production of mRNA, RNAi, or even gRNA, all of which have important applications in genetic screens and synthetic biology53-57.
In principle, the evolved proximity-dependent split RNAP system lays the foundation for the generation of a new set of RNAP tags that encode multidimensional PPIs in RNA signals. The key to realizing such a technology is developing the approaches to deploy the tools in mammalian cells and the creation of a set of orthogonal proximity-dependent split RNAPs that drive transcription from unique DNA promoters based on a target PPI, thereby producing a unique RNA output. In this report, we develop a series of orthogonal split RNAP tags for interrogating PPIs in mammalian systems, and apply the new tools to probe Bcl-2-family interactions and their pharmacological engagement. Application of the tools in “one-by-four” interactome analyses of the Bcl-2 pathway reveals endogenous ligand competition and selectivity profiling of both clinical and preclinical drug candidates. Together, this work establishes split RNAPs as a versatile new addition to the PPI analysis toolbox, with particular advantages for multidimensional and competitive PPI interrogation, and future synthetic biology applications.
RESULTS AND DISCUSSION
Engineering split RNAP tags to detect Bcl-2 family PPIs
We first tested whether the proximity-dependent split RNAP tags can detect Bcl-2 family PPIs (Figure. 1A). To validate and optimize the protein sensors, we deployed an E. coli-based transcriptional reporter system43,58. We cloned expression vectors that constitutively express the BH3 binding domains of tBID and NOXA each fused to the evolved RNAPN tag through a flexible linker. We also cloned a negative control, deadBID, that consists of tBID devoid of its necessary BH3 domain59,60. In conjunction, we cloned a series of anti-apoptotic proteins including Bcl-2, Bcl-W, Bcl-XL and Mcl-1, each fused to the T7 RNAPC tag, into an arabinose-inducible vector. To monitor in vivo transcriptional output of the assembled T7 RNAP, we used a reporter vector that produces luciferase in response to transcription from the T7 promoter. Reporter E. coli cells were then cotransformed with the three vectors, induced with arabinose, and analyzed for luminescence output (Figure 1B and S1, Table S1). As expected, because all four Bcl-2-family proteins are known to interact with tBID61, interactions of Bcl-2, Bcl-W, Bcl-XL, and Mcl-1 with tBID each produced a robust enhancement of RNAP activity compared to the deadBID control, with between a 24-fold and 207-fold dynamic range (Figure 1C). More striking was that the NOXA-fused RNAPN only showed significant transcriptional output (34-fold) when combined with Mcl-1-fused RNAPC, confirming NOXA selectively binds to Mcl-1 over the other anti-apoptotic proteins assayed61,62. These experiments indicate that the split RNAP tags can reproduce known in vitro affinity measurements in live E. coli with a robust RNA output signal.
Figure 1.
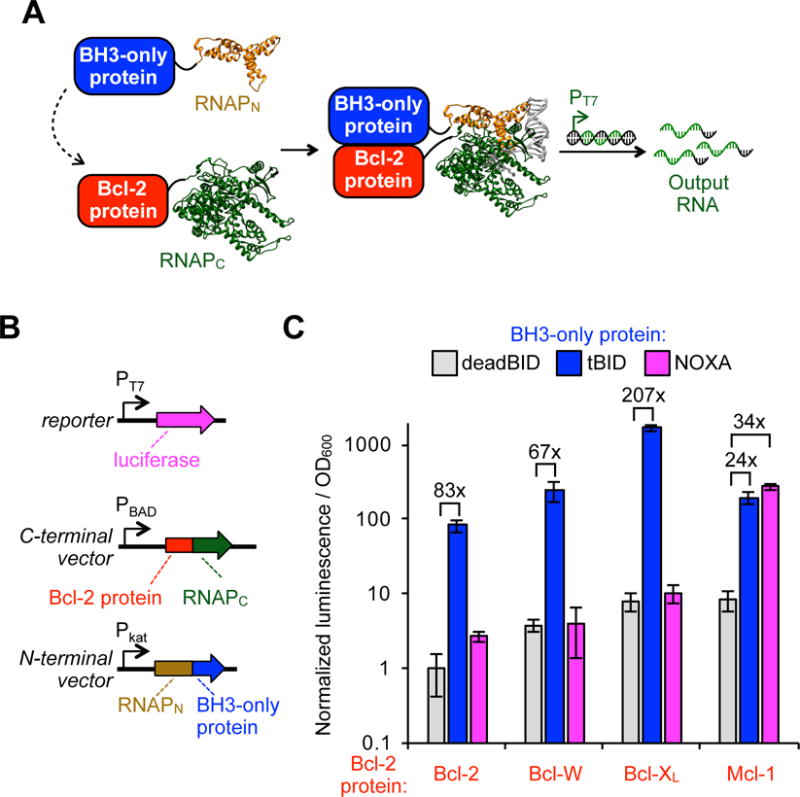
Split RNAP biosensors can detect Bcl-2 family PPIs in E. coli. (A) Schematic of split T7 RNAP tags to monitor Bcl-2-family PPIs. A target Bcl-2 protein is fused to a C-terminal T7 RNAP variant (RNAPC) and a target BH3-only protein ligand is fused to an evolved proximity-dependent N-terminal T7 RNAP variant (RNAPN). Interaction between the fusion proteins results in assembly of the RNAP and a transcriptional output signal. (B) Vector system to test Bcl-2 split RNAP detection system in E. coli. (C) Transcriptional output of split RNAPs with a series of Bcl-2 proteins interaction with a set of BH3-only peptides assayed in E. coli using the vectors shown in (B). Cells were induced for 3 h with arabinose and then analyzed for luminescence. Error bars are ± s.e.m., n = 4. “deadBID” is a modified tBID BH3-only peptide with the key binding portions removed as a negative control.
Monitoring binary Bcl-2 family PPIs in mammalian cells with split RNAP tags
Upon validating that the split RNAP tags can detect Bcl-2-family PPIs in E. coli, we sought to measure the performance of the system in a more physiologically relevant setting, mammalian HEK293T cells. We adapted the expression vectors for mammalian cell deployment by cloning a CMV-driven vector for each of the RNAPN-tagged BH3-only protein fusions (Figure 2A). Next, we cloned a series of vectors that each featured a CMV-driven RNAPC tagged anti-apoptotic protein fusion, as well as a T7 promoter-driven GFP mRNA circuit52,63. PPI-dependent RNAP assembly could therefore easily be measured by imaging GFP fluorescence, which is resultant from RNAPN and RNAPC reassembly, mRNA transcription, and GFP translation. Due to the signal amplification of this output, the data is qualitative, but the ease of analysis permits facile system optimization and validation. Cotransfection of any anti-apoptotic-RNAPC construct with the RNAPN-deadBID vector resulted in cells with very low GFP fluorescence (Figure 2B, C, S2–S5). However, cotransfection of RNAPN-tBID with each Bcl-2 family RNAPC fusion resulted in a dramatic enhancement of GFP fluorescence. Similar to the results in E. coli, and as expected based on in vitro measurements, RNAPN-NOXA only induced robust GFP fluorescence when cotransfected with Mcl-1-RNAPC (Figure 2B, C, S2–S5). These imaging experiments indicate that the split RNAP tags are capable of measuring binary PPIs between Bcl-2-family proteins in mammalian cells.
Figure 2.
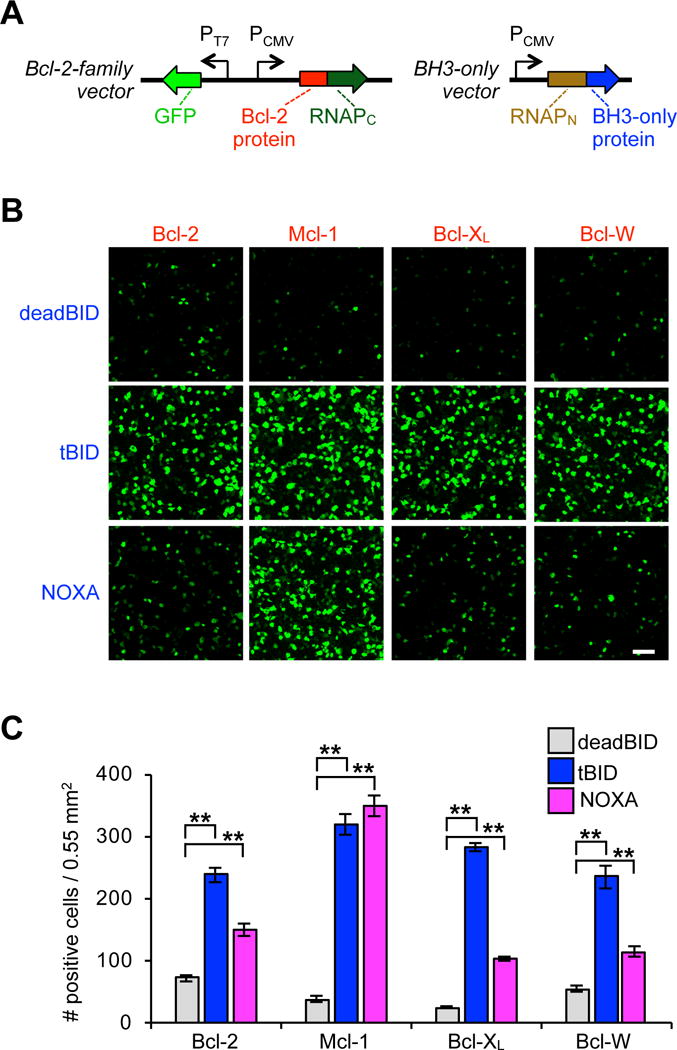
Split RNAP biosensors can detect Bcl-2 family PPIs in mammalian cells. (A) Vector system to test binary Bcl-2 split RNAP PPI detection system in mammalian cells using PPI driven GFP mRNA as the RNA output. (B) HEK293T cells cotransfected with the plasmids shown in (A). 30 h after transfection, the cells were analyzed for GFP expression by fluorescence microscopy. Interactions between tBID and Bcl-2, Mcl-1, Bcl-XL, and Bcl-W, are readily detected compared to control, while NOXA is much more selective for interaction with Mcl-1. (C) Quantification of (B) (error bars are ± s.e.m, n = 5). Student’s t-test; **P < 0.001. 100 μm scale bar shown.
One-by-two PPI analysis with orthogonal RNAP tags
Once we validated that split RNAP tags could measure single binary PPIs in mammalian cells, we sought to test whether we could detect two competing PPIs simultaneously. The goal would be to allow one BH3-only protein to interact with two different anti-apoptotic proteins in the same cell, such that each interacting pair drives a different RNA output. Previously, we utilized an orthogonal RNAPC43, RNAPC(CGG), that selectively transcribes from the “CGG” promoter upon assembly with the RNAPN. In order to deploy this orthogonal system in conjunction with the T7-based system, we cloned a mammalian expression vector that produces Mcl-1-RNAPC(CGG) and RFP expression via the CGG promoter. We first deployed this other reporter vector system with the Bcl-2-RNAPC(T7) reporter and each of the RNAPN-BH3-only proteins by cotransfecting cells with all three vectors and measuring GFP and RFP fluorescence (Figure 3A). This way, the interaction between the BH3-only protein and Bcl-2 would produce GFP fluorescence, while the interaction between the BH3-only protein and Mcl-1 would produce RFP fluorescence. Cells with RNAPN-deadBID displayed low fluorescence in both channels, as expected (Figure 3B, C, S6). However, RNAPN-tBID produced robust fluorescent signal in both the RFP and GFP channels. Moreover, RNAPN-NOXA produced more RFP than GFP fluorescence, recapitulating the in vitro and E. coli measurements in live mammalian cells. Intriguingly, in this competitive experiment tBID gave less signal than NOXA with Mcl-1, which is not what was observed in the individual PPI assays (Figure 2). This type of competitive binding effect can only be observed by monitoring multiple PPIs simultaneously.
Figure 3.
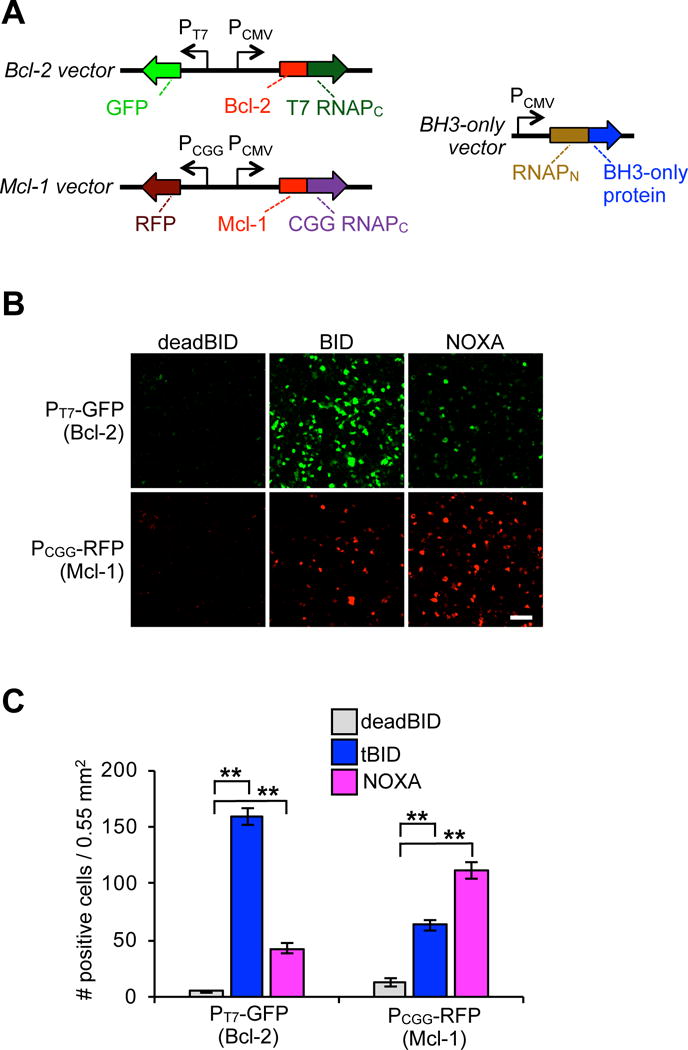
Split RNAP biosensors can monitor two Bcl-2 family PPIs simultaneously in mammalian cells. (A) Vector system to simultaneously monitor competitive interactions between Bcl-2 and Mcl-1 with BH3-only proteins in mammalian cells using mRNA for GFP and RFP as the two orthogonal RNA outputs from each PPI. (B) HEK293T cells cotransfected with the plasmids shown in (A). 30 h after transfection, the cells were analyzed for GFP and RFP expression by fluorescence microscopy. Bcl-2 and Mcl-1 were both found to interact with tBID, while NOXA was found to interact more with Mcl-1. (C) Quantification of (B) (error bars are ± s.e.m, n = 5). Student’s t-test; **P < 0.001. 100 μm scale bar shown.
We also tested the binary PPI monitoring system with a second competitive PPI pair, Bcl-XL and Mcl-1. Similarly, tBID interactions could be detected with both anti-apoptotic proteins, while NOXA interactions were more selective for Mcl-1 (Figure S7, S8). These results confirm that two PPIs can be monitored simultaneously in mammalian cells using this set of orthogonal split RNAP tags.
Monitoring target engagement and selectivity of PPI inhibitors in live cells
With the system to simultaneously measure two competitive PPIs in hand, we next sought to detect pharmacological engagement of PPI inhibitors in live cells. We tested the recently FDA approved Bcl-2 inhibitor ABT-19964,65 (Venetoclax) and the preclinical Mcl-1 inhibitor A-121044766 (A121). Treatment with ABT-199 blocked the interaction between Bcl-2 and tBID (Figure 4A, B, S9), while treatment with A-1210477 blocked the interaction between Mcl-1 and tBID. Coadministration of both inhibitors in a combination therapy experiment blocked both PPIs, though the level of Mcl-1 inhibition was less than in the single treatment conditions.
Figure 4.
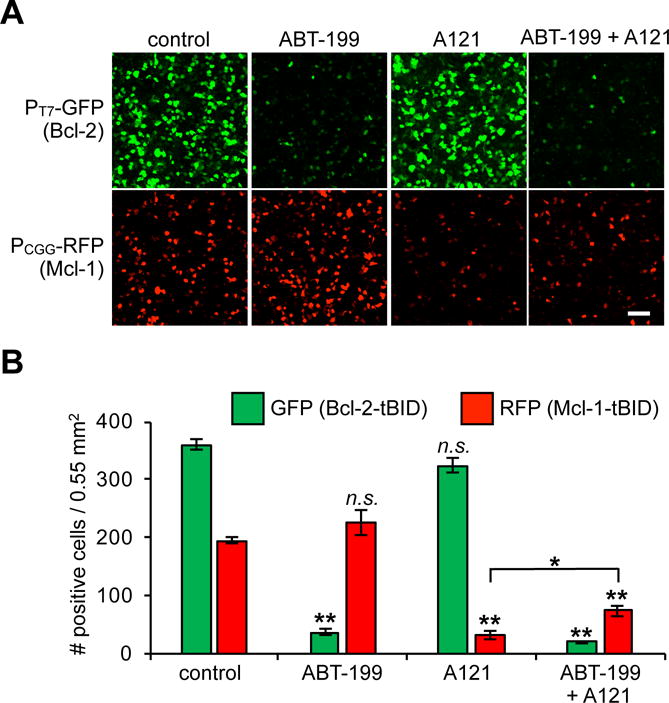
Spit RNAP biosensors can simultaneously monitor pharmacological engagement of two PPIs in live cells. (A) HEK293T cells cotransfected with the plasmids shown in Figure 3A. Upon transfection, DMSO carrier control, 0.5 μM ABT-199, 10 μM A1210477, or a combination of both 0.5 μM ABT-199 and 10 μM A1210447 were added to the cells. After 30 h, the cells were analyzed for GFP and RFP expression by fluorescence microscopy. (B) Quantification of (A) (error bars are ± s.e.m, n = 5). Student’s t-test; *P < 0.01, **P < 0.001 (relative to control when not otherwise indicated). n.s. = not significant. 100 μm scale bar shown.
Intriguingly, the measured signal between Mcl-1 and tBID in the presence of competition with either Bcl-2 or Bcl-XL was lower than when measured as a single binary interaction without competition (Figure 3 vs Figure 2). This is in-principle expected, because Mcl-1 is a roughly equal (Bcl-2) or lower (Bcl-XL) affinity binder to tBID61, and suggests that our dual-monitoring approach can recapitulate endogenous competitive interactions. Moreover, coadministration of both an Mcl-1 and Bcl-2 inhibitor in the competitive environment caused the Mcl-1 inhibitor to be less effective (Figure 4B, S9), presumably because more tBID was accessible in the system when not bound to Bcl-2 and may reflect differences in affinity between NOXA and tBID67, or differences in pharmacological effectiveness. Collectively, these data illustrate why multidimensional PPI analysis in live cells is critical for understanding pharmacological engagement of competitively-interacting PPI networks.
Generation of a series of orthogonal RNAPC tags
Unlike fluorescent reporters, RNAP-based reporter systems are not limited by spectral overlap, and should therefore be capable of measuring higher-order PPI networks. In our spit RNAP system, orthogonality comes from the RNAPC tags that each drive transcription from unique DNA promoters upon RNAP assembly, thereby permitting the transcription of unique output RNA signals. Thus far, only T7 and CGG RNAPC tags have been developed as proximity-dependent split RNAP reporters. Therefore, we next sought to expand the approach by generating a panel of orthogonal tags for deployment in PPI network analysis experiments.
We mined the literature for mutations within our RNAPC tags that alter its DNA promoter specificity68,69, and cloned these variants into our E. coli luciferase reporter system to assay their ability to function as proximity-dependent split RNAP reporters. First, we tested orthogonality by measuring a series of eight putative RNAPC variants on a panel of five different DNA promoters, T7, CGG, K1F, CTGA, and T3. All of the variants displayed robust activity on their target promoter, but the variants differed in terms of overall activity and off-target activity on the other promoters (Figure 5A, B). Based on overall activity and selectivity, we selected T7, K1F-b, CTGA, and T3 RNAPC variants, along with their respective promoter sequences, as a series of four orthogonal RNAPC tags to pursue further. We omitted the CGG variant used in the one-by-two interaction analysis because it showed crosstalk with both the CTGA and T3 promoters. Critically, all of the variants, especially those selected for further study, maintained very good dynamic range for PPI detection when paired with the evolved RNAPN and split isoleucine zipper peptides used previously43,70, displaying a 134-fold to 300-fold dynamic range (Figure 5C).
Figure 5.
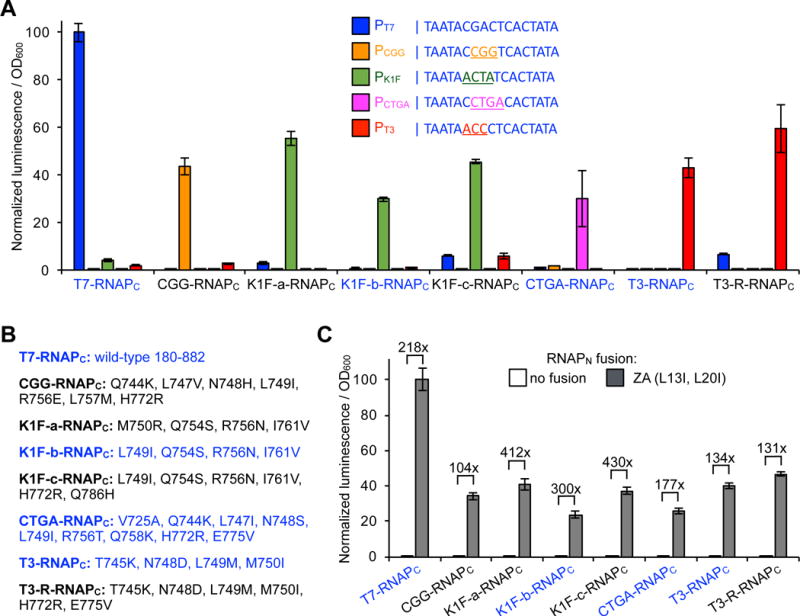
Uncovering a series of orthogonal proximity-dependent C-terminal split RNAPs. (A) E. coli transcriptional reporter assay for DNA promoter orthogonality of a series of split C-terminal T7 RNAP variants fused to ZB (ZB-RNAPC) on putative DNA promoter sequences. Sequences for the five target DNA promoters are shown (T7-blue, CGG-orange, K1F-green, CTGA-magenta, T3-red). E. coli transformed with the vector system shown in Figure 1B: 1) an expression vector for an evolved N-terminal RNAP fused to ZA (RNAPN-ZA), 2) an expression vector for a target RNAPC variant to be tested, and 3) a reporter vector that drives luciferase based on a target DNA promoter sequence. Cells were induced for 3 h with arabinose and then analyzed for luminescence. Error bars are ± s.e.m., n = 4. (B) Mutations shown for each RNAPC variant tested. (C) Proximity-dependent assembly of the series of ZB-RNAPC variants. E. coli transformed with the vector system shown in Figure 1B: 1) an expression vector for RNAPN fused to either nothing as a negative control or to ZA, 2) an expression vector for a target RNAPC variant to be tested, fused to ZB, and 3) a reporter vector that drives luciferase based on the target DNA promoter sequence of the variant being tested. Cells were induced for 3 h with arabinose and then analyzed for luminescence. Error bars are ± s.e.m., n = 4. The variants that were selected for further study are color-coded in blue.
One-by-four interactome analysis by RT-qPCR
With the four orthogonal RNAPC variants and four orthogonal promoters validated, we next sought to test whether we could monitor four Bcl-2 family PPIs simultaneously in mammalian cells. We envisioned a system in which one vector would express a target binding partner fused to RNAPN, and a series of four other vectors would each express different protein targets fused to the four orthogonal RNAPC variants (T7, K1F-b, CTGA, and T3) (Figure 6A). The RNAPC expression vectors would also each contain gene circuits that drive transcription of unique RNA outputs from the four orthogonal promoters, thereby “encoding” each PPI in a separate RNA signal (Figure 6B).
Figure 6.
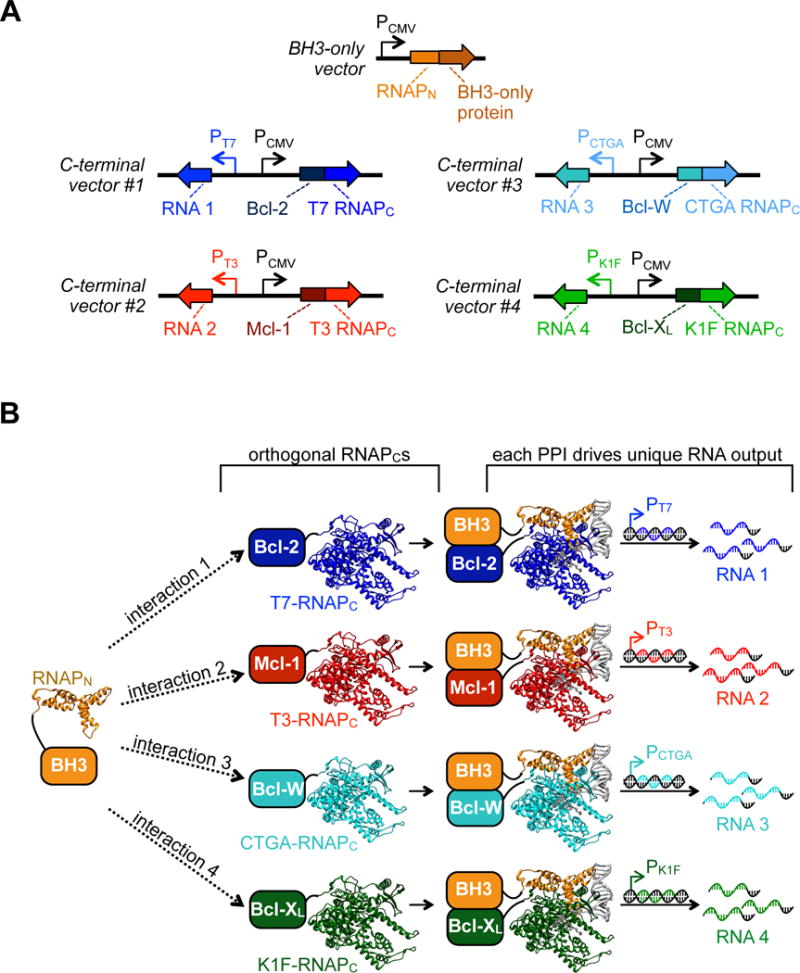
Schematic of a system to detect 4 multidimensional PPIs simultaneously. (A) Design of mammalian vector system to measure a one-by-four PPI network. Expression vectors for a series of anti-apoptotic-RNAPC using the orthogonal RNAPC variants developed in Figure 5 were engineered with a gene circuit that produces an orthogonal RNA output signal from transcription on the orthogonal DNA promoter. Four orthogonal vectors were created, which can then be deployed along with a BH3-only-RNAPN expression vector. (B) Schematic of one-by-four RNAP tagging system for multidimensional PPI analysis. The BH3-only peptide fusion on the RNAPN tag can competitively bind to a series of Bcl-2-family proteins, each themselves fused to orthogonal RNAPC tags. Interactions between each Bcl-2-family protein with the target results in the assembly of an orthogonal RNAP, which in turn transcribes an output RNA signal from an orthogonal DNA promoter. The relative amount of each PPI can be measured by RT-qPCR analysis of the output RNA signals.
To test the feasibility of a 1×4 interactome analysis with the split RNAP system, we cloned the orthogonal RNAPC variants into each of the Bcl-2 fusion vectors and swapped out the T7 promoter for the corresponding orthogonal promoter sequence matching each RNAPC variant. Rather than attempting to use four orthogonal fluorescent proteins, we aimed instead to measure each split RNAP assembly by direct quantitative RNA analysis. Aside from opening up future possibilities of doing even higher-order interaction analysis where spectral overlap of fluorescent proteins becomes prohibitive, we postulated that analyzing the RNA directly would also offer kinetic advantages and a more quantitative assay. Degradation of most fluorescent proteins is quite slow60, meaning that even once we inhibit a PPI, the time it takes for that inhibition to change the intracellular fluorescent protein concentration is long. However, RNA turnover is much faster, meaning we should be able to detect inhibition of a target PPI in a more dynamic manner. To enable this approach, we then changed the RNA output on each orthogonal RNAPC fusion vector to a unique, arbitrary sequence, and designed corresponding unique qPCR primers to analyze each output RNA. It should be noted that due to differences in primer efficiencies and promoter strengths, differences in outputs can only be measured in a relative manner between different conditions.
We transfected the set of 1×4 interactome vectors into HEK293T cells, allowed the cells to grow for 40 h, isolated total RNA from the cells, and then analyzed each unique PPI-dependent RNA output signal by quantitative reverse transcription PCR (RT-qPCR). We used analysis of GAPDH and the RNAPC gene using primers that do not bind to any mutation sites that change promoter specificity, and therefore report on total RNAPC levels, as a control for transfection and RNA isolation (Figure S10). First, we assayed whether we could recapitulate the deadBID, tBID, and NOXA binding interactions. As observed in the binary PPI analysis, we detected increased RNA production between all four Bcl-2 family proteins when the RNAPN was fused to tBID compared to dBID, but the NOXA fusion only showed enhanced RNA synthesis from the Mcl-1 fusion (Figure 7A). Furthermore, we again observed increased signal from the NOXA-Mcl-1 interactions compared to the tBID-Mcl-1 interactions in this competitive environment. Next, we assayed whether we could monitor competitive inhibition upon ABT-199 treatment in the context of tBID binding. We delivered the 1×4 interactome vectors using the RNAPN-tBID binding partner and tested whether loading the cells with ABT-199 for 3 h, 24 h, or 40 h resulted in measurable PPI inhibition, and which PPIs were affected. We observed selective Bcl-2/tBID PPI inhibition at 3 h with enhanced inhibition at 24 h. At 3 h, we detected no other off-target inhibition. However, we observed Bcl-XL/tBID PPI inhibition at the longer, 24 h, time point. This off-target activity has not previously been observed at the concentration used64, but has been noted at higher concentrations. Because the kinetics of the inhibition differ between targets, this off-target effect could be due to pharmacological cross-talk, or biological effects such as changes in endogenous Bcl-2-family binding partner levels. Collectively, these experiments indicate that the split RNAP PPI detection system can simultaneously monitor four PPIs in mammalian cells and detect competitive PPI inhibition on relatively short time scales using RNA analysis techniques.
Figure 7.
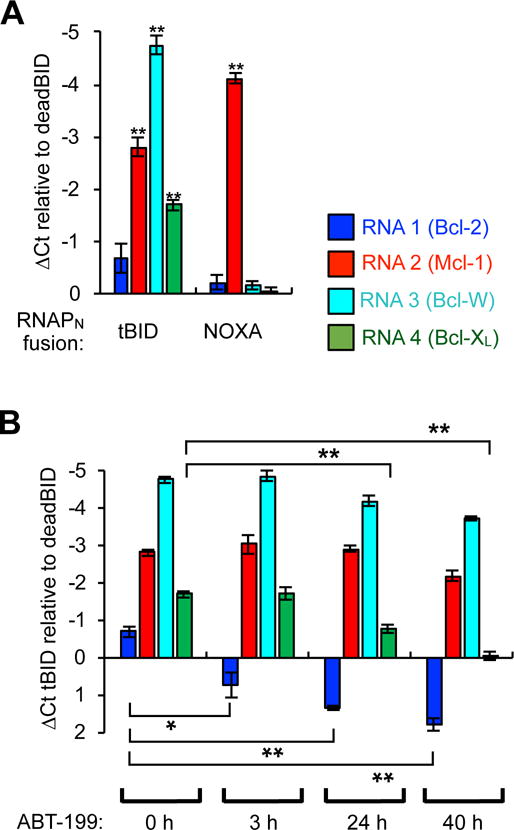
Detection of 1×4 Bcl-2 family PPIs simultaneously in mammalian cells by RT-qPCR analysis of the unique RNA outputs. (A) HEK293T cells were cotransfected with the plasmids shown in Figure 6A with the fusions indicated, grown for 40 h, lysed, and then total RNA was isolated and quantified by RT-qPCR. Separate PCR primers were used for each of the four unique RNA outputs to measure split RNAP assembly with each target. The data displayed is the delta-Ct value in comparison to cells transfected with the RNAPN-deadBID “negative control”. Therefore, a more negative value indicates more of a particular RNA is generated, and therefore more of a given interaction was present. (B) HEK293T cells were cotransfected with the plasmids shown in (Figure 6A) with the tBID-RNAPN fusion, grown for 40 h with 0.5 μM ABT-199 added at different time points, lysed, and then total RNA was isolated and quantified by RT-qPCR as described in (A). The data displayed is the delta-Ct value in comparison to cells transfected with the RNAPN-deadBID “negative control”. Error bars are ± s.e.m., n = 4. Student’s t-test; *P < 0.05, **P < 0.0005.
CONCLUSION
In conclusion, we developed and validated a series of split RNAP biosensors to monitor multidimensional PPI networks in live mammalian cells. Measuring endogenous biomolecular interactions in mammalian systems remains a substantial challenge, but an increasingly important problem as PPI modulators continue to gain momentum in the laboratory and clinic. The split RNAP biosensor approach described here can synergize with advances in nucleic acid sequencing technologies, by encoding new types of biological information in the high-throughput sequencing (HTS) data flow. Coupling the tools disclosed here with cell identifying bar-coding sequences in the RNA outputs or with direct in situ RNA sequencing technologies45 would permit single-cell analysis of multidimensional PPI networks. By leveraging advances in nucleic acid computation71,72, the RNA outputs of the split RNAP sensors could be adapted for complex genetic screens, in which a series of interactions in a network are screened in parallel for specific effects. The RNA outputs could easily be engineered to mediate cell viability, leading to a very simple and inexpensive mammalian genetic screening platform. Moreover, proteins of interest could be genomically-tagged, improving physiological relevance and single-cell consistency. This would also allow even higher-order networks to be probed and screened for. Although we found that transient transfection worked well for the experiments presented here, removing the complications due to heterogeneity in transfection efficiency will likely decrease variability and sensitivity to subtle effects. Finally, for synthetic biology applications, multiple split RNAP biosensors could analyze individual cells’ interactomes to determine cell type or disease state and drive a therapeutic outcome, as has been done for microRNA detection systems73.
We selected Bcl-2 family PPIs as a test bed to develop our split RNAP biosensor technology due to the availability of known small molecule inhibitors, which provided us with robust positive controls. Our observations of competitive effects of PPIs for the same target ligand upon pharmacological engagement of the Bcl-2 proteins is an excellent illustration as to why targeting PPIs therapeutically is so challenging. In future work, we will explore whether other therapeutically-relevant PPIs with known off-targets can also be assayed in our system. Integrating our rapid endogenous multidimensional PPI analysis approaches in the drug discovery process could substantially benefit the selection of pre-clinical molecules for both in cellulo efficacy and off-target interactions, thereby improving the drug discovery pipeline. Moreover, rather than measuring secondary treatment effects, direct PPI disruption measurements can be used to validate mechanisms of new therapeutic agents to gain enhanced understanding of targeted therapies prior to further preclinical development.
EXPERIMENTAL DETAILS
Cloning
All plasmids were constructed by Gibson Assembly74 from PCR products generated using Q5 DNA Polymerase (NEB) or Phusion Polymerase. All plasmids were sequenced at the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility. All used vectors are described in Table S1 and maps for each plasmid are shown in Figure S1. Full vector sequences and annotated vector maps are available upon request.
Luciferase-based transcription assays of Bcl-2 proteins in E. Coli
Experiments were conducted as previously described43. Briefly, S1030 cells75 were transformed by electroporation with three plasmids: (i) a T7 RNAPN-linker-BH3 only expression plasmid, (ii) an Bcl-2 family protein-linker-T7 RNAPC expression plasmid, and (iii) a reporter plasmid that encodes luciferase under control of a T7 RNA polymerase promoter. Single colonies were then grown to saturation overnight at 37 °C, and then each well of a 96-well deep well plate containing 0.54 mL of LB with antibiotics and 10 mM arabinose was inoculated with 60 μL of the overnight culture. After growth with shaking at 37 °C for 3 h, 150 μL of each culture was transferred to a 96-well black wall, clear bottom plate (Nunc), and luminescence and OD600 was measured on a Synergy Neo2 Hybrid Multi-Mode Reader (BioTek). The data were analyzed by dividing the luminscence values by the background-corrected OD600 value, then subtracting out the background from the reporter vector alone. All values were then normalized to the transcription from Bcl-2/deadBID, which was assigned an arbitrary value of 1, allowing the values from each luminescence plot to be compared to one another.
Luciferase-based transcription assays of orthogonal C-terminal RNAP variants in E. Coli
Experiments were conducted as previously described43 and similar to as noted above. Briefly, S1030 cells75 were transformed by electroporation with three plasmids: (i) a T7 RNAPN-linker-ZA expression plasmid, (ii) a ZB-linker-RNAPC expression plasmid with the listed mutations, and (iii) a reporter plasmid that encodes luciferase under control of a given target promoter sequence. Single colonies were then analyzed as described above. The data were analyzed by dividing the luminescence values by the background-corrected OD600 value, then subtracting out the background from the reporter vector alone. The experiment was then repeated, but this time only assaying each variant on its target promoter and assessing proximity-dependence by comparing assembly with RNAPN-linker-ZA with a plasmid that has no ZA fusion. For both experiments, values were then normalized to the transcription from the T7 RNAPC on the T7 promoter, which was assigned an arbitrary value of 100. Each variant was screened across all five promoters in this way. The sequence of split RNAP fusions were shown in Table S2.
Cell culture
HEK293T cells (ATCC) were maintained in DMEM (high glucose, L-glutamine, phenol red, sodium pyruvate; obtained from Gibco or Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technologies, Qualified US origin) and 1% penecillin/streptomycin (P/S, Gibco/Life Technologies). Multiple biological replicates were performed with cells from different passages and freshly thawed aliquots.
Mammalian fluorescence imaging and inhibitor assay
HEK293T cells were cultured in DMEM (high glucose, glutamine, phenol red, pyruvate; Gibco/Life Technologies) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technologies, Qualified US origin). The cells were plated on an 8-well coverglass slide (Labtek) and transfected in the next day with 300 ng of Bcl2-family vector and 300 ng of a BH3-only vector (Table S1 and Figure S1) using 1.8 μL of Lipofectamine 3000 (ThermoFisher Scientific) following the standard protocol. For the inhibitor experiments, 500 nM ABT-199, 10 μM A-1210477, or DMSO control was added during cell culture and post transfection. The cells were imaged on an Olympus BX53 microscope using a GFP and RFP filter set and a 10× objective.
Image processing and quantification
For image analysis of fluorescent protein outputs, we counted the number of positive cells in each channel. Each image for a given condition was processed using identical conditions to adjust brightness and contrast to a level where background fluorescence was observed for control samples in ImageJ (Wayne Rasband, NIH). The fluorescent spots in each GFP image were counted as fluorescent cell number in ImageJ for quantification analysis. A macro batch script for each analysis was used to ensure the settings for each group were identical. The following script was used in the “Batch Process” of ImageJ: “setThreshold(239, 5000); setOption(“BlackBackground”, false); run(“Convert to Mask”); run(“Watershed”); run(“Analyze Particles…”, “size=100-Infinity pixel include summarize in_situ” “. The “Count” results were then used for quantification analysis. We found that counting the number of GFP-positive cells yielded the most reproducible results.
RT-qPCR analysis
HEK293T cells were transfected with split RNAP vectors. 400 ng for each of the five plasmids showed in Fig. 6A. 6 μL of Lipofectamine 3000 (ThermoFisher Scientific) was used for the transfection in 12-well plates (Corning) and biological replicates were performed in quadruplicate for each condition. The inhibitor ABT-199 was omitted, added during transfection, or added 16 h or 37 h after transfection, and the cells were harvested at 40h after transfection. RNA was purified using an RNeasy Kit (Qiagen) and was reverse-transcribed using PrimeScript TM RT reagent Kit (TaKaRa). The transcribed cDNA was used as the qPCR template and the PCR reactions were performed on a LightCycler 96 Instrument (Roche) using FastStart Essential DNA Green Master (Roche). The DNA templates for RNA 1~4 and corresponding Q-PCR primers are listed in Table S3.
Supplementary Material
Acknowledgments
This work was supported by the University of Chicago, the National Institute of General Medical Sciences of the National Institutes of Health (R35 GM119840) to B.C.D., the Cancer Research Foundation, the University of Chicago Medicine Comprehensive Cancer Center (P30CA14599), and the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR000430). We thank Chuan He (University of Chicago) and Yamuna Krishnan (University of Chicago) for supplying materials and equipment.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting tables and figures (PDF)
Notes
J.P. and B.C.D. have filed a provisional patent application on proximity-dependent split RNAPs.
References
- 1.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Nature. 2005;437:1173. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 2.Chatr-aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen DC, Stark C, Breitkreutz A, Kolas N, O’Donnell L, Reguly T, Nixon J, Ramage L, Winter A, Sellam A, Chang C, Hirschman J, Theesfeld C, Rust J, Livstone MS, Dolinski K, Tyers M. Nucleic Acids Res. 2015;43:D470. doi: 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. Nucleic Acids Res. 2015;43:D447. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makley LN, Gestwicki JE. Chem Biol Drug Des. 2013;81:22. doi: 10.1111/cbdd.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modell AE, Blosser SL, Arora PS. Trends Pharmacol Sci. 2016;37:702. doi: 10.1016/j.tips.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj M, Bullock BN, Arora PS. Bioorg Med Chem. 2013;21:4051. doi: 10.1016/j.bmc.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tavassoli A. Curr Opin Chem Biol. 2017;38:30. doi: 10.1016/j.cbpa.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Delbridge AR, Strasser A. Cell Death Differ. 2015;22:1071. doi: 10.1038/cdd.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. Mol Cell. 2010;37:299. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czabotar PE, Lessene G, Strasser A, Adams JM. Nat Rev Mol Cell Biol. 2014;15:49. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 11.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Enschede SH, Humerickhouse RA, Wierda WG, Seymour JF. New Engl J Med. 2016;374:311. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perciavalle RM, Opferman JT. Trends Cell Biol. 2013;23:22. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. Biochim Biophys Acta. 2011;1813:508. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 14.Ivanov AA, Khuri FR, Fu H. Trends Pharmacol Sci. 2013;34:393. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Besbes S, Billard C. Cell Death Dis. 2015;6:e1810. doi: 10.1038/cddis.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. Nat Rev Drug Discovery. 2017;16:273. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 17.Newman RH, Fosbrink MD, Zhang JChem. Rev. 2011;111:3614. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welch CM, Elliott H, Danuser G, Hahn KM. Nat Rev Mol Cell Biol. 2011;12:749. doi: 10.1038/nrm3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wehr MC, Rossner M. J Drug Discovery Today. 2016;21:415. doi: 10.1016/j.drudis.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Miller KE, Kim Y, Huh WK, Park HO. J Mol Biol. 2015;427:2039. doi: 10.1016/j.jmb.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerppola TK. Chem Soc Rev. 2009;38:2876. doi: 10.1039/b909638h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Banerjee D, Liu YS, Chen XY, Liu XG. Analyst. 2010;135:1839. doi: 10.1039/c0an00144a. [DOI] [PubMed] [Google Scholar]
- 23.Handly LN, Yao J, Wollman R. J Mol Biol. 2016;428:3669. doi: 10.1016/j.jmb.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao Z, Petschnigg J, Ketteler R, Stagljar I. Nat Chem Biol. 2015;11:387. doi: 10.1038/nchembio.1809. [DOI] [PubMed] [Google Scholar]
- 25.Slavoff SA, Liu DS, Cohen JD, Ting AY. J Am Chem Soc. 2011;133:19769. doi: 10.1021/ja206435e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW. Nature. 2017;545:505. doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksoz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE. Cell. 2005;122:957. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 28.Hoppe AD, Scott BL, Welliver TP, Straight SW, Swanson JA. PLoS One. 2013;8:e64760. doi: 10.1371/journal.pone.0064760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuchenov D, Laketa V, Stein F, Salopiata F, Klingmuller U, Schultz C. Cell Chem Biol. 2016;23:1550. doi: 10.1016/j.chembiol.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabantous S, Nguyen HB, Pedelacq JD, Koraichi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS. Sci Rep. 2013;3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, Huang B. Nat Commun. 2016;7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blakeley BD, Chapman AM, McNaughton BR. Mol Biosyst. 2012;8:2036. doi: 10.1039/c2mb25130b. [DOI] [PubMed] [Google Scholar]
- 33.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Proc Natl Acad Sci U S A. 2004;101:12288. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jester BW, Cox KJ, Gaj A, Shomin CD, Porter JR, Ghosh I. J Am Chem Soc. 2010;132:11727. doi: 10.1021/ja104491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martell JD, Yamagata M, Deerinck TJ, Phan S, Kwa CG, Ellisman MH, Sanes JR, Ting AY. Nat Biotechnol. 2016;34:774. doi: 10.1038/nbt.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wehr MC, Laage R, Bolz U, Fischer TM, Grunewald S, Scheek S, Bach A, Nave KA, Rossner MJ. Nat Methods. 2006;3:985. doi: 10.1038/nmeth967. [DOI] [PubMed] [Google Scholar]
- 37.Gray DC, Mahrus S, Wells JA. Cell. 2010;142:637. doi: 10.1016/j.cell.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thaminy S, Miller J, Stagljar I. Methods Mol Biol. 2004;261:297. doi: 10.1385/1-59259-762-9:297. [DOI] [PubMed] [Google Scholar]
- 39.Hu CD, Kerppola TK. Nat Biotechnol. 2003;21:539. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galperin E, Verkhusha V, Sorkin A. Nat Methods. 2004;1:209. doi: 10.1038/nmeth720. [DOI] [PubMed] [Google Scholar]
- 41.Shekhawat SS, Ghosh I. Curr Opin Chem Biol. 2011;15:789. doi: 10.1016/j.cbpa.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furman JL, Badran AH, Shen S, Stains CI, Hannallah J, Segal DJ, Ghosh I. Bioorg Med Chem Lett. 2009;19:3748. doi: 10.1016/j.bmcl.2009.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pu J, Zinkus-Boltz J, Dickinson BC. Nat Chem Biol. 2017;13:432. doi: 10.1038/nchembio.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filonov GS, Moon JD, Svensen N, Jaffrey SR. J Am Chem Soc. 2014;136:16299. doi: 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, Peters DT, Tu5czyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM. Science. 2014;343:1360. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Nat Biotechnol. 2015;33:495. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moffitt JR, Hao JJ, Wang GP, Chen KH, Babcock HP, Zhuang XW. Proc Natl Acad Sci U S A. 2016;113:11046. doi: 10.1073/pnas.1612826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S, Xu LG, Ma W, Wu XL, Sun MZ, Kuang H, Wang LB, Kotov NA, Xu CL. J Am Chem Soc. 2016;138:306. doi: 10.1021/jacs.5b10309. [DOI] [PubMed] [Google Scholar]
- 49.Wabuyele MB, Soper S. A Single Molecules. 2001;2:13. doi: 10.1002/1522-2683(200110)22:18<3939::AID-ELPS3939>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Piepenburg O, Williams CH, Stemple DL, Armes NA. PLOS Biol. 2006;4:1115. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamft BM, Marblestone AH, Kording K, Schmidt D, Martin-Alarcon D, Tyo K, Boyden ES, Church G. PLoS One. 2012;7:e43876. doi: 10.1371/journal.pone.0043876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pu J, Chronis I, Ahn D, Dickinson BC. J Am Chem Soc. 2015;137:15996. doi: 10.1021/jacs.5b10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Church GM, Elowitz MB, Smolke CD, Voigt CA, Weiss R. Nat Rev Mol Cell Biol. 2014;15:289. doi: 10.1038/nrm3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nunez JK, Harrington LB, Doudna JA. ACS Chem Biol. 2016;11:681. doi: 10.1021/acschembio.5b01019. [DOI] [PubMed] [Google Scholar]
- 55.Chappell J, Watters KE, Takahashi MK, Lucks JB. Curr Opin Chem Biol. 2015;28:47. doi: 10.1016/j.cbpa.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 56.Kalhor R, Mali P, Church GM. Nat Methods. 2017;14:195. doi: 10.1038/nmeth.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Cell. 2014;159:647. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meighen EA. Microbiol Rev. 1991;55:123. doi: 10.1128/mr.55.1.123-142.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zha HB, AimeSempe C, Sato T, Reed JC. J Biol Chem. 1996;271:7440. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- 60.Li HL, Zhu H, Xu CJ, Yuan JY. Cell. 1998;94:491. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 61.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Mol Cell. 2005;17:393. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 61.Lee EF, Czabotar PE, Van Delft MF, Michalak EM, Boyle MJ, Willis SN, Puthalakath H, Bouillet P, Colman PM, Huang DCS, Fairlie WD. J Cell Biol. 2008;180:341. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemphill J, Chou C, Chin JW, Deiters A. J Am Chem Soc. 2013;135:13433. doi: 10.1021/ja4051026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang HC, Humerickhouse RA, Rosenberg SH, Elmore SW. Nat Med. 2013;19:202. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 65.Soderquist RS, Eastman A. Mol Cancer Ther. 2016;15:2011. doi: 10.1158/1535-7163.MCT-16-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJ, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers A. J Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Cancer Cell. 2006;9:351. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 68.Meyer AJ, Ellefson JW, Ellington AD. ACS Synth Biol. 2015;4:1070. doi: 10.1021/sb500299c. [DOI] [PubMed] [Google Scholar]
- 69.Segall-Shapiro TH, Meyer AJ, Ellington AD, Sontag ED, Voigt CA. Mol Syst Biol. 2014;10:742. doi: 10.15252/msb.20145299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L. J Am Chem Soc. 2005;127:146. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 71.Ran T, Douek Y, Milo L, Shapiro E. Sci Rep. 2012;2:641. doi: 10.1038/srep00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farzadfard F, Lu TK. Science. 2014;346:1256272. doi: 10.1126/science.1256272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Science. 2011;333:1307. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- 74.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Nat Methods. 2009;6:343. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 75.Carlson JC, Badran AH, Guggiana-Nilo DA, Liu DR. Nat Chem Biol. 2014;10:216. doi: 10.1038/nchembio.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.