

Abstract
Systems to control Cas9 with spatial and temporal precision offer opportunities to decrease side effects, protect sensitive tissues, and create gene therapies that are only activated at defined times and places. Here, we present the design of new Cas9 controllers based on RNA polymerase (RNAP)-based biosensors that produce gRNAs, thereby regulating target knockout. After development and validation of a new abscisic acid-inducible biosensor to control Cas9, we lowered the background of the system using continuous evolution. To showcase the versatility of the approach, we designed biosensors that measure medically-relevant PPIs to drive knockout. Finally, to test whether orthogonal RNAP biosensors could integrate multiple input signals to drive multiple gRNA-based outputs with a single Cas9 protein, we designed an “on-switch/off switch” controller. Addition of one input activates the “on switch” and induces knockout, while addition of a second input activates the “off switch” and produces a gRNA that directs the Cas9 protein to degrade the “on switch” gRNA vector, thereby deactivating it. This combined activation and deactivation system displayed very low background and inducible target knockout using different combinations of small molecule treatment. Our results establish engineered RNAP biosensors as deployable Cas9 control elements and open up new opportunities for driving genetic editing technologies by diverse input signals.
The RNA-guided endonuclease Cas9 from type II clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) systems has revolutionized the practicality and therapeutic potential of genome engineering1-3. Cas9 is programmed through complex formation with a single guide RNA (gRNA) that directs the protein to a specific DNA locus by base pair complementarity4. Deployment of Cas9-based systems in the laboratory or the clinic could be substantially improved by general methods to activate the gene editing function of Cas9 only in target cells and tissues with spatial and temporal control, as well as methods to shut the system down if side-effects begin to emerge due to off-target activities5.
The conditional regulation of Cas9 through external activation signals has been shown to lessen off-target cleavage events compared to constitutively active Cas96. To-date, approaches to regulate Cas9/gRNA genome editing activity have generally focused on engineering the Cas9 proteins. For example, Cas9 has been split and each half fused to dimerization domains7-10, allosteric regulatory domains have been inserted into the Cas9 protein6, 11, 12, Cas9 has been fused to small molecule-regulated protein degrons13, and unnatural amino acids have be inserted into Cas9 for control purposes14. One issue with these approaches is that they each need to be carefully optimized for a specific Cas9 variant. As new Cas9 systems continue to be discovered and optimized15, 16, each of these control systems in turn needs to be reinvented. More importantly, controlling Cas9 at the protein level does not permit leveraging the potential for multidimensionality through different activation signals driving different editing responses, which is trivial for Cas9 through the use of different gRNAs. Therefore, systems to control Cas9/gRNA genome editing activity at the gRNA level could offer advantages in terms of generality with any Cas9 variant and the opportunity for multidimensional responses. Thus far, gRNAs have been engineered with strand displacement-based riboswitches17 and ribozymes that respond to ligands18.
In this work, we develop new small molecule control systems for Cas9 using evolved RNA polymerase (RNAP)-based biosensors that drive target gRNAs when activated (Figure 1A). We show that the biosensors can detect interactions between medically-relevant proteins, such as the B cell lymphoma 2 (Bcl-2) and truncated Bcl-2 homology 3 (BH3)-interacting domain death agonist (tBID), and drive gene knockout based on those interactions. We go on to deploy two small molecule-activated biosensors in a logic gate, with one small molecule input activating targeted genome engineering and the other small molecule input deactivating the system. The “turn on”, “turn off” system maintained reasonable levels of targeted genome engineering but displayed very low background editing in the absence of “turn on” activation. Together, these results establish that regulating Cas9 at the gRNA level using engineered RNAPs is a viable strategy for engineering multidimensional, responsive genome engineering control systems. The ease of engineering the new ABA biosensor further solidifies the “plug-and-play” nature of our evolved split RNAP scaffold and presages future utility of these approaches for sensing and responding to diverse inputs.
Figure 1.

Design and application of an abscisic acid (ABA) inducible biosensor to control Cas9. (A) Schematic of Cas9 split RNAP biosensor control concept. Evolved proximity-dependent split RNAP halves (RNAPN and RNAPC) are fused to sensor domains, such that when the domains interact, the RNAP assembles and produces a gRNA, which then assembles with Cas9 and knocks out a target gene. (B) Vector system to test ABA-induced split RNAP detection system in E. coli. (C) Transcriptional output of split RNAPs upon addition of varying concentrations of ABA assayed in E. coli using the vectors shown in (B). Cells were induced for 3 h with arabinose and then analyzed for luminescence. Error bars are ± s.e.m., n = 5. (D) Vector system to deploy ABA biosensors in mammalian cells. (E) Knockout efficiency of ABA biosensor with N-29-1 RNAPN compared to a constitutive gRNA. HEK293-GFP cells transfected with varying combinations of the vectors shown in (D), grown for 6 days, and then analyzed for GFP knockout by flow cytometry. Addition of 10 μM ABA for 5 days induces knockout. Error bars are ± s.e.m., n = 3.
Generation of an abscisic acid-inducible RNAP biosensor for Cas9 control
Recently, we reported the generation of a versatile biosensor platform based on evolved, proximity-dependent split RNAPs19, 20. In order to generate a new Cas9 control system, we aimed to leverage the evolved split RNAP system to create a ligand-inducible transcriptional controller for gRNA production in mammalian systems. We chose to generate an RNAP biosensor for abscisic acid (ABA), due to its lack of targets in the human proteome, using previously reported ABA-inducible dimerization systems based on the ABI and PYL proteins21. We fused ABI and PYL onto the N-terminal split RNAP (RNAPN) and C-terminal split RNAP (RNAPC) halves of our evolved split RNAP system and measured ABA-induced transcriptional activation of the biosensor in E. coli (Figure 1B, Supporting Information Table S1 and Figure S1). Induction with ABA resulted in a robust, dose-dependent increase in RNA synthesis, with a 468-fold dynamic range after just three hours of induction (Figure 1C).
Given that the new ABA biosensor performed well in E. coli, we next tested whether we could use the ABA biosensor to control Cas9/gRNA genome editing in mammalian cells. We cloned a GFP-targeting gRNA controller gene circuit using the engineered ABA split RNAP system (Figure 1D), which we deployed along with Staphylococcus aureus Cas9 (saCas9)22. Assaying the system in GFP-expressing HEK293 (HEK293-GFP) cells showed that the background level of genome editing in the absence of inducer was 14%, compared to 1.7% experimental background for cells transfected with an RFP vector alone as a transfection control. However, induction with 10 μM ABA for 5 days resulted in a knockout efficiency of 29%, compared with 69% knockout from a control vector with constitutive gRNA production under these experimental conditions (Figure 1E). These results demonstrate the viability of using RNAP-based biosensors to control Cas9 at the gRNA level, but the high background knockout activity in the absence of inducer precludes utility of the approach. Although the evolved split RNAP biosensor platform appeared to have a very low background in E. coli and our previous mammalian experiments, the irreversible nature of genome engineering accentuates any level of leaky activation, revealing a deficit and an opportunity for improvement of the split RNAP biosensors. Therefore, we next turned our attention to lowering the background activity of the split RNAP-based biosensor platform.
Evolution and deployment of RNAP biosensors with lower background
In our original work to create the proximity-dependent split RNAPs, we used Phage-Assisted Continuous Evolution (PACE)23 to evolve proximity dependent assembly of the split RNAP using leucine zipper peptides as a model dimerization system20. In brief, phage carried an evolving RNAPN fused to one leucine zipper, and E. coli cells housed two RNAPCs fused to either a binding peptide or a non-binding peptide. Each RNAPC drove either positive or negative selection pressure. At the conclusion of the reported continuous evolutions, we found that we could not enhance the negative selection any further without extinction of the phage. Upon further analysis of the evolved gene products subsequent to the published studies, we discovered that the RNAPN leucine zipper peptide had evolved a low but measurable affinity for the off-target control peptide. Therefore, we reasoned that we could improve the negative selection by simply using an off-target RNAPC without any fusion protein. To test this concept, we cloned a new negative accessory plasmid (APneg) with stronger ribosome binding sites (RBSs) controlling the RNAPC and dominant negative gIII (gIIIneg), without any fusion protein (Figure S1, Table S1). We initiated PACE with libraries of phage from the end of our previous evolutions20, and found that the phage could now replicate with the stronger APnegs. Full evolutionary details are outlined in Figure S2. Briefly, after five days of PACE with sequentially stronger selection pressure using combinations of posAP and negAPs (Figure 2A), the populations converged on several new RNAPN mutations (Figure S3). Several of the new mutations exist at the split protein interface, but some are in unstructured regions of the protein (Figure S4), illustrating why unbiased evolution is a powerful design strategy for this type of protein engineering challenge.
Figure 2.
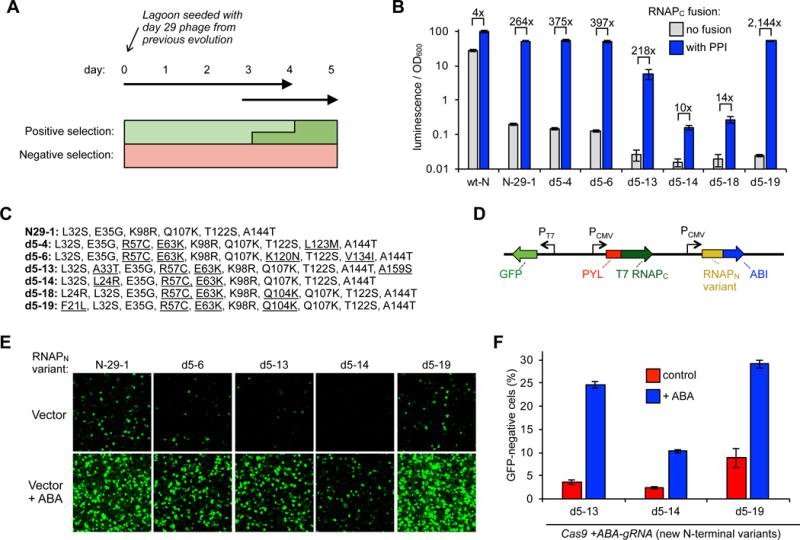
Evolution of improved split RNAP biosensors and deployment as Cas9 controllers. (A) Overview of PACE design to evolve split RNAP biosensors with a lower background. (B) Transcriptional reporter assays of a series of RNAPN variants that emerged from the evolution. E. coli cells were transformed with, 1) a vector expressing a zipper peptide-fused RNAPN variant, 2) a vector expressing RNAPC either fused to a zipper peptide or without a fusion, and 3) a T7 promoter-driven luciferase reporter vector. The cells were induced for 3 h with arabinose and then analyzed for luminescence. Error bars are ± s.e.m., n = 5. (C) Mutations of the RNAPN variants assayed in (B). (D) Vectors designed to test new RNAPN variants in a mammalian GFP expression assay. (E) HEK293T cells were transfected with the vectors shown in (D) containing different RNAPN variants, stimulated with either nothing or 10 μM ABA 7 h post transfection, grown for 23 h, and then analyzed for GFP expression by fluorescence microscopy (quantification and full imaging shown in Figure S5 and S6). (F) Knockout efficiency of new RNAPN variants in ABA biosensor system. HEK293-GFP cells were transfected with the vector system shown in Figure 1D. 10 μM ABA or DMSO control were added one day after transfection and grown for 5 days more, and then analyzed for GFP knockout by flow cytometry. Error bars are ± s.e.m., n = 3.
Transcriptional reporter assays revealed that most of the newly-evolved variants had a lower background than the starting evolved variant (N-29-1), with some variants having up to a 2,100-fold dynamic range based on the fused protein-protein interaction (Figure 2B), indicating the evolution dramatically improved the split RNAP platform. We selected a subset of variants (Figure 2C) to assay in a mammalian cell GFP transcriptional reporter assay (Figure 2D), which generally reproduced the trends observed in E. coli, further demonstrating that the evolved variants had a lower background and robust ABA responsiveness in mammalian cells (Figure 2E, Figure S5 and S6). We also assayed the optimal inducer concentration, and found that induction with 1-100 μM ABA resulted in similar levels of activation, similar to the 10 μM concentration used thus far (Figure S7, Table S2). Finally, we cloned the new variants into the gRNA controller gene circuit (Figure 1D), and found that they overall maintained reasonable genome engineering activation upon induction with ABA (10% - 34%), but in the absence of ABA the background level of knockout was decreased (2.3% - 8.8%), in some cases near the background of the assay (1.7%) (Figure 2F). Collectively, these results demonstrate that the newly-evolved RNAP biosensors perform better as ABA-inducible Cas9 controllers.
Using Bcl-2 protein-protein interactions (PPIs) to drive Cas9
One advantage of the protein-based split-RNAP biosensor scaffold is that one could, in principle, detect medically relevant PPIs and drive responses based on those interactions. The eventual concept would then be to use measurements about endogenous biochemical events to control Cas9, thereby selecting cell types based on molecular markers. To explore this possibility, we designed RNAP-based biosensors that detect interactions between the apoptotic regulatory protein, Bcl-2, and its ligand, tBID24, using methods we recently developed25. We designed vectors that express the RNAPC fused to Bcl-2 and RNAPN fused to either the binding domain of BID (tBID) or a control peptide with key binding components omitted (dBID) (Figure 3A). Delivery of only the Bcl-2-RNAPC vector along with Cas9 resulted in very low knockout of 1.9%, while adding in the control, RNAPN-dBID vector resulted in a background knockout without the PPI of 7.2% (Figure 3B). However, when the interacting RNAPN-tBID vector is deployed, the knockout efficiency rises to 21.6%. To confirm that this effect is due to the Bcl-2/tBID PPI, we treated the cells with a Bcl-2 PPI inhibitor, ABT19926, which lowered the level of knockout to 11.9%. Additionally, we observed some dose-dependent alterations in gRNA output levels due to ABT199 inhibition as assayed by RT-qPCR (Table S3). Collectively, these results highlight the versatility of the RNAP biosensor platform and illustrate the possibility of using biosensors of disease-relevant, regulated PPIs as endogenous molecular markers to control Cas9.
Figure 3.
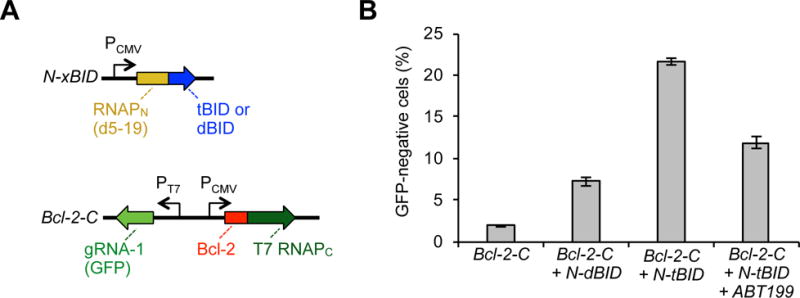
Biosensors that detect Bcl-2 PPIs and drive Cas9 knockout. (A) Vectors designed to express Bcl-2 fused to T7 RNAPC and the evolved RNAPN fused to either an interacting BID (tBID) or a non-interacting BID (dBID), along with T7 promoter-drive gRNA to GFP. (B) HEK293-GFP cells were transfected with different combinations of the vector system shown in (A), grown for 6 days, and then analyzed for GFP knockout by flow cytometry. Addition of 500 nM ABT199 6 h after transfection and maintained for the following 2 days lowered the PPI-dependent knockout. Error bars are ± s.e.m., n = 3.
Deploying dual biosensors for small molecule logic gate Cas9 control
Although the newly evolved split RNAP variants displayed lower levels of knockout in the absence of induction, there was still a measurable background activity for the most active variants. To remedy this, we envisioned that a second, “off switch” gRNA controller gene circuit, which produces a gRNA that targets and degrades the “on switch” gRNA controller upon activation by a different small molecule inducer, could lower the background and “protect” cells by turning off the system prior to induction. We had previously generated a rapamycin-inducible split RNAP biosensor and an orthogonal RNAPC that drives transcription from a unique DNA promoter sequence20. Using these tools, we generated a rapamycin-inducible gRNA controller gene circuit that instead produces a gRNA that targets saCas9 to the ABA-inducible gRNA controller gene circuit (Figure 4A). In this way, the “off switch” can be activated to shut down the entire system prior to activation of the “on-switch”, thereby lowering the background knockout in target cells. Both controllers utilize the same saCas9 protein, so all of the control emerges from differential gRNA generation.
Figure 4.
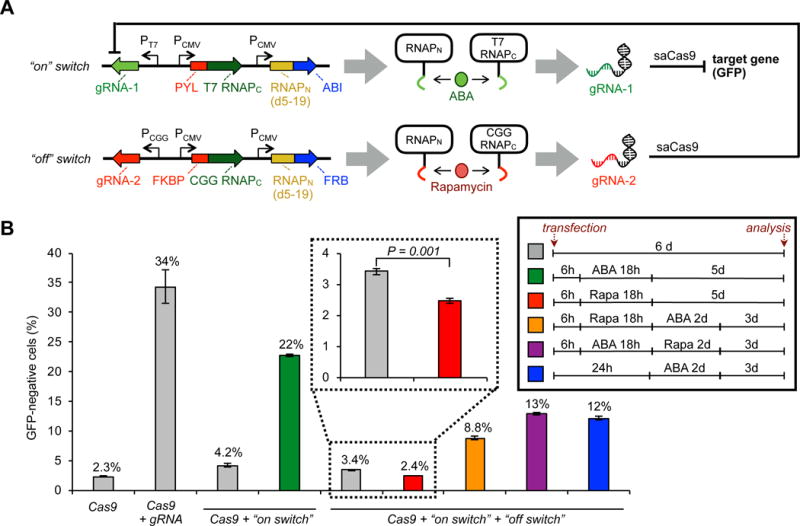
Dual control of Cas9 using two small molecule biosensors. (A) Schematic of “on switch” vector that produces an ABA-inducible RNAP biosensor system and triggers gRNA production for a target gene (GFP), and “off switch” vector that produces a rapamycin-inducible RNAP biosensor system and triggers gRNA production that targets the “on switch” vector. (B) HEK293-GFP cells were transfected with a Cas9 expression vector and the vectors shown in (A), grown for 6 d, then analyzed for GFP knockout by flow cytometry. Treatment with 10 μM ABA and/or 10 nM rapamycin (“Rapa”) for the given times shown in the inset induces either the “on switch” or “off switch” vectors and corresponding target knockout or system deactivation. Cells were washed between each treatment change to remove the molecule from the previous condition. Error bars are ± s.e.m., n = 3.
To test the dual controller system, we delivered the “on switch” and “off switch” vectors, along with an saCas9 expression vector, into mammalian cells, and treated the cells with ABA and/or rapamycin at various time points (Figure 4B). Under the experimental conditions, a constitutive gRNA vector induced knockout at 34% relative to 2.3% background without a gRNA. The “on-switch” vector alone, without activation, raised the background knockout to 4.2%, but was readily activated by 18h of ABA treatment to 22% knockout. However, with both the “on switch” and “off switch” vectors, rapamycin treatment lowered the background to 2.4%, which is equivalent to the background of the experimental cells treated with RFP transfection control vectors, thereby demonstrating the ultra-low background in the dually controlled system. ABA treatment in the absence of rapamycin still induced reasonable knockout (12%), though to a lower level than the “on-switch” alone, possibly due to leaky activation of the “off switch”. Pretreatment with rapamycin and then ABA still induced knockout (8.8%), but to a lower extent, showcasing the ability of the “off switch” to protect cells. The incomplete deactivation of the rapamycin-induced “off switch” is possibly due to heterogeneity of DNA plasmid uptake in the transient transfections, leading to incomplete degradation. In any case, as a proof-of-concept, these observations demonstrate the ability of multi-controlled Cas9 systems to protect cells from subsequent Cas9 knockout, while also lowering the background in a small molecule-dependent manner.
Our current system displays a very low background, but also suffers from a diminished level of activation, which has been observed in other reported Cas9 control systems. Because our system uses the full-length, wild-type Cas9 protein, all of our performance loss is due to gRNA production inefficiencies from the RNAP biosensors. The heterogeneity in response could be due to the transient transfections used in this study. In future work, using viral delivery systems or direct protein delivery could offer better control over the Cas9 response. Additionally, the localization of the DNA substrate for the split RNAP biosensors may be heterogeneous and suboptimal. Interestingly, we found that the split RNAPs actually produce more gRNA than a constitutive PU6–driven gRNA, which is commonly used for constitutive Cas9 knockout, indicating further optimization of our system is certainly possible (Figure S8). For example, using small, chemically-modified, gRNA-generating DNA substrates could increase potency and result in a more homogenous response.
Recently, a “Self-Limiting Cas9 circuit” was developed27, in which a self-targeting gRNA regulates the levels of Cas9 protein, thereby decreasing off-target activities. Our “on switch/off switch” approach described here is conceptually similar, but has the added benefit of temporal regulation afforded by small molecule control. Finally, we note that we chose the rapamycin biosensor for validation in cell culture, but since rapamycin has targets in human cells, it would likely not be the best biosensor system for therapeutic applications.
In summary, this work demonstrates that engineered RNAP-based biosensors can control Cas9 in a multidimensional manner using exogenously added small molecules or endogenous PPI measurements. Moreover, these studies validate that multidimensional control of Cas9 at the gRNA level opens up new opportunities for engineering Cas9 activation responses using multiple input signals. An advantage of the RNAP-based scaffold is that biosensors for a range of physiological activities can be easily created by leveraging the wealth of precedent on how to engineer fluorescent protein-based biosensors based on split GFP and FRET probes28, 29. Finally, the newly-evolved RNAPN variants generated here will likely find utility in other synthetic biology applications due to their exceptionally low background activity and very large dynamic range.
Methods
Cloning
All plasmids were constructed by Gibson Assembly from PCR products generated using Q5 DNA Polymerase (NEB) or Phusion Polymerase. pSPgRNA was a gift from Professor Charles Gersbach (Addgene plasmid #47108)30, which was used to construct the constitutive gRNA vector. The gene for saCas9 was a gift from Professor Feng Zhang, which was cloned into a custom CMV-driven mammalian expression vector. The genes for ABI and PYL were gifts from Professor Fu-Sen Liang, which were cloned into our custom vectors. Phage used in this research were constructed and evolved in previous work20. Briefly, the RNAPN-ZA fusion had been cloned into an SP phage backbone and underwent 29 days evolution before the evolution in this current work. All plasmids were sequenced at the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility. All used vectors are described in Table S1 and maps for each plasmid are shown in Figure S1. The constructed gRNA sequences for GFP knockout (gRNA-1) and “off switch” (gRNA-2) are listed in Table S4. Full vector sequences and annotated vector maps are available upon request.
Phage-assisted continuous evolution (PACE)
PACE was carried out using previously described methods20 with slight modifications. The key difference was that the negative AP contained no fusion protein on the RNAPC. During the evolution, phage samples were collected every 24 h, then boiled for 10 min to lyse the phage and release the genomes. PCR was then used to amplify the DNA library containing the RNAPN variants, which was then subcloned into vector p3-7. Single colonies were picked from the transformation and subjected to analysis by Sanger sequencing. The results of the sequence analysis during the course of the evolution are shown in Figure S3.
Luciferase-based in vivo transcription assays of split RNAPs
Experiments were conducted as previously described20. Briefly, S1030 cells were transformed with three plasmids: (i) a constitutive RNAPN-expression plasmid, (ii) an arabinose induced RNAPC-expression plasmid, (iii) a T7 promoter driven luciferase expression plasmid. Single colonies were then grown in a 96-deep well plate overnight at 37 °C, and 50 μL of the culture was transferred to a new 96-deep well plate containing 0.5 mL of LB with antibiotics and 10 mM arabinose. After growth with shaking at 37 °C for 3 h, 150 μL of each culture was transferred to a 96-well black wall, clear bottom plate (Nunc), and luminescence and OD600 was measured on a Synergy Neo2 Hybrid Multi-Mode Reader (BioTek). The data were analyzed by dividing the luminescence values by the background-corrected OD600 value. All values were then normalized to the wild-type split RNAP fused to ZA and ZB, which was assigned an arbitrary value of 100. For the ABA induced system, the experiment was performed identically, except upon outgrowth, different concentrations of ABA were added together with the arabinose.
Mammalian fluorescence imaging and quantification
HEK293T cells (ATCC) were maintained in DMEM (glucose, GlutaMAX™, phenol red, sodium pyruvate, obtained from Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco/Life Technologies, Qualified US origin) and 1% penicillin/streptomycin (P/S, Gibco/Life Technologies). For the data shown in Figure 2E, S5, HEK293T cells were plated on an 8-well coverglass slide (Cellvis) and transfected the next day with 500 ng of split RNAP GFP expression vector (Table S1 and Figure S1) using 1.5 μL of Lipofectamine 3000 (ThermoFisher Scientific) following the standard protocol. 10 μM ABA was added 7 h after transfection. The cells were imaged 23 h later on an Olympus BX53 microscope using a GFP filter set and a 10× objective. For the data shown in Figure S7, HEK293T cells were plated on an 8-well coverglass slide and transfected the next day with 500 ng of pJin 278 (Table S1 and Figure S1) using 1.5 μL of Lipofectamine 3000 (ThermoFisher Scientific) following the standard protocol. 10 μM ABA was added during transfection. The cells were imaged 46 h later on a Leica DMi8 microscope using a GFP filter set and a 20× objective. For quantification, identical settings were used for a given condition to adjust brightness and contrast in ImageJ (Wayne Rasband, NIH). The fluorescent spots in each GFP image were counted as fluorescent cell numbers in ImageJ for quantification analysis and macro batch scripts were used for each analysis in the “Batch Process” of ImageJ. The data for Figure S5 was obtained using the script, “setThreshold(1000, 5000); setOption(“BlackBackground”, false); run(“Convert to Mask”); run(“Watershed”); run(“Analyze Particles…”, “size=100-Infinity pixel include summarize in_situ” ”. For Figure S7, the threshold was set to (6000, 65535) instead as images were taken as 16-bit rather than 8-bit. The “Count” results were then used for quantification analysis.
GFP knockout
HEK293-GFP cells (GenTarget), which genomically encode GFP, were used to test knockout efficiencies, and were maintained in the same DMEM media as described above for HEK293T cells. 7×104 cells were plated on a 48-well plate (NEST) for each experiment. One day after plating, the cells were transfected with 1.8 μL Lipofectamine 3000 (Life Technologies) and corresponding vector DNAs. In the assays shown in Figure 1E and 2F, 20 ng RFP expression plasmid, 300 ng saCas9 expression plasmid22, and 300 ng GFP-knockout gRNA plasmid were used. In the assays shown in Figure 3B, 20 ng RFP expression plasmid, 200 ng saCas9 expression plasmid22, 300 ng of RNAPN-xBID, and 300 ng Bcl-2-RNAPC vector were used. In the assays shown in Figure 4B, 20 ng RFP expression plasmid, 250 ng saCas9 expression plasmid, 150 ng GFP-knockout gRNA(gRNA-1) plasmid and 250 ng GFP knockout switch-off gRNA(gRNA-2) plasmid were used. For single small molecule treatment, 10 μM ABA was added 24 h after transfection (Fig. 1E and 2F). For the double small molecule treatment experiments (Fig. 4B), ABA or Rapamycin were added sequentially and the cells were washed between each treatment. For all assays, 6 days after transfection, the cells were trypsinized and resuspended in DMEM supplemented with 10% FBS and analyzed on a LSR Fortessa 4-15 flow cytometer (BD digital instrument, 488 nm laser with 530/30 nm filter for GFP, 561 nm laser with 610/20 nm filter for RFP). GFP negative or positive cells were analyzed on RFP-gated cells as a transfection control.
Quantitative reverse transcription PCR (RT-qPCR) analysis of gRNA production
HEK293T cells were transfected with 1 μg DNA of corresponding vectors for each sample in 12-well plates (Denville). Lipofectamine 3000 (ThermoFisher Scientific) was used for the transfection following standard protocol. For the ABA-induced gRNA production test in Table S2, 1 μg of pJin 264 was used for each sample and 0, 1, 10, or 100 μM ABA was added when transfecting cells. For the concentration-dependent test of ABT199 in Table S3, 500 ng of each plasmid (pJin 310 and p12-34) was used for each sample and 0, 50, 250, 500 or 1,000 nM ABT199 was added when transfecting cells. The cells were harvested 40 h after transfection for these assays. For analysis of amount of gRNA produced by split RNAP in Fig S8, 1 μg of plasmid was used and DMSO or corresponding small molecules were added 15 h after transfection, and the cells were harvested 26 h later. After cell lysis, RNA was purified using an RNeasy Kit (Qiagen), then reverse-transcribed using PrimeScript TM RT reagent Kit (TaKaRa). The Transcribed cDNA libraries were analyzed by qPCR on a LightCycler 96 Instrument (Roche) using FastStart Essential DNA Green Master (Roche). The primer sequences for qPCR are listed in Table S4.
Supplementary Material
Acknowledgments
This work was supported by the University of Chicago, the National Institute of General Medical Sciences of the National Institutes of Health (R35 GM119840) to B.C.D., and the University of Chicago Medicine Comprehensive Cancer Center (P30CA14599). We thank F. Zhang (MIT), C. Gersbach (Duke University), F.S. Liang (University of New Mexico), C. He (University of Chicago), and Y. Krishnan (University of Chicago) for supplying materials and equipment.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at XXX. Supplementary Tables and Figures.
COMPETING FINANCIAL INTERESTS
B.C.D. and J.P. have filed a provisional patent on split RNAP-based biosensors.
References
- 1.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 2.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nunez JK, Harrington LB, Doudna JA. Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem Biol. 2016;11:681–688. doi: 10.1021/acschembio.5b01019. [DOI] [PubMed] [Google Scholar]
- 6.Davis KM, Pattanayak V, Thompson DB, Zuris JA, Liu DR. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright AV, Sternberg SH, Taylor DW, Staahl BT, Bardales JA, Kornfeld JE, Doudna JA. Rational design of a split-Cas9 enzyme complex. Proc Natl Acad Sci U S A. 2015;112:2984–2989. doi: 10.1073/pnas.1501698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zetsche B, Volz SE, Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen DP, Miyaoka Y, Gilbert LA, Mayerl SJ, Lee BH, Weissman JS, Conklin BR, Wells JA. Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity. Nat Commun. 2016;7:12009. doi: 10.1038/ncomms12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee M, Li J, Liang Y, Ma G, Zhang J, He L, Liu Y, Li Q, Li M, Sun D, Zhou Y, Huang Y. Engineered Split-TET2 Enzyme for Inducible Epigenetic Remodeling. J Am Chem Soc. 2017;139:4659–4662. doi: 10.1021/jacs.7b01459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, Savage DF. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. 2016;34:646–651. doi: 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richter F, Fonfara I, Bouazza B, Schumacher CH, Bratovic M, Charpentier E, Moglich A. Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res. 2016;44:10003–10014. doi: 10.1093/nar/gkw930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maji B, Moore CL, Zetsche B, Volz SE, Zhang F, Shoulders MD, Choudhary A. Multidimensional chemical control of CRISPR-Cas9. Nat Chem Biol. 2017;13:9–11. doi: 10.1038/nchembio.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo J, Liu QY, Morihiro K, Deiters A. Small-molecule control of protein function through Staudinger reduction. Nat Chem. 2016;8:1027–1034. doi: 10.1038/nchem.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC, Doudna JA, Banfield JF. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–241. doi: 10.1038/nature21059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Zhan Y, Chen Z, He A, Li J, Wu H, Liu L, Zhuang C, Lin J, Guo X, Zhang Q, Huang W, Cai Z. Directing cellular information flow via CRISPR signal conductors. Nat Methods. 2016;13:938–944. doi: 10.1038/nmeth.3994. [DOI] [PubMed] [Google Scholar]
- 18.Ferry QR, Lyutova R, Fulga TA. Rational design of inducible CRISPR guide RNAs for de novo assembly of transcriptional programs. Nat Commun. 2017;8:14633. doi: 10.1038/ncomms14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pu J, Chronis I, Ahn D, Dickinson BC. A Panel of Protease-Responsive RNA Polymerases Respond to Biochemical Signals by Production of Defined RNA Outputs in Live Cells. J Am Chem Soc. 2015;137:15996–15999. doi: 10.1021/jacs.5b10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pu J, Zinkus-Boltz J, Dickinson BC. Evolution of a split RNA polymerase as a versatile biosensor platform. Nat Chem Biol. 2017;13:432–438. doi: 10.1038/nchembio.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang FS, Ho WQ, Crabtree GR. Engineering the ABA plant stress pathway for regulation of induced proximity. Sci Signal. 2011;4:rs2. doi: 10.1126/scisignal.2001449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pu J, Dewey J, Hadji A, LaBelle JL, Dickinson BC. RNA Polymerase tags to monitor multidimensional protein-protein interactions reveal pharmacological engagement of Bcl-2 proteins. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b06152. in press. http://dx.doi.org/10.1021/jacs.7b06152. [DOI] [PMC free article] [PubMed]
- 26.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 27.Petris G, Casini A, Montagna C, Lorenzin F, Prandi D, Romanel A, Zasso J, Conti L, Demichelis F, Cereseto A. Hit and go CAS9 delivered through a lentiviral based self-limiting circuit. Nat Commun. 2017;8:15334. doi: 10.1038/ncomms15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frommer WB, Davidson MW, Campbell RE. Genetically encoded biosensors based on engineered fluorescent proteins. Chem Soc Rev. 2009;38:2833–2841. doi: 10.1039/b907749a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oldach L, Zhang J. Genetically encoded fluorescent biosensors for live-cell visualization of protein phosphorylation. Chem Biol. 2014;21:186–197. doi: 10.1016/j.chembiol.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, Guilak F, Crawford GE, Reddy TE, Gersbach CA. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.