

Abstract
The spliceosome undergoes dramatic changes in a splicing cycle. Structures of B, Bact, C, C*, and ILS complexes revealed mechanisms of 5′ splice site (ss) recognition, branching, and intron release, but lacked information on 3′ ss recognition, exon ligation and release. Here, we report a cryoEM structure of the post-catalytic P complex at 3.3Å resolution, revealing that 3′ ss is mainly recognized through non-Watson-Crick basepairing with the 5′ ss and branch point. Furthermore, an unidentified protein becomes stably associated with the P complex, securing the 3′ exon and potentially regulating Prp22 activity. Prp22 binds nucleotides 15–21 in the 3′ exon, enabling it to pull the intron-exon or ligated exon in a 3′ to 5′ direction to achieve 3′ ss proofreading or exon release, respectively.
Introduction
Introns are spliced out through two sequential transesterification steps catalyzed by the spliceosome, a huge RNA-protein complex made of five snRNAs and over one hundred proteins. Spliceosomal remodeling by ATP-dependent DExD/H-box helicases during each splicing cycle generates at least seven distinct states known as the B (1), Bact (2), B*, C (3, 4), C* (5, 6), P, and ILS (7) complexes. Following the first transesterification reaction (also referred to as the branching reaction) catalyzed by B*, the C complex emerges encompassing the newly freed 3′ OH group of 5′ exon and a lariat intermediate. The C complex further rearranges to form the C* complex, primed for the second transesterification reaction (also referred to as the ligation reaction). In the ligation reaction, the two exons join together, forming the post-catalytic P complex that contains the ligated exons and the lariat. The exons are then released through the action of helicase Prp22, generating the ILS complex, which only contains the lariat.
High-resolution cryo electron microscopy (cryoEM) structures of almost all these complexes of the splicing cycle have been determined (1–7), leaving two missing pieces of the puzzle—atomic descriptions of the B* and P complexes. Several important questions—all related to the P complex—remain to be answered for the complete molecular mechanism of splicing, including how the 3′ ss is recognized and docked to the proximity of 5′ exon, how exon ligation is catalyzed, and how the DEAH-box ATPase/helicase Prp22 promotes 3′ ss proofreading and exon release.
Here, we report a cryoEM structure of S. cerevisiae (yeast) P complex at 3.3 Å resolution. Our atomic model of the P complex reveals that the 3′ ss recognition is driven by the interaction between 3′ and 5′ ss, likely facilitated by a stem-like structure formed between the intronic regions close to the branch site and 3′ ss. Our structure reveals that one or more unidentified proteins become stably associated with the core components of the P complex around Prp8 and Prp22, securing the 3′ exon and potentially regulating the activity of Prp22. The structure demonstrates that Prp22 binds nucleotides 15–21 in the 3′ exon, enabling it to pull the intron-exon spanning the 3′ ss or the ligated exons in a 3′ to 5′ direction to fulfill its function in 3′ ss proofreading or exon release, respectively. By providing insights into the molecular mechanism of 3′ ss recognition, exon ligation, and the action of RNA helicase Prp22, the atomic model of the P complex fills a major gap in our understanding of the splicing cycle.
Results
Overall structure
We purified the P complex using a protein A tag on Cef1 and a CBP tag on Prp22 from yeast carrying the Prp22H606A mutant, a mutant in the DEAH box that blocks exon release after ligation at non-permissive temperature (8). We observed a significant accumulation of ligated ACT1 exons in spliceosome purified from the Prp22H606A strain under non-permissive temperature, compared to the wild-type (WT) strain, while intron lariats are at similar levels in both strains (fig. S1A). We determined the cryoEM structure of the P complex to an average resolution of 3.3 Å based on the “gold-standard” Fourier shell correlation (FSC) 0.143 cutoff criterion (9, 10). We built models for ligated exons (21 nucleotides downstream of the exon-exon junction) and interacting splicing factors including Prp22, Prp8, Slu7, and Prp18 (fig. S1–S4, Table S1). We also modeled U2, U5, U6 snRNAs, the intron lariat and other proteins that are nearly identical to those in the C* complex structures (6, 7), though with clearly better resolved density features. In addition, there are well-defined densities for multiple helices around Prp22 and Prp8, although the identity of their belonging protein(s) cannot be established based on possible sequences, known spliceosomal structures and Prp22 or Prp8-interacting partners. We will refer to this unidentified protein as UNK, noting that these helices can belong to multiple proteins or are connected to a known protein in the P complex through a flexible linker.
The overall architecture of the P complex is similar to the structures of the C* complex (5, 6) (Fig. 1, Movie S1) with several major differences. Of the two near-atomic resolution structures of the yeast C* complex, one (6) was assembled using a pre-mRNA substrate with the 2′-OH group of the G nucleotide at 3′ ss removed which inhibits exon ligation (11), while the other (5) was based on computational classification of wild-type spliceosome complexes. We therefore will make most of our detailed structural comparison with the former structure. First, the most significant difference between the two complexes is that the 3′ intron-exon (see below for definition) is not visible in the C* complex, while exons are clearly ligated and the 3′ intron (see below for definition) remain docked in the active site in the P complex (Fig. 2A–B). To facilitate our description, we refer to the intron region upstream of the branch point (BP) as the 5′ intron, the region downstream of BP as the 3′ intron, and the 3′ intron covalently linked (at the 3′ ss) to the 3′ exon as the 3′ intron-exon. Second, the 1585 loop of Prp8 [residues 1576–1599, also referred to as the α finger (12–14)] and the C-terminal tail of Prp22 (residues 1106–1145) become ordered in the P complex and interact with both the 3′ intron and 3′ exon. Third, several regions in the Slu7/Prp18 heterodimer (aa. 30 to 165 and 241 to 292 in Slu7 and aa. 189–221 in Prp18) are ordered in the P complex while absent in the C* complex structure. Finally, an unknown protein(s) made of mostly helices interact with Prp22, Prp8, Cef1, and Syf1 in the P complex.
Figure 1. Overall structure of the P complex.

(A–C) Three different views of the P complex with each subunit colored according to subunit identity. The view orientation in (C) is the same as those used in (5) to facilitate comparison with the structures of other spliceosomal complexes. The inset in (C) depicts the RNA elements only and is used as a landmark to orient subsequent figures. (D) List of modeled subunits in different functional sub-complexes.
Figure 2. RNA components in the P complex.
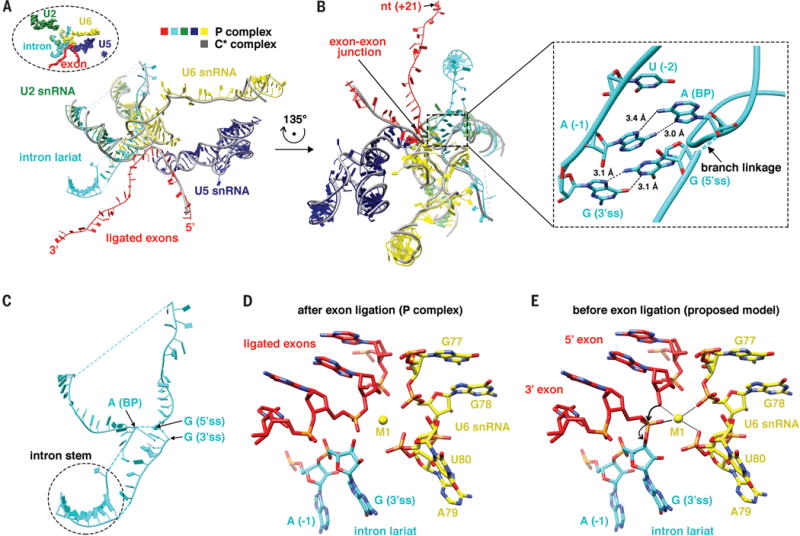
(A) Pre-mRNA (ligated exon and intron lariat), U2, U5, and U6 snRNAs in the P complex, with each RNA colored differently. Grey represents pre-mRNA, U2, U5, and U6 snRNAs in the C* complex, noting that the 3′ intron-exon of pre-mRNA is not present in the C* complex. The inset demonstrates the orientation of the figure.
(B) A different view of the RNA components in (A). The inset shows the interaction between the 3′ ss and the 5′ ss with non-Watson-Crick basepairing and pi-pi stacking.
(C) The intron forms a stem-like structure that brings the 3′ ss close to the 5′ ss.
(D) The structure of the active site in the P complex.
(E) A model of the active site right before exon ligation, illustrating the mechanism of exon ligation.
RNA components in the active site
Comparison of the RNA components in the active site of the C* and P complexes reveals how the 3′ intron-exon is loaded. In the C* complex, the 3′ intron-exon spanning the 3′ ss is not docked to the active site yet and is in fact entirely missing in the structure. The region that would accommodate the presumably disordered 3′ intron-exon is exposed, consistent with biochemical observations that the 3′ intron-exon is susceptible to RNase H cleavage after Prp16-mediated conformational changes but before exon ligation (i.e., the C* complex) (15). In the P complex, the 3′ intron-exon is docked into the active site through the recognition of both the 3′ ss and 3′ exon (Fig. 2A–B).
The 3′ ss is recognized through two main structural features. The first is the interaction between the 3′ ss and 5′ ss (inset of Fig. 2B). The 3′ ss is located in close proximity to the 2′–5′ linkage formed between the 5′ ss and BP A nucleotide in the P complex. The last nucleotide in the intron (the underlined G in the UAG sequence of 3′ ss) forms a G-G non-Watson-Crick basepair with the first nucleotide in the intron (GU in the 5′ ss), consistent with previous biochemical and genetic analyses indicating an essential non-Watson-Crick interaction between the first and last nucleotide of intron (16). The second to last nucleotide in the intron (UAG in 3′ ss) forms an A-A non-Watson-Crick basepair with the BP A nucleotide, as well as a pi-pi stacking with the 5′ ss G nucleotide. The third to last nucleotide in the intron (UAG in the 3′ ss) forms a pi-pi stacking with the BP A (inset of Fig. 2B). In addition, the last nucleotide in the intron (UAG in the 3′ ss) interacts with A51, C58, and A59 of U6 snRNA. Furthermore, the last several nucleotides in the intron interact extensively with the 1585 loop of Prp8 which likely facilitates the recognition of the 3′ ss. For example, nucleotides U (−2) and A (−1) (UAG in the 3′ ss) both form hydrogen bonds with Prp8 Q1594. The fourth to the last nucleotide in the intron also forms a pi-pi interaction with F1581 of Prp8.
A second structural feature that could facilitate 3′ ss recognition is a stem-like structure formed close to the BP and 3′ ss (Fig. 2C). The density of the stem is relatively weak and not defined enough for individual base assignment, potentially because multiple introns with different sequences form a similar stem-like structure around this region. An examination of yeast intron sequences indicates that many of them could form stem-like secondary structures close to the BP and 3′ ss (fig. S5). These stem-like structures could bring the 3′ ss close to the BP and the active site, facilitating the recognition of the 3′ ss through interactions with the 5′ ss.
The 3′ exon is recognized through interactions with U5 snRNA. In the C* structure, the 5′ exon forms Watson-Crick basepairs and additional hydrogen bonds with Loop1 (96–100) of U5 snRNA. In our structure of the P complex, the 3′ exon mainly interacts with U5 snRNA through pi-pi stacking between the first nucleotide in the 3′ exon and nucleotide U96 in U5 snRNA, consistent with previous studies that suggest that U5 Loop1 interacts with both the 5′ and 3′ exon and facilitating the alignment of both exons (17, 18).
In the P complex, the 3′ ss is close to the ligated exons (Fig. 2D) even though the second transesterification reaction has already occurred, allowing us to envision how exons are ligated (Fig. 2E). The last nucleotide of the 5′ exon is in close proximity to the 3′ ss. The catalytic Mg2+ (M1) is coordinated by G78 and U80 of U6 snRNA as well as the first nucleotide of the 3′ exon and is in close proximity to the 3′ OH at the end of the 5′ exon. M1 is in a perfect position to activate the 3′ OH group for nucleophilic attack on the 3′ ss, ligating the two exons and cleaving off the 3′ intron, as suggested by biochemical analyses (19, 20). The M2 ion proposed to stabilize the leaving group in exon ligation (19) is absent in the P complex, which could be reflective of a post-catalytic state.
Protein UNK, Prp8, Prp22 and Slu7/Prp18 jointly stabilize the 3′ intron and 3′ exon
The major differences in protein components (UNK, Prp22, Prp8, the Slu7/Prp18 heterodimer) between the C* and P complex structures are around the 3′ intron and 3′ exon. Protein UNK is composed mostly of helices and is located near Prp22 and Prp8 (Fig. 3). Helix A in protein UNK is positioned like a “door bolt” between the linker domain of Prp8 and RecA2 of Prp22, locking the 3′ exon in place (Fig. 3). Helices B and C in UNK starts from the RecA2 region of Prp22, contacts the RNaseH domain, goes under the stem-like region of the 3′ intron, and ends around the RT domain of Prp8, securing the 3′ intron (Fig. 3) [helices C and D of UNK are present in the C* complex, but with less well-defined densities and were not connected (6)]. Therefore, protein UNK likely plays a major role in ensuring the correct positioning of 3′ intron and 3′ exon, potentially for both exon ligation and release.
Figure 3. Protein UNK, Prp8, Prp22, and Slu7/Prp18 jointly stabilize the 3′ intron and 3′ exon in the P complex.

The bulk of the proteins are shown in surface model. Four helices (A–D) in protein UNK and regions in each of the other proteins that are present in the P complex structure but absent in the C* complex structure are shown in ribbons, including the 1585 loop of Prp8, the C-terminal tail (designated as C-tail) of Prp22, the 189–221 loop of Prp18, and residues 30–165 and 241–292 of Slu7. The insect is a zoomed-in view to better illustrate the relationship among the 1585 loop of Prp8, the C-tail of Prp22, the 189–221 loop of Prp18 and the 3′ intron/exon.
Prp22, the DEAH-box RNA helicase with an extended C-terminal tail that interacts with Prp8, wraps around the exon and contacts the intron together with Prp8 (Fig. 3). The 1585 loop of Prp8 and the C-terminal tail of Prp22 both become ordered in the P complex. The 1585 loop of Prp8 interacts with U2 snRNA, the 3′ exon, the 3′ intron, and the C-terminal tail of Prp22. The C-terminal end of Prp22 inserts into the cavity formed by the RT domain of Prp8, the 1585 loop of Prp8, U2 snRNA, the 3′ intron, and the 3′ exon, and interacts with all these components. Both the 1585 loop of Prp8 and the C-terminal tail of Prp22 are likely important for stabilizing the conformation of the 3′ intron and exon for exon ligation and/or release. As a consequence, the 3′ exon in the P complex makes extensive interactions with Prp8 and extends all the way to the core of Prp22 interacting with its C-terminal domain (CTD) and the two RecA domains. This is consistent with biochemical studies demonstrating that the 3′ exon is completely accessible to RNase H digestion in the C* complex, but more than 20 nucleotides of the 3′ exon are protected immediately after exon ligation, but before exon release (15).
Several regions of Prp18/Slu7 (residues 189–221 in Prp18 as well as residues 30 to 165 and 241 to 292 in Slu7) become ordered in the P complex (Fig. 3). Loop 189–221 in Prp18 inserts deeply into the N and RNase H domains of Prp8, interacts with the nucleotides close to the 3′ ss. Slu7 directly interacts with the 3′ intron through residues R287 and R289, consistent with crosslinking observed between Slu7 and the 3′ ss (21, 22). In addition, region 165–292 in Slu7 wraps around multiple domains of Prp8 (including the linker domain where the 1585 loop resides) (Fig. 3), potentially helping the 1585 loop becomes ordered in the P complex. The combined effect of protein UNK, the 1585 loop of Prp8, the C-terminal tail of Prp22, as well as the loops in Prp18 and Slu7 help secure the 3′ intron and 3′ exon to the right position for nucleophilic attack of the 3′ ss by the 3′ OH group at the end of the 5′ exon.
The structural basis of Prp22-mediated exon release and 3′ ss proofreading
Prp22 plays at least two distinct roles in the splicing cycle. The first is to release ligated exons in an ATPase and helicase dependent manner (23). The second is to proofread the 3′ ss for exon ligation, allowing for alternative 3′ ss choice through an ATP-dependent mechanism (24, 25). There have been some discrepancies on whether Prp22 plays a third, ATP-independent role in facilitating the ligation reaction (23, 26).
Our structure illustrates how Prp22 participates in exon release. In the P complex, Prp22 attaches to the spliceosome at the periphery through interaction with Prp8, protein UNK, and the 3′ exon (Figs. 1 and 4A). Prp22 binds to nt +15 to +21 in the 3′ exon (with respect to the exon-exon junction), using its two RecA domains and the CTD. It can perceivably use a winching motion to pull exon out of the spliceosome, consistent with previous biochemical observations (25). It also explains why at least 18 nucleotides are required for efficient exon release, even though only three nucleotides in the 3′ exon are required for exon ligation (23). The interaction between Prp22 and the 3′ exon is likely important for Prp22 to stay on the spliceosome so when the exon is pulled out, Prp22 consequently falls off the spliceosome (27). Therefore, Prp22 binds to ss RNA and RNA translocation rather than unwinding drives the release of the ligated exon, consistent with the observation that exon release by Prp22 is a less demanding requirement than unwinding (8). The ligated exon is mostly surrounded by the N, RT, thumb, and linker domains of Prp8, with additional interactions with helix A of protein UNK, the C-terminal tail of Prp22, and a loop region from Prp18. The Prp8 domains are highly modular and are connected by linkers, which presumably allow the domains to loosen up and let the exon pass.
Figure 4. Structure and function of Prp22.
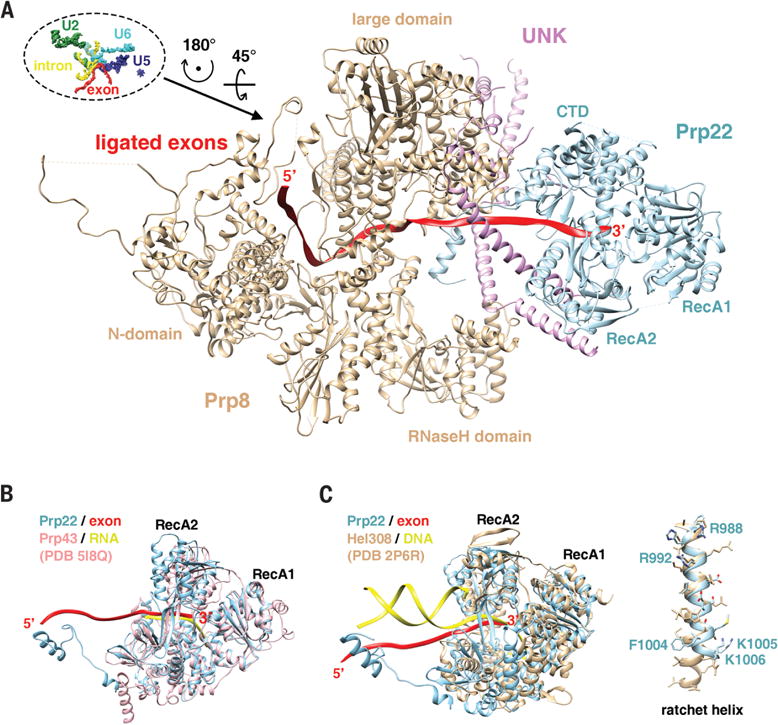
(A) Prp22 attaches to the spliceosome through its interaction with Prp8, protein UNK, and the 3′ exon.
(B) Prp22 has a similar structure as Prp43, both are DEAH-box helicases involved in splicing.
(C) The structure of Prp22 resembles DNA helicase Hel308. The ratchet helix of Hel308 superimposes well with helix 988–1006 of Prp22 and multiple residues (labeled) on this helix may be important for the translocation activity of Prp22 critical for exon release and 3′ ss proofreading.
The structures of Prp22 bound to the 3′ exon in the P and C* complexes also shed light on how Prp22 proofreads 3′ ss. Although the 3′ intron-exon is not visible in the C* complex, there is density for 3 nt that is likely part of the 3′ exon in the center of Prp22 (6). This suggests that Prp22 could pull the 3′ intron-exon after the 3′ intron-exon is docked close to the end of 5′ exon, but before exon ligation. This pulling destabilizes the 3′ ss and spliceosome interaction, competing against the exon ligation event, providing an opportunity to discard sub-optimal 3′ ss or to select alternative 3′ ss (25). The branch formation at the intron lariat, the interaction between 5′ ss and U6 snRNA, the interaction between BP sequence and U2 snRNA, together with the proteins that stabilize these interactions, potentially make it hard for Prp22 to completely pull out 3′ intron-exon. In addition, our density of the CTD of Prp22 is much better defined than that in the C* complex (6), suggesting that Prp22 is more stably associated with the core components in the P complex than in the C* complex. Indeed, helices A and B (absent in the C* complex) in protein UNK both serve to fasten Prp22 onto the P complex by binding to Prp22 on one end of the helices and binding to Prp8 on the other end of the helices (Fig. 3). An additional short helix-loop-helix from protein UNK binds to the Oligonucleotide-Binding (OB) domain within the CTD of Prp22. Protein UNK may stimulate the helicase activity of Prp22, just as two G-patch proteins (Spp2 and Ntr1) stimulate the helicase activity of Prp2 and Prp43, presumably by binding to their OB domains (28–32).
The well-defined density of Prp22 in our P complex structure allowed us to compare Prp22 with other helicases. The structure of Prp22 is similar to Prp43 (Fig. 4B), both of which resemble DNA helicase Hel308 (33, 34) (Fig. 4C). Nucleotide strands in all three structures go into the same channel formed by the two RecA domains and the CTD. The crystal structure of Hel308 in complex with a partially unwound DNA duplex and biochemical analyses revealed a ratchet helix (residues 586–611) critical for DNA translocation (34) (Fig. 4C). Residues R592 and W599 on the ratchet helix interact with ss DNA, while the N-terminus of the ratchet helix interacts with motif IVa of RecA2. ATP-dependent movement of RecA2 is therefore thought to modulate the position of the ratchet helix and facilitate strand translocation. Helix 987–1006 in Prp22 is well superimposed with the ratchet helix in Hel308 (Fig. 4C). R992Prp22 is equivalent to R592Hel308. While there is no Trp equivalent to W599Hel308, F1004Prp22 may form a comparable pi-pi stacking with the RNA strand. There are several other basic residues (R988Prp22, K1005 Prp22 and K1006Prp22) on the ratchet helix. The 987–1006 helix and multiple residues on this helix (R992Prp22, R988Prp22, F1004Prp22, K1005Prp22, and K1006Prp22) are likely important for Prp22-mediated exon release and possibly 3′ ss proofreading.
Discussion
Our structure of the P complex reveals that the 3′ ss is recognized mainly through non-Watson-Crick basepairing between the 3′ ss and the 5′ ss as well as the BP (Fig. 2B), which is further stabilized by their interactions with surrounding proteins. Although Watson-Crick basepairs are crucial for RNA structures, non-Watson-Crick basepairs are often involved in protein-RNA or RNA-RNA interactions (35, 36). For example, non-Watson-Crick basepairs are used for ss recognition in self-splicing group I introns (37). The 5′ ss (GU) and 3′ ss (AG) are conserved among all species, testifying the importance of non-Watson-Crick basepairing in 3′ ss recognition.
On the other hand, what brings the 3′ ss close to the 5′ ss and BP in order to form these non-Watson-Crick basepairing has remained elusive. It is usually the first YAG after BP that is recognized as the 3′ ss, leading to the scanning hypothesis which assumes that the spliceosome reads the intron from the BP in a 5′ to 3′ direction until it encounters the first YAG. However, the only moving part in the pre-ligation C* complex is the 3′ exon being pulled by Prp22 in a 3′ to 5′ direction. Therefore, the YAG further away from the BP (instead of the first YAG) would have been encountered by the spliceosome active site first. Instead, the stem-like structure we observed close to the BP and 3′ ss (Fig. 2C) suggest that secondary structures formed within the 3′ intron plays an important role in bringing the 3′ ss close to the active site for basepairing with the 5′ ss. This is consistent with previous bioinformatics and experimental observations (38–40). The presence and stability of these secondary structures can potentially (38)be a way to modulate splicing efficiency at the 3′ ss. It also implies that intronic secondary structures may play a much more prevalent role in 3′ ss recognition and alternative splicing in higher eukaryotes than what have been previously appreciated.
The Prp22 structure bound to the 3′ exon in our structure (made possible by the high quality map in this region) together with the C* complex structure provide a vivid image of how Prp22 fulfills its function in exon release and 3′ ss proofreading (Fig. 5). Prp22 binds to the +15 to +21 nucleotides (with respect to the exon-exon junction, Fig. 4A), enabling it to pull on the 3′ intron-exon (in C*) or ligated exon (in P) using a winching motion from this remote location, achieving 3′ ss proofreading or exon release activity, respectively, in agreement with previous biochemical data (23, 25).
Figure 5. A schematic representation of the splicing cycle from the C* the P complex based on our structure and the structures of the C* complex.
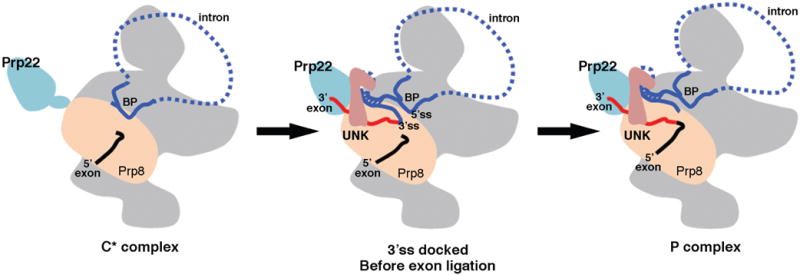
In C* (left), the branching reaction has occurred, but the 3′ intron-exon has not been loaded to the active site. In the spliceosomal complex immediately before ligation (middle), protein UNK and several other proteins help load the 3′ intron-exon into the active site, where the 3′ ss is recognized through interactions with the 5′ ss facilitated by the stem-like structure formed in the intron. Prp22 pulls the 3′ intron-exon to fulfill its function in 3′ ss proofreading. In the P complex (right), the two exons are ligated but the intron lariat remains in the complex. Prp22 pulls the ligated exon to release it from the spliceosome.
Our structure revealed a yet to be identified protein UNK mostly made of helices that is likely important for the function of Prp22 (Fig. 5). None of the known second step factors present in our sample fits the density. On the other hand, the density suggest that UNK could be composed of multiple proteins that await further experimental confirmation, including the N-terminal domain of Prp22 and FYV6, a protein with unknown function. Protein UNK provides a “door bolt” between Prp22 and Prp8 to lock the 3′ exon in position (Fig. 3). It also contacts the OB domain of Prp22, potentially stimulates its activity similar to G-patch proteins, offering a possible way to regulate Prp22 activity so that it efficiently pulls out the ligated exon, but not the 3′ intron-exon. With the atomic models of nearly all other major complexes already in the literature, the P complex structure reported here highlights the exciting prospect that a full mechanistic understanding of the entire splicing cycle may just be around the corner.
Supplementary Material
Acknowledgments
We thank Dr. Beate Schwer for the p358-PRP22 and p358-H606A plasmids as well as yeast strain YBST1. This work was supported by NIH grants GM114178 (R. Z.), GM071940 and AI094386 (Z.H.Z.). We acknowledge the use of instruments at the Electron Imaging Center for Nanomachines (supported by UCLA and by grants from the NIH (1S10OD018111, 1U24GM116792) and NSF (DBI-1338135 and DMR-1548924)). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The model has been deposited in the Protein Data Bank with ID 6BK8 and the EM map has been deposited in EMD with ID EMD-7109.
Footnotes
Author Contributions
X.L., R.Z. and Z.H.Z. conceived the project; X.L. and L.Z. prepared, optimized, and performed biochemical analyses of the sample; S.L., J.J., Y.C. and Z.H.Z. recorded and processed the EM data; R.H. and K.C.H. performed mass spectrometry analyses; S.L. built the atomic models; S.L., R.Z. and Z.H.Z. analyzed and interpreted the models; S.L., X.L. and R.Z. prepared the illustrations; R.Z., S.L. and Z.H.Z. wrote the paper; and all authors contributed to the editing of the manuscript.
References and Notes
- 1.Plaschka C, Lin PC, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–621. doi: 10.1038/nature22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast activated spliceosome at 3.5 A resolution. Science. 2016;353:904–911. doi: 10.1126/science.aag0291. [DOI] [PubMed] [Google Scholar]
- 3.Wan R, Yan C, Bai R, Huang G, Shi Y. Structure of a yeast catalytic step I spliceosome at 3.4 A resolution. Science. 2016;353:895–904. doi: 10.1126/science.aag2235. [DOI] [PubMed] [Google Scholar]
- 4.Galej WP, et al. Cryo-EM structure of the spliceosome immediately after branching. Nature. 2016;537:197–201. doi: 10.1038/nature19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast step II catalytically activated spliceosome. Science. 2017;355:149–155. doi: 10.1126/science.aak9979. [DOI] [PubMed] [Google Scholar]
- 6.Fica SM, et al. Structure of a spliceosome remodelled for exon ligation. Nature. 2017 doi: 10.1038/nature21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan R, Yan C, Bai R, Lei J, Shi Y. Structure of an Intron Lariat Spliceosome from Saccharomyces cerevisiae. Cell. 2017;171:120–132 e112. doi: 10.1016/j.cell.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Campodonico E, Schwer B. ATP-dependent remodeling of the spliceosome: intragenic suppressors of release-defective mutants of Saccharomyces cerevisiae Prp22. Genetics. 2002;160:407–415. doi: 10.1093/genetics/160.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen THD, et al. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 A resolution. Nature. 2016;530:298–302. doi: 10.1038/nature16940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galej WP, Nguyen TH, Newman AJ, Nagai K. Structural studies of the spliceosome: zooming into the heart of the machine. Curr Opin Struct Biol. 2014;25:57–66. doi: 10.1016/j.sbi.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y. The Spliceosome: A Protein-Directed Metalloribozyme. J Mol Biol. 2017;429:2640–2653. doi: 10.1016/j.jmb.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Schwer B. A conformational rearrangement in the spliceosome sets the stage for Prp22-dependent mRNA release. Mol Cell. 2008;30:743–754. doi: 10.1016/j.molcel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker R, Siliciano PG. Evidence for an essential non-Watson-Crick interaction between the first and last nucleotides of a nuclear pre-mRNA intron. Nature. 1993;361:660–662. doi: 10.1038/361660a0. [DOI] [PubMed] [Google Scholar]
- 17.Newman AJ, Norman C. U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell. 1992;68:743–754. doi: 10.1016/0092-8674(92)90149-7. [DOI] [PubMed] [Google Scholar]
- 18.Sontheimer EJ, Steitz JA. The U5 and U6 small nuclear RNAs as active site components of the spliceosome. Science. 1993;262:1989–1996. doi: 10.1126/science.8266094. [DOI] [PubMed] [Google Scholar]
- 19.Fica SM, et al. RNA catalyses nuclear pre-mRNA splicing. Nature. 2013;503:229–234. doi: 10.1038/nature12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yean SL, Wuenschell G, Termini J, Lin RJ. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature. 2000;408:881–884. doi: 10.1038/35048617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umen JG, Guthrie C. Prp16p, Slu7p, and Prp8p interact with the 3′ splice site in two distinct stages during the second catalytic step of pre-mRNA splicing. Rna. 1995;1:584–597. [PMC free article] [PubMed] [Google Scholar]
- 22.McPheeters DS, Schwer B, Muhlenkamp P. Interaction of the yeast DExH-box RNA helicase prp22p with the 3′ splice site during the second step of nuclear pre-mRNA splicing. Nucleic Acids Res. 2000;28:1313–1321. doi: 10.1093/nar/28.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. Embo J. 1998;17:2086–2094. doi: 10.1093/emboj/17.7.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayas RM, Maita H, Staley JP. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol. 2006;13:482–490. doi: 10.1038/nsmb1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semlow DR, Blanco MR, Walter NG, Staley JP. Spliceosomal DEAH-Box ATPases Remodel Pre-mRNA to Activate Alternative Splice Sites. Cell. 2016;164:985–998. doi: 10.1016/j.cell.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohrt T, et al. Molecular dissection of step 2 catalysis of yeast pre-mRNA splicing investigated in a purified system. RNA. 2013;19:902–915. doi: 10.1261/rna.039024.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozgur S, et al. The conformational plasticity of eukaryotic RNA-dependent ATPases. The FEBS journal. 2015;282:850–863. doi: 10.1111/febs.13198. [DOI] [PubMed] [Google Scholar]
- 28.Silverman EJ, et al. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol Cell Biol. 2004;24:10101–10110. doi: 10.1128/MCB.24.23.10101-10110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fourmann JB, Tauchert MJ, Ficner R, Fabrizio P, Luhrmann R. Regulation of Prp43-mediated disassembly of spliceosomes by its cofactors Ntr1 and Ntr2. Nucleic Acids Res. 2017;45:4068–4080. doi: 10.1093/nar/gkw1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warkocki Z, et al. The G-patch protein Spp2 couples the spliceosome-stimulated ATPase activity of the DEAH-box protein Prp2 to catalytic activation of the spliceosome. Genes Dev. 2015;29:94–107. doi: 10.1101/gad.253070.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka N, Aronova A, Schwer B. Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev. 2007;21:2312–2325. doi: 10.1101/gad.1580507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walbott H, et al. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. Embo J. 2010;29:2194–2204. doi: 10.1038/emboj.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y, Staley JP, Andersen GR, Nielsen KH. Structure of the DEAH/RHA ATPase Prp43p bound to RNA implicates a pair of hairpins and motif Va in translocation along RNA. RNA. 2017;23:1110–1124. doi: 10.1261/rna.060954.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 35.Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermann T, Westhof E. Non-Watson-Crick base pairs in RNA-protein recognition. Chemistry & biology. 1999;6:R335–343. doi: 10.1016/s1074-5521(00)80003-4. [DOI] [PubMed] [Google Scholar]
- 37.Michel F, Hanna M, Green R, Bartel DP, Szostak JW. The guanosine binding site of the Tetrahymena ribozyme. Nature. 1989;342:391–395. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 38.Gahura O, Hammann C, Valentova A, Puta F, Folk P. Secondary structure is required for 3′ splice site recognition in yeast. Nucleic Acids Res. 2011;39:9759–9767. doi: 10.1093/nar/gkr662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plass M, Codony-Servat C, Ferreira PG, Vilardell J, Eyras E. RNA secondary structure mediates alternative 3′ss selection in Saccharomyces cerevisiae. RNA. 2012;18:1103–1115. doi: 10.1261/rna.030767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer M, Plass M, Perez-Valle J, Eyras E, Vilardell J. Deciphering 3′ss selection in the yeast genome reveals an RNA thermosensor that mediates alternative splicing. Mol Cell. 2011;43:1033–1039. doi: 10.1016/j.molcel.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 41.Schwer B, Meszaros T. RNA helicase dynamics in pre-mRNA splicing. EMBO J. 2000;19:6582–6591. doi: 10.1093/emboj/19.23.6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knop M, et al. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 43.Carragher B, et al. Leginon: an automated system for acquisition of images from vitreous ice specimens. J Struct Biol. 2000;132:33–45. doi: 10.1006/jsbi.2000.4314. [DOI] [PubMed] [Google Scholar]
- 44.Zheng SQ, Palovcak E, Armache J-P, Cheng Y, Agard DA. bioRxiv. 2016 [Google Scholar]
- 45.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 47.Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nature methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nature methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6:4459–4462. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 51.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Keating KS, Pyle AM. RCrane: semi-automated RNA model building. Acta Crystallogr D Biol Crystallogr. 2012;68:985–995. doi: 10.1107/S0907444912018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nature methods. 2013;10:74–76. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.