

To the Editor
Burkitt leukemia/lymphoma (BL) is a rare hematological malignancy that accounts for 1-5% of acute lymphoblastic leukemia and non-Hodgkin lymphoma.[1] Patients with BL have an aggressive disease course and high propensity for central nervous system involvement, with pathogenic chromosomal translocations leading to dysregulation of the MYC oncogene. High-grade B-cell leukemia/lymphoma (HGBCL) is a group of rapidly proliferative B-cell neoplasms that lacks some of the characteristic morphologic, immunophenotypic, or molecular cytogenetic findings of BL but have a similarly aggressive clinical course.[2] HGBCL was adopted by the 2016 revision of the World Health Organization (WHO) classification and replaced the term “B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and BL.”[3]
BL is highly curable with chemoimmunotherapy, with a 3-year overall survival (OS) rate of over 80%.[4, 5] Despite the high cure rate in the frontline setting, patients who are refractory to initial treatment failure have a poor prognosis.[6] Few studies however have systematically evaluated outcomes of adults with relapsed/refractory disease or the efficacy of salvage chemotherapy in this setting.
We retrospectively identified 145 adults with BL or HGBCL treated with frontline hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone)-based regimens approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center. Patients received frontline therapy between January 1992 and July 2015. Forty-six patients were treated prior to the development of rituximab; however, beginning in November 1999, all subsequent patients received an anti-CD20 monoclonal antibody as part of frontline treatment. All patients provided informed consent according to institutional guidelines and the Declaration of Helsinki. Patients fulfilled WHO classification criteria for BL or HGBCL.[3]
Twelve patients died before initial response assessment and 2 were unevaluable due to inadequate records. Among the 131 patients evaluable for response, 35 patients (27%) had relapsed (n=32) or refractory (n=3) BL or HGBCL and are the subject of this analysis. Early and late relapse were defined as relapse <6 months and ≥6 months from the time of first remission, respectively. Overall response rate (ORR), which was defined as the composite of complete remission (CR) and partial remission (PR), relapse-free survival (RFS), and OS were evaluated.
Characteristics of the evaluable population are shown in Supplemental Table 1. The median age was 51 years (18-76 years). One patient with BL had the variant translocation t(8;22)(q24;q11), and 4/7 with HGBCL had MYC rearrangements. Initial presentation was leukemia in 27 patients (77%) and lymphoma in 8 (23%). Frontline treatment was hyper-CVAD alone in 10 patients, hyper-CVAD plus rituximab in 24, and hyper-CVAD plus ofatumumab in 1. Three patients were refractory to frontline treatment. Among the 32 relapsed patients, 29 had achieved CR and 3 had achieved PR to frontline therapy. The median time from first remission to relapse was 6.8 months (0.7-75.3 months). Twenty-one patients (60% of the entire cohort) experienced late relapse after first remission. Six patients relapsed after >1 year of initial remission, with 3 patients having exceptionally late relapses after >2 years (one patient each with relapse at 26, 41 and 75 months).
Twenty-eight patients with relapsed/refractory disease received at least one salvage regimen and are evaluable for response (early relapse/refractory, n=10; late relapse, n=18). The backbone salvage regimens received were hyper-CVAD in 12 patients, ICE (ifosfamide, carboplatin, etoposide) in 6, EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin) in 2, MOAD (methotrexate, vincristine, pegylated L-asparaginase, dexamethasone) in 2, and miscellaneous regimens in 6. Of the 12 patients who were re-induced with hyper-CVAD-based regimens, 10 had experienced late relapse after first remission. Of the 2 patients who experienced early relapse and received salvage hyper-CVAD, one received augmented hyper-CVAD (i.e. addition of L-asparaginase) and one received the same hyper-CVAD regimen again; notably, this latter patient was treated in the early 1990s when fewer salvage options were available. Subsequent stem cell transplant (SCT) was performed in 6 patients (allogeneic, n=3; autologous, n=3). The ORR to salvage chemotherapy was 39% (CR, n=8; PR, n=3); 17 patients were refractory. The ORR for patients with late relapse and with refractory disease/early relapse was 61% and 0%, respectively (P=0.002).
The median OS for the entire cohort (measured from time of treatment failure or first relapse) was 2.8 months, with a 1-year OS rate of 11% (Figure 1A). Among responding patients, the median RFS was 3.3 months, with a 1-year RFS rate of 18% (Figure 1B). Only 2 patients were still alive without relapse at last follow-up. One of these patients relapsed 12 months after initial diagnosis, achieved CR with hyper-CVAD plus rituximab and then underwent salvage autologous SCT. The other patient had late relapse 75 months after initial diagnosis, achieved CR with EPOCH plus rituximab and did not undergo SCT consolidation.
Figure 1.
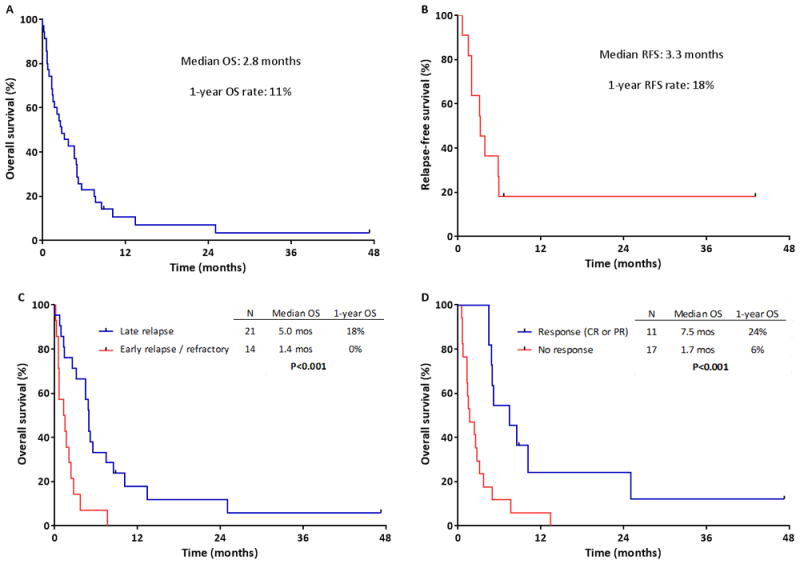
Survival of patients with relapsed/refractory Burkitt or high-grade B-cell leukemia or lymphoma. A) Overall survival measured from time of first treatment failure (N=35) and B) Relapse-free survival measured from time of second remission (N=11). C) Overall survival of patients with refractory disease or early relapse (initial remission duration <6 months) versus late relapse (initial remission duration ≥6 months). D) Overall survival of patients who responded to salvage chemotherapy versus those who did not respond.
Patients with late relapse had a median OS of 5.0 months, compared to only 1.4 months for those with refractory disease/early relapse (P<0.001; Figure 1C). Initial diagnosis did not influence OS. The median OS for patients with leukemia versus lymphoma was 3.7 months and 2.3 months, respectively (P=0.23). Patients with BL versus HGBCL had a median OS of 3.7 months and 2.7 months, respectively (P=0.74). Patients who responded to salvage treatment had superior OS compared to those who did not (median OS: 7.5 months and 1.7 months, respectively; P<0.001; Figure 1D). Those who achieved CR had the best outcomes, with a median OS of 8.8 months and 1-year OS rate of 33%.
In pediatric studies of relapsed/refractory BL, long-term survival of approximately 30-35% has been achieved with salvage chemotherapy with subsequent SCT.[7-9]. Little information is available about long-term survival of adults with relapsed/refractory disease. In one older study on the role of autologous SCT for BL, the 3-year OS rate for patients with chemosensitive relapse was 37%.[10] This study however did not account for patients who were not able to undergo SCT due to rapid disease progression despite salvage chemotherapy. This likely accounts for the relatively poorer outcomes observed in our cohort.
In the present study, the ORR of salvage chemotherapy for patients with relapsed/refractory disease was 39% (CR rate: 29%). Survival at last follow-up was observed in only 2 of 35 patients (6%) and only in those with late relapse who subsequently achieved CR with salvage therapy. The poor long-term survival observed in the present study highlights that effective therapies for relapsed/refractory BL are needed. These patients should be considered for clinical trials with novel agents such as monoclonal antibody constructs or targeted therapies, followed by SCT for responding patients. Given the rarity of this disease, multicenter collaborations will be needed.
Interestingly, we observed three patients who relapsed >2 years after initial remission, which could theoretically represent a different clone and therefore not a “true” relapse. Based on the available pathological studies, all of these relapsed samples resembled the initial diagnostic sample and therefore appear to represent a true, late BL relapse. However, further testing (such as next-generation sequencing) has not been performed to more definitively address this issue. These exceptionally late relapses comprised only 9% of the total relapses observed in this cohort, which suggests that while very late relapses can occur, they appear to be relatively uncommon.
In conclusion, patients with relapsed/refractory BL or HGBCL have very poor outcomes, with an ORR to salvage therapy of 39% and a median OS of only 2.8 months. Patients with a remission duration after frontline therapy of ≥6 months and who achieve CR with salvage chemotherapy have superior outcomes, although the long-term survival of this group is still poor. Novel treatment strategies are needed for patients with relapsed/refractory disease.
Supplementary Material
Acknowledgments
Funding source: Supported by the MD Anderson Cancer Center Support Grant CA016672
Footnotes
Authorship Contributions
N.J.S. designed the study, collected and analyzed the data, and wrote the manuscript; E.J. and H.M.K. designed the study, collected and analyzed the data, treated patients, and wrote the manuscript; H.K., M.K. and S.P. collected and analyzed the data; J.D.K performed pathologic interpretation; F.R., D.A.T, G.G-M., J.E.C, W.W., S.V., Z.E. A.F., P.A.T. and S.M.O. treated patients. All authors reviewed and approved the manuscript.
Disclosure of Conflicts of Interest: The authors report no conflicts of interest.
References
- 1.Ferry JA. Burkitt’s lymphoma: clinicopathologic features and differential diagnosis. The oncologist. 2006;11:375–383. doi: 10.1634/theoncologist.11-4-375. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DA, O’Brien S, Faderl S, et al. Burkitt lymphoma and atypical Burkitt or Burkitt-like lymphoma: should these be treated as different diseases? Current hematologic malignancy reports. 2011;6:58–66. doi: 10.1007/s11899-010-0076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106:1569–1580. doi: 10.1002/cncr.21776. [DOI] [PubMed] [Google Scholar]
- 5.Ribrag V, Koscielny S, Bosq J, et al. Rituximab and dose-dense chemotherapy for adults with Burkitt’s lymphoma: a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:2402–2411. doi: 10.1016/S0140-6736(15)01317-3. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson C, LaCasce A. How I treat Burkitt lymphoma in adults. Blood. 2014;124:2913–2920. doi: 10.1182/blood-2014-06-538504. [DOI] [PubMed] [Google Scholar]
- 7.Anoop P, Sankpal S, Stiller C, et al. Outcome of childhood relapsed or refractory mature B-cell non-Hodgkin lymphoma and acute lymphoblastic leukemia. Leukemia & lymphoma. 2012;53:1882–1888. doi: 10.3109/10428194.2012.677534. [DOI] [PubMed] [Google Scholar]
- 8.Kim H, Park ES, Lee SH, et al. Clinical outcome of relapsed or refractory burkitt lymphoma and mature B-cell lymphoblastic leukemia in children and adolescents. Cancer research and treatment : official journal of Korean Cancer Association. 2014;46:358–365. doi: 10.4143/crt.2013.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jourdain A, Auperin A, Minard-Colin V, et al. Outcome of and prognostic factors for relapse in children and adolescents with mature B-cell lymphoma and leukemia treated in three consecutive prospective “Lymphomes Malins B” protocols. A Societe Francaise des Cancers de l’Enfant study. Haematologica. 2015;100:810–817. doi: 10.3324/haematol.2014.121434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sweetenham JW, Pearce R, Taghipour G, et al. Adult Burkitt’s and Burkitt-like non-Hodgkin’s lymphoma--outcome for patients treated with high-dose therapy and autologous stem-cell transplantation in first remission or at relapse: results from the European Group for Blood and Marrow Transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996;14:2465–2472. doi: 10.1200/JCO.1996.14.9.2465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.