

Abstract
The ubiquitin proteasome pathway was discovered in the 1980s to be a central component of the cellular protein degradation machinery with essential functions in homeostasis, which include preventing the accumulation of misfolded or deleterious proteins. Cancer cells produce proteins that promote both cell survival and proliferation, and/or inhibit mechanisms of cell death. This notion set the stage for preclinical testing of proteasome inhibitors as a means to shift this fine equilibrium towards cell death. Since the late 1990s, clinical trials have been conducted for a variety of malignancies, leading to regulatory approvals of proteasome inhibitors to treat multiple myeloma and mantle cell lymphoma. First-generation and second-generation proteasome inhibitors can elicit deep initial responses in patients with myeloma, in whom these drugs have dramatically improved outcomes, but relapses are frequent and acquired resistance to treatment eventually emerges. In addition, promising preclinical data obtained with proteasome inhibitors in models of solid tumours have not been confirmed in the clinic, indicating a role for primary resistance. Investigation of the mechanisms of resistance is, therefore, essential to further maximize the utility of this class of drugs in the era of personalized medicine. Herein, we discuss the advances and challenges resulting from the introduction of proteasome inhibitors into the clinic.
Introduction
The ubiquitin proteasome pathway (UPP) is an extensive and complex protein degradation pathway present in all eukaryotic cells1–3. The UPP is the most important regulated intracellular protein degradation system, and is involved in processes such as apoptosis, cell survival, cell-cycle progression, DNA repair, and antigen presentation, among others. To ensure appropriate destruction of damaged or misfolded proteins, or of proteins that are no longer needed, the components of this system must act in a highly coordinated manner through different steps that include polyubiquitination, deubiquitination, and degradation of the target protein. To facilitate this coordination, the components of the UPP associate in various physical structures, including the proteasome itself4. A single ubiquitin activating enzyme 1 (E1), and multiple ubiquitin-conjugating enzymes (E2) and ubiquitin-protein ligases (E3) mediate initial polyubiquitination of the target. The proteasome regulatory subunits, which include both the 19S particle in the constitutive proteasome and the 11S particle in the immunoproteasome, mediate deubiquitination. Once the target proteins have been deubiquitinated, the proteasome degrades them via the function of the 20S core particle catalytic sites, thereby releasing oligopeptides. The three catalytic sites present in the 20S core particle are named after enzymes that have a similar proteolytic activity, and include sites with chymotrypsin-like (β5), trypsin-like (β2), and post-glutamyl peptide hydrolyzing, or caspase-like (β1) activities. In some cases, the 20S proteasome core can act alone through ubiquitin-independent degradation pathways. Alternative forms of the proteasome, such as the immunoproteasome and thymoproteasome, are involved in routine proteolytic functions, antigen processing, and T-cell selection5. Thus, the total capacity of a cell to digest proteins through the UPP encompasses the contribution of both ubiquitin-dependent and ubiquitin–independent pathways. Nevertheless, the pool of proteins destined for degradation through the UPP is referred to as the load on the proteasome. Further digestion by aminopeptidases and carboxypeptidases then turns the oligopeptides into amino acids that can be reused by the cell1–3. Similarly, deubiquitination yields monomeric ubiquitin as part of a recycling mechanism that maintains a free pool of ubiquitin within the cell and avoids ubiquitin degradation. Sensitivity to proteasome inhibition has been linked to an imbalance between cellular proteasome load and capacity. For example, lower levels of proteasome activity and higher proteasome workload can result in accumulation of polyubiquitinated proteins, which could potentially lead to proteasomal stress and increased sensitivity to proteasome inhibition. Proteins can also be destroyed within the cell through alternative pathways, for example, by aggregation in microtubule-based cellular structures called aggresomes with subsequent degradation via the autophagy pathway6. Readers interested in an in-depth summary of the assembly, regulation, and biology of the UPP are referred to a review on this topic4 that was published in 2014.
Deregulation of the UPP can result in either enhanced or reduced degradation of key targets that contribute to oncopathogenesis7. Notable examples include down-regulation of cell-cycle and tumour-suppressor proteins, such as p53, and cyclin-dependent kinase inhibitor 1B (p27), or up-regulation of oncogenic proteins, including activation of the nuclear factor kappa-B (NF-κB)8–10. In this Review, we describe the current status of UPP inhibition as a therapeutic strategy in cancer.
Inhibitors of the UPP
Proteasome inhibitors were initially developed as agents with potential benefit in preventing cancer-related cachexia, owing to the role of the UPP in protein turnover. Many preclinical studies then emerged showing that small-molecule proteasome inhibitors could induce apoptosis in cultured cell lines and murine models of cancer, at which point their utility as chemotherapeutics was postulated. One of the early hypotheses driving the study of these agents was that they could inhibit NF-κB signaling by blocking degradation of the NF-κB inhibitor IκB, thereby preventing nuclear translocation of NF-κB. This rationale led to the development of bortezomib, a first-generation proteasome inhibitor, and later to that of second-generation agents, including carfilzomib, ixazomib, and oprozomib, which were developed, in part, in attempts to improve upon the benefits observed with bortezomib.
First-generation proteasome inhibitors
Inhibitors of the 20S proteolytic core of the proteasome are the most extensively studied proteasome inhibitors to date, and three of these agents (bortezomib, ixazomib and carfilzomib) are currently approved for the treatment of multiple myeloma or mantle-cell lymphoma (MCL). Bortezomib was the first proteasome inhibitor to be brought into clinical use.
Bortezomib is a slowly reversible inhibitor, which binds with the catalytic site of the 26S proteasome, enabling inhibition of the β5/chymotrypsin-like and, to a lesser extent, the β2/trypsin-like and β1/post-glutamyl peptide hydrolyzing activities11. This agent is commonly used in the first-line and relapsed and/or refractory settings in patients with multiple myeloma, and in those with MCL12 (Table 1). About a decade ago, bortezomib showed impressive clinical activity as a single agent in the treatment of relapsed and/or refractory multiple myeloma initially in a phase I study13, and then in phase II clinical trials14,15. These findings supported the accelerated approval of bortezomib by the FDA in 2003 as a salvage treatment, initially for patients with treatment-refractory disease. Importantly, and for the first time, bortezomib prolonged progression-free survival (PFS) in this patient population compared with the previous line of therapy (regardless of the prior treatment used), and some patients were able to achieve their first complete remissions (Table 1). Full regulatory approval occurred in 2005, following the publication of results from a large phase III trial16 in 669 patients with relapsed multiple myeloma after 1–3 prior lines of therapy. In comparison with high-dose dexamethasone, which was then a standard of care in this setting, improvements in time to progression (TTP), overall response rate (ORR), and overall survival were demonstrated in the patients who received bortezomib16 (Table 1).
Table 1.
Key clinical trials with bortezomib in multiple myeloma
| Study characteristics | Number of patient and disease setting | Results |
|---|---|---|
|
Richardson et al.14,15 Phase II Single arm Bortezomib standard schedule* Dexamethasone: 20 mg on days 1, 2, 4, 8, 11 and 12 added if suboptimal response Primary outcome: efficacy |
n = 202 RRMM to last therapy received |
CR: 4% nCR: 6% PR: 18% MR: 7% Adverse events: thrombocytopenia, fatigue, peripheral neuropathy and neutropenia Median OS: 17 months Median TTP: 7 months |
|
Richardson et al.16 Phase III Bortezomib versus HDD Bortezomib standard schedule* HDD: 40 mg orally daily days 1–4, 9–12 and 17–20 of 35-day cycles Primary outcome: TTP |
Bortezomib: n = 333 HDD: n = 336 RRMM |
ORR: 38% (bortezomib) versus 18% (HDD; P <0.001) Adverse events: higher rates of GI toxicities, cytopenias and fatigue with bortezomib versus HDD (P <0.05) Median TTP: 6.2 months (bortezomib) versus 3.5 months (HDD) |
|
Orlowski et al.22 Phase III Bortezomib versus bortezomib plus peg-dox Bortezomib standard schedule* Combination arm: peg-dox 30 mg/m2 i.v. after bortezomib on day 4 of each cycle Primary outcome: TTP |
Bortezomib: n = 322 Peg-dox: n = 324 RRMM |
ORR: 41% (bortezomib), versus 44% (peg-dox; P >0.05) Adverse events: increased rates of cytopenias, fatigue, diarrhoea and hand-foot syndrome with peg-dox versus bortezomib alone Median TTP 6.5 months vs. 9.3 months peg-dox, 15-month OS 65% vs. 76% peg-dox |
|
San Miguel et al.17 Phase III VMP versus MP Melphalan 9 mg/m2 and prednisone 60 mg/m2 on days 1–4 of nine 6-week cycles, either alone or with bortezomib 1.3 mg/m2 on days 1, 4, 8, 11, 22, 25, 29 and 32 during cycles 1–4 and on days 1, 8, 22 and 29 during cycles 5–9 Primary outcome: TTP |
VMP: n = 344 MP: n = 338 NDMM |
PR: 71% (VMP), versus35% (MP) CR: 30% (VMP) versus 4% (MP) Adverse events: higher rate of grade 3 events with VMP (53%) versus MP (44%; P = 0.02); similar rates of grade 4 events and treatment-related deaths TTP: 24 months (VMP) versus 16.6 months (MP; P <0.01) OS hazard ratio: 0.61 (VMP vs MP) . At median follow up of 16.3 months, 13% of patients in VMP group and 22% of patients in MP group had died (P = 0.008) |
|
Harousseau et al.140 Phase III VAD ± DCEP versus VD ± DCEP VAD: vincristine 0.4 mg daily; doxorubicin 9 mg/m2 continuous infusion on days 1–4; and dexamethasone 40 mg on days 1–4, 9–12 and 17–20 of cycles 1 and 2 of 28-day cycles VD: bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11; and dexamethasone 40 mg on days1–4 of all cycles and days 9–12 of cycles 1 and 2 DCEP: dexamethasone 40 mg on days 1–4 of 2 cycles(4-week duration); cyclophosphamide 400 mg/m2; etoposide 40 mg/m2; and cisplatin 15 mg/m2/day on days 1–4 (continuous infusion) Primary outcome: CR/nCR rate (postinduction) |
VAD: n = 121 patients VAD + DCEP: n = 121 patients VD: n = 121 patients VD + DCEP: n = 119 patients NDMM |
nCR/CR postinduction: 14.8% (VD) versus 6.4% (VAD) (P <0.05) Adverse events: haematological toxicity and toxicity-related deaths more frequent with VAD than VD (7 versus 0 toxicity-related deaths; P = 0.02); higher rate of peripheral neuropathy (grade≥2) with VD (29.7%) versus VAD (13%; P <0.05) Median PFS: 36 months (VD) versus 29.7 months (VAD; P = 0.064) 3-year OS 81.4% (VD) versus 77.4% (VAD; P = 0.05) |
|
Moreau et al.29 Phase III Bortezomib subcutaneous versus bortezomib intravenous Bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 of 21-day cycles, by subcutaneous injection or intravenous infusion Primary outcome: non-inferiority of ORR |
s.c.: n = 148 patients i.v.: n = 74 patients RRMM |
ORR: 42% (both groups; P = 0.002 for non-inferiority of response) Adverse events: peripheral neuropathy with s.c. versus i.v. of any grade (38% versus 53%), ≥grade 2 (24% versus 41%), ≥grade 3 (6% versus 16%) ;P <0.05 for all groups TTP: 10.4 months (s.c.) versus 9.4 months (i.v.; P = 0.3) 1-year OS: 72.6% (s.c.) versus 76.7% (i.v.; P = 0.5) |
|
Cavo et al.141 Phase III VTD versus TD VTD: thalidomide 100/200 mg daily; bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11; dexamethasone 40 mg daily on 8 out of first 12 days but not consecutively TD: thalidomide and dexamethasone (same as in other arm) Primary outcome: rate of CR after induction therapy |
VTD: n = 241 patients TD: n = 239 patients NDMM |
CR/nCR: 31% (VTD) versus 11% (TD) after 3 cycles of induction therapy (P <0.0001) Adverse events: Rate of grade 3/4 skin rash and peripheral neuropathy: 10% (VTD) versus 2% (TD; P <0.05); discontinuation rate due to disease progression: 5% (TD) versus 0% (VTD; P <0.05) 3-year PFS: 68% (VTD) and 56% (TD) (P <0.05) 3-year OS: 86% (VTD) versus 84% (TD; P = 0.3) |
|
Rosiñol et al.142 Phase III VTD versus TD versus VBMP/VBAD/B VTD: bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11; thalidomide 200 mg daily (escalating doses in first cycle); and dexamethasone 40 mg o days 1–4, 9–13 for 24 weeks TD: thalidomide and dexamethasone (same as in other arm) VBMP/VBAD/B: 4 cycles if alternating VBMP/VBAD () followed by 2 cycles bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 of 21-day cycles Primary outcome: CR rate post-induction and post-autologous transplant |
VTD: n = 130 patients TD: n = 127 patients VBMCP/VBAD/B: n = 129 patients NDMM |
Postinduction CR: 35% (VTD), 21% (VBMCP/VBAD/B) and 14% (TD) Post-autologous transplant CR: 46% (VTD), 38% (VBMCP/VBAD/B) and 24% (TD) Adverse events: grade 3/4 neutropenia: 22% (VBMCP/VBAD/B) versus 14% (TD) and 10% (VTD); DVT/PE and peripheral neuropathy: 12% and 14% (VTD) versus 5% and 5% (TD) and 6% and 9% (VBMCP/VBAD/B) PFS: 56.2 months (VTD) versus 35.3 months (VBMCP/VBAD/B) versus 28.2 months (TD) 4-year OS:74% (VTD) versus 70% (VBMCP/VBAD/B) and 65% (TD) |
|
Sonneveld et al.21 Phase III VAD versus PAD VAD: vincristine 0.4 mg daily; doxorubicin 9 mg/m2 continuous infusion on days 1–4; and dexamethasone 40 mg on days 1–4, 9–12 and 17–20 of 28-day cycles for 3 cycles PAD: bortezomib 1.3 mg/m2 on days 1,4,8 and 11; doxorubicin and dexamethasone (same as in other arm) Primary outcome: PFS adjusted by ISS stage |
VAD: n = 414 patients PAD: n = 413 patients NDMM |
Postinduction nCR/CR: 11% (PAD) versus 5% (VAD); P <0.05 Best response nCR/CR: 49% (PAD) versus 34% (VAD); P <0.05 Adverse events: peripheral neuropathy grade ≥2: 40% (PAD) versus 18% (VAD); P <0.05 Median PFS: 35 months (PAD) versus 28 months (VAD); P <0.05 PFS improvement with PAD maintained after adjustment by ISS stage (P <0.05) 5-year OS: 61% (PAD) versus 55% (VAD); P >0.05 when adjusted for ISS |
|
Kumar et al.143 Phase II VRD versus VCD versus VCD mod versus VRCD Bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11; and dexamethasone 40 mg on days 1, 8 and 15, with either cyclophosphamide 500 mg/m2 on days 1 and, 8 (in VRD) or on days 1, 8 and 15 (in VCD-mod) and/or lenalidomide 15 mg on days 1–14 (in VRCD) or 25 mg on days 1–14 in 3-week cycles (maximum 8 cycles), followed by maintenance with bortezomib 1.3 mg/m2 on days 1, 8, 15 and 22 of 6-week cycles for 4 cycles (all arms) Primary outcome: safety and efficacy |
VRD: n = 42 patients VCD: n = 33 patients VCD-mod: n = 17 patients VDCR: 48 patients NDMM |
ORR: 80% (VRCD), 73% (VRD), 63% (VCD) and 82% (VCD-mod) ≥VGPR: 58% (VRCD), 51% (VRD), 41% (VCD) and 53% (VCD-mod) Adverse events: higher rates of haematological events observed with VCDR than in other groups 1-year PFS: 86% (VRCD), 83% (VRD), 93% (VCD), 100% (VCD-mod) 1-year OS: 100% all groups |
|
Petrucci et al.26 Phase II Bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 of 21-day cycles with or without dexamethasone at investigator’s discretion for up to 8 cycles Primary outcome: safety and efficacy in bortezomib retreatment |
n = 130 patients with RRMM who previously tolerated bortezomib 1.3 mg/m2 alone or in combination, achieved PR with previous bortezomib regimen and received last bortezomib dose ≥6 months before | ORR: 40% CR: 1% Adverse events: Thrombocytopenia (35% grade 3), peripheral neuropathy (9% grade 3), anaemia, diarrhoea, constipation Median TTP: 18.9 months |
|
San Miguel et al.23 Phase III Bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11; and dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11 and 12, with placebo or panobinostat 20 mg on days 1, 3, 5, 8, 10 and 12 Primary outcome: PFS |
n = 768 patients (n = 387 to panobinostat, bortezomib, dexamethasone and n = 381 to placebo, bortezomib, dexamethasone) NDMM |
ORR: 60.7% (panobinostat) versus 54.6% (placebo); P = 0.09 nCR: 27.6% (panobinostat) versus 15.7% (placebo); P = 0.00006 Adverse events: grade 3 and 4 thrombocytopenia: 67% (panobinostat) versus 31% (placebo), lymphopenia: 53% (panobinostat) versus 31% (placebo), diarrhoea: 26% (panobinostat) versus 8% (placebo), fatigue: 24% (panobinostat) versus 12% (placebo), neuropathy: 18% (panobinostat) versus 15% (placebo) Median PFS: 12.0 months (panobinostat) versus 8.1 months (placebo); P <0.0001 Median OS: 33.6 months (panobinostat) versus 30.4 months (placebo); P = 0.26 |
|
Durie et al.19 Phase III RD versus VRD RD: lenalidomide 25 mg on days 1–21; dexamethasone 40 mg on days 1, 8, 15 and 22 of 28-day cycles VRD: lenalidomide 25 mg on days 1–14; dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11 and 12; and bortezomib 1.3 mg/m2 i.v. on days 1, 4, 8 and 11 of 21-day cycles Primary outcome: PFS |
n = 474 patients with NDMM (n = 232 RD and n = 242 VRD) | ORR: 81.5% (VRD) versus 71.5% (RD) VGPR: 27.8% (VRD) versus 23.4% (RD) CR: 15.7% (VRD) versus 8.4% (RD) Adverse events: ≥grade 3 lymphopenia: 23% (VRD) versus 18% (RD), neutropenia: 19% (VRD) versus 21% (RD), thrombocytopenia 18% (VRD) versus 14% (RD), sensory neuropathy: 23% (VRD) versus 3% (RD), thrombosis and/or embolism: 8% (VRD) versus 9% (RD), diarrhoea: 8% (VRD) versus 2% (RD), second primary malignancies: 3% (VRD) versus 4% (RD) Median PFS: 43 months (VRD) versus 31 months (RD); P < 0.05 Median OS: not reached (VRD) versus 63 months (RD); P <0.05 |
|
Palumbo et al.27 Phase III DaVD vs VD DaVD: daratumumab 16 mg/kg IV on days 1, 8 and 15 during cycles 1–3, once every 3 weeks during cycles 4–8, and once every 4 weeks thereafter until disease progression or unacceptable toxic effects; bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 of cycles 1–8; dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11 and 12 every 21 days VD: bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 of cycles 1–8; dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11 and 12 every 21 days Primary outcome: PFS |
n = 498 with RRMM (n = 251 DaVD and n = 247 VD) |
ORR: 83% (DaVD) versus 63% (VD) CR: 46% (DaVD) versus 21% (VD) P <0.05 Adverse events: ≥grade 3 lymphopenia: 9.5% (DaVD) versus 2.5% (VD), neutropenia: 13% (DaVD) versus 4% (VD), thrombocytopenia 45% (DaVD) versus 32% (VD), anaemia 14.4% (DaVD) versus 16% (VD), sensory neuropathy: 4.5% (DaVD) versus 6.8% (VD), diarrhoea: 3.7% (DaVD) versus 1.3% (VD), second primary malignancies: 2.5% (DaVD) versus 0.4% (VD) Median PFS: not reached (DaVD) versus 7.2 months (VD); P <0.05 Median OS: not reached in either treatment group owing to short median follow up duration of 7.4 months |
The results of these trials led to the adoption of new standards of care. *Bortezomib standard schedule: 1.3 mg/m2 i.v. on days 1, 4, 8 and 11 of 21-day cycles. CR, complete response; DaVD, daratumumab, bortezomib, dexamethasone; DCEP, dexamethasone, cyclophosphamide, etoposide, cisplatin; DVT/PE: deep venous thrombosis/pulmonary embolism;; GI, gastrointestinal; HDD, high-dose dexamethasone; ISS, International Staging System; i.v., intravenous; MM, multiple myeloma; MP, melphalan, prednisone; MR, minor response; MTD, maximum tolerated dose; NA, not available; nCR, near-complete response; NDMM, newly diagnosed myeloma; ORR, overall response rate; OS, overall survival; PAD, bortezomib, doxoruicin, dexamethasone; peg-dox, pegylated doxorubicin; PFS, progression-free survival; PR, partial response; RD, lenalidomide, dexamethasone; RRMM, relapsed and/or refractory multiple myeloma; s.c., subcutaneous; TD, thalidomide, dexamethasone; TTP, time to progression; VAD, vincristine, doxorubicin, dexamethasone; VBMCP/VBAD/B, regimen that alternates vincristine, doxorubicin, dexamethasone followed by 2 cycles of bortezomib, melphalan, cyclophosphamide, prednisone and vincristine; VCD, bortezomib, cyclophosphamide, dexamethasone; VCD-mod, modified VCD; VD, bortezomib, dexamethasone; VGPR, very good partial response; VMP, bortezomib, melphalan, prednisone; VRCD, bortezomib, lenalidomide, cyclophosphamide, dexamethasone; VRD, bortezomib, lenalidomide, dexamethasone; VTD, bortezomib, thalidomide, dexamethasone.
Once the efficacy of bortezomib was established in patients with relapsed and/or refractory multiple myeloma, attention turned to those with newly diagnosed disease. In 2008, a randomized phase III trial17 comparing melphalan plus prednisone (MP) alone or with bortezomib (VMP) in newly diagnosed patients considered ineligible for transplantation; increased TTP, PFS and overall survival were observed in the patients who received VMP (Table 1). Since then, the efficacy of bortezomib has been evaluated in many different combinations, perhaps most notably with immunomodulatory drugs (IMiDs), resulting in active regimens associated with high overall and complete response rates17,18. For example, one of the most widely used combination-treatment options for patients with newly diagnosed multiple myeloma comprises bortezomib, lenalidomide and dexamethasone (RVD)18 (Table 1). Notably, in 2015, Durie et al.19 reported results of the SWOG S0777 phase III trial, in which the efficacy of an induction regimen consisting of RVD was compared with that of lenalidomide plus dexamethasone. The results of this trial confirmed the superiority of the RVD regimen, which in many patients led to remission of a prolonged duration and improved overall survival19 (Table 1). The results of other key clinical trials demonstrated the efficacy of bortezomib as part of induction therapy in patients with newly diagnosed multiple myeloma (Table 1), which was further established by a meta-analysis of the results from randomized phase III trials that were available at the time20. This meta-analysis revealed that the median PFS with bortezomib-based therapy was 35.9 months compared with 28.6 months without bortezomib (P <0.001), and the overall survival at 3 years was 79.7% compared with 74.7%.
Bortezomib might also be effective as a maintenance therapy, according to data from a phase III trial21 in patients with newly diagnosed multiple myeloma, in which bortezomib was used in both the induction and maintenance regimens. Patients were randomly assigned to receive bortezomib, doxorubicin and dexamethasone (PAD), or vincristine, doxorubicin and dexamethasone (VAD), followed by autologous stem-cell transplantation in both groups, and then maintenance with either bortezomib (PAD group) or thalidomide (VAD group). Patients who received PAD–bortezomib showed improved responses and PFS compared with those treated with VAD–thalidomide (Table 1), especially those with high-risk multiple myeloma harbouring chromosome 17p deletions21.
Bortezomib has also been successfully combined with other agents to improve clinical outcomes in the relapsed and/or refractory settings. For example, the efficacy of bortezomib plus pegylated doxorubicin was compared with that of bortezomib monotherapy in patients with relapsed multiple myeloma, and the combination regimen prolonged TTP and improved overall survival, and these benefits were statistically significant22 (Table 1). In a study published in 201423, bortezomib and dexamethasone were combined with the oral pan-deacetylase inhibitor panobinostat, partly on the basis that the latter agent would inhibit aggresome formation, a potential mechanism of inducible or acquired resistance to proteasome inhibitors24. This phase III trial revealed a median PFS of 12.0 months with the three-drug regimen versus 8.1 months with bortezomib plus dexamethasone25 (P <0.0001; Table 1). On the basis of a subgroup analysis showing improved PFS for the triplet combination23, the FDA and European Medicines Agency (EMA) approved panobinostat in combination with bortezomib and dexamethasone for the treatment of patients with relapsed and/or refractory myeloma who had received at least two prior lines of therapy (including bortezomib and an IMiD). Panobinostat is now being studied in combination with RVD as a first-line treatment for myeloma26; after four cycles of treatment, the ORR was 93%, including a complete response rate of 44%. Of note, patients with multiple myeloma who have had a previous response to bortezomib can be re-treated with the same agent if they had a relapse at least 6 months after completion of a prior course of bortezomib treatment26, further demonstrating the therapeutic value of this drug.
Bortezomib has also been successfully combined with the anti-CD38 monoclonal antibody daratumumab. Palumbo et al.27 conducted a randomized phase III trial in patients with relapsed and/or refractory myeloma using bortezomib and dexamethasone with or without daratumumab. The 12-month PFS improved from 27% to 60% when daratumumab was added to the combination of bortezomib and dexamethasone, providing evidence of another effective therapeutic strategy for patients with myeloma.
The development of peripheral neuropathy can be a dose-limiting factor in patients receiving bortezomib. Up to 80% of patients with newly diagnosed multiple myeloma treated with bortezomib develop any grade neuropathy18, and long-term exposure and/or higher doses can increase the risk of neuropathy. Changes in the route of bortezomib administration, from intravenous to subcutaneous, or in the dosing schedule, from biweekly to weekly, seem to reduce the incidence of neuropathy while maintaining efficacy28,29. Indeed, owing to the results of a large randomized study28, the subcutaneous administration of bortezomib was approved in 2012 by both the FDA and EMA, and the intravenous administration of this agent has, to some extent, fallen out of favour among clinicians. Neuropathy might be a result of the off-target effects of bortezomib30, and is, to some extent, a class effect of proteasome inhibitors, but the exact underlying mechanisms are not fully understood. In addition to neuropathy, bortezomib can induce thrombocytopenia, which is cyclical and, to some extent, predictable, with a 60% reduction from baseline during each treatment cycle and recovery between cycles31. Other adverse events associated with bortezomib include nausea and diarrhoea, fatigue, and neutropenia. Finally, the use of bortezomib and, indeed, of other proteasome inhibitors, both alone and in combinations, has been associated with an increased risk of herpes zoster virus reactivation32, indicating the need for prophylaxis with antivirals, which is recommended for all patients.
Second-generation proteasome inhibitors
A number of different pharmacophores have been described as having the ability to interact with the N-terminal threonine residues in the catalytic proteasome subunits that are responsible for the nucleophilic attacks involved in the cleavage of peptide bonds. Similarly to boronic acids (which include bortezomib and ixazomib), epoxyketones can also bind to these N-terminal threonines; however, unlike boronates, which bind reversibly, epoxyketones form irreversible bonds that can prolong the duration of proteasome inhibition. The possibility of a long duration of inhibition provided the rationale for phase I studies of carfilzomib (previously known as PR-171), which revealed that this agent is both tolerable and active against relapsed and/or refractory multiple myeloma, even in some patients who had received prior bortezomib therapy33,34. Subsequently, carfilzomib received accelerated approval in 2012 by the FDA as a single agent for the treatment of multiple myeloma in patients who have received at least two prior lines of therapy and with disease that was refractory to the most-recent line of treatment. The approval decision was based on results from a single-arm study in 266 patients34, in whom carfilzomib was given on days 1, 2, 8, 9, 15 and 16 of 28-day cycles at 20 mg/m2 during the first cycle, and then at 27 mg/m2, starting in cycle two, for up to 12 cycles (20/27 schedule). An ORR of 23.7% was reported, including one complete response and 13 very good partial responses (VGPR; the criteria for determining responses have been defined elsewhere35), with a median duration of response (DOR) of 7.8 months36.
A confirmatory phase III trial37 compared standard-dose carfilzomib (20/27 schedule) with best supportive care (in the form of low-dose steroids with optional cyclophosphamide) in patients with treatment-refractory multiple myeloma. Interestingly, this trial did not meet its primary end point of improving overall survival (hazard ratio (HR) 0.98, 95% confidence interval (CI) 0.76–1.2), owing, in part, to the substantial activity of the control regimen38. A second confirmatory study38 in a cohort of patients with multiple myeloma who had received 1–3 lines of prior treatment compared lenalidomide plus dexamethasone (RD; control) with RD plus carfilzomib (KRD). In this phase III trial, Stewart et al.38 randomly assigned 792 patients to receive RD or KRD with carfilzomib at 20 mg/m2 on days 1 and 2 of cycle one, and then at 27 mg/m2 for the remainder of cycle one and beyond. The use of KRD improved PFS compared with that achieved with RD (26.3 months versus 17.6 months; P = 0.0001)39. An interim analysis showed that the 24-month overall survival was 73.3% and 65% for the KRD and RD groups, respectively, and, although the median survival was not reached in either group, a statistically significant benefit favouring KRD was observed (P = 0.04). Dose adjustments were made in both groups; however, in the KRD arm, the lenalidomide dose was reduced more frequently than that of carfilzomib (in 43.4% and 11% of patients, respectively). Furthermore, disease progression was the most frequent cause of treatment discontinuation, and discontinuation was less likely with KRD than with RD (39.8% versus 50.1%). Adverse events of any rate were more frequent with KRD, including cardiopulmonary events, such as dyspnoea (2.8% versus 1.8%), cardiac failure (3.8% versus 1.8%), ischaemic heart disease (3.3% versus 2.1%) and hypertension (4.3% versus 1.8%). The frequency of other events, such as peripheral neuropathy, was similar in both groups. Despite this higher rate of certain types of adverse events, patients reported superior health-related quality of life and derived substantial added benefit from KRD, owing to the effectiveness of this treatment, which has been established as a new standard of care for patients with relapsed multiple myeloma.
In 2014, a study tested high-dose carfilzomib, administered as a 30-minute infusion, and given initially at 20 mg/m2 on days 1 and 2 of cycle one, and then escalated to 56 mg/m2 (20/56 schedule) for the remaining doses, along with dexamethasone. This regimen showed evidence of potentially enhanced activity compared with the standard-dose regimen40. This observation supported the design and initiation of a large phase III trial41, in a cohort of 929 patients with relapsed multiple myeloma after 1–3 prior lines of therapy, comparing high-dose carfilzomib plus dexamethasone (KD) with bortezomib and dexamethasone (VD). In this trial, which is one of only a few completed studies comparing two agents with similar mechanisms of action in myeloma, KD was found to be superior to VD (PFS 18.7 versus 9.4 months; HR 0.53, 95% CI 0.44–0.65; P <0.0001)42. Serious adverse events were more common in the carfilzomib arm (48% versus 36%), although grade ≥2 peripheral neuropathy was considerably more common with bortezomib (32% versus 6%; P <0.0001). Cardiac adverse events of any grade were, however, more common with KD, including hypertension (25% versus 9%), cardiac failure (8% versus 2%) and shortness of breath (28% versus 13%). Acute renal failure (all grades) was also more common with carfilzomib plus dexamethasone (8% versus 4%). Despite a higher number of adverse events in the carfilzomib group, treatment discontinuation and treatment-related deaths were comparable in both groups (5%)41. Beyond the 20/56 dosing schedule, interest has emerged in administering carfilzomib at an even higher dose given weekly instead of twice weekly. A phase I study42 has identified 70 mg/m2 carfilzomib (or 20/70 dosing) as the maximum-tolerated dose when given with dexamethasone, with updated efficacy data demonstrating a 72% ORR in patients who had received 1–3 prior lines of therapy43. Of note, a phase III trial comparing once weekly with twice weekly administration of carfilzomib plus dexamethasone in patients with relapsed and/or refractory multiple myeloma is currently underway (NCT02412878)44.
In the setting of newly diagnosed disease, data from two early phase uncontrolled studies43,45 have shown that KRD is well-tolerated and effective45,46. Similar to other treatment studies47, the depth of response of patients with multiple myeloma to treatment improved with continued therapy. KRD also was also well tolerated in patients deemed ineligible for transplantation48. Notably, the National Clinical Trials Network (National Cancer Institute, USA) is performing a study comparing KRD and RVD in patients with newly diagnosed multiple myeloma (NCT01863550)49 that is expected to shed further light on the comparative efficacy and toxicity of these powerful combination regimens. Carfilzomib has also been evaluated in combination with melphalan and prednisone (CMP) as induction therapy in patients with newly diagnosed multiple myeloma who are deemed ineligible for transplantation50. The results of this phase I/II study established a maximum-tolerated dose of 20 mg/m2 on days 1 and 2 of the first cycle, followed by carfilzomib up to 36 mg/m2 on days 8, 9, 22, 23, 29 and 30 of a 42-day cycle, and on days 1, 2, 8, 9, 22, 23, 29 and 30 every 42 days for the remaining cycles, along with 9 mg/m2 melphalan and 60 mg/m2 prednisone on days 1–450. In this study, dose-limiting toxicities included deep vein thrombosis, febrile neutropenia, fever and hypotension, while adverse events of any grade, as observed in at least 25% of patients included anaemia, fatigue, neutropenia, infections, nausea, elevated levels of liver enzymes and peripheral neuropathy. An ORR of 90% was noted, which included VGPR and complete response rates of 58% and 12%, respectively. These encouraging data led to a phase III trial comparing CMP with VMP, but a press release indicated that no statistically significant difference was observed between the two arms, and formal presentation of the data is pending51. Several clinical trials of proteasome inhibitors in patients with multiple myeloma that might be relevant for the field are also underway (Table 2).
Table 2.
Selected ongoing clinical trials with proteasome inhibitors in multiple myeloma
| Proteasome inhibitor under evaluation | Trial phase, interventions, and primary end point | Schedule | Patient population | Reference |
|---|---|---|---|---|
| Bortezomib | Phase III VRD versus RD PFS |
VRD: bortezomib s.c. or i.v. on days 1, 4, 8 and 11; lenalidomide on days 1–14; dexamethasone p.o.on days 1, 2, 4, 5, 8, 9, 11 and 12 of 21-day cycles for 8 cycles (in the absence of disease progression or unacceptable toxicities) RD: lenalidomide p.o. on days 1–21; dexamethasone on days 1, 8, 15 and 22 of 28-day cycles for 6 cycles (in the absence of disease progression or unacceptable toxicities) |
NDMM without an intent for immediate aSCT | NCT01530594144 |
| Bortezomib and carfilzomib | Phase III VRD versus CRD 2 years of lenalidomide maintenance versus indefinite maintenance OS |
VRD: bortezomib s.c. or i.v. on days 1, 4, 8 and 11 of cycles 1–8 and on days 1 and 8 of cycles 9–12; lenalidomide p.o. on days 1–14 of all cycles; dexamethasone p.o. on days 1, 2, 4, 5, 8, 9, 11 and 12 of cycles 1–8 and on days 1, 2, 8 and 9 of cycles 9–12 of 21-day cycles CRD: carfilzomib i.v. on days 1, 2, 8, 9, 15 and 16; lenalidomide p.o.on days 1–21; dexamethasone p.o. on days 1, 8, 15 and 22 of 28-day cycles for 9 cycles 2 years of lenalidomide: lenalidomide p.o. on days 1–21 of 28-day cycles for 24 cycles (in the absences of disease progression or unacceptable toxicities) Indefinite maintenance: lenalidomide p.o. on days 1–21 of 28-day cycles (in the absence of disease progression or unacceptable toxicities) |
NDMM | NCT0186355049 |
| Bortezomib and carfilzomib | Phase III CMP versus VMP OS |
CMP: carfilzomib 20/36 mg/m2 i.v. on days 1, 2, 8, 9, 22, 23, 29 and 30; melphalan p.o.; prednisone 60 mg/m2 p.o. days 1–4 for 9 cycles VMP: bortezomib 1.3 mg/m2 i.v. or s.c. on days 1, 4, 8, 11, 22, 25, 29 and 32 of cycles 1–4, and days 1, 8, 22 and 29 of cycles 5–9; melphalan 9 mg/m2 on days 1–4; prednisone p.o. 60 mg/m2 on days 1–4 of all cycles |
NDMM | NCT0181875251 |
| Carfilzomib | Phase III Addition of carfilzomib to dexamethasone once weekly versus twice weekly ORR, PFS and OS |
Carfilzomib once weekly: carfilzomib 20 mg/m2 i.v. on day 1 of cycle 1, then 70 mg/m2 on days 8 and 15 of cycle 1 and days 1, 8 and 15 of other cycles; dexamethasone 40 mg i.v. or orally on days 1, 8, 15 and 22 of cycles 1–9 and days 1, 8 and 15 of all other cycles Carfilzomib twice weekly: carfilzomib 20 mg/m2 i.v. on days 1 and 2 of cycle 1, then 27 mg/m2 on days 8, 9, 15 and 16 of cycle 1 and days 1, 2, 8, 9, 15 and 16 of other cycles; dexamethasone p.o. (same as other arm) |
RRMM | NCT0241287844 |
| Ixazomib | Phase III IRD versus RD PFS |
IRD: ixazomib 4 mg p.o. on days 1,8 and 15; lenalidomide 25 mg p.o. on days 1–21; and dexamethasone 40 mg p.o. on days 1, 8, 15 and 22 of 28-day cycles (until disease progression) RD: placebo p.o. on days 1, 8 and 15; lenalidomide and dexamethasone p.o. (same as other arm) on 28-day cycles (until disease progression) |
NDMM | NCT01850524145 |
| RRMM | NCT01564537146 | |||
| Ixazomib | Phase III Ixazomib versus placebo PFS |
Ixazomib 3 mg p.o. on days 1, 8 and 15 of cycles 1–4, then 3–4 mg on days 1, 8 and 15 of cycles 5–26 of 28-day cycles | NDMM, maintenance without aSCT | NCT02312258147 |
| NDMM, maintenance after aSCT | NCT02181413128 |
aSCT, autologous stem cell transplantation; CMP, carfilzomib, melphalan, prednisone; CRD, carfilzomib, lenalidomide, dexamethasone; IRD, ixazomib, lenalidomide, dexamethasone; i.v., intravenous; NDMM, newly diagnosed multiple myeloma; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; p.o, by mouth; RD, lenalidomide, dexamethasone; RRMM, relapsed and/or refractory myeloma; s.c., subcutaneous; VMP, bortezomib, melphalan, prednisone; VRD, bortezomib, lenalidomide, dexamethasone.
Carfilzomib seems to have a distinctly different adverse-event profile compared to that of bortezomib. Whereas the rates of peripheral neuropathy are lower with carfilzomib, a few patients have developed cardiovascular complications, including hypertension and heart failure with decreased left ventricular ejection fraction46. Endothelial dysfunction has been proposed to occur after carfilzomib administration through inhibition of endothelial nitric oxide synthase activity52. An echocardiography assessment is recommended for all patients before starting carfilzomib treatment, though utility of this evaluation in predicting those at risk for cardiac events has not been proven, and additional studies are needed in this area. Close monitoring for shortness of breath, lower-extremity oedema or paroxysmal nocturnal dyspnoea is also warranted. Furthermore, carfilzomib can infrequently cause renal impairment, which is even rarer with bortezomib. These differences highlight the need for additional studies of the apparently distinct downstream effects of these two drugs53.
Other novel proteasome inhibitors
The efficacy of other proteasome inhibitors has been evaluated in clinical trials, including the orally bioavailable reversible peptide boronate ixazomib, the irreversible epoxyketone oprozomib, the intravenous β-lactone marizomib, and the boronate delanzomib (Tables 2 and 3). Among these oral proteasome inhibitors, ixazomib is most advanced in clinical development54–56, which started with several phase I studies in patients with relapsed and/or refractory myeloma, in whom treatment-associated toxicities were mostly related to gastrointestinal adverse events or skin rash. The overall rate of neuropathy was 20%, but because the response rates were under 20%, development was moved forward in combination regimens, especially with RD55; among 64 evaluable patients with newly diagnosed disease, 58% had a VGPR or better after 12 cycles of treatment with this regimen54, and this supported a randomized phase III study of ixazomib plus RD compared with RD56. Treatment with ixazomib plus RD improved PFS (20.6 months versus 14.7 months with RD) in patients with relapsed or refractory myeloma after 1–3 prior lines of therapy57. These results supported the regulatory approval of ixazomib by the FDA in 2015. Additional studies of this agent in several settings, including as induction and maintenance therapy, are underway. Another oral proteasome inhibitor, oprozomib, is being evaluated in phase I/II clinical trials as a single agent58,59, or in combination with other drugs57,60, with encouraging early data, but later-phase trials have not yet been initiated. Finally, marizomib, which might target the proteasome more broadly, as it seems to inhibit all three major proteolytic activities of the proteasome61, is also being studied, and has shown early signs of activity in combination regimens62.
Table 3.
Selected inhibitors of the 20S proteasome
| Name and manufacturer | Description | Administration | Regulatory status | Response in representative trials (IMWG criteria)35 | Common adverse effects |
|---|---|---|---|---|---|
| Bortezomib (Millennium) | Boronate Reversible action Targets β5 > β1 >β2 |
Intravenous or subcutaneous | FDA-approved for frontline therapy for RRMM, and MCL after one prior line of therapy | Single-agent bortezomib versus high-dose dexamethasone in RRMM: ORR 38% versus 18% 1-year OS 80% versus 66%16 Bortezomib, lenalidomide and dexamethasone in NDMM: ORR 100% 21-month OS 97%143 |
Peripheral neuropathy Nausea Vomiting Diarrhoea Constipation Cytopenias Infection |
| Carfilzomib (ONYX) | Epoxyketone Irreversible action Targets β5 >β2/β1 |
Intravenous | FDA-approved for RRMM | RRMM previously treated with an immunomodulatory drug proteasome inhibitor: ORR 23%36 |
Shortness of breath Heart failure Reduced rates of peripheral neuropathy compared with bortezomib |
| Ixazomib (Millennium) | Boronate Reversible action Targets β5 > β1 |
Oral | FDA-approved for RRMM Early-phase clinical trials in AML, follicular lymphoma and peripheral T-cell lymphoma |
IRD versus RD in RRMM: PFS 20.6 versus 14.7 months; CR 11.7% versus 6% IRD in NDMM: 93% ≥PR; 67% ≥VGPR; 24% ≥CR; 14% ≥sCR Ixazomib plus lenalidomide as maintenance post ASCT: safety profile established, other responses not yet reported Ixazomib alone in RRMM with no or little exposure to bortezomib: 34% ≥PR; 6% sCR149 |
Skin rash Peripheral oedema Peripheral neuropathy Fatigue Cytopenia Infection |
| ONX 0912 (oprozomib; ONYX) | Epoxyketone Irreversible action Targets β5 |
Oral | Early-phase clinical trials in haematological malignancies and solid tumours | Single-agent oprozomib in RRMM and WM: 25% CBR and80% CBR (in one dose), respectively150 | Nausea Vomiting Diarrhoea |
| NPI 0052 (marizomib; Nereus) | β-lactone Irreversible action Targets β5 >β2 >β1 |
Intravenous | Early-phase clinical trials in RRMM, NSCLC, pancreatic cancer, melanoma, lymphoma and advanced-stage solid tumours | Single-agent marizomib in RRMM: ORR 20% (by EBMT criteria)151 |
Not reported |
|
CEP-18770 (delanzomib;) |
Boronate Reversible action Targets β5 >β1 |
Intravenous Oral |
Early-phase clinical trials in RRMM (terminated), solid tumours and lymphoma | Not reported | Not reported |
AML, acute myeloid leukaemia; ASCT, autologous stem-cell transplantation; CBR, clinical benefit rate; CR, complete response; EBMT, European Group for Blood and Marrow Transplant; IMWG, International Myeloma Working Group; IRD, ixazomib, lenalidomide, dexamethasone; NDMM, newly diagnosed multiple myeloma; NSCLC, non-small cell lung cancer; ORR, overall response rate; OS, overall survival; PR, partial response; RD, lenalidomide, dexamethasone; RRMM, relapsed and/or refractory multiple myeloma; sCR, stringent complete response; VGPR, very good partial response; WM, Waldenström’s macroglobulinaemia.
Lymphoma and myeloma-related disorders
Bortezomib has also been approved by the FDA and EMA for the treatment of MCL, initially in the relapsed and/or refractory setting for patients who have received at least one prior line of therapy. MCL is a heterogeneous B-cell non-Hodgkin lymphoma that can have an aggressive course and is regarded as incurable. In phase II clinical trials in patients with relapsed or refractory MCL, bortezomib showed signs of effectiveness over previous standard therapies, with a response rate of up to 50%, including a complete response rate of 4–8%63–68. The most common high-grade adverse events described in these studies included fatigue, peripheral neuropathy and cytopenias63–68. In 2006, the FDA granted approval of bortezomib for patients with relapsed and/or refractory MCL, based on the results of a multicentre phase II study in 155 patients66. In this trial, the ORR and median DOR observed in patients who received bortezomib were 31% and 6.3 months, respectively66. In 2015, an open-label phase III study12 was conducted in 487 patients with previously untreated MCL who were ineligible for a bone marrow transplant and were randomly allocated to receive R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) or VR-CAP (bortezomib, rituximab, cyclophosphamide, doxorubicin and prednisone). An improved median PFS (24.7 months versus 14.4 months; P <0.001) and four-year overall survival (64.4% versus 53.9%; P = 0.17) were observed in patients who received VR-CAP12. The results of this study led to the approval of bortezomib in 2014 for patients with previously untreated MCL. Bortezomib might also benefit patients with MCL as a maintenance therapy, as suggested by the results of a phase II trial68, in which R-CHOP was administered as induction therapy, followed by bortezomib as maintenance therapy. This treatment was well tolerated, and the reported toxicities were mainly haematological and grade 3 or higher peripheral neuropathy (in 5% of patients)69. With this regimen, the 5-year overall survival and 2-year PFS were 66% and 62%, respectively, indicating a more favourable response than that observed in previous studies in patients with MCL treated only with R-CHOP68,70. Proteasome inhibitors have also been used for the treatment of MCL in other trials (Table 4).
Table 4.
Selected clinical trials with proteasome inhibitors in mantle cell lymphoma
| Study characteristics | Patient number and characteristics | Results |
|---|---|---|
| Fisher et al.56 Phase II (single arm) Bortezomib: 1.3 g/m2 on days 1, 4, 8 and 11 of 21-day cycles for up to 17 cycles Primary outcome: safety and efficacy |
n = 155 Relapsed and/or refractory disease | ORR: 33% CR: 8% Median DOR: 9.2 months Median TTP: 6.2 months Adverse events: peripheral neuropathy, fatigue and thrombocytopenia |
| Robak et al.12 Phase III R‐CHOP versus VR-CAP R-CHOP: rituximab 375 mg/m2, cyclophosphamide 750 mg/m2; doxorubicin 50 mg/m2; vincristine 1.4 mg/m2 on day 1; and prednisone 100 mg on days 1–5 of 21-day cycles VR-CAP: rituximab 375 mg/m2; cyclophosphamide 750 mg/m2; doxorubicin 50 mg/m2 on day 1; bortezomib 1.3 mg/m2 on days 1, 4, 8; and 11, and prednisone 100 mg on days 1–5 of 21-day cycles Primary outcome: PFS |
n = 487 Newly diagnosed disease |
4-year OS: 54% (R-CHOP) versus 64% (VR-CAP) Median PFS: 14.4 months (R‐CHOP) versus 24.7 months (VR-CAP) Adverse events: lower rates of thrombocytopenia, and neutropenia and/or infection with R-CHOP compared with VR-CAP; similar rates of peripheral neuropathy with resolution to baseline after treatment in most cases |
| Till et al.59 Phase II (single arm) VR-CHOP: rituximab 375 mg/m2; cyclophosphamide 750 mg/m2; doxorubicin 50 mg/m2; vincristine 1.4 mg/m2 on day 1; bortezomib 1.3 mg/m2 on days 1 and 4; and prednisone 100 mg on days 1–5 on 21-day cycles for 6 cycles followed by vincristine 1.3 mg/m2 on days 1, 4, 8 and 11 of 3-month cycles for 2 years Primary outcome: efficacy |
n = 65 Newly diagnosed disease |
Median PFS: 29.5 months 5-year OS: 66% Adverse events: neutropenia, thrombocytopenia and peripheral neuropathy |
CR: complete response; DOR, duration of response; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone; TTP, time to progression; VR-CAP: bortezomib, rituximab, cyclophosphamide, doxorubicin and prednisone; VR-CHOP: bortezomib, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone.
Bortezomib has been shown to be effective for the treatment of Waldenström’s macroglobulinaemia, another type of non-Hodgkin lymphoma. The use of bortezomib is not approved for this indication, but this agent is often used in clinical practice in the first-line and relapsed and/or refractory settings. Investigators conducting three phase II studies69,71,72 testing rituximab and bortezomib combinations in patients with newly diagnosed Waldenström’s macroglobulinaemia reported response rates of 66–85%, with responses occurring in the first 2–3 months of treatment71–73. A randomized phase III trial74 evaluating the addition of bortezomib to a dexamethasone, rituximab and cyclophosphamide regimen is currently ongoing, and results from this study should be reported in the next few years, and will hopefully help to clarify the role of bortezomib in the first-line setting (Table 5). Bortezomib can aggravate pre-existing neuropathies, and therefore, the substitution of this agent with carfilzomib to potentially spare the onset of neuropathy has been tested in a trial for patients with Waldenström’s macroglobulinaemia. In this study73, 28 newly diagnosed patients received carfilzomib together with rituximab plus dexamethasone, with an observed ORR of 87%, and only one patient developed neuropathy75. Early phase trials with carfilzomib are planned in the setting of relapsed Waldenström’s macroglobulinaemia, as well as with ixazomib in the first-line setting (Table 5).
Table 5.
Ongoing clinical trials with proteasome inhibitors in myeloma-related cell dyscrasias
| Study characteristics | Patient population | Results |
|---|---|---|
| Bortezomib | ||
| Kastritis et al.79 Phase III MD versus VMD MD: melphalan 0.22 mg/kg; and dexamethasone 40 mg on days 1–4 of 28-day cycles for 9 cycles VMD: bortezomib 1.3 mg/m2 on days 1, 4, 8 and 11 in cycles 1 and 2 and days 1, 8, 15 and 22 in following cycles; melphalan and dexamethsanone (same as other arm) Primary outcome: ORR |
n = 70 patients with AL amyloidosis transplant ineligible (enrolment ongoing up to 110 patients) | VGPR after 8 cycles: 35% (MD) versus 65% (VMD); P = 0.036 Adverse events: grade 3/4 events comparable between arms (P = 0.15) Median OS and PFS not reached for either arm |
| Sanchorawala et al.76 Phase II Bortezomib 1.3 mg/m2 i.v. and dexamethsaone 20 mg i.v. on days 1, 4, 8 and 11 of 21-day cycles for 2 cycles followed by bortezomib-conditioning regimen 1 mg/m2 i.v. on days −6,−3,+1 and +4 and high-dose melphalan (either 140 mg/m2 or 200 mg/m2) Primary outcome: ORR and survival |
Patients with newly diagnosed AL amyloidosis | 63% haematological CR at 6 months post-transplantation Adverse events: diarrhoea, infection, skin rash, GVHD, splenic rupture (no statistically significant differences) Median OS and PFS not reached |
|
NCT0178802074 Phase III DRC ± bortezomib DRC: dexamethasone 20 mg on days 1, 8 and 15; rituximab 375 mg/m2 on day 1; and cyclophosphamide 200 mg/m2 on days 1–5 of 29-day cycles Bortezomib 1.6 mg/m2 on days 1, 8 and 15 of 29 day-cycles Primary outcome: OS |
Patients with newly diagnosed WM | NA |
| Ixazomib | ||
|
NCT0165965880 Phase III Ixazomib versus physician’s choice Ixazomib 4 mg on days 1, 8 and 15; and dexamethasone 20 mg on days 1, 8, 15 and 22 of 28-day cycles) Primary outcome: OS |
Patients with relapsed and/or refractory AL amyloidosis | NA |
|
NCT02400437152 Phase II Ixazomib on days 1, 8 and 15; rituximab on day 1; and dexamethasone on days 1, 8 and 15 of 28-day cycles Primary outcome: Rate of VGPR |
Patients with newly diagnosed WM | NA |
| Carfilzomib | ||
|
NCT01813227153 Phase II Carfilzomib 20/56 mg/m2 on days 1, 2, 8, 9, 15 and 16; rituximab 375 mg/m2 on day 16; and dexamethasone on days 1, 2, 8, 9, 15 and 16 of 28-day cycles Primary outcome: safety and efficacy |
Patients with relapsed WM | N/A |
AL amyloidosis, light-chain amyloidosis; DRC, dexamethasone, rituximab and cyclophosphamide; GVHD, graft-versus-host disease; MD, dexamethasone, melphalan; NA, not available; OS, overall survival; PFS, progression-free survival; VGPR, very good partial response; VMD, bortezomib, dexamethasone, melphalan; WM, Waldenström’s macroglobulinaemia.
Light-chain (AL) amyloidosis is a plasma-cell dyscrasia that can be associated with multiple myeloma or Waldenström’s macroglobulinaemia, and is caused by the abnormal deposition of amyloidogenic monoclonal light chains secreted by neoplastic clonal B cells or plasma cells. AL amyloidosis can be treated effectively with proteasome inhibitors76, and a combination of bortezomib, cyclophosphamide and dexamethasone is currently a widely accepted initial standard of care for the treatment of this condition, in part, owing to a report by Mikhael et al.76 In their study76, 17 patients with AL amyloidosis received cyclophosphamide, bortezomib and dexamethasone before stem-cell transplantation, resulting in a haematological response rate of 94%, including a complete response rate of 71%, and a median DOR of 22 months. In a larger retrospective collaborative European study investigating first-line cyclophosphamide, bortezomib and dexamethasone in 230 patients with AL amyloidosis77,78, a 60% haematological response rate (including a 23% complete response rate) was observed. Importantly, preliminary results of a phase III trial78 evaluating the effect of bortezomib in patients newly diagnosed with AL amyloidosis who were not eligible for autologous transplantation showed higher response rates with the combination of bortezomib, melphalan and dexamethasone compared with melphalan plus dexamethasone (ORR 76% versus 58%, P = 0.17; and VGPR 65% versus 35%, P = 0.036)79. In addition, the oral proteasome inhibitor ixazomib is being evaluated in a study currently registering patients with relapsed and/or refractory AL amyloidosis (NCT01659658)80, results of which are expected to be reported in the next few years. Other clinical trials testing proteasome inhibitors in patients with AL amyloidosis and Waldenström’s macroglobulinaemia are currently ongoing (Table 5).
Other malignancies
The effect of proteasome inhibitors, especially bortezomib, has been studied in clinical trials involving patients with other haematological malignancies, including acute myeloid leukaemia79,81,82, myelodysplastic syndrome81 and acute lymphoblastic leukaemia83, but the responses observed in these settings did not merit further exploration81–84. Similarly, despite highly encouraging preclinical data, clinical studies in patients with solid tumours failed to demonstrate any efficacy of this agent85. A mechanism proposed to account for this resistance involves the pharmacokinetic and pharmacodynamic profile of bortezomib, with impaired distribution to solid tumours and increased availability in the blood and/or bone marrow. Some authors have suggested that the administration of bortezomib doses higher than those approved for the treatment of myeloma (1.3 mg/m2 per dose) might help to overcome this problem. This theory, however, is not widely accepted, and the use of doses of bortezomib at >1.3 mg/m2 might be limited by the toxic effects of this agent86. Nevertheless, the use of novel proteasome inhibitors, such as carfilzomib or ixazomib, might enable higher and better-tolerated treatment doses to be administered to patients with solid tumours (NCT00531284, NCT01949545)87,88.
Plasma-cell sensitivity
Cells depend on the correct functioning of the UPP for survival, and must balance proteasome load and capacity to maintain homeostasis (Figure 1a). Plasma cells, which produce high quantities of immunoglobulins, are among the most sensitive cell types to the deregulation of protein-degradation systems. For example, bortezomib can be used to prevent acute antibody-mediated rejection of solid organ transplants by abrogating the function of nonmalignant plasma cells89. The use of bortezomib is also under investigation for the treatment of antibody-mediated autoimmune diseases, and in experimental model systems, including systemic lupus erythematosus74. Interestingly, in a preclinical model of the latter disease, both short-lived and long-lived nonmalignant plasma cells were sensitive to proteasome inhibition90–93. In these models, plasma-cell death occurs, at least partially, through the activation of programmed cell death when the unfolded protein response (UPR), which normally helps to protect plasma cells from apoptosis, is overwhelmed by a large load of ubiquitin–protein conjugates88–92. The important role of UPR balance is also supported by prior observations of decreased proteasomal capacity93 — despite increased immunoglobulin secretion — which leads to apoptosis triggered by an unfavourable proteasome load–capacity ratio94. The UPR is a cellular stress response that is activated as a result of the accumulation of unfolded or misfolded proteins in the lumen of the endoplasmic reticulum. The main function of the UPR, initially, is to restore cellular homeostasis, which is accomplished by reducing protein synthesis and increasing the production of molecular chaperones to assist protein folding. Temporarily, the UPR enables cell survival in an adverse environment by balancing proteasome load and capacity. If proteotoxic stress persists, however, through mechanisms such as prolonged proteasome inhibition — which reduces proteasome capacity while increasing proteasome load (Figure 1b) — the main role of the UPR is then to enable the activation of cell-cycle arrest and apoptotic processes95,96.
Figure 1.

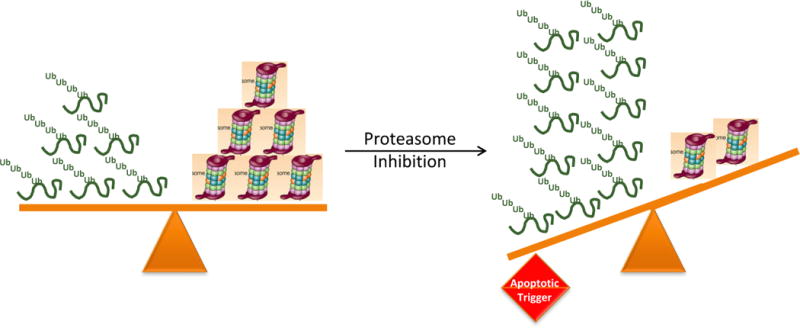
Balance between proteasomal load and capacity. a. Under normal conditions, cells maintain a balance between proteasome load and capacity via modulation of factors such as the protein-synthesis rate, chaperone capacity, deubiquitination activity, and the synthesis and assembly of proteasome subunits. b. Multiple factors can enhance or mitigate the load and capacity of the proteasome, thereby disturbing this balance. If compensatory mechanisms cannot be activated, this disturbance leads to proteotoxic stress and to the activation of apoptotic pathways.
Malignant plasma cells are thought to be even more exquisitely sensitive to proteasome inhibition than non-malignant plasma cells. This selectivity might be explained, in part, by the constitutive activation of NF-κB described in myeloma cells97. NF-κB mediates survival and resistance to chemotherapeutic agents and radiotherapy by inducing the expression of cytokines, such as IL-6, anti-apoptotic factors and adhesion molecules. Usually, NF-κB is bound to inhibitory proteins (IκBs) that tether NF-κB in the cytoplasm, thus blocking its activity as a nuclear transcription factor; when IκBs are degraded through the UPP, NF-κB is released to activate transcription of its target genes in the nucleus. Importantly, blockade of the function of IκBs with an inhibitor of IκB kinase resulted in only a 20–50% decrease in cell proliferation98. This result suggests that mechanisms independent of NF-κB function also contribute to the increased sensitivity of malignant plasma cells to proteasome inhibition. For example, malignant plasma cells are under considerably more stress to produce immunoglobulins than normal plasma cells. This increased protein production is positively correlated with proteasome sensitivity, such that an increase in the proteasomal load-versus-capacity ratio increases sensitivity to proteasome inhibition, whereas a decrease in protein synthesis renders plasma cells more resistant99 (Figure 1a). Decreased levels of proteasome activity in myeloma cells have also been shown to result in the accumulation of polyubiquitinated proteins at the expense of a free ubiquitin pool, a mechanism that results in induction of apoptosis100. Interestingly, the absolute number of immunoproteasomes in multiple-myeloma cell lines has also been positively correlated with increased resistance to treatment with bortezomib99, presumably because such proteasomes add to proteasome capacity. Myeloma cells might also be more sensitive to proteasome inhibition through the accumulation of defective ribosomal products, which trigger the activation of pro-apoptotic factors, such as C/EBP homologous protein (CHOP)-10, which is induced in response to stress and leads to UPR activation101. Myeloma cells have been reported to constitutively express the endoplasmic reticulum chaperones glucose-regulated protein (GRP)-78 and GRP-94, the functions of which are to allow for increased immunoglobulin production and assembly as a way of protecting from endoplasmic reticulum stress and activation of pro-apoptotic factors, such as CHOP-10102,103.
Resistance to proteasome inhibitors
Several preclinical studies have been published that examined mechanisms of resistance to proteasome inhibitors, most of which have focused on bortezomib. In initial studies, investigators reported that cell lines with resistance to bortezomib harboured mutations in the highly conserved binding pocket targeted by such drugs within the proteasome subunit β5104,105. The authors of these studies hypothesized that these substitutions affect the reversible binding of bortezomib, and suggested that screening for mutations in the gene that encodes this protein, PSMB5, should be considered106. When bortezomib-resistant patient samples were analyzed, however, no PSMB5 mutations were identified107–109. This finding is consistent with early studies showing that mutations in genes encoding proteasome subunits tend to result in a lethal phenotype in yeast110. The overexpression of proteasome subunit β5 and other subunits, such as β2 and β1, has also been detected in cell lines derived from Burkitt lymphoma, myeloid leukaemia, plasmacytoid lymphoma, and myeloma, and has been reported to act as possible mechanisms of resistance to bortezomib111–113. Studies in myeloma cell lines, however, suggest that the induction of these proteins is modest and might contribute only minimally to resistance111,112. Moreover, free β5 subunits are catalytically inactive by themselves and cannot typically bind inhibitors unless they are assembled into functional proteasomes6.
Transcriptional blockade of proteasome-mediated protein degradation113 leads to induction of heat shock proteins (HSPs) and related chaperones114, which function to maintain protein folding and, therefore, might contribute to drug resistance115. Indeed, the results of many preclinical studies indicate that inhibition of various HSPs, especially HSP90, can enhance the efficacy of proteasome inhibitors116. Results from early phase trials combining HSP90 inhibitors with proteasome inhibitors have led to the identification of safe dose ranges for both drugs; however, randomized studies evaluating the possible superiority of these combinations over other standards have not been reported to date. Proteasome inhibition can also force cells to rely on other pathways for either sequestration or proteolysis of toxic proteins; one such pathway might be, as mentioned earlier, the aggresome–autophagy pathway. Supporting evidence for this pathway as a contributor to drug resistance was provided by a phase II study of panobinostat, an inhibitor of aggresome formation, in combination with bortezomib and dexamethasone in patients with multiple myeloma who had relapsed and/or refractory disease after bortezomib treatment117. Among 55 patients, 34.5% achieved a partial response or better, and another 18% had a reduction in disease burden of ~25–50%. Of note, many novel agents, such as daratumumab118, have been shown to be effective in a proportion of patients with multiple myeloma harbouring resistance to proteasome inhibitors, but whether these data are sufficient to validate molecular mechanisms of resistance remains unclear. This lack of validation is partly caused by clinical resistance not being uniformly defined: patients who are resistant to an agent or regimen at one particular time point might recover sensitivity to the same agent(s) at a later point, given the ‘clonal tides’ hypothesis of myeloma biology119. Moreover, the ideal trial design to prove the ability of one agent to resensitize disease to another agent, for example, bortezomib, would involve enrolment only of patients with progressive myeloma after receiving a bortezomib-containing regimen as their most recent line of therapy. These patients would then be randomly assigned to receive the novel agent or regimen either without or with bortezomib, with the aim of determining whether the cohort receiving the bortezomib-containing combination would have a higher response rate and/or DOR; such studies have not been performed.
Activation of a number of alternative mechanisms has been implicated in the proteasome-inhibitor-resistant phenotype. One preclinical study using bortezomib-resistant myeloma cell lines addressed the induction of the IGF-1/IGF-1R pathway. In these cell lines, blockade of IGF-1 downstream effectors re-sensitized cell lines to bortezomib. Furthermore, in the presence of bortezomib, treatment with the IGF-1R inhibitor OSI-906 induced higher rates of myeloma cell death than bortezomib alone, both in vitro and in vivo113. A downstream target of IGF-1/IGF-1R is AKT (protein kinase B), which has been shown to be activated by proteasome inhibitors in preclinical studies in models of myeloma120. These data are relevant in light of early phase clinical trial data published in 2015 suggesting that the inhibition of AKT might overcome resistance to bortezomib in clinical settings121.
In another interesting line of investigation, Stessman et al.122 used a mouse myeloma model to identify gene-expression signatures associated with resistance or sensitivity to bortezomib. Resistance-related gene signatures were enriched for expression of nuclear factor (erythroid-derived 2)-like 2 (NFE2L2), which is activated as part of an antioxidant-response pathway122. Interestingly, high baseline expression of antioxidant-related pathway genes has been associated with resistance to treatment with bortezomib in patients with leukaemic MCL123. Thus, cancer cells that have an elevated antioxidant capacity before treatment might be resistant to bortezomib. The downstream NFE2L2 gene target POMP encodes the proteasome maturation protein proteassemblin, a chaperone responsible for the assembly of active proteasome particles from inactive precursor subunits. In 2015, Li and collaborators108 found that POMP is a mediator of the bortezomib-resistant phenotype, providing a potential novel target for chemosensitization124.
In 2013, using RNA-interference screens, Leung-Hagesteijn and colleagues125 identified that the expression of serine/threonine-protein kinase/endoribonuclease (IRE1) and its downstream transcription factor, effector X-box-binding protein 1 (XBP-1), are required for bortezomib sensitivity. XBP-1 is also a transcription factor involved in plasma-cell differentiation and immunoglobulin production126; these researchers found that, prior to treatment, the myeloma tumour contained subpopulations with different expression levels of IRE1 and XBP-1, which were associated with different sensitivities to bortezomib. Cells with low IRE1 and XBP-1 levels were less-differentiated plasma cells with lower levels of immunoglobulin synthesis, lower proteasome load and lower endoplasmic reticulum stress compared with other subpopulations, and with an inherent resistance to bortezomib (Table 6). In another study111, MCL-derived cells resistant to bortezomib were also found to have plasmacytic differentiation with decreased immunoglobulin production127. These findings suggest that some myeloma cell subpopulations are inherently resistant to proteasome inhibitors, and could lead to identification of biomarkers that might predict sensitivity to bortezomib. These findings, however, would also suggest that all patients harbouring resistance to proteasome inhibitors should have non-secretory myeloma, but only a small minority of patients have this disease phenotype.
Table 6.
Mechanisms of acquired resistance to proteasome inhibition
| Key components of resistance | Resistance mechanism | |
|---|---|---|
| Mutations | Mutations in PSMB5 (gene encoding proteasome subunit β5): G322A, C323T, or both C322A/C326T A49T substitution in β5 binding pocket |
Reduction in the affinity of proteasome inhibitors for the catalytically active N-terminal threonine in proteasome subunits |
| Overexpression of ubiquitin-proteasome pathway components | β5 and other proteasome subunits | Increase in number of binding sites for proteasome inhibitors, reducing their ability to suppress proteolysis |
| Activation of the aggresome–autophagy pathway | Histone deacetylase 6 | Sequestration of toxic proteins in aggresomes, and activation of autophagy to promote cell survival |
| Heat shock protein (HSP) induction | HSP70, HSP90 and other HSP family members | Enhanced protein chaperone capabilities, thereby maintaining protein homeostasis and reducing proteotoxic stress; increased threshold for apoptosis |
| Growth factor induction | Overexpression of IGF‐1/IGF-1R pathway components | Activation of antiapoptotic signalling through AKT |
| Antioxidant response pathway induction | Overexpression of NFE2L2 (gene encoding nuclear factor erythroid 2-related factor 2) | Promotes proteasome assembly through induction of proteasome maturation protein (POMP) |
| Plasma cell differentiation | Reduced expression of IRE1 and XBP-1 | Decreased immunoglobulin synthesis and proteotoxic stress, thereby reducing proapoptotic activity of proteasome inhibitors |
| EGFR/JAK/STAT signaling | Expression levels of tight junction protein 1 (ZO-1), which suppresses EGFR signaling | Induction of signalling linked with increased expression of proteasome subunits |
Finally, the authors of a study published in 2016127 reported that the expression levels of tight junction protein 1 (ZO-1) are strongly and directly associated with high sensitivity to bortezomib in both myeloma and MCL cell line models and primary myeloma cells128. Interestingly, the downstream mechanism implicated in sensitivity was activation of EGFR signaling, which enhanced the levels of proteasome subunit synthesis in a signal transducer and activator of transcription 3 (STAT3)-dependent manner127. The use of therapies targeting EGFR, such as erlotinib, and/or its downstream signaling intermediates could, therefore, be a promising approach that might overcome the activation of this pathway in patients with suppression of ZO‐1 and EGFR activation. These data also further underscore the importance of the ratio of proteasome load to capacity (Figure 1) as a determinant of drug sensitivity, and suggest that the effect of other resistance mechanisms in the load–capacity balance might be discovered in the future. Continued research to investigate other — as yet undescribed — mechanisms of resistance and possible strategies to target them will be necessary to maximize the role of proteasome inhibitors as successful therapies for multiple myeloma.
Conclusions
The UPP is a major mechanism of protein turnover in eukaryotic cells, and deregulation of this pathway contributes to the pathobiology of a myriad of malignancies. Therapeutic targeting of the UPP through the use of inhibitors of the 20S proteasome core proteolytic activities has constituted an important advance in the treatment of patients with haematological malignancies, such as MCL and especially multiple myeloma. Indeed, the ability of many patients to achieve complete remissions, long PFS and overall survival durations, and even reach a status of no detectable minimal residual disease after being treated with these agents is a clear triumph of translational medicine. Moreover, the availability of novel, chemically distinct drugs with an irreversible mode of action and/or an oral formulation, indicates that the benefits of proteasome inhibitors could be further extended in the treatment of myeloma. Challenges remain, however, as both primary (or innate) and secondary (or acquired) resistance mechanisms increasingly compromise the effectiveness of proteasome-inhibitor therapy. The identity of these mechanisms remained elusive for some time, but studies carried out in the past 10 years have begun to unravel many of the pathways involved, suggesting the feasibility of a biomarker-driven approach that will enable the identification of patients most likely to benefit from proteasome inhibitor-based therapies. Future approaches also need to examine the effectiveness of novel combination regimens as strategies to achieve chemosensitization and overcome resistance. The optimization of combination regimens might help to broaden the applicability of this class of drugs to other haematological malignancies and possibly also to solid tumours, for which the efficacy of proteasome inhibitors has so far been unimpressive, despite a strong preclinical rationale. A greater understanding of the mechanisms by which toxicities manifest (in particular, peripheral neuropathy and cardiotoxicity) could help to improve the safety profile of these agents — for example, via the development of specific and active immunoproteasome inhibitors129.
Beyond the proteasome, the UPP has multiple other potential targets that are suitable for pharmacological intervention. For example, new compounds targeting deubiquitinases and other upstream regulatory components of the protein-turnover machinery might show anti-tumour activity alone, and/or synergistic cytotoxic efficacy in combinations with proteasome inhibitors. In this regard, the neural precursor cell expressed developmentally down-regulated 8 (NEDD8)-activating enzyme inhibitor pevonedistat130,131, and the E3 ubiquitin-protein ligase MDM2 inhibitor RG7112132 have demonstrated clinical activity in patients with acute myeloid leukaemia. Also, the anti-tumour activity of proteolysis-targeting chimeric molecules (PROTACs)133,134 that induce proteasome-mediated degradation of specific protein targets is being evaluated preclinically, and hopefully these agents will soon be incorporated into clinical trials. The general mode of action of PROTACs is based, in part, on the use of a phthalimide moiety conjugated with other protein-binding moieties to bring specific targets protein into close proximity with the E3 ligase Cereblon, resulting in target polyubiquitination and subsequent proteasomal degradation135. Of note, this mechanism of action builds on that hypothesized to at least partially explain the activity of IMiDs135,136, such as thalidomide, lenalidomide and pomalidomide, one of the major classes of agents used to treat patients with multiple myeloma and other haematological malignancies, including B-cell lymphomas and myelodysplastic syndrome. Molecular studies have revealed that these IMiDs are able to bind Cereblon136,137, resulting in polyubiquitination of the DNA-binding proteins Ikaros and Aiolos138,139, demonstrating that these drugs also affect the UPP. Thus, proteasome inhibition is just one of several approaches that can already be leverage to alter UPP function in anti-tumour therapeutic approaches; our ability to target the UPP will likely continue expanding in years to come.
Key Points.
The proteasome is a central component of the protein degradation machinery in eukaryotic cells
Both transformed and normal cells depend on proteasome function to control the expression of proteins linked to cell survival and proliferation
Clinical trials using proteasome inhibitors in myeloma, lymphoma and amyloidosis have transformed the treatment of these diseases by establishing new standards of care
Three proteasome inhibitors have received regulatory approval and are routinely used in clinical settings, including bortezomib, carfilzomib and ixazomib, which have transformed the care of patients with myeloma and MCL
Primary proteasome inhibitor resistance remains a challenge in patients with solid tumors, and acquired resistance can develop even after initial responses in myeloma and MCL, the mechanisms of which are beginning to be understood
Clinical evaluation of compounds targeting the upstream regulatory components of the proteasome is underway; in the future, compounds that target proteasome-mediated degradation of specific proteins might also become available
Acknowledgments
The work of the authors is supported by the MD Anderson Cancer Center SPORE in Multiple Myeloma (P50 CA142509) and the MD Anderson Cancer Center Support Grant (P30 CA016672). R.Z.O., the Florence Maude Thomas Cancer Research Professor, would also like to acknowledge support from the National Cancer Institute (U10 CA032102, R01 CA184464 and CA194264), and thank the Brock Family Myeloma Research Fund, the Diane & John Grace Family Foundation, the Jay Solomon Myeloma Research Fund, and the Yates Ortiz Myeloma Fund.
Biographies
Elisabet E. Manasanch is an Assistant Professor at the Department of Lymphoma/Myeloma at the University of Texas MD Anderson Cancer Center (Houston, Texas, USA), where her work is focused on the evaluation of myeloma precursor disease. In particular, her research is focused on biomarker discovery for early myeloma, novel prediction models for progression to multiple myeloma, treatment of smouldering myeloma and multiple myeloma. She graduated with an MD degree from the University of Barcelona (Spain) in 2005 and received a MHSc in Clinical Research from Duke University (Durham, North Carolina, USA) in 2014. She pursued internal medicine and medical oncology training at the University of Massachusetts (Boston, Massachusetts, USA) and National Institutes of Health (Bethesda, Maryland, USA), respectively, and is board-certified in both specialties.
Robert Z. Orlowski, PhD, MD, is the Florence Maude Thomas Cancer Research Professor in the Departments of Lymphoma/Myeloma and Experimental Therapeutics at the University of Texas MD Anderson Cancer Center. He currently serves as the Chair Ad Interim of the Department of Lymphoma/Myeloma, and Director of the Section, Multiple Myeloma. Dr Orlowski has published more than 180 peer-reviewed articles and has been awarded multiple research grants. He was named the Leukemia and Lymphoma Society Man of the Year in 2006, was the recipient of Emil Frei III Award for Excellence in Translational Research in 2010 and Waun Ki Hong MD, Division of Cancer Medicine 2015 Mentor of the Year Award. Dr Orlowski is the lead principal investigator of the MD Anderson Cancer Center SPORE in Multiple Myeloma, and the leader of the MD Anderson Cancer Center Moon Shot Program in High-Risk Multiple Myeloma, and the Director of the Hematological Malignancies Program of the NIH Cancer Center Support Grant (CCSG) at the same institution. Dr Orlowski is also a Chair of the SWOG Barlogie/Salmon Myeloma Committee, one of the cooperative groups that are a part of the US National Clinical Trials Network. He is a member of the American Society for Biochemistry and Molecular Biology/FASEB. He serves on National Cancer Institute Myeloma Steering Committee, the American Cancer Society Cancer Drug Discovery Peer Review Committee, and the Editorial Board of Clinical Cancer Research, among others.
Footnotes
Competing interests statement
R.Z.O. has served on advisory boards for Amgen, which developed and markets carfilzomib, and for Takeda Pharmaceuticals, which developed and markets bortezomib and ixazomib, and has received research support from these firms for clinical and laboratory projects. E.E.M. declares no competing interests.
Subject ontology terms
Biological sciences/Molecular biology/Proteolysis/Proteasome
[URI/631/337/474/2085]
Health sciences/Oncology/Cancer/Haematological cancer
[URI/692/4028/67/1990]
Biological sciences/Cell biology/Cell death/Apoptosis
[URI/631/80/82/23]
Biological sciences/Cancer/Cancer therapy
[URI/631/67/1059]
Health sciences/Health care/Therapeutics/Drug therapy/Combination drug therapy
[URI/692/700/565/1436/1437]
By preventing the accumulation of misfolded or damaged proteins, the ubiquitin-proteasome pathway has essential functions in cell homeostasis. Cancer cells produce proteins that promote cell survival and proliferation, and inhibit mechanisms of cell death, and thus, clinical trials have tested the therapeutic effect of proteasome inhibitors on patients with a variety of cancer types, mainly haematological malignancies. Herein, Manasanch and Orlowski discuss the advances and challenges derived from the introduction of proteasome inhibitors in the clinic, including therapeutic resistance.
References
- 1.Orlowski M, Michaud C. Pituitary multicatalytic proteinase complex. Specificity of components and aspects of proteolytic activity. Biochemistry. 1989;28(24):9270–8. doi: 10.1021/bi00450a006. [DOI] [PubMed] [Google Scholar]
- 2.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 4.Manasanch EE, Korde N, Zingone A, Tageja N, Fernandez de Larrea C, Bhutani M, et al. The proteasome: mechanisms of biology and markers of activity and response to treatment in multiple myeloma. Leukemia & lymphoma. 2014;55(8):1707–14. doi: 10.3109/10428194.2013.828351. [DOI] [PubMed] [Google Scholar]
- 5.Murata S, Takahama Y, Tanaka K. Thymoproteasome: probable role in generating positively selecting peptides. Current opinion in immunology. 2008;20(2):192–6. doi: 10.1016/j.coi.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. The Journal of cell biology. 1999;146(6):1239–54. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orlowski RZ, Dees EC. The role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancer. Breast Cancer Res. 2003;5(1):1–7. doi: 10.1186/bcr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orlowski RZ, Baldwin AS., Jr NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 9.Lu Z, Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. 2010;9:2342–2352. doi: 10.4161/cc.9.12.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love IM, Shi D, Grossman SR. p53 ubiquitination and proteasomal degradation. Methods Mol Biol. 2013;962:63–73. doi: 10.1007/978-1-62703-236-0_5. [DOI] [PubMed] [Google Scholar]
- 11.Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys. 2000;383(1):1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- 12.Robak T, Huang H, Jin J, Zhu J, Liu T, Samoilova O, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. The New England journal of medicine. 2015;372(10):944–53. doi: 10.1056/NEJMoa1412096. [DOI] [PubMed] [Google Scholar]
- 13.Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2002;20(22):4420–7. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 14.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin DH, et al. Extended follow-up of a phase II trial in relapsed, refractory multiple myeloma:: final time-to-event results from the SUMMIT trial. Cancer. 2006;106(6):1316–9. doi: 10.1002/cncr.21740. [DOI] [PubMed] [Google Scholar]
- 15.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. The New England journal of medicine. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 16.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. The New England journal of medicine. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 17.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. The New England journal of medicine. 2008;359(9):906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 18.Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116(5):679–86. doi: 10.1182/blood-2010-02-268862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durie B, Hoering A, Rajkumar SV, Muneer A, Epstein J, Kahanic S, et al. Bortezomib, Lenalidomide and Dexamethasone Vs. Lenalidomide and Dexamethasone in Patients (Pts) with Previously Untreated Multiple Myeloma without an Intent for Immediate Autologous Stem Cell Transplant (ASCT): Results of the Randomized Phase III Trial SWOG S0777. Blood. 2015;126(23) doi: 10.1038/s41408-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sonneveld P, Goldschmidt H, Rosinol L, Blade J, Lahuerta JJ, Cavo M, et al. Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: a meta-analysis of phase III randomized, controlled trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(26):3279–87. doi: 10.1200/JCO.2012.48.4626. [DOI] [PubMed] [Google Scholar]
- 21.Sonneveld P, Schmidt-Wolf IG, van der Holt B, El Jarari L, Bertsch U, Salwender H, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/GMMG-HD4 trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(24):2946–55. doi: 10.1200/JCO.2011.39.6820. [DOI] [PubMed] [Google Scholar]
- 22.Orlowski RZ, Nagler A, Sonneveld P, Blade J, Hajek R, Spencer A, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(25):3892–901. doi: 10.1200/JCO.2006.10.5460. [DOI] [PubMed] [Google Scholar]
- 23.San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. The Lancet Oncology. 2014;15(11):1195–206. doi: 10.1016/S1470-2045(14)70440-1. [DOI] [PubMed] [Google Scholar]
- 24.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(24):8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah J, Feng L, Manasanch E, Weber D, Thomas S, Turturro F, et al. Phase I/II Trial of the Efficacy and Safety of Combination Therapy with Lenalidomide/Bortezomib/Dexamethasone (RVD) and Panobinostat in Transplant-Eligible Patients with Newly Diagnosed Multiple Myeloma. Blood. 2015;126(23) [Google Scholar]
- 26.Petrucci MT, Giraldo P, Corradini P, Teixeira A, Dimopoulos MA, Blau IW, et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. British journal of haematology. 2013;160(5):649–59. doi: 10.1111/bjh.12198. [DOI] [PubMed] [Google Scholar]
- 27.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. The New England journal of medicine. 2016;375(8):754–66. doi: 10.1056/NEJMoa1606038. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Ai L, Cui G, Gowrea B, Li M, Hu Y. Once- versus twice-weekly Bortezomib induction therapy with dexamethasone in newly diagnosed multiple myeloma. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2012;32(4):495–500. doi: 10.1007/s11596-012-0086-7. [DOI] [PubMed] [Google Scholar]
- 29.Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. The lancet oncology. 2011;12(5):431–40. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 30.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(9):2734–43. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 31.Lonial S, Waller EK, Richardson PG, Jagannath S, Orlowski RZ, Giver CR, et al. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood. 2005;106(12):3777–84. doi: 10.1182/blood-2005-03-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chanan-Khan A, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau JL, et al. Analysis of herpes zoster events among bortezomib-treated patients in the phase III APEX study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(29):4784–90. doi: 10.1200/JCO.2007.14.9641. [DOI] [PubMed] [Google Scholar]
- 33.O’Connor OA, Stewart AK, Vallone M, Molineaux CJ, Kunkel LA, Gerecitano JF, et al. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(22):7085–91. doi: 10.1158/1078-0432.CCR-09-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alsina M, Trudel S, Furman RR, Rosen PJ, O’Connor OA, Comenzo RL, et al. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(17):4830–40. doi: 10.1158/1078-0432.CCR-11-3007. [DOI] [PubMed] [Google Scholar]
- 35.Durie BG, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–73. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 36.Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–25. doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludwig T, et al. Carfilzomib vs low-dose corticosteroids and optional cyclophosphamide in patients with relapsed and refractory multiple myeloma (RRMM): results from a phase 3 study (focus) Annals of Oncology. 2014;25:v1–v41. [Google Scholar]
- 38.Hajek R, Bryce R, Ro S, Klencke B, Ludwig H. Design and rationale of FOCUS (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM) BMC cancer. 2012;12:415. doi: 10.1186/1471-2407-12-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. The New England journal of medicine. 2015;372(2):142–52. doi: 10.1056/NEJMoa1411321. [DOI] [PubMed] [Google Scholar]
- 40.Lendvai, et al. A phase 2 single-center study of carfilzomib 56 mg/m2 with or without dexamethasone in relapsed multiple myeloma. Blood. 2014;124:899–906. doi: 10.1182/blood-2014-02-556308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lendvai N, Hilden P, Devlin S, Landau H, Hassoun H, Lesokhin AM, et al. A phase 2 single-center study of carfilzomib 56 mg/m2 with or without low-dose dexamethasone in relapsed multiple myeloma. Blood. 2014;124(6):899–906. doi: 10.1182/blood-2014-02-556308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hajek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. The Lancet Oncology. 2016;17(1):27–38. doi: 10.1016/S1470-2045(15)00464-7. [DOI] [PubMed] [Google Scholar]
- 43.Berenson JR, Cartmell A, Bessudo A, Lyons RM, Harb WA, Tzachanis D, et al. Updated results from CHAMPION-1, a phase I/II study investigating weekly carfilzomib with dexamethasone for patients (Pts) with relapsed or refractory multiple myeloma (RRMM) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(Suppl) abstr 8527. [Google Scholar]
- 44.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT02412878?term=NCT02412878&rank=1.
- 45.Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S, et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012;120(9):1801–9. doi: 10.1182/blood-2012-04-422683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA oncology. 2015;1(6):746–54. doi: 10.1001/jamaoncol.2015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benboubker, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. NEJM. 2014;371:906–917. doi: 10.1056/NEJMoa1402551. [DOI] [PubMed] [Google Scholar]
- 48.Dytfeld D, Jasielec J, Griffith KA, Lebovic D, Vesole DH, Jagannath S, et al. Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99(9):e162–4. doi: 10.3324/haematol.2014.110395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01863550?term=NCT01863550&rank=1.
- 50.Moreau P, Kolb B, Attal M, Caillot D, Benboubker L, Tiab M, et al. Phase 1/2 study of carfilzomib plus melphalan and prednisone in patients aged over 65 years with newly diagnosed multiple myeloma. Blood. 2015;125(20):3100–4. doi: 10.1182/blood-2015-02-626168. [DOI] [PubMed] [Google Scholar]
- 51.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01818752?term=NCT01818752&rank=1.
- 52.Chari A, Hajje D. Case series discussion of cardiac and vascular events following carfilzomib treatment: possible mechanism, screening, and monitoring. BMC cancer. 2014;14:915. doi: 10.1186/1471-2407-14-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegel D, Martin T, Nooka A, Harvey RD, Vij R, Niesvizky R, et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica. 2013;98(11):1753–61. doi: 10.3324/haematol.2013.089334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar SK, Bensinger WI, Zimmerman TM, Reeder CB, Berenson JR, Berg D, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–55. doi: 10.1182/blood-2014-01-548941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar SK, Berdeja JG, Niesvizky R, Lonial S, Laubach JP, Hamadani M, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. The Lancet Oncology. 2014;15(13):1503–12. doi: 10.1016/S1470-2045(14)71125-8. [DOI] [PubMed] [Google Scholar]
- 56.Richardson PG, Baz R, Wang M, Jakubowiak AJ, Laubach JP, Harvey RD, et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124(7):1038–46. doi: 10.1182/blood-2014-01-548826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moreau P, Masszi T, Grzasko N, Bahlis N, Hansson M, Pour L, et al. Ixazomib, an Investigational Oral Proteasome Inhibitor (PI), in Combination with Lenalidomide and Dexamethasone (IRd), Significantly Extends Progression-Free Survival (PFS) for Patients (Pts) with Relapsed and/or Refractory Multiple Myeloma (RRMM): The Phase 3 Tourmaline-MM1 Study. Blood. 2015;126(23) [Google Scholar]
- 58.Shah J, Niesvizky R, Stadtmauer E, Rifkin R, Berenson JR, Berdeja J, et al. Oprozomib, Pomalidomide, and Dexamethasone (OPomd) in Patients (Pts) with Relapsed and/or Refractory Multiple Myeloma (RRMM): Initial Results of a Phase 1b Study. Blood. 2015;126(23) [Google Scholar]
- 59.Vij R, Savona M, Siegel D, Kaufman JL, Badros A, Ghobrial I, et al. Clinical Profile of Single-Agent Oprozomib in Patients (Pts) with Multiple Myeloma (MM): Updated Results from a Multicenter, Open-Label, Dose Escalation Phase 1b/2 Study. Blood. 2014 Abstract. ASH. [Google Scholar]
- 60.Parameswaran H, Shain K, Voorhees P, Gabrail NY, Muneer A, Zonder J, et al. Oprozomib and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma: Initial Results from the Dose Escalation Portion of a Phase 1b/2, Multicenter, Open-Label Study. Blood. 2014 Abstract. ASH. [Google Scholar]
- 61.Potts B, Albitar M, Anderson K, Baritaki S, Berkers C, Bonavida B, et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets. 2011;11(3):254–84. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investigational new drugs. 2012;30(6):2303–17. doi: 10.1007/s10637-011-9766-6. [DOI] [PubMed] [Google Scholar]
- 63.Goy A, Younes A, McLaughlin P, Pro B, Romaguera JE, Hagemeister F, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin's lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(4):667–75. doi: 10.1200/JCO.2005.03.108. [DOI] [PubMed] [Google Scholar]
- 64.O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin's lymphoma and mantle cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(4):676–84. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 65.Strauss SJ, Maharaj L, Hoare S, Johnson PW, Radford JA, Vinnecombe S, et al. Bortezomib therapy in patients with relapsed or refractory lymphoma: potential correlation of in vitro sensitivity and tumor necrosis factor alpha response with clinical activity. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(13):2105–12. doi: 10.1200/JCO.2005.04.6789. [DOI] [PubMed] [Google Scholar]
- 66.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(30):4867–74. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 67.Belch A, Kouroukis CT, Crump M, Sehn L, Gascoyne RD, Klasa R, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol. 2007;18(1):116–21. doi: 10.1093/annonc/mdl316. [DOI] [PubMed] [Google Scholar]
- 68.Gerecitano J, Portlock C, Moskowitz C, Hamlin P, Straus D, Zelenetz AD, et al. Phase 2 study of weekly bortezomib in mantle cell and follicular lymphoma. British journal of haematology. 2009;146(6):652–5. doi: 10.1111/j.1365-2141.2009.07775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Till BG, Li H, Bernstein SH, Fisher RI, Burack WR, Rimsza LM, et al. Phase II trial of R-CHOP plus bortezomib induction therapy followed by bortezomib maintenance for newly diagnosed mantle cell lymphoma: SWOG S0601. British journal of haematology. 2016;172(2):208–18. doi: 10.1111/bjh.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lenz, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG) JCO. 2005;23:1984–92. doi: 10.1200/JCO.2005.08.133. [DOI] [PubMed] [Google Scholar]
- 71.Dimopoulos MA, Garcia-Sanz R, Gavriatopoulou M, Morel P, Kyrtsonis MC, Michalis E, et al. Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN) Blood. 2013;122(19):3276–82. doi: 10.1182/blood-2013-05-503862. [DOI] [PubMed] [Google Scholar]
- 72.Ghobrial IM, Xie W, Padmanabhan S, Badros A, Rourke M, Leduc R, et al. Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenstrom Macroglobulinemia. American journal of hematology. 2010;85(9):670–4. doi: 10.1002/ajh.21788. [DOI] [PubMed] [Google Scholar]
- 73.Treon SP, Ioakimidis L, Soumerai JD, Patterson CJ, Sheehy P, Nelson M, et al. Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(23):3830–5. doi: 10.1200/JCO.2008.20.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.US National Library of Medicine. ClinicalTrials.gov. 2015 https://www.clinicaltrials.gov/ct2/show/NCT01788020?term=NCT01788020&rank=1.
- 75.Treon SP, Tripsas CK, Meid K, Kanan S, Sheehy P, Chuma S, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenstrom's macroglobulinemia. Blood. 2014;124(4):503–10. doi: 10.1182/blood-2014-03-566273. [DOI] [PubMed] [Google Scholar]
- 76.Sanchorawala V, Brauneis D, Shelton AC, Lo S, Sun F, Sloan JM, et al. Induction therapy with bortezomib followed by bortezomib-high dose melphalan and stem cell transplantation for AL amyloidosis: Results of a prospective clinical trial. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2015 doi: 10.1016/j.bbmt.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 77.Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. 2012;119(19):4391–4. doi: 10.1182/blood-2011-11-390930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612–5. doi: 10.1182/blood-2015-01-620302. [DOI] [PubMed] [Google Scholar]
- 79.Kastritis E, Leleu X, Arnulf B, Zamagni E, Mollee P, Cibeira MT, et al. A Randomized Phase III Trial of Melphalan and Dexamethasone (MDex) Versus Bortezomib, Melphalan and Dexamethasone (BMDex) for Untreated Patients with AL Amyloidosis Blood. 2014 2014: Abstract Presented ASH. [Google Scholar]
- 80.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01659658?term=NCT01659658&rank=1.
- 81.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(7):923–9. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Attar EC, Amrein PC, Fraser JW, Fathi AT, McAfee S, Wadleigh M, et al. Phase I dose escalation study of bortezomib in combination with lenalidomide in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) Leukemia research. 2013;37(9):1016–20. doi: 10.1016/j.leukres.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119(25):6025–31. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Messinger YH, Gaynon PS, Sposto R, van der Giessen J, Eckroth E, Malvar J, et al. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood. 2012;120(2):285–90. doi: 10.1182/blood-2012-04-418640. [DOI] [PubMed] [Google Scholar]
- 85.Huang Z, Wu Y, Zhou X, Xu J, Zhu W, Shu Y, et al. Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future oncology. 2014;10(10):1795–807. doi: 10.2217/fon.14.30. [DOI] [PubMed] [Google Scholar]
- 86.Piperdi B, Ling YH, Liebes L, Muggia F, Perez-Soler R. Bortezomib: understanding the mechanism of action. Molecular cancer therapeutics. 2011;10(11):2029–30. doi: 10.1158/1535-7163.MCT-11-0745. [DOI] [PubMed] [Google Scholar]
- 87.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT00531284?term=NCT00531284&rank=1.
- 88.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01949545?term=NCT01949545&rank=1.
- 89.Woodle ES, Alloway RR, Girnita A. Proteasome inhibitor treatment of antibody-mediated allograft rejection. Current opinion in organ transplantation. 2011;16(4):434–8. doi: 10.1097/MOT.0b013e328348c0e5. [DOI] [PubMed] [Google Scholar]
- 90.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nature medicine. 2008;14(7):748–55. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 91.Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nature reviews Rheumatology. 2011;7(3):170–8. doi: 10.1038/nrrheum.2011.1. [DOI] [PubMed] [Google Scholar]
- 92.Gomez AM, Vrolix K, Martinez-Martinez P, Molenaar PC, Phernambucq M, van der Esch E, et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. Journal of immunology. 2011;186(4):2503–13. doi: 10.4049/jimmunol.1002539. [DOI] [PubMed] [Google Scholar]
- 93.Bontscho J, Schreiber A, Manz RA, Schneider W, Luft FC, Kettritz R. Myeloperoxidase-specific plasma cell depletion by bortezomib protects from anti-neutrophil cytoplasmic autoantibodies-induced glomerulonephritis. Journal of the American Society of Nephrology : JASN. 2011;22(2):336–48. doi: 10.1681/ASN.2010010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cenci S, Mezghrani A, Cascio P, Bianchi G, Cerruti F, Fra A, et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. The EMBO journal. 2006;25(5):1104–13. doi: 10.1038/sj.emboj.7601009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & development. 1998;12(7):982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(23):12625–30. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer cell. 2007;12(2):115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa B as a therapeutic target in multiple myeloma. The Journal of biological chemistry. 2002;277(19):16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 99.Cenci S, Oliva L, Cerruti F, Milan E, Bianchi G, Raule M, et al. Pivotal Advance: Protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J Leukoc Biol. 2012;92(5):921–31. doi: 10.1189/jlb.1011497. [DOI] [PubMed] [Google Scholar]
- 100.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113(13):3040–9. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 101.Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer research. 2007;67(4):1783–92. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 102.Melnick J, Dul JL, Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370(6488):373–5. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- 103.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu S, Yang J, Song X, Gong S, Zhou H, Guo L, et al. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J Pharmacol Exp Ther. 2008;326(2):423–31. doi: 10.1124/jpet.108.138131. [DOI] [PubMed] [Google Scholar]
- 105.Lu S, Yang J, Chen Z, Gong S, Zhou H, Xu X, et al. Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Experimental hematology. 2009;37(7):831–7. doi: 10.1016/j.exphem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 106.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112(6):2489–99. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 107.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lichter DI, Danaee H, Pickard MD, Tayber O, Sintchak M, Shi H, et al. Sequence analysis of beta-subunit genes of the 20S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood. 2012;120(23):4513–6. doi: 10.1182/blood-2012-05-426924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang L, Kumar S, Fridley BL, Kalari KR, Moon I, Pelleymounter LL, et al. Proteasome beta subunit pharmacogenomics: gene resequencing and functional genomics. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(11):3503–13. doi: 10.1158/1078-0432.CCR-07-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fujiwara T, Tanaka K, Orino E, Yoshimura T, Kumatori A, Tamura T, et al. Proteasomes are essential for yeast proliferation. cDNA cloning and gene disruption of two major subunits. The Journal of biological chemistry. 1990;265(27):16604–13. [PubMed] [Google Scholar]
- 111.Fuchs D, Berges C, Opelz G, Daniel V, Naujokat C. Increased expression and altered subunit composition of proteasomes induced by continuous proteasome inhibition establish apoptosis resistance and hyperproliferation of Burkitt lymphoma cells. Journal of cellular biochemistry. 2008;103(1):270–83. doi: 10.1002/jcb.21405. [DOI] [PubMed] [Google Scholar]
- 112.Ruckrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M, et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia. 2009;23(6):1098–105. doi: 10.1038/leu.2009.8. [DOI] [PubMed] [Google Scholar]
- 113.Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012;120(16):3260–70. doi: 10.1182/blood-2011-10-386789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(22):14374–9. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lianos GD, Alexiou GA, Mangano A, Mangano A, Rausei S, Boni L, et al. The role of heat shock proteins in cancer. Cancer letters. 2015;360(2):114–8. doi: 10.1016/j.canlet.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 116.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006;107(3):1092–100. doi: 10.1182/blood-2005-03-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Richardson PG, Schlossman RL, Alsina M, Weber DM, Coutre SE, Gasparetto C, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122(14):2331–7. doi: 10.1182/blood-2013-01-481325. [DOI] [PubMed] [Google Scholar]
- 118.Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016 doi: 10.1016/S0140-6736(15)01120-4. [DOI] [PubMed] [Google Scholar]
- 119.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120(5):1060–6. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, Mitsiades C, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107(10):4053–62. doi: 10.1182/blood-2005-08-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, et al. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014;124(14):2190–5. doi: 10.1182/blood-2014-03-559963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stessman HA, Baughn LB, Sarver A, Xia T, Deshpande R, Mansoor A, et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Molecular cancer therapeutics. 2013;12(6):1140–50. doi: 10.1158/1535-7163.MCT-12-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weniger MA, Rizzatti EG, Perez-Galan P, Liu D, Wang Q, Munson PJ, et al. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(15):5101–12. doi: 10.1158/1078-0432.CCR-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li B, Fu J, Chen P, Ge X, Li Y, Kuiatse I, et al. The Nuclear Factor (Erythroid-derived 2)-like 2 and Proteasome Maturation Protein Axis Mediates Bortezomib Resistance in Multiple Myeloma. The Journal of biological chemistry. 2015 doi: 10.1074/jbc.M115.664953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer cell. 2013;24(3):289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–7. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 127.Perez-Galan P, Mora-Jensen H, Weniger MA, Shaffer AL, 3rd, Rizzatti EG, Chapman CM, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. 2011;117(2):542–52. doi: 10.1182/blood-2010-02-269514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X-D, Baladandayuthapani V, Lin H, Mulligan G, Li B, Esseltine D-L, et al. Tight junction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma through EGFR/JAK1/STAT3 signaling. Cancer cell. 2016;29 doi: 10.1016/j.ccell.2016.03.026. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kimura H, Caturegli P, Takahashi M, Suzuki K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. Journal of immunology research. 2015;2015:541984. doi: 10.1155/2015/541984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(1):34–43. doi: 10.1158/1078-0432.CCR-15-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. British journal of haematology. 2015;169(4):534–43. doi: 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- 132.Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(4):868–76. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8554–9. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Molecular & cellular proteomics : MCP. 2003;2(12):1350–8. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 135.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376–81. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–50. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 137.Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2012;118(18):4771–9. doi: 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN) British journal of haematology. 2014;164(6):811–21. doi: 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Harousseau JL, Attal M, Avet-Loiseau H, Marit G, Caillot D, Mohty M, et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(30):4621–9. doi: 10.1200/JCO.2009.27.9158. [DOI] [PubMed] [Google Scholar]
- 141.Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–85. doi: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]
- 142.Rosinol L, Oriol A, Teruel AI, Hernandez D, Lopez-Jimenez J, de la Rubia J, et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood. 2012;120(8):1589–96. doi: 10.1182/blood-2012-02-408922. [DOI] [PubMed] [Google Scholar]
- 143.Kumar S, Flinn I, Richardson PG, Hari P, Callander N, Noga SJ, et al. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood. 2012;119(19):4375–82. doi: 10.1182/blood-2011-11-395749. [DOI] [PubMed] [Google Scholar]
- 144.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01530594?term=NCT01530594&rank=1.
- 145.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01850524?term=NCT01850524&rank=1.
- 146.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT01564537?term=NCT01564537&rank=1.
- 147.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT02312258?term=NCT02312258&rank=1.
- 148.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT02181413?term=NCT02181413&rank=1.
- 149.Moreau, et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016;374:1621–1634. doi: 10.1056/NEJMoa1516282. [DOI] [PubMed] [Google Scholar]
- 150.Ghobrial, et al. Clinical Profile Of Single-Agent Modified-Release Oprozomib Tablets In Patients (Pts) With Hematologic Malignancies: Updated Results From a Multicenter, Open-Label, Dose Escalation Phase 1b/2 Study. (abstract) Blood. 2013;122(21):3184. [Google Scholar]
- 151.Richardson, et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood. 2016;127:2693–2700. doi: 10.1182/blood-2015-12-686378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.US National Library of Medicine. ClinicalTrials.gov. 2016 https://www.clinicaltrials.gov/ct2/show/NCT02400437?term=NCT02400437&rank=1.
- 153.US National Library of Medicine. ClinicalTrials.gov. 2015 https://www.clinicaltrials.gov/ct2/show/NCT01813227?term=NCT01813227&rank=1.