

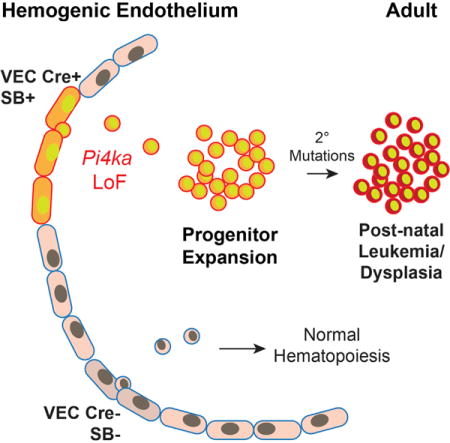
SUMMARY
Given its role as the source of definitive hematopoietic cells, we sought to determine whether mutations initiated in the hemogenic endothelium would yield hematopoietic abnormalities or malignancies. Here, we find that endothelium-specific transposon mutagenesis in mice promotes hematopoietic pathologies that are both myeloid and lymphoid in nature. Frequently mutated genes included previously recognized cancer drivers and additional candidates, such as Pi4ka, a lipid kinase whose mutation was found to promote myeloid and erythroid dysfunction. Subsequent validation experiments showed that targeted inactivation of the Pi4ka catalytic domain or reduction in mRNA expression inhibited myeloid and erythroid cell differentiation in vitro and promoted anemia in vivo through a mechanism involving deregulation of AKT, MAPK, SRC, and JAK-STAT signaling. Finally, we provide evidence linking PI4KAP2, previously considered a pseudogene, to human myeloid and erythroid leukemia.
Graphical abstract
INTRODUCTION
The hematopoietic lineage emerges during a narrow developmental window from a specialized subset of endothelial cells: the hemogenic endothelium (HemEnd) (Dzierzak and de Pater, 2016). Hematopoietic stem progenitor cells (HSPCs) enter the circulation from HemEnd sites to first seed and expand the fetal liver and later occupy the bone marrow.
The specification of the HemEnd requires Etv2 and Runx1, while HSPCs budding from the HemEnd involves Gata2, Jagged1-Notch, and Hedgehog (Clements and Traver, 2013; Eliades et al., 2016). In recent years, the field has come to appreciate the diversity, repopulation capacity, and plasticity of single hematopoietic stem cells (HSCs) at the HemEnd stage, meaning before their seeding in hematopoietic organs (Guibentif et al., 2017). For example, VE-Cadherin-expressing HemEnd gives rise to myeloid-erythroid biased HSPCs (Chen et al., 2011), while TGF-β and BMP signaling differentially activate myeloid versus lymphoid-biased HSCs (Challen et al., 2010; Crisan et al., 2015). It is well known that lineage-specific transcription factors that drive specification of blood cells (CEBPa, Ikaros, MLL, SCL, Etv6, etc.) can lead to leukemia when deregulated (Orkin and Zon, 2008). Similarly, genes involved in the regulatory process that control budding of HSCs from the HemEnd might also promote neoplastic transformation when disrupted.
Seeking to expand our current understanding of the genes that regulate hematopoiesis (and their potential transformation) starting as early as HSPC budding, we performed a forward genetic screen using Sleeping Beauty (SB) transposon mutagenesis to target the HemEnd. On insertion into the genome, transposons disrupt splicing and expression of targeted genes, causing both gain- and loss-of-function events and facilitating the discovery of oncogenes and tumor suppressors in a variety of solid and blood cancers (Moriarity and Largaespada, 2015). HemEnd-initiated SB transposon mutagenesis yielded myeloid, erythroid, and lymphoid malignancies with mutations in both well-known regulators of those lineages and candidate genes uncovered in this study. Among these candidates, we identified a previously unknown role for phosphatidyl inositol lipid kinase (Pi4ka) in erythroid and myelopoiesis. Recurrent Pi4ka mutations were previously identified in histiocytic sarcomas driven by SB mutagenesis in cells expressing a myeloid-specific Lyz-Cre transgene, but no causative mechanisms were reported (Been et al., 2014). A lipid kinase that phosphorylates phosphatidyl-inositols at the D4 position, the Pi4ka protein may be important in a broad array of biological processes, including signaling complexes, ion channel activity, lipid transfer, vesicle transport, and actin binding (Balla et al., 2009; Minogue and Waugh, 2012). Here, we validated Pi4ka’s biological significance in hematopoiesis and demonstrated its link to Akt and Erk signaling, the former classically known to regulate hematopoietic differentiation. Furthermore, we identified the human PI4KA “pseudogene,” PI4KAP2, as a dominant-negative inhibitor of the PI4KA signaling pathway.
RESULTS
HemEnd Mutagenesis Promotes Hematopoietic Malignancies
We targeted mutagenesis to the endothelium using a conditional SB transposon strategy (Dupuy et al., 2009) (Figures 1A and 1B). VE-Cadherin-Cre (VEC-Cre) recombinase (Alva et al., 2006) was used to drive expression of the transposase enzyme specifically in endothelial cells, where it could cut and paste transposons randomly into TA dinucleotides distributed throughout the genome (Riordan et al., 2014). VEC-Cre is first expressed in the HemEnd by embryonic day (E) 9.5 in a salt-and-pepper manner with progressive penetration and homogeneous expression by E12.5 (Alva et al., 2006). Due to this mosaic expression pattern in the HemEnd (transient phase lasting from E10.5–E12.5) by E10.5, some cells were targeted by mutagenesis while others were not, creating a competitive mixture of mutated and non-mutated populations.
Figure 1. Initiating Mutagenesis in the Hemogenic Endothelium Generates Hematopoietic Malignancies.
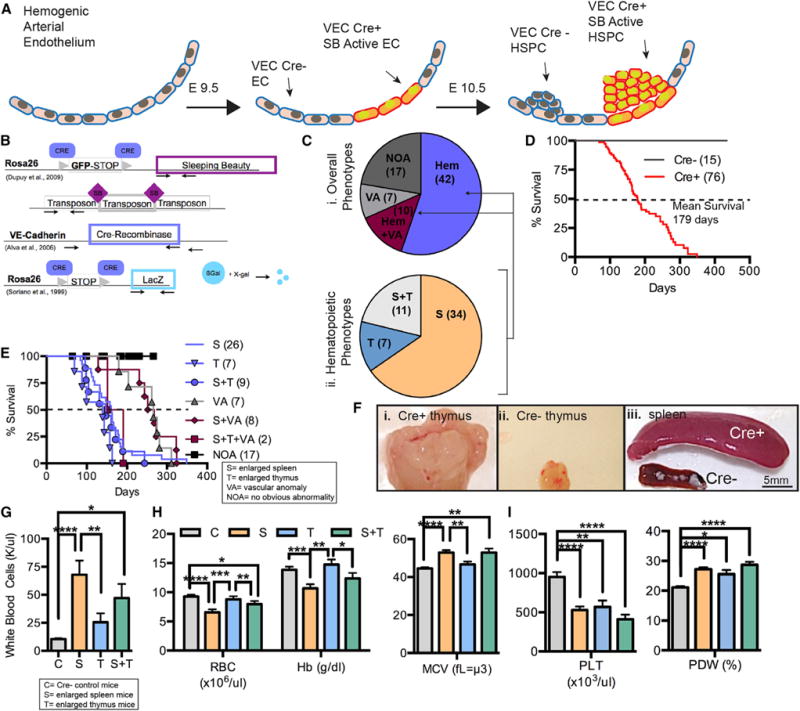
(A and B) Onset of VE-Cadherin-Cre (VEC-Cre) expression, and therefore SB Transposase, at E9.5 in the progeny of (B) SB T2/Onc2; VEC-Cre/Rosa26-LacZ mice.
(C) Frequencies of abnormalities in these mice (i). Relative occurrence of enlarged spleens and thymus (ii).
(D) Overall survival of Cre+ and CrE− mice (number of mice in parentheses).
(E) Kaplan-Meier curve breakdown of animals with indicated maladies.
(F) Cre+ enlarged thymus (i), Cre− normal thymus (ii), Cre+ enlarged spleen and Cre− normal spleen (iii). Scale bar, 5 mm.
(G) White blood cell counts for Cre+ animals with enlarged spleens (S; n = 24), enlarged thymus (T; n = 5), or a combination of both (S+T; n = 10), Cre− littermates (n = 10).
(H) Red blood cell (RBC) concentration, hemoglobin (Hb) concentration, and mean cell volume (MCV) for Cre+ animals with an enlarged spleen (S; n = 24), thymus (T; n = 5), or both (S+T; n = 10) compared with Cre− controls (Cs; n = 10).
(I) Platelet concentrations (PLT) and platelet distribution width (PDW%) in S, T, S+T, and C animals.
(G–I) Data are represented as mean ± SEM, Student’s t test (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001) S, enlarged spleen; T, enlarged thymus; C, Cre negative; VT, vascular tumors; NOA, no obvious abnormalities; Hem, hematopoietic malignancy; HSPC, hematopoietic stem progenitor cell; Hb, hemoglobin; MCV, mean corpuscular volume; PLT, platelet count; PDW%, size distribution of platelet width.
Whereas a previous SB screen targeting HSCs using Vav-Cre (Berquam-Vrieze et al., 2011) yielded only lymphoid leukemia, the VEC-Cre screen generated both myeloid and lymphoid malignancies. A total of 76 Cre+ and 15 Cre− (non-mutagenized) mice were evaluated, with 59 Cre+ and 0 Cre− mice presenting with pathology. From this cohort, 55.3% (n = 42) developed hematopoietic abnormalities alone, 9.2% (n = 7) developed vascular anomalies, and 13.2% (n = 10) developed a combination of both (Figure 1Ci). Mice with hematopoietic abnormalities were further categorized into those with an enlarged spleen (65.4%, n = 34), an enlarged thymus (13.5%, n = 7), or both (21.2%, n = 11) (Figure 1Cii). Overall, mutagenized mice had a mean survival of 179 days (Figure 1D). An enlarged thymus was associated with faster disease kinetics compared with splenomegaly (mean survival of 139 days versus 161 days, respectively) (Figure 1E). Representative images of pathology are shown in Figure 1F. Affected mice had ~4- to 7-fold increased white blood cell counts compared with Cre− littermates (Figure 1G) and frequently exhibited anemia (reduced red blood cell [RBC] count and hemoglobin [Hg] concentration) and increased RBC size (mean corpuscular volume [MCV]) (Figure 1H). Although affected mice commonly had abnormal platelet counts compared with Cre− animals, no differences were observed based on the primary affected site (Figure 1I). Overall, the findings indicate that targeted mutagenesis initiated in the HemEnd results in hematopoietic malignancies.
Spleen and Thymus Malignancies Have Distinct Mutation Signatures
The SB mutagenesis system enables precise genomic coordinates of transposon-induced mutations to be determined using linker-mediated PCR and Illumina next-generation sequencing (Brett et al., 2011). Subsequent statistical analyses identified recurrently mutated regions containing clonally expanded transposon insertions at a higher rate than would be predicted in the absence of selective pressure. Because these analyses assumed a random transposition pattern, we first confirmed the unbiased distribution of insertions across all chromosomes in affected spleen and thymus DNA samples (Figures S1A–S1C). Indeed, our data were consistent with the well-established unbiased nature of SB screens in general (Bard-Chapeau et al., 2014; Dupuy et al., 2009; Keng et al., 2009; Riordan et al., 2014). We next used gene-centric common insertion site (gCIS) analysis to identify clusters of clonally expanded insertions enriched near protein coding regions. Interestingly, the number of gCISs associated with the thymus phenotype was double that of the spleen phenotype, despite the same average number of total insertions per sample (Figures S1D and S1E), supporting the concept that the cell of origin influences gCIS mutations, which in turn influences malignancy (Berquam-Vrieze et al., 2011).
Our screen activated transposon-mediated mutagenesis in the HemEnd and thus early definitive HSCs. A comparison of the gCIS list with recurrently mutated genes identified in blood cancers arising from global (non-tissue specific) and Vav-Cre-(HSPC)-driven mouse mutagenesis screens revealed substantial overlap, highlighting the ability of this approach to identify genes relevant to lymphoid malignancies (Figure 2A; Tables S1 and S2; Figure S1F). Of note, previous HSPC-targeting screens were not also able to generate myeloid malignancies without mutant JAK sensitization.
Figure 2. Thymus and Spleen Malignancies Have Distinct Gene Insertion Signatures.
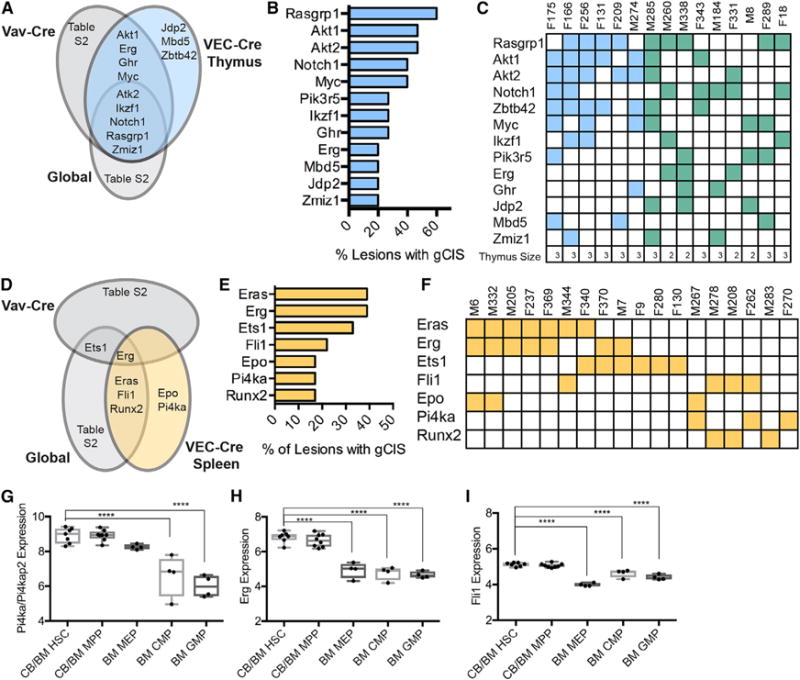
(A) gCIS associated with thymic abnormalities in this screen when compared with others.
(B) Most commonly mutated genes in enlarged thymus.
(C) Distribution of mutated genes (rows) in each mouse (column). F, female; M, male. The number relates to the ID of the individual mouse. Enlarged thymus phenotype is shown in blue, enlarged spleen and thymus are depicted in green. Size of the thymus is shown at the bottom as a reference (3 > 500 mg, 500 > 2 > 100 mg, 1 < 100 mg).
(D) gCIS associated with splenic abnormalities in this screen compared with other screens.
(E) Most common gCISs in enlarged spleens.
(F) Several genes (rows) were often found together in the same lesion (column).
(G–I) mRNA expression as measured by microarray of myeloid-associated gCIS Pi4ka (G), Erg (H), and Fli1 (I) in different compartments from human bone marrow (BloodChIP); one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). n = 4–8 per group.
See also Figures S1 and S2.
The most commonly mutated genes for the enlarged thymus phenotype were Rasgrp1, Akt1, and Akt2, with insertions often occurring concurrently in the same lesion (Figures 2B and 2C). Additional frequently mutated genes included Notch1 and Myc (Figure 2B). Many of the genes identified in this cohort have been previously associated with T cell malignancy (Manabe et al., 2006; Oki et al., 2011; Rasmussen et al., 2009).
Frequently mutated genes in enlarged spleens were Eras, Erg, and Ets1, which occurred in 40% of samples (Figures 2D and 2E). Other recurrently mutated genes included Fli1, Epo, and Runx2 (Figure 2E). Interestingly, several genes were commonly found together in a single spleen, suggestive of a cooperative function in transformation, and included: Eras, Erg, and Epo; Erg and Ets1; and Fli1 and Runx2 (Figure 2F). These genes have been implicated in pathological myelo-erythropoiesis and hematopoiesis in general, providing strong validation to the screen (Athanasiou et al., 2000; Huang et al., 2009; Zochodne et al., 2000). Interestingly, malignancies resulting from HemEnd-initiated mutagenesis frequently contained mutations in genes like Pi4ka, Erg, and Fli1, which are more highly expressed in HSCs compared with other hematopoietic progenitor cells (Figures 2G–2I) (Chacon et al., 2014). Most splenomegaly associated mutations were also correlated with abnormal blast-like cells in the blood and reduction in polymorphonuclear cells in the bone marrow (Figures S2A and S2B).
Pi4ka Insertion Is Associated with Progenitor Accumulation and Reduced RBCs
We next focused on Pi4ka, which, unlike most of the genes identified by the screen, had not been previously associated with hematopoietic abnormalities or hematopoiesis in general. The three Pi4ka transposon insertions (each from a distinct individual) were distributed throughout the gene in both transcriptional orientations (Figure 3A), a pattern suggestive of inactivating mutations (Copeland and Jenkins, 2010). Consistent with this hypothesis, the mutations were associated with decreased Pi4ka mRNA (Figures 3B and 3C), and immunohistochemical analyses indicated a significant decrease of Pi4ka protein compared with controls (Figure 3D). Histological evaluation of affected spleens also revealed expanded red pulp zones compared with Cre− spleens (Figure 3D). May-Grunwald stain of white blood cells from mice with Pi4ka insertions showed a prevalence of myeloid lineage, nucleated erythroid lineage, and blast-like immature cells (Figure 3D, bottom). Strikingly, when the Pi4ka insertion occurred in the absence of additional gCIS mutations (F270), the predominant cell type was blast-like and immature, which was also associated with anemia (Figures 3D [bottom] and 3E). On the other hand, when the Pi4ka mutation occurred in the same lesion as either the Epo or Fli1 mutation, lesions appeared myelodysplastic and were characterized by an abundance of nucleated erythroid lineage cells (Figure 3D, bottom).
Figure 3. Transposon Insertions in the Pi4ka Gene Are Associated with Blast-like Phenotype and Decreased Red Blood Cells.
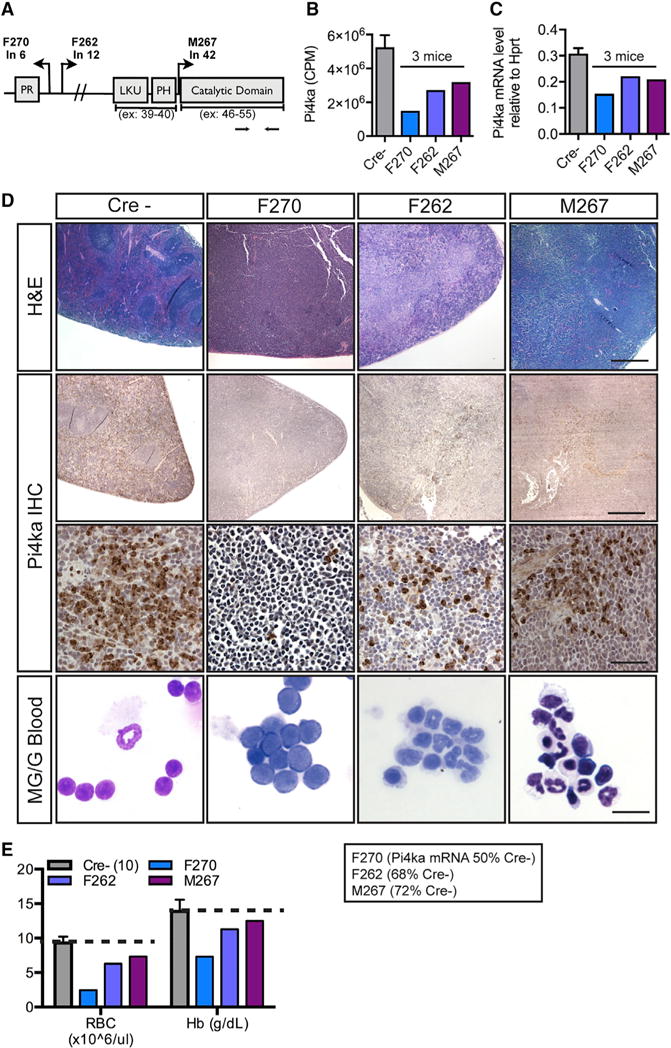
(A) Transposon insertions throughout the Pi4ka gene.
(B) Pi4ka RNA sequencing counts per million for affected and C spleens.
(C) Pi4ka expression by qPCR of the individual spleens from affected mice compared to Cre− spleens (n = 6).
(D) H&E of Cre− spleens and those with Pi4ka insertions (scale bar, 600 mm). Pi4ka immunohisto-chemistry (IHC) in spleens of C and affected mice (top: scale bar, 600 μm; middle: scale bar, 60 μm). Bottom: Cytospins of RBC-lysed blood from C and affected animals (scale bar, 15 μm)
(E) CBC analysis in C and Pi4ka-affected animals. For Cs (Cre−), data are represented as mean ± SEM of n = 10 animals.
Pi4ka Insertions Are Associated with Impaired Myelo- and Erythropoiesis
To assess the cellular composition and clonality of spleens with Pi4ka mutations, we first performed transcriptome analysis of both mutant and control spleens (Figure 4A). Based on the expression of cell-type-specific markers (Zhu and Emerson, 2002), Pi4ka mutant spleens had less lymphocytic, more HSCs, and more common myeloid progenitor (CMP) features when compared with controls. Animal F270 had the least mature monocyte-macrophage character, both F270 and F262 had increased granulocyte-monocyte progenitor (GMP) features, and both F270 and M267 had decreased mature erythroid hemoglobin transcripts. These results led us to hypothesize that Pi4ka might have a role in erythroid and myeloid maturation. Consistently, downregulated genes were enriched for Gene Ontology categories related to mature blood cells markers (Figure 4B), and upregulated genes were enriched for categories related to cell proliferation (Figure 4C). Expression of the genes defined in Figure 4A revealed strong similarity between two independent regions of each affected and control spleen (Pearson correlation; diagonal) (Figure 4D), indicating that the anomalies were clonal. As anticipated, comparisons between individuals showed much lower correlation.
Figure 4. Molecular Characterization of Pi4ka Mutant Spleens.
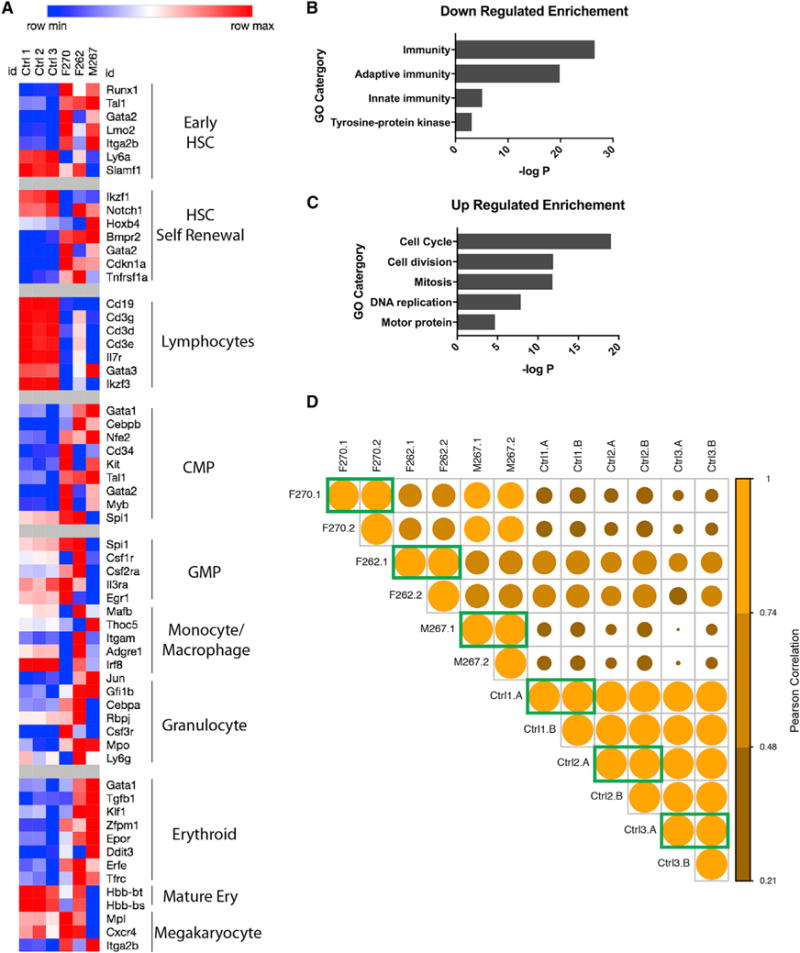
(A) Hematopoietic gene transcriptional signatures for the three Pi4ka affected mice compared to three controls.
(B and C) Gene Ontology categories enriched by genes downregulated (B) and upregulated (C) greater than 2-fold.
(D) Clonal analysis comparing transcript counts for hematopoietic genes (as in A) between two regions of spleen removed from the same mouse (green boxes). Pearson correlations are color coded and visualized by circle size.
See also Figure S3.
Pi4ka Is Expressed in HSPC Budding from the HemEnd and in Adult Mouse Lineage-Negative Bone Marrow Cells
Next, we sought to evaluate the expression profile of Pi4ka in the hematopoietic compartment. Immunofluorescence staining in E9.5 mouse aortas captured HSPC, budding from HemEnd (CD31+), expressing Pi4ka at the cell membrane (Figure S3A). Pi4ka was also expressed in a subset of CD45+, lineage-cocktail-negative cells in the mouse adult bone marrow, but not in the associated vasculature (Cdh5+) (Figures S3B and S3C). To confirm these findings, we performed qPCR for Pi4ka on sorted adult bone marrow cell populations, detecting elevated expression in Lin Sca1+cKit+ HSPCs compared with other progenitor populations and the Lin+ cell fraction (Figure S3D). In addition, a microarray data from BloodChIP (Chacon et al., 2014) demonstrates the highest PI4KA expression in human HSC, multipotential progenitor (MPP), and megakarycocyte/erythrocyte progenitor (MEP) populations in addition to acute myeloid leukemia (AML) (Figure S3E).
Loss of pi4kaa Function in Zebrafish Inhibits Erythroid Differentiation
To further explore the biological relevance of Pi4ka in an independent system, we evaluated the role of zebrafish homolog pi4kaa in hematopoiesis. A splice inhibitory morpholino targeting the catalytic domain prevented splicing of pi4kaa exons 49 and 50 in a dose-dependent manner (Figure S4A). o-Dianisidine staining indicated lower hemoglobin content in the 48 hr post-fertilization (hpf) morphant embryos compared with controls (Figure S4B). To quantify differences in erythroid lineage cells, flow cytometry was performed on control and morpholino-treated gata1:DsRED (erythroid cells); fli1:GFP (endothelial and hematopoietic progenitor cells); or lcr:GFP (erythroid cells) embryos (≥100 embryos per biological replicate) at 48 hpf. Morphant (MO) animals were treated with an additional p53 morpholino to enhance embryo viability, whereas control (C) animals were treated with just the p53 morpholino. Due to its long half-life, DsRED measurement was equivalent to total erythroid lineage cells. Pi4kaa inhibition resulted in significantly less erythroid lineage cells in both fish lines (Figures 5A and 5B). fli1:GFP; gata1:DsRED fish were then used to assess the differentiation of erythroid line age cells in MOs. In C embryos, a prominent fli1:GFP;gata1:DsRED double-positive population was observed at 24 hpf, characteristic of immature hematopoietic cells (Figure S4C). As development proceeded and cells differentiated, this population was decreased. By 48 hpf, gata1:DsRED cells had lost fli1:GFP expression in C embryos (Figure S4D). In contrast, the fli1:GFP;gata1:DsRED double-positive population was partially retained in pi4kaa MOs (~3.5 higher median fli1:GFP expression) (Figure 5C; Figure S4E).
Figure 5. Loss of Pi4ka Decreases Erythroid Differentiation In Vivo and In Vitro.
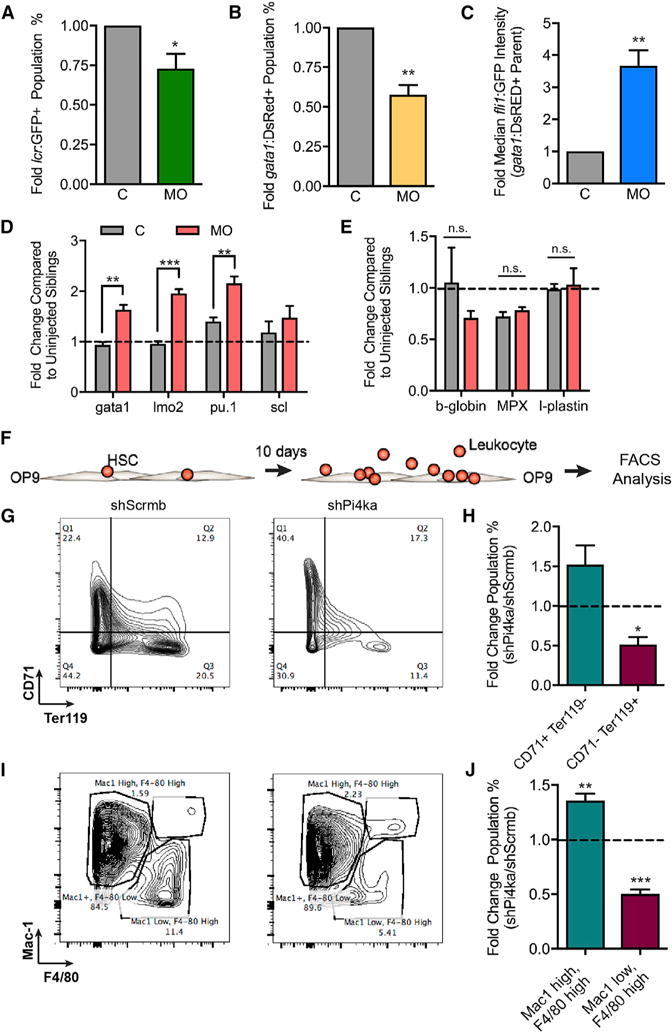
(A) Percent of LCR-GFP cells in MO fish compared with C for three independent experiments. For each data point in (A)–(C), >100 embryos were pooled and data are represented as mean ± SEM.
(B) Percent Gata1-DsRED cells in MO fish compared with C for four independent experiments.
(C) Graph of median Fli1:GFP intensity of the Gata1:DsRED parent population for four independent experiments.
(D) RNA expression in 24 hpf whole C and morpholino-injected embryos for gata1, lmo2, pu.1, and scl transcripts for three to four independent experiments. For each data point in (D) and (E), 20 embryos were pooled and data are represented as mean ± SEM.
(E) RNA expression in whole C and morpholino injected fish for b-globin, MPX, and l-plastin.
(F) Illustration of in vitro mouse HSPC differentiation on an OP9 stromal layer for 10 days.
(G) Three independent experiments in which shScrmb- and shPi4ka-infected HSPCs grown on OP9 stromal cells were probed for CD71 and Ter119 expression by flow cytometry.
(H) Quantification of three independent experiments plotted as the fold difference of shScrmb and shPi4ka.
(I) Mac1+/− (Mac1 high/low) and F4/80+/− (F4/80 high/low) expression and quantification in the same cells described in (G).
(J) Quantification of four independent experiments.
Bars indicate SEM. Student’s t test and ANOVA were used to calculate significance between two groups (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). See also Figure S4.
We performed qPCR on whole pi4kaa MO and C embryos to assess markers of stem and mature blood cells. Indicators of hematopoiesis gata1 (erythroid progenitor marker), lmo2 (erythroid-myeloid progenitor marker), and pu.1 (myeloid progenitor marker) were higher in MOs at 24 hpf (Figure 5D). While beta-globin was slightly lower than in Cs, the mature myeloid markers mpx and l-plastin were not affected in pi4kaa MOs (Figure 5E).
Pi4ka Knockdown in Mouse HSPC Impairs Progression of Mouse Erythro- and Myelopoiesis In Vitro in a Cell-Autonomous Manner
Given the evidence for a role in zebrafish erythropoiesis, we sought to determine the importance of Pi4ka in murine myelo- and erythropoiesis by evaluating HSPC differentiation in vitro. As the zebrafish experiments involved “whole-body” pi4kaa targeting, we sought to test whether Pi4ka loss functioned in a hematopoietic-cell-autonomous manner. We sorted adult mouse bone marrow HSPCs (Lin− cKit+Sca1+), treated with either lentivirus (lenti)-small hairpin RNA against Pi4ka or scrambled sequence for 24 hr, washed to remove virus, and subsequently co-cultured on an OP9 stromal layer in the presence of cytokines (Figure 5F). After 10 days of culture, flow cytometry revealed significantly less CD71− (immature erythroid marker), Ter119+ (mature eythroid marker) erythroid linage cells in cultures derived from shRNA for Pi4ka (shPi4ka)-treated HSPCs (Figures 5G and 5H). Similarly, an increase in Mac1+ (Mac1 high), F4/80+ (F4/80 high) cells with a complementary decrease in Mac1− (Mac1 low), F4/80+ (F4/80 high) cells was observed in the shPi4ka-treated condition (Figures 5I and 5J), reflecting the phenotype seen in mouse F270 from the original screen. A differentiation assay in the G1E-ER4 cell line also demonstrated an arrest in progressive differentiation (Ter119 level) with shPi4ka treatment (Figure S4F).
Akt and Erk Signaling Is Altered Downstream of Pik4a Knockdown In Vitro
To gain mechanistic clarity on pathways regulated downstream of Pi4ka, we explored the effect of loss of function on a panel of relevant signaling effectors. Pi4ka knockdown by lentiviral shRNA in 32D mouse myeloid lineage cells (Figure 6A, right) significantly increased phospho-ERK (p-Erk) and depressed interleukin-3 (IL-3)-induced p-Akt, as determined by western blot and flow cytometry (Figure 6A; Figure S5A). We validated the signaling effects in another cell type (HEK293), where Pi4ka small interfering RNA (siRNA) enhanced stem cell factor (SCF)-induced p-Erk levels (Figure 6B). Together, these results suggest that Pi4ka is important for regulating the balance between p-Akt and p-Erk signaling downstream of the cytokine receptors interleukin-3 receptor (IL-3R) and cKit.
Figure 6. Loss of Pi4ka In Vitro Blunts Akt Signaling and Enhances ERK Signaling.
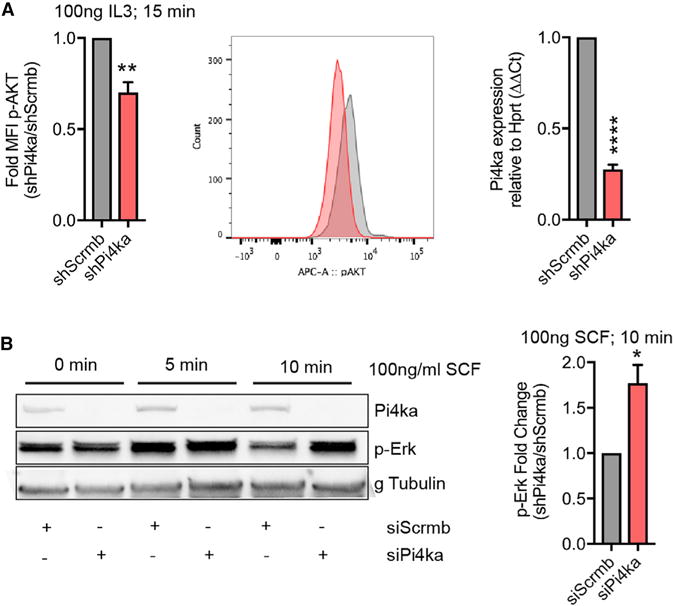
(A) Phospho-flow cytometry on 32D cells treated with 100 ng/mL IL-3 for 15 min. Fold change in MFI ratio of shPi4ka:shScrmb. Representative FACS plot (middle). Quantification of Pi4ka knockdown in 32D cells by qPCR (right) (summary of three independent experiments ± SEM).
(B) HEK293 cells treated with siScrmb and siPi4ka were stimulated with 100 ng/mL SCF for 0, 5, and 10 min. Protein lysates were probed for Pi4ka, p-ERK, and gamma tubulin, as loading C. Quantification (right) (summary of three independent experiments ± SEM).
Student’s t test and ANOVA were used to compare two conditions with statistical significance defined as *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. See also Figure S5.
Human PI4KAP2 Protein Lacks Kinase Activity and It Is Upregulated in Myelo- and Erythroleukemia Cell Lines
According to the COSMIC genome database, PI4KA mutations have been found in 363 unique human cancer samples, including somatic frameshift mutation p.T1995fs*4 in three lymphoid neoplasms (COSMIC Study COSU440) and many missense mutations, some occurring in primary and cell line leukemia. Interestingly, the human genome encodes pseudogenes, absent in mice, that could further affect PI4KA function. We hypothesized that human PI4KAP2 (PI4KA pseudogene 2), which encodes an N-terminally truncated, kinase-domain-deleted version of the PI4KA protein (Figures 7A and 7B), could act in a dominant-negative manner. The BloodChIP database indicates that the PI4KA promoter is similarly primed by activation marks in both CD34+ HSPCs and K562 eythroleukemia cells (Figure S6A), while the predicted PI4KAP2 promoter has relatively higher activation marks in K562 (Figure S6B). Furthermore, K562 cells have both PI4KA and PI4KAP2 promoter binding by the hematopoietic transcription factor Erg, while normal CD34+ HSPCs do not (Figures S6C and S6D).
Figure 7. The Human PI4KAP2 Gene Yields a Protein Product and Has Higher mRNA Expression Relative to PI4KA in Erythro- and Myelo-Leukemia Cell Lines.
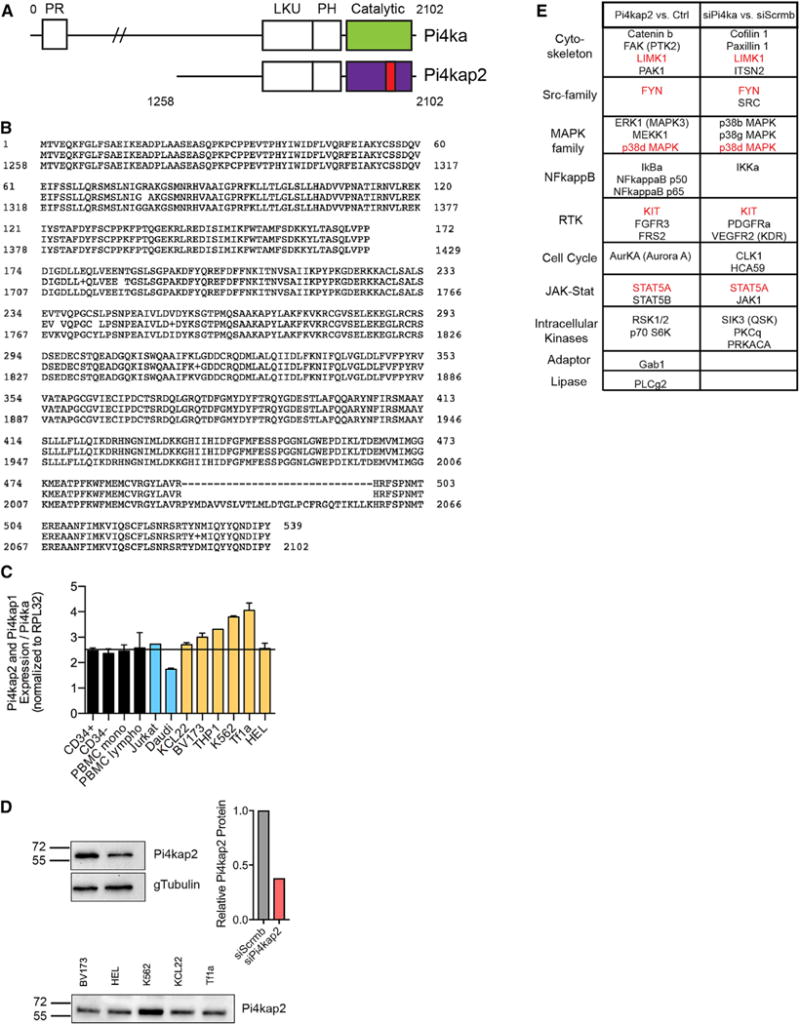
(A) Schematic comparing PI4KA and PI4KAP2 proteins. PI4KAP2 lacks the N-terminal domain and has a deletion in the kinase domain (red). PR, proline-rich domain; LKU, lipid kinase unique domain; PH, plekstrin homology domain.
(B) Alignment of PI4KA and PI4KAP2 amino acid sequences shows major homology starting at amino acid 1,258 of PI4KA, except for missing amino acids in the kinase domain of PI4KAP2.
(C) Ratio of mRNA expression of PI4KAP2 and PI4KAP1 compared with PI4KA in a panel of normal cord and peripheral blood cells (black), human lymphoid leukemia (blue), and myeloid leukemia (orange) cell lines.
(D) Top: Validation of an antibody probing cells subjected to siRNA targeting PI4KAP2 and quantification. Bottom: Western blot showing endogenous PI4KAP2 in myeloid leukemia cell lines.
(E) Antibody array summary of proteins that change when lysates from cells overexpressing PI4KAP2 were compared with vector C (left) and of proteins that change when lysates from cells knocked down for PI4KA were compared with C cells (right). Proteins in common between the two comparisons are highlighted in red. See also Figures S6 and S7.
Given the apparent increased PI4KAP2 promoter accessibility/priming and concomitant hematopoietic transcription factor binding in malignant cells, we assessed differential expression in myeloid leukemia, lymphoid leukemia, normal hematopoietic progenitor cells, and normal blood mononuclear cells (monocytes and lymphocytes) (Figure 7C). We designed primers to distinguish the PI4KA transcript (located in the region deleted in PI4KAP2) from the PI4KAP1 and PI4KAP2 transcripts (reverse primer spanning the region flanking the deleted kinase domain in PI4KAP1/2), although we were unable to distinguish between the two pseudogene transcripts. While PI4KAP1/2 and PI4KA were similarly expressed in CD34+ cord blood cells, peripheral blood mononuclear cells (PBMCs), and a T cell leukemia cell line, the ratio of PI4KAP1/2 to PI4KA expression was generally increased in myelo- and erythroleukemia cell lines, consistent with the BloodChIP data. We confirmed expression of PI4KAP2 protein in the myelo- and erythroleukemia cell lines using an antibody validated through decreased signal after siRNA knockdown in HEK293 cells (Figure 7D). These results demonstrate that PI4KAP2 codes for an expressed protein and is not a pseudogene, a conclusion further validated by fusing the cDNA sequence to a C-terminal hemagglutinin (HA)-tag. Both the exogenously expressed (HA-tagged) and endogenous protein were detected by western blot with a PI4KAP2 antibody (Figure S7A).
Given the lack of the kinase domain in PI4KAP2, we explored its potential role as an antagonistic regulator of PI4KA. As expected, PI4KAP2 protein purified from HEK293 cells using immuno-precipitation of the HA-tag displayed no in vitro kinase activity, in contrast to purified PI4KA protein (Figure S7B). We compared the impact of PI4KAP2 gain of function to PI4KA loss of function by probing an antibody microarray with lysates from HEK293 cells transfected with PI4KAP2 expression vector, C vector, siRNA against PI4KA, or C siRNA (Figures S7C and S7D). Protein categories affected by altering PI4KA signaling included cytoskeleton regulation, Src-family kinases, MAPK family, NFkappaB, receptor tyrosine kinases, cell cycle regulators, JAK-STAT signaling members, intracellular kinases, adaptors, and lipases (Figure 7E). Although there were some differences in the effects of PI4KAP2 expression and PI4KA knockdown, commonly affected proteins included: LIMK1 FYN, KIT, p38d MAPK, and STAT5A. The entire array can be found in Table S3.
DISCUSSION
Our results are consistent with literature reporting a pleiotropic requirement for Pi4ka in normal morphogenesis. In zebrafish, pi4kaa was shown to be necessary for pectoral fin development downstream of fibroblast growth factor receptor (FGFR) signaling through regulation of PI3K-Akt signaling (Ma et al., 2009). In Drosophila, it was shown to be required for smoothened activation during imaginal wing disc development (Yavari et al., 2010). Furthermore, global deletion of Pi4ka in adult mice uncovered its requirement for gastrointestinal stability (Bojjireddy et al., 2014; Vaillancourt et al., 2012). Our data add to this body of knowledge and implicate Pi4ka in hematopoiesis.
We postulate that Pi4ka is likely to regulate hematopoiesis in several ways in addition to its effect in Akt signaling. Pi4ka is a lipid kinase that phosphorylates the D4 position of the phosphatidyl-inositol ring (Minogue and Waugh, 2012). The resulting phosphatidyl-inositol, 4-phosphate (PIP4) provides a docking point for other proteins to bind to the inner leaflet of the plasma membrane (Balla et al., 2009). Once docked, additional lipid kinases can phosphorylate the ring at other positions, creating more complex phosphatidyl-inositols that can become substrates for phospholipases. Interestingly, phospholipase gamma 1 has been shown to have a role in primitive zebrafish hematopoiesis (Ma et al., 2007). In fact, we observe defects of the primitive erythroid lineage when we inhibit pi4kaa in our zebrafish model. Furthermore, oxysterol-binding proteins, which insert sterols into the plasma membrane (Villasmil et al., 2012), require PIP4 to dock. Oxysterols themselves can inhibit the proliferation of hematopoietic cell progenitors (Gregorio-King et al., 2002). Plasma membrane fluidity, as well as the specific constituency of lipids, influences cell-surface receptor signaling (Sunshine and Iruela-Arispe, 2017). Indeed, sterols have been shown to affect smoothened activation and hedgehog signaling, which are known to regulate HemEnd - HSCs (Crisan et al., 2016). Finally, there are early reports correlating changes in phosphatidyl-inositol lipid composition and hematopoietic cell proliferation and differentiation (Michell et al., 1990).
PI4KAP2, previously thought to not encode a functional protein, is the result of gene duplication in humans and does not exist in mice (Szentpetery et al., 2011). However, we demonstrated that this gene is not only transcribed, but also translated into a protein that has signaling consequences. Based on its lack of a kinase domain, we hypothesized that the PI4KAP2 protein could act in a dominant-negative fashion (similar to the effects of losing expression of PI4KA). The ratio of PI4KAP2 to PI4KA is higher in myeloid and erythroid cell lines compared with other cell types tested. Strikingly, the ERG transcription factor was documented to bind to the promoter region, specifically in malignant (K562) cells. One could speculate of a scenario in which higher levels of ERG (such as in Down syndrome) could enhance PI4KAP2 expression and deregulate the PI4KA pathway in the context of in utero leukemia development. Our findings indicate that PI4KAP2 overexpression or PI4KA knockdown induces similar alterations in MAPK, Srk-family kinases, and JAK-STAT signaling pathways. We also documented the effect of PI4KAP2 and PI4KA on proteins like FAK, PAK1, STAT5, and KIT, which are known leukemia drivers (Chatterjee et al., 2014). In addition, pi4kaa is known to regulate the PI3K-AKT pathway downstream of FGFR signaling (Ma et al., 2009). Our findings demonstrate similar effects in mouse 32D myeloid progenitor cells. FGFR1 signaling appears to be required for HSC repopulation, and increased expression of FGFR3 has been reported in CD34+ myeloid leukemia cells (de Haan et al., 2003; Dvorak et al., 2003). In this study, PI4KAP2 overexpression altered FGFR3 protein levels as per findings in the antibody array.
In conclusion, this forward genetic screen supports the concept that mutations initiated at the hemogenic endothelium stage can carry consequences for the hematopoietic lineage. We identified Pi4ka as an important cell-autonomous regulator of hematopoiesis, which in turn pointed to PI4KAP2, found to be dysregulated in human myeloid and erythroid leukemia cell lines.
EXPERIMENTAL PROCEDURES
Mice
VEC-Cre; ROSA26-LacZ transgenic mice (Alva et al., 2006) were crossed to conditional ROSA26-LsL-SB transposase T2/Onc2 mice (Dupuy et al., 2005) to initiate mutagenesis in hemogenic endothelial cells starting at E9.5. VEC-Cre-negative and wild-type C57BL/6J mice were used as controls. iCdh5-Cre recombinase; Rosa26-TdTomato mouse femurs and tibia were flushed to isolate marrow strands (Lizama et al., 2015). Animal protocols were reviewed and approved by the University of California, Los Angeles (UCLA) Institutional Animal Care and use Committee (ARC#2005-223-33G).
Sequencing of Transposon Insertion Sites and Identification of Gene-Centric Common Insertion Sites (gCISs)
Genomic DNA from tumors was analyzed by ligation-mediated PCR (LM-PCR) to identify transposon integration sites, as previously described (Berquam-Vrieze et al., 2011). Briefly, genomic DNA was digested with either AluI or NlaIII restriction enzymes. Double-stranded adaptor oligonucleotides were ligated to free DNA ends, followed by two rounds of PCR with nested primers to specifically amplify transposon/genome junctions and add on sequences necessary for sequencing. Amplified junctions were purified and sequenced using Illumina HiSeq. Clonal insertion sites were defined and gene-centric CIS (gCIS) analyses were performed to identify candidate genes implicated in tumorigenesis, as previously described (Brett et al., 2011).
Hematology
Complete blood count (CBC) analysis was performed using a Hemavet machine (Drew Scientific). After RBC lysis, leukocytes were spun onto slides using a Shandon Cytospin 4 (Themo Fisher Scientific). Slides were stained with May-Grunwald and Giemsa stains (Sigma-Aldrich).
Immunohistochemistry
Paraformaldehyde-fixed, paraffin-embedded tissue sections were deparaffinized, subjected to heat-mediated antigen retrieval, blocked with normal serum, and stained with antibody against Pi4ka. Biotinylated anti-rabbit secondary antibody was followed by Avidin-Biotin Complex Elite and DAB Peroxidase Kit (Vector Laboratories). See antibodies in Supplemental Experimental Procedures. An Olympus DP73 camera and cellSens software were used to image non-fluorescent stains.
RNA Sequencing
RNA was purified using an RNeasy mini kit (QIAGEN), and libraries were prepared with a TruSeq polyA selection kit using 1 mg of RNA manually or a TruSeq stranded polyadenylation (poly-A) selection kit with 50 ng of RNA using the NeoPrep system (Illumina). Libraries were sequenced on a HiSeq 4000 system (Illumina). For the clonal analysis, we sequenced single-end 50 bp. For the cell-subtype expression analysis, we sequenced paired-end 100 bp.
Fish
Zebrafish lines were maintained in accordance with the UCLA Department of Laboratory Animal Medicine’s Animal Research Committee guidelines. The following lines were used: Tg(gata1:DSRED; fli1:GFP), Tg(lcr:GFP) (Ganis et al., 2012), and wild-type AB fish. lcr:GFP fish were purchased from the UCLA Zebrafish Core Facility. All embryos were treated with 1 × 1-phenul-2-thiorea (PTU) (to inhibit pigment formation) at 24 hpf. Eight pg or 12 pg of the splice-inhibitory pi4kaa morpholino was injected with 2 pg or 3 pg of p53 morpholino, respectively. Splicing efficiency was examined with previously published primers. See oligonucleotide sequences in Supplemental Experimental Procedures. o-Dianisidine stain was used to stain hemoglobin.
qPCR Transcriptional Analysis
RNA was isolated using RNeasy Mini and Micro kits (QIAGEN). Mouse tissue cDNA was made using a Superscript III system (Invitrogen). Zebrafish and human cDNA were generated with an iScript cDNA synthesis kit (BioRad). Twenty whole zebrafish per treatment were used for RNA isolation. Zebrafish, mouse, and human qPCR primers are listed in Supplemental Experimental Procedures. SYBR-Green-based qPCR (BioRad) was performed as previously described (Briot et al., 2014).
Cells
OP9 cells (a gift from the Mikkola Laboratory, UCLA) were cultured in alpha minimum essential medium (aMEM) with 2 mM L-glutamine, 1% pen-strep, 20% Hyclone (Thermo Fisher Scientific) fetal bovine serum (FBS). For OP9/leukocyte co-cultures, media was supplemented with 5 ng/mL thrombopoietin (TPO), 50 ng/mL SCF, 10 ng/mL FMS-like tyrosine kinase 3 ligand (Flt3L), 5 ng/mL interleukin-6 (IL-6), and 5 ng/mL IL-3 (Peprotech). 32D cells (CRL-11346 American Type Culture Collection [ATCC]) were cultured according to ATCC recommendations with 10 ng/mL IL-3. Lenti-X HEK293 cells (632180 Clonetech) were cultured in DMEM 10% FBS. G1E-ER4 cells (a gift from the Ganz Laboratory, UCLA) were cultured with tamoxifen according to Rylski et al. (2003). BV173, KCL22, and K562 cells (a gift from the Colicelli Laboratory, UCLA), Tf1a (ATCC CRL-2451) and HEL 92.1.7 (ATCC TIB-180) were all cultured according to ATCC recommendations.
Human PBMC and CD34+ cord blood cells were purchased from the UCLA/Core Facility Research (CFAR) Virology Core Laboratory. Mononuclear cells were isolated using a Ficoll gradient. Monocytes (PBMC fraction adhering to a TC plate) and lymphocytes (non-adhered portion) were used for RNA. For cord blood, CD34+ (positive selected) and CD34− (negative selected) blood cells were isolated using human Miltenyi MACS CD34 MicroBead Kit Ultra Pure and used for RNA.
Flow Cytometry and Cell Sorting
Flow cytometry was performed using BD Fortessa and LSRII machines (BD Biosciences). FACS was performed using BD Aria instruments. For RNA isolation of bone marrow subpopulations, Lin+, HSC (Lin−, cKit+, Sca1+, CD34−), GMP (Lin−, cKit+, Sca1−, CD34−, FcgR+), MEP (Lin−, cKit+, Sca1−, CD34−, FcgR−), and CMP (Lin−, cKit+, Sca1−, CD34+, FcgRlow) cells were sorted from RBC-lysed bone marrow.
For OP9 cultures, HSC (Lin−, cKit+, Sca1+) cells were sorted after lineage depletion of bone marrow using mouse lineage depletion kit and LS columns (Miltenyi). Mouse hematopoietic cell differentiation on OP9 cultures was assessed from a single-cell suspension generated from cultured cells using antibodies against CD45, Mac1 (CD11b), F4/80, Ter119, and CD71.
Zebrafish flow cytometry and sorting was based on fli1:GFP, lcr:GFP or gata1:DsRED fluorescent signal (n = 100 per treatment). Dechorionated embryos were digested with 5 mg/mL Liberase-TM (Roche) for 1 hr at 33°C as in Bertrand et al. (2007).
Molecular Cloning
The Pi4kap2 coding sequence was amplified from pDONR223-PI4KAP2 (23601 Addgene) using primers (Supplemental Experimental Procedures) that introduced an HA-tag at the 3′ end and flanked the fragment with PstI and AgeI restriction enzyme sequences. The fragment was ligated into the pJet cloning vector using CloneJET kit instructions (K1231 Thermo Fisher Scientific) and amplified in Stbl3 bacteria (C737303 Thermo Fisher Scientific). The pLV-Ef1a-MCS-IRES-RFP-puro plasmid and the Pi4kap2HA-pJet vector were linearized with PstI and AgeI. The Pi4kap2-HA fragment was gel purified and ligated into the pLV vector using T4 ligase. For the shRNA vector purchased from Origene (TL510615), the MND-GFP cassette (a gift from the Kohn Laboratory) was used to replace the CMV-GFP reporter.
Lenti shRNA Transduction
Pi4ka or scrambled shRNA plasmids along with VSG-G and ?8.2 packaging plasmids were transfected into 293T cells using Lipofectamine 2000 (Life Technologies). Virus was collected and concentrated by centrifugation. For primary HSPCs, high concentration virus was used to doubly infect cells using Retronectin-coated (Clontech) plates (40 μg/mL) after an overnight pre-stimulation in serum-free StemSPAN (StemCell Technologies, Inc.) or StemMACS (Miltenyi) supplemented with four times the cytokine concentration used in OP9 co-culture over the course of 24 hr before being washed and moved to OP9 stromal cells. For 32D and G1E-ER4 cells, the same virus was used to doubly infect cells using Retronectin-coated plates as above for 24 hr in the presence of culture medium.
Cell Transfection
For siRNA studies, HEK293 cells were treated with 60 pmol non-targeted (4390843 Invitrogen), Pi4ka-targeted siRNA (4392420 ID:s224264, Invitrogen) or Pi4kap2-targeted siRNA (4390771 ID: n310610) in the presence of Lipofectamine RNAi Max (Invitrogen) for two days. Similarly, for overexpression of Pi4kap2, cells were treated with 3 μg of pLV-Ef1a-Pi4kap2-HA-RFP in the presence of Lipofectamine2000 (Invitrogen) for two days.
Western Blot
32D cells and HEK293 cells were lysed in modified radioimmunoprecipitation assay (mRIPA) buffer (50 mM Tris, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 0.15M NaCl, and 10mM beta-glycerophosphate) in the presence of 200 mM Na3VO4 and protease inhibitor cocktail (11873580001, Sigma). Lysates were run on 4%–20% gradient acrylamide gels (BioRad) and transferred to nitrocellulose membranes. See antibodies in Supplemental Experimental Procedures.
Antibody Array
Cell lines expressing Ef1a-Pi4kap2-HA-RFP, Ef1a-RFP empty vector, non-targeted siRNA, or Pi4ka-targeted RNA were lysed in protein lysis buffer (5 mM EDTA, 2 mM EGTA, 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.0, 20 mM NaF, 20 mM Na4P207, 1 mM Na3VO4, 60 mM beta-glycerophosphate, 50 nM phenylarsine oxide, 1% Triton X-100) containing protease inhibitor cocktail and phosphatase inhibitors. Samples were sonicated and then centrifuged for 30 min at 14,000 × g. Protein was quantified and pooled at equal concentrations. Lysates were then probed using the Kinexus KAM-900P array (Kinexus Bioinformatics Corporation), which contains 613 phospho-site-specific antibodies and 265 pan-specific antibodies (targeting 878 cell signaling proteins).
Statistical Analysis
For every dataset, it was first determined wither parametric or non-parametric analysis was appropriate and the use of either Student’s t test or Mann Whitney to assess significance. Log-rank test was used for survival curve statistics. Paired Student’s t test and one-way ANOVA were used to assess the significance of transcriptional differences between sorted hematopoietic sub-populations with the null assumption that they were the same. Paired Student’s t test was used to assess significance between experimental and C conditions for zebrafish and OP9 co-culture experiments. Unpaired Student’s t test was used for western blot and MFIR calculations. Statistical analyses were performed in Prism 7.0 according to the manufacturer’s recommendations (GraphPad Software) (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
DATA AND SOFTWARE AVAILABILITY
The accession number for all datasets reported in this paper is GEO: GSE108355.
Supplementary Material
Highlights.
Initiation of mutagenesis in the hemogenic endothelium yields hematopoietic malignancy
Pi4ka is expressed in hematopoietic stem progenitor cells
Pi4ka has a regulatory role in myelo- and erythropoiesis
PI4KAP2 is a protein-coding negative regulator of Pi4ka signaling
Acknowledgments
The authors wish to thank Ms. Michelle Steel, Dr. Vincenzo Calvanese, Ms. Felicia Codrea, Ms. Jessica Scholes, Ms. Valerie Rezek, Ms. Deborah Anisman-Posner, and Mr. Ha Neul Lee for valuable technical assistance; Drs. Don Kohn and Hanna Mikkola for guidance; the UCLA Broad Stem Cell Institute Flow Cytometry Core; the UCLA Tissue Procurement Core Laboratory; the UCLA Zebrafish Core Facility; the UCLA Vector Core supported by CURE/P30 DK041301; and the UCLA/CFAR Virology Core Laboratory (grant 5P30 AI028697). This work was supported by NIH grant CA197943 to M.L.I.-A. and N.R.S.A. (T32 HL69766) and a United Negro College Fund (UNCF)/Merck Graduate Science Research Dissertation Fellowship to S.Z (grant 20145117).
Footnotes
DECLARATION OF INTERESTS
We have no conflicts of interest to report.
AUTHOR CONTRIBUTIONS
S.Z. designed and performed the experiments, analyzed the data, and wrote the paper. J.D.R. designed and performed the experiments, analyzed the data, and assisted in manuscript preparation. A.M.C. designed and performed the experiments and analyzed the data. G.E.H. performed the experiments and analyzed the data. K.H., M.M., T.S., K.C.W., and G.H. performed the experiments. J.-N.C. and A.J.D. provided critical reagents and guidance. L.I.-A. designed the study, analyzed the data, and wrote the paper.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.017.
References
- Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- Athanasiou M, Mavrothalassitis G, Sun-Hoffman L, Blair DG. FLI-1 is a suppressor of erythroid differentiation in human hematopoietic cells. Leukemia. 2000;14:439–445. doi: 10.1038/sj.leu.2401689. [DOI] [PubMed] [Google Scholar]
- Balla T, Szentpetery Z, Kim YJ. Phosphoinositide signaling: new tools and insights. Physiology (Bethesda) 2009;24:231–244. doi: 10.1152/physiol.00014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Nguyen AT, Rust AG, Sayadi A, Lee P, Chua BQ, New LS, de Jong J, Ward JM, Chin CKY, et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet. 2014;46:24–32. doi: 10.1038/ng.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Been RA, Linden MA, Hager CJ, DeCoursin KJ, Abrahante JE, Landman SR, Steinbach M, Sarver AL, Largaespada DA, Starr TK. Genetic signature of histiocytic sarcoma revealed by a sleeping beauty transposon genetic screen in mice. PLoS ONE. 2014;9:e97280. doi: 10.1371/journal.pone.0097280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berquam-Vrieze KE, Nannapaneni K, Brett BT, Holmfeldt L, Ma J, Zagorodna O, Jenkins NA, Copeland NG, Meyerholz DK, Knudson CM, et al. Cell of origin strongly influences genetic selection in a mouse model of T-ALL. Blood. 2011;118:4646–4656. doi: 10.1182/blood-2011-03-343947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Kim AD, Violette EP, Stachura DL, Cisson JL, Traver D. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development. 2007;134:4147–4156. doi: 10.1242/dev.012385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A, Wilson S, et al. Pharmacological and genetic targeting of pPI4KA reveals its important role in maintaining plasma membrane PtdIns4p and PtdIns(4,5)p2 levels. J Biol Chem. 2014;289:6120–6132. doi: 10.1074/jbc.M113.531426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett BT, Berquam-Vrieze KE, Nannapaneni K, Huang J, Scheetz TE, Dupuy AJ. Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors. PLoS ONE. 2011;6:e24668. doi: 10.1371/journal.pone.0024668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot A, Jaroszewicz A, Warren CM, Lu J, Touma M, Rudat C, Hofmann JJ, Airik R, Weinmaster G, Lyons K, et al. Repression of Sox9 by Jag1 is continuously required to suppress the default chondrogenic fate of vascular smooth muscle cells. Dev Cell. 2014;31:707–721. doi: 10.1016/j.devcel.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon D, Beck D, Perera D, Wong JWH, Pimanda JE. BloodChIP: a database of comparative genome-wide transcription factor binding profiles in human blood cells. Nucleic Acids Res. 2014;42:D172–D177. doi: 10.1093/nar/gkt1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Ghosh J, Ramdas B, Mali RS, Martin H, Kobayashi M, Vemula S, Canela VH, Waskow ER, Visconte V, et al. Regulation of Stat5 by FAK and PAK1 in oncogenic FLT3- and KIT-driven leukemogenesis. Cell Rep. 2014;9:1333–1348. doi: 10.1016/j.celrep.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Li Y, De Obaldia ME, Yang Q, Yzaguirre AD, Yamada-Inagawa T, Vink CS, Bhandoola A, Dzierzak E, Speck NA. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. 2011;9:541–552. doi: 10.1016/j.stem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WK, Traver D. Signalling pathways that control vertebrate haematopoietic stem cell specification. Nat Rev Immunol. 2013;13:336–348. doi: 10.1038/nri3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer. 2010;10:696–706. doi: 10.1038/nrc2916. [DOI] [PubMed] [Google Scholar]
- Crisan M, Kartalaei PS, Vink CS, Yamada-Inagawa T, Bollerot K, van IJcken W, van der Linden R, de Sousa Lopes SM, Monteiro R, Mummery C, Dzierzak E. BMP signalling differentially regulates distinct haematopoietic stem cell types. Nat Commun. 2015;6:8040. doi: 10.1038/ncomms9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Solaimani Kartalaei P, Neagu A, Karkanpouna S, Yamada-Inagawa T, Purini C, Vink CS, van der Linden R, van Ijcken W, Chuva de Sousa Lopes SM, et al. BMP and Hedgehog regulate distinct AGM hematopoietic stem cells ex vivo. Stem Cell Reports. 2016;6:383–395. doi: 10.1016/j.stemcr.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan G, Weersing E, Dontje B, van Os R, Bystrykh LV, Vellenga E, Miller G. In vitro generation of long-term repopulating hematopoietic stem cells by fibroblast growth factor-1. Dev Cell. 2003;4:241–251. doi: 10.1016/s1534-5807(03)00018-2. [DOI] [PubMed] [Google Scholar]
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, Largaespada DA, Scheetz TE, Jenkins NA, Copeland NG. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009;69:8150–8156. doi: 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak P, Dvorakova D, Doubek M, Faitova J, Pacholikova J, Hampl A, Mayer J. Increased expression of fibroblast growth factor receptor 3 in CD34+ BCR-ABL+ cells from patients with chronic myeloid leukemia. Leukemia. 2003;17:2418–2425. doi: 10.1038/sj.leu.2403152. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, de Pater E. Regulation of blood stem cell development. Curr Top Dev Biol. 2016;118:1–20. doi: 10.1016/bs.ctdb.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Eliades A, Wareing S, Marinopoulou E, Fadlullah MZH, Patel R, Grabarek JB, Plusa B, Lacaud G, Kouskoff V. The hemogenic competence of endothelial progenitors is restricted by Runx1 silencing during embryonic development. Cell Rep. 2016;15:2185–2199. doi: 10.1016/j.celrep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganis JJ, Hsia N, Trompouki E, de Jong JLO, DiBiase A, Lambert JS, Jia Z, Sabo PJ, Weaver M, Sandstrom R, et al. Zebrafish globin switching occurs in two developmental stages and is controlled by the LCR. Dev Biol. 2012;366:185–194. doi: 10.1016/j.ydbio.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio-King CC, Collier FM, Bolton KA, Ferguson M, Hosking JB, Collier GR, Kirkland MA. Effect of oxysterols on hematopoietic progenitor cells. Exp Hematol. 2002;30:670–678. doi: 10.1016/s0301-472x(02)00833-0. [DOI] [PubMed] [Google Scholar]
- Guibentif C, Rönn RE, Böiers C, Lang S, Saxena S, Soneji S, Enver T, Karlsson G, Woods NB. Single-cell analysis identifies distinct stages of human endothelial-to-hematopoietic transition. Cell Rep. 2017;19:10–19. doi: 10.1016/j.celrep.2017.03.023. [DOI] [PubMed] [Google Scholar]
- Huang H, Yu M, Akie TE, Moran TB, Woo AJ, Tu N, Waldon Z, Lin YY, Steen H, Cantor AB. Differentiation-dependent interactions between RUNX-1 and FLI-1 during megakaryocyte development. Mol Cell Biol. 2009;29:4103–4115. doi: 10.1128/MCB.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KAT, Sarver A, Starr TK, Akagi K, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27:264–274. doi: 10.1038/nbt.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizama CO, Hawkins JS, Schmitt CE, Bos FL, Zape JP, Cautivo KM, Borges Pinto H, Rhyner AM, Yu H, Donohoe ME, et al. Repression of arterial genes in hemogenic endothelium is sufficient for haematopoietic fate acquisition. Nat Commun. 2015;6:7739. doi: 10.1038/ncomms8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma ACH, Liang R, Leung AYH. The role of phospholipase C gamma 1 in primitive hematopoiesis during zebrafish development. Exp Hematol. 2007;35:368–373. doi: 10.1016/j.exphem.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Ma H, Blake T, Chitnis A, Liu P, Balla T. Crucial role of phosphatidylinositol 4-kinase IIIalpha in development of zebrafish pectoral fin is linked to phosphoinositide 3-kinase and FGF signaling. J Cell Sci. 2009;122:4303–4310. doi: 10.1242/jcs.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe N, Kubota Y, Kitanaka A, Ohnishi H, Taminato T, Tanaka T. Src transduces signaling via growth hormone (GH)-activated GH receptor (GHR) tyrosine-phosphorylating GHR and STAT5 in human leukemia cells. Leuk Res. 2006;30:1391–1398. doi: 10.1016/j.leukres.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Michell RH, Conroy LA, Finney M, French PJ, Brown G, Creba JA, Bunce CM, Lord JM. Inositol lipids and phosphates in the regulation of the growth and differentiation of haemopoietic and other cells. Philos Trans R Soc Lond B Biol Sci. 1990;327:193–207. doi: 10.1098/rstb.1990.0054. [DOI] [PubMed] [Google Scholar]
- Minogue S, Waugh MG. The phosphatidylinositol 4-kinases: don’t call it a comeback. Subcell Biochem. 2012;58:1–24. doi: 10.1007/978-94-007-3012-0_1. [DOI] [PubMed] [Google Scholar]
- Moriarity BS, Largaespada DA. Sleeping Beauty transposon insertional mutagenesis based mouse models for cancer gene discovery. Curr Opin Genet Dev. 2015;30:66–72. doi: 10.1016/j.gde.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki T, Kitaura J, Watanabe-Okochi N, Nishimura K, Maehara A, Uchida T, Komeno Y, Nakahara F, Harada Y, Sonoki T, et al. Aberrant expression of RasGRP1 cooperates with gain-of-function NOTCH1 mutations in T-cell leukemogenesis. Leukemia. 2011;26:1038–1045. doi: 10.1038/leu.2011.328. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MH, Wang B, Wabl M, Nielsen AL, Pedersen FS. Activation of alternative Jdp2 promoters and functional protein isoforms in T-cell lymphomas by retroviral insertion mutagenesis. Nucleic Acids Res. 2009;37:4657–4671. doi: 10.1093/nar/gkp469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JD, Drury LJ, Smith RP, Brett BT, Rogers LM, Scheetz TE, Dupuy AJ. Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens. BMC Genomics. 2014;15:1150. doi: 10.1186/1471-2164-15-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylski M, Welch JJ, Chen YY, Letting DL, Diehl JA, Chodosh LA, Blobel GA, Weiss MJ. GATA-1-mediated proliferation arrest during erythroid maturation. Mol Cell Biol. 2003;23:5031–5042. doi: 10.1128/MCB.23.14.5031-5042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine H, Iruela-Arispe ML. Membrane lipids and cell signaling. Curr Opin Lipidol. 2017;28:408–413. doi: 10.1097/MOL.0000000000000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentpetery Z, Szakacs G, Bojjireddy N, Tai AW, Balla T. Genetic and functional studies of phosphatidyl-inositol 4-kinase type IIIα. Biochim Biophys Acta. 2011;1811:476–483. doi: 10.1016/j.bbalip.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt FH, Brault M, Pilote L, Uyttersprot N, Gaillard ET, Stoltz JH, Knight BL, Pantages L, McFarland M, Breitfelder S, et al. Evaluation of phosphatidylinositol-4-kinase IIIα as a hepatitis C virus drug target. J Virol. 2012;86:11595–11607. doi: 10.1128/JVI.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasmil ML, Bankaitis VA, Mousley CJ. The oxysterol-binding protein superfamily: new concepts and old proteins. Biochem Soc Trans. 2012;40:469–473. doi: 10.1042/BST20120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell. 2010;19:54–65. doi: 10.1016/j.devcel.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Emerson SG. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene. 2002;21:3295–3313. doi: 10.1038/sj.onc.1205318. [DOI] [PubMed] [Google Scholar]
- Zochodne B, Truong AH, Stetler K, Higgins RR, Howard J, Dumont D, Berger SA, Ben-David Y. Epo regulates erythroid proliferation and differentiation through distinct signaling pathways: implication for erythropoiesis and Friend virus-induced erythroleukemia. Oncogene. 2000;19:2296–2304. doi: 10.1038/sj.onc.1203590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.