

Abstract
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is an inherited myocardial disease characterized by fibro‐fatty replacement of the right ventricular myocardium, and associated with paroxysmal ventricular arrhythmias and sudden cardiac death (SCD). It is currently the second most common cause of SCD after hypertrophic cardiomyopathy in young people <35 years of age, causing up to 20% of deaths in this patient population. This condition has a male preponderance and is more commonly found in individuals of Italian and Greek descent. To date, there is no single diagnostic test for ARVC/D and the diagnosis is made based on clinical, electrocardiographic, and radiological findings according to the Revised 2010 Task Force Criteria. In this review, we will discuss the mainstay treatment which includes pharmacotherapy, implantable cardioverter‐defibrillator insertion for abortion of sudden cardiac death, and in the advanced stages of the disease cardiac transplantation.
Keywords: arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic right ventricular dysplasia
1. INTRODUCTION
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is an uncommon inherited cardiac disease characterized by progressive right ventricular (RV) dysfunction due to fibro‐fatty replacement of the myocardium and associated with high risk of ventricular arrhythmias and sudden cardiac death (SCD).1, 2 ARVC/D has a predominantly autosomal dominant inheritance, although recessive forms associated with a cutaneous phenotype, such as Naxos disease and Carvajal syndrome, are also observed.3, 4 Despite RV abnormalities being the predominant finding, it has been recently appreciated that patients with ARVC/D may also have some degree of left ventricular (LV) involvement5 and indeed severe LV impairment can sometimes be the initial manifestation of the disorder.6 An LV‐predominant form of ARVC/D has recently been described.7 Independently of which ventricle is initially or predominantly affected, during the later stages, advanced disease can result in biventricular heart failure, which may closely resemble dilated cardiomyopathy (DCM). LV dysfunction is observed more frequently with greater RV dysfunction, worse functional class, and leads to an increased tendency to cardiovascular adverse events relating to heart failure.8 The same study showed no clear relationship between LV involvement and an increased rate of arrhythmic events.8 Clinical manifestations vary with the age of the patient and stage of disease.9 In this article, we will review the pathophysiology or ARVC/D, the main diagnostic modalities used clinically aiding the diagnosis and patient management.
2. PATHOPHYSIOLOGY OF ARVC/D
The pathophysiological mechanisms in ARVC/D involve desmosomal abnormalities that can arise from mutations in cell adhesion proteins or intracellular signaling components.10 A number of genes have been implicated in the pathogenesis of ARVC/D,11 as illustrated in Table 1. Particularly, reduced cardiac desmoglein‐2 and desmocollin‐2 levels appear to be specifically associated with ARVC/D, independent of the gene mutations found.12 The desmosome normally maintains cell‐to‐cell adhesion and confers mechanical strength to tissues (Figures 1 and 2). In the extracellular space, desmosomal cadherins (desmocollin and desmoglein) bind strongly to each other. Cadherins span the plasma membrane and attach to linker proteins (plakoglobin, desmoplakin, and plakophilin‐2) in the intracellular space.1 Plakoglobin and desmoplakin are intracellular proteins anchoring desmosomes to desmin intermediate filaments. Moreover, plakoglobin contributes to interlinking adherens junctions with the actin cytoskeleton and participates in cellular signaling to the nucleus and desmosome organization.13, 14 Defects in linking sites of these proteins can interrupt cell adhesion, especially under conditions of increased mechanical stress or stretch, leading to cell death, progressive loss of myocardium, and fibro‐fatty replacement.15 As such, surviving myocardial fibers within the fibro‐fatty tissue from zones of slow conduction provide a medium for re‐entry ventricular arrhythmias.16, 17, 18, 19 The degeneration‐inflammation model posits that the resulting cellular damage is found in tissues under high mechanical stress.1 Indeed, this notion is in keeping with the observations that exercise increases age‐related penetrance and risk of arrhythmias in ARVC/D‐associated mutation carriers.20 The potential role of calcium‐sensitive pathways in the pro‐arrhythmia mechanism of ARVC/D has been proposed.21 In a recent meta‐analysis, however, the presence of desmosomal gene mutations was not associated with global or regional structural and functional alterations, epsilon wave, or VT of left bundle branch morphology.22
Table 1.
Updated gene list responsible for ARVC/D pathology
| Subtype | Gene | Location | Reference |
|---|---|---|---|
| ARVC1 | TGFB3 | 14q24.3 | 89 |
| ARVC2 | RYR2 | 1q43 | 90 |
| ARVC3 | Unknown | 14q12‐q22 | 91 |
| ARVC4 | TTN | 2q32.1‐q32.3 | 92 |
| ARVC5 | TMEM43 | 3p25.1 | 93 |
| ARVC6 | Unknown | 10p14‐p12 | 94 |
| ARVC7 | DES | 2q35 | 95 |
| ARVC8 (Carvajal) | DSP | 6p24.3 | 96 |
| ARVC9 | PKP2 | 12p11 | 97 |
| ARVC10 | DSG2 | 18q12.1 | 98 |
| ARVC11 | DSC2 | 18q12.1 | 99 |
| ARVC12 (Naxos) | JUP | 17q21.2 | 15 |
| Others | PLN | 6q22.1 | 100 |
| LMNA | 1q22 | 101 | |
| SCN5A | 3p21 | 102 | |
| CTNNA3 | 10q22.2 | 103 | |
| CDH2 | – | 104 |
Figure 1.
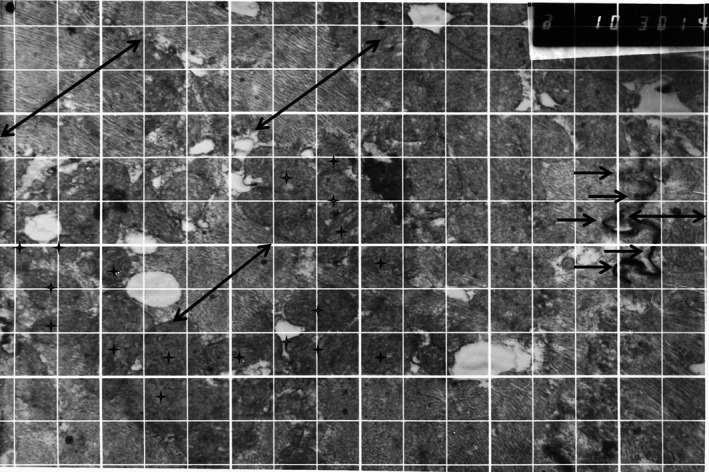
Desmosomes (arrows) connect cytoskeletons of adjacent cells (myofibrils, double arrows) and contain centrally dense lamellae composed of polypeptides, plakoglobin, and other proteins. Clusters of mitochondria (asterisks) can be seen
Figure 2.
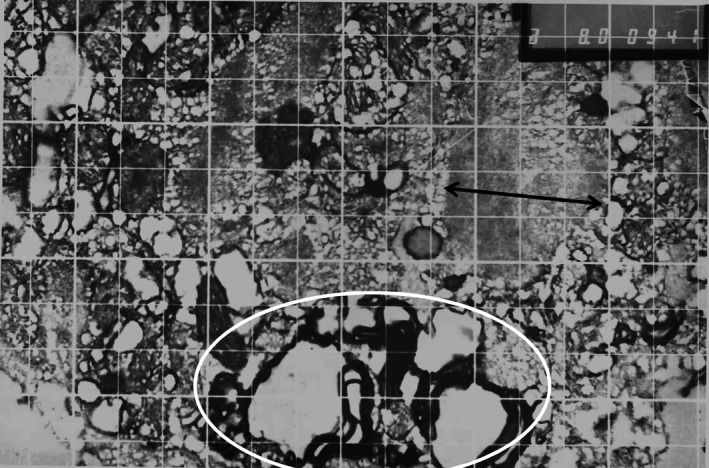
The desmosomes (ellipse) show dense cell borders and glycoproteins in the intercellular space. Plakoglobin is a plaque protein of intercellular junctions
3. CLINICAL PRESENTATION
Classically, ARVC/D usually presents between the second and fourth decades of life with syncope, symptomatic arrhythmias, or SCD. An example of monomorphic VT in a patient with ARVC/D is shown in Figure 3 (reproduced from23 with permission). Chest pain can be the presenting finding of the disorder.24 One‐third of the patients become symptomatic before the 30th year of life. ARVC/D can lead to deleterious consequences, such as ventricular arrhythmias, pump failure, and death. Competitive sports have been associated with a twofold increased risk of ventricular arrhythmias and mortality, and earlier presentation of symptoms comparing with inactive patients and patients who participated in recreational sport.25 Another interesting finding is the relation between meteorological factors and outcomes in patients with ARVC/D. Particularly, higher temperature and larger variation in humidity within 3 days of events were independently associated with the development of ventricular arrhythmic and sudden mortality events.26 Intracardiac thrombosis may occur in certain patients with ARVC/D.27 Atrial arrhythmias are also common in ARVC/D and present at a younger age than in the general population.28 Atrial arrhythmias are associated with male gender, increasing age, and left atrial dilation and clinically important, and they are associated with inappropriate implantable cardioverter‐defibrillator shocks29 and increased risk of both heart failure and death.28 In addition to tachy‐arrhythmias, brady‐arrhythmias are also observed in this condition.30
Figure 3.
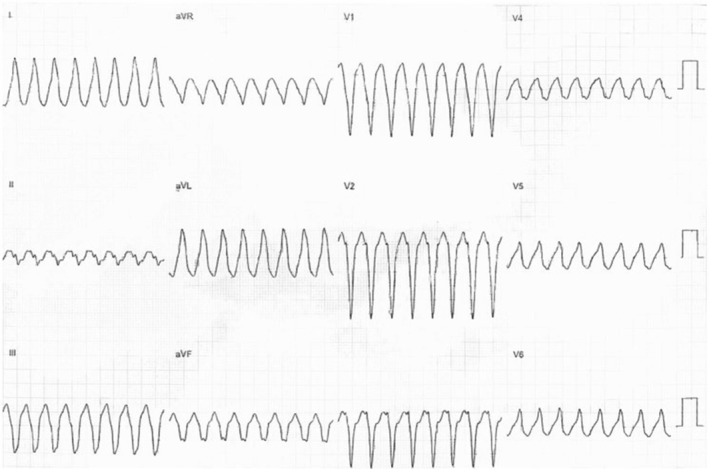
An electrocardiogram (ECG) showing monomorphic ventricular tachycardia (VT) with a left bundle branch block pattern with superior axis from a patient with arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). Figure reproduced from ref. 23 with permission
4. DIAGNOSING ARVC/D: THE CHALLENGES
There is no single diagnostic test for ARVC/D. The diagnosis is made based on major and minor clinical, electrical, and imaging criteria that have been devised by expert consensus of the Task Force Criteria (TFC) originally proposed in 1994 and further revised in 2010 (Table 2).31 In the original 1994 TFC, the clinical diagnosis was based strongly on symptomatic index cases and SCD victims—those with overt and severe phenotypes. Consequently, the 1994 criteria were highly specific, but they lacked sensitivity for early and familial disease.32, 34, 35 A systematic review and meta‐analysis of five retrospective studies compared the diagnostic concordance between the 1994 and 2010 criteria.36 8.6% and 3.6% satisfied the 1994 and 2010 major criteria, whereas 29.2% and 1.9% satisfied their minor criteria. Therefore, the 2010 revised TFC have resulted in a significant reduction in the number of patients that satisfy the cardiovascular magnetic resonance criteria and in a statistically significant increase in the number of patients diagnosed with definite ARVC/D compared to the 1994 criteria.36 Since then, modifications of the original criteria have been proposed to facilitate clinical diagnosis in first‐degree relatives, who often have an incomplete disease phenotype.37 According to these recommendations, familial ARVC/D is said to occur when the following conditions are met: (i) T‐wave inversion in the right precordial leads in individuals older than 14 years of age; (ii) late potentials by signal‐averaged ECG (SAECG); and (iii) ventricular tachycardia with left bundle branch block morphology on the ECG or exercise testing or >200 premature ventricular contractions in 24 hours.
Table 2.
International Task Force Criteria modified in 2010 (reproduced from 31 with permission)
| Domain | Major criteria | Minor criteria |
|---|---|---|
| Family history |
|
|
| ECG abnormalities |
|
|
| Arrhythmias |
|
|
| Tissue characteristics |
|
|
| Global or regional functional or structural abnormalities | ||
| Echocardiography |
|
|
| MRI |
|
|
| RV angiography |
|
|
4.1. Electrocardiography
In the ECG, epsilon waves, which are late potentials occurring between the end of the QRS complex and the onset of the T‐wave, and T‐wave inversion in the right precordial leads of V1 to V3 may be observed. Epsilon waves are specific for ARVC/D although it is only observed in 30% of patients and are best seen in the right precordial leads V1‐V3 (Figure 4). Particularly, epsilon waves in lead aVR in patients with arrhythmogenic right ventricular cardiomyopathy are rare electrocardiographic findings with a specificity of 100%.38 The detection of epsilon waves on 12‐lead ECG has been associated with higher episodes of sustained VT, but reassuringly, this did not lead to increased SCD incidence.39 A case of a child with extensive involvement of both right and left ventricular walls and epsilon waves in all precordial leads has been reported.40 However, interobserver variability in the assessment of epsilon waves is high.41 As a result, the assessment of the epsilon waves must be performed cautiously particularly in patients with who would not otherwise meet diagnostic criteria. The sensitivity of epsilon waves on the ECG is low between 25% and 38%, and therefore, a normal ECG does not exclude the diagnosis of ARVC/D.42, 43 The use of Fontaine bipolar precordial lead electrocardiography (F‐ECG) increased the sensitivity to 50%.43 Electrical abnormalities in ARVC/D are important as these changes precede structural changes.44
Figure 4.
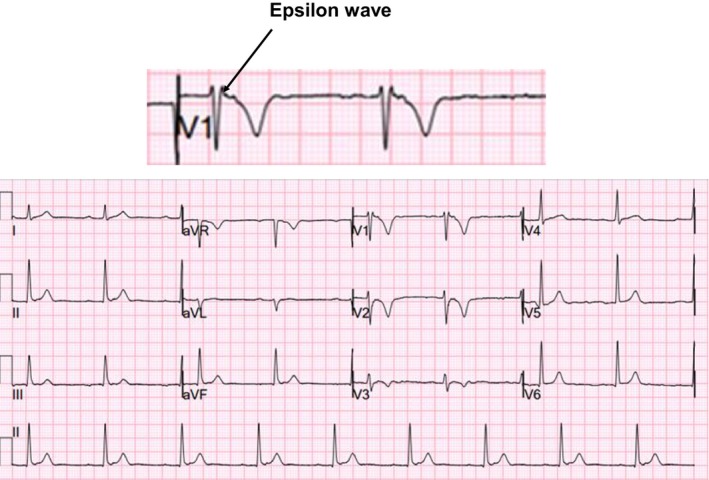
An electrocardiogram (ECG) showing the presence of an epsilon wave (black arrow) that is pathognomonic of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D)
A more specialized electrocardiographic technique, known as signal‐averaged electrocardiography (SAECG), can also be utilized. It aims to filter interference, unmask any microvariations, and display late potentials (if any) within the QRS complex by averaging the multiple electrical signals generated by the heart.45 Late potentials are thought to represent electrical depolarization abnormalities and are defined within the minor depolarization criteria in the 2010 TFC (Table 2). The criteria include the following:
Filtered QRS duration ≥114 ms
Duration of terminal QRS <40 μV: ≥38 ms
Root‐mean‐square voltage of terminal 40 ms: ≤20 μV
As long as any of the above is satisfied, the minor criterion in the 2010 TFC is fulfilled. Even though SAECG can be useful, it must be noted that an abnormal SAECG is not specific to ARVC as it can also be seen in other conditions such as myocarditis and scarring in ischemic heart disease.
4.2. Imaging: echocardiography and cardiac magnetic resonance (CMR)
Noninvasive imaging modalities such as echocardiography and CMR can be used to identify structural and functional abnormalities described in the revised TFC. It is also possible to investigate these invasively using RV angiography. Echocardiographic features of ARVC/D include global ventricular dilatation (Figure 5), reduced RV ejection fraction with normal LV, or mild segmental dilatation of the RV or regional RV hypokinesis.31 CMR similarly demonstrates these abnormalities but at a higher resolution (Figure 6). There are several advantages of CMR over echocardiography. Firstly, it provides gold standard values for both LV and RV volumes. Secondly, it provides clear delineation of RV anatomy for micro‐aneurysms and trabeculations. Thirdly, it can clearly visualize RV hypokinesis and akinesis.46 All of these features are part of the 2010 criteria, making CMR an excellent modality for diagnosis. Moreover, CMR can help to identify conditions that can mimic ARVC/D.46 For example, Khaji et al47 presented a patient with mega‐epsilon wave, right ventricular dilatation, and inducible VT that was initially diagnosed as ARVC/D by the Task Force Criteria. Further examination with endomyocardial biopsy revealed the diagnosis of sarcoidosis.47 A study which compared clinical and electrophysiological parameters between patients with ARVC/D and cardiac sarcoidosis showed that patients with sarcoidosis had reduced left ventricular ejection fraction, a significantly wider QRS, right‐sided apical VT, and more inducible forms of monomorphic VT.48 Furthermore, cardiovascular magnetic resonance and specifically the cardiac volume, in addition to the degree and location of cardiac involvement, can be used to distinguish between these two disease entities.49 The combined use of signal and wall motion parameters of cardiovascular magnetic resonance has been proposed to be contemporaneously considered for achieving a better diagnostic accuracy for the diagnosis of ARVC/D.50
Figure 5.
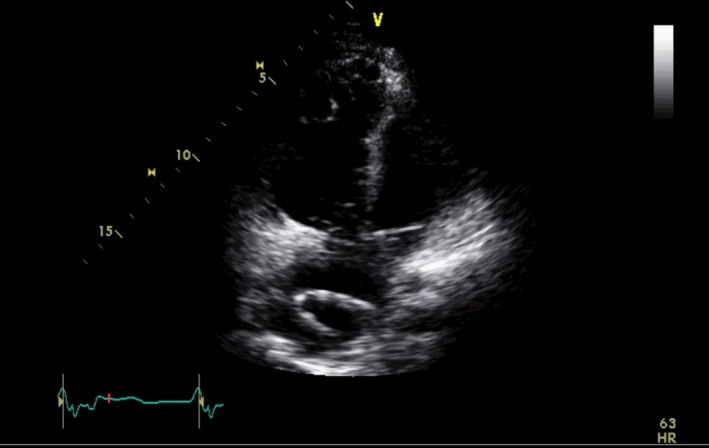
Modified four‐chamber view during transthoracic echocardiography demonstrating a dilated right ventricle
Figure 6.
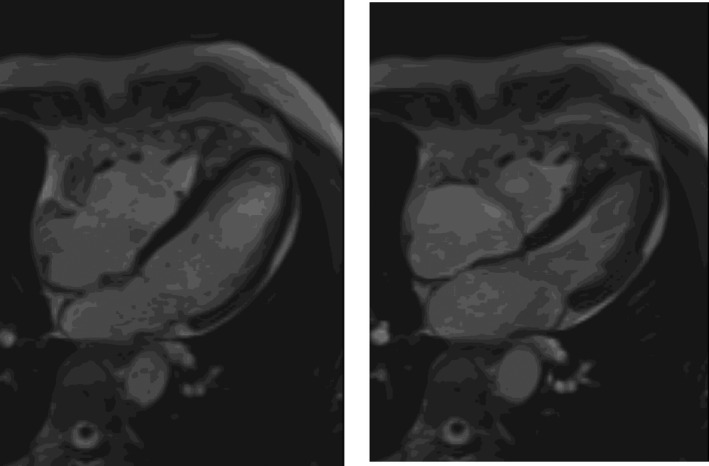
Four‐chamber view of a cardiovascular magnetic resonance study in diastole (left panel) and systole (right panel) showing a very dilated RV
4.3. Genetic studies, biopsy, and histology
Genetic testing can identify desmosomal mutations in approximately 30%‐60% of ARVC/D cases.51, 52 Of these, PKP2 mutations are most frequently observed, with an estimated prevalence of 10%‐47% among unrelated probands and 70%‐82% among familiar ARVC/D cases.10, 53 The second commonest mutated gene is desmoplakin (DSP).1 Endomyocardial biopsy guided by voltage mapping may provide ARVC/D diagnosis confirmation.54
The main macroscopical features of ARVC/D are shown in a short‐axis section of an explanted heart at the time of heart transplantation for ARVC/D (Figure 7). This demonstrates a grossly dilated right ventricle. The ventricular wall is widely replaced by fat tissue. There are prominent right ventricular trabeculations, and the muscular tissue of the numerous right ventricular trabecules appears to be preserved.
Figure 7.
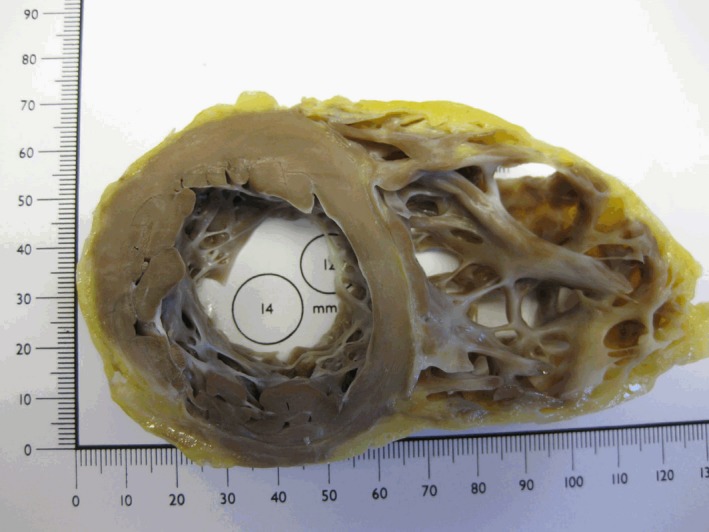
Short‐axis section of an explanted heart at the time of heart transplantation for ARVC/D demonstrates the right ventricular wall to be composed of fat tissue, and there is prominent right ventricular trabeculation. The myocardium of the numerous prominent trabecles appears macroscopically unaffected, which has been confirmed histologically
In terms of histology, the hallmark of the disease is fibro‐fatty replacement of the right ventricular myocardium and right ventricular part of the interventricular septum. Figure 8 shows full‐thickness sections of anterior right ventricular wall and interventricular septum submitted from the explanted heart shown in Figure 7. On hematoxylin and eosin stain (left), there is an abrupt transition between small areas of preserved myocardium and areas of transmural fibro‐fatty replacement in the anterior right ventricular wall and right ventricular myocardium of interventricular septum. Collagen stains, such as Sirius red (right), are useful to visualize areas of fibrosis in the RV wall.
Figure 8.
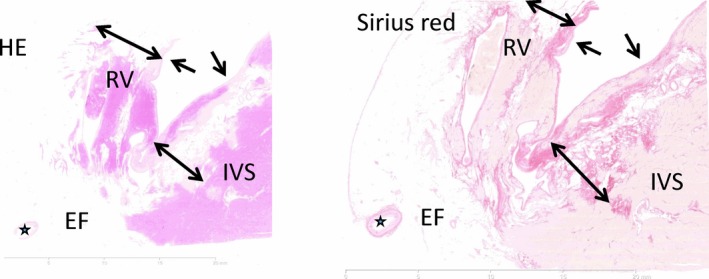
Full‐thickness section of the anterior right ventricular (RV) wall and interventricular septum (IVS) including epicardial fat (EF) using conventional hematoxylin and eosin (H&E) (left) and collagen stains (Sirius red) (right) at ×0.42 magnification. There is patchy fibro‐fatty replacement of the muscular wall affecting the anterior right ventricular wall and the right interventricular septum. The myocardial architecture of the left IVS is unaffected. Asterisk: coronary artery. The single‐headed arrow points toward endocardium. The double‐headed arrow marks the areas affected by fibro‐fatty replacement
Immunohistochemistry for plakoglobin is often equivocal and shows, as in this confirmed case of ARVC/D, a variegated staining pattern with focal normal staining reaction in section submitted from interventricular septum (IVS) (left), and a decreased staining reaction in a transmural tissue section from RV, which corresponds to an abrupt transition to an area of fibro‐fatty replacement (right) (Figure 9).
Figure 9.
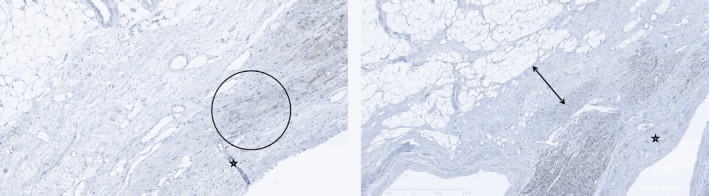
Plakoglobin staining from different areas of right‐sided interventricular septum, at ×10 magnification. There is a variegated staining reaction with focal positive staining, and abrupt transition to an area of fibro‐fatty replacement showing negative staining reaction for Plakoglobin in rare cardiomyocytes (encircled) (left). There is patchy mild decreased staining reaction of heart muscle cells adjacent to hyalinized mid‐wall fibrosis. The endocardium is marked by an asterisk. The double‐headed arrow marks the area of subendocardial fibrous replacement
4.4. Differential diagnosis
It is important to differentiate ARVC/D from other right ventricular disorders, such as Brugada syndrome, as overlapping features may be found.55 Moreover, cardiovascular conditions such as peripartum cardiomyopathy46 or athlete's heart can present with similar clinical and imaging findings.56 A correct diagnosis is important because unlike ARVC/D, athlete's heart would not justify disqualification from competitive sports.56 Distinguishing between ARVC/D and athlete's heart remains a diagnostic challenge. High‐level endurance training is associated with RV elongation, dilation, and hence enlargement compared to isometric physical activities.57 As such, an enlarged RV dimension alone is not a reliable diagnostic criterion for ARVC/D in elite athletes. A large proportion of athletes also express echocardiographic morphological findings often evident in documented ARVC/D, including rounded RV apex, and prominent RV trabeculations and moderator band.58 By contrast, impaired RV systolic function is associated only with ARVC/D and not with athlete's heart. The use of both RV dilation and systolic dysfunction might serve as a useful diagnostic tool to separate between the two. Despite all the imaging parameters, and new normal specific ranges for athletes,58 reaching a diagnosis of ARVC/D in an athlete can sometimes remain challenging and a short period of detraining with subsequent assessment usually with cardiac MRI can be helpful in resolving this ambiguity.59
5. MANAGEMENT
In ARVC/D, the main goal is to avoid the high‐risk events of malignant arrhythmias and SCD and slow the progression of heart failure.2, 60 Competitive sports are discouraged.2, 61 While patients with ARVC are allowed to perform exercise including sports as part of a healthy lifestyle, they should not exercise to maximal capacity and be vigilant to any symptoms of palpitations.20 Frequent endurance exercise increases the risk for VT/VF and heart failure.20The management of patients with ARVC/D in specific situations such as pregnancy is beyond the scope of this review, but is directed to the following reference.62 Anti‐arrhythmic medications, such as beta‐blockers and class‐III agents, are advised.2 Sotalol and amiodarone with or without the need for conventional beta‐blockers are potent (effective is used in 3 sentences in a row).16, 63 Calcium channel blockers may be effective in selected patients.64 The addition of flecainide in combination with sotalol/metoprolol may be an adequate strategy for the control of ventricular arrhythmias in patients with ARVC/D refractory to single‐agent therapy and/or catheter ablation.65 In general, the most sufficient combinations appear to be sotalol or flecainide and amiodarone/beta‐blockers.64
The American College of Cardiology, the American Heart Association and the European Society and Cardiology recommended ICD implantation for the prevention of SCD events.66 Risk stratification and indication to ICD implantation in ARVC/D has been proposed by an international task force consensus statement.67 One study reported that the annual cardiac mortality in patients with ARVC/D who were implanted with an ICD was 0.9%.68 Finally, VT ablation targeting late potentials abolition seems to be effective in preventing VT recurrence in patients with or without RV structural abnormalities.69 In the multicenter registry, clinical response (freedom from SCD, VT requiring hospitalization, or heart transplantation) after the last ablation (predominantly endocardial) was 86% at 1 year, 69% at 5 years, and 60% at 10 years.70 On the other hand, the combined endocardial and epicardial approach resulted in better procedural success and long‐term VT‐free survival compared with the endocardial approach in ARVC/D patients with recurrent VTs.71, 72 In fact, the combined endocardial and epicardial VT ablation eliminated all clinical and induced VTs, and the addition of scar dechanneling resulted in noninducibility in all cases.72 Identification of conducting channels (CCs) inside or between the scars can be achieved via endocardial high‐density substrate mapping.73
However, another single‐center study showed that the vast majority of critical VT circuits were epicardial while epicardial ablation of VT appeared to be both safe and effective in achieving arrhythmia control in ARVC/D.74 A recent meta‐analysis showed better outcomes with the combined endocardial and epicardial ablation approach compared with the endocardial approach only.75 However, a stepwise approach with an endocardial ablation first and additional epicardial ablation only if VT episodes are still inducible has been proposed.76, 77 Furthermore, an inducibility‐guided catheter ablation strategy of VT in patients with ARVC/D has been proposed to prevent unnecessary epicardial ablation procedures.78 Electrical regression of SAECG after catheter ablation in ARVC/D has been found to be associated with fewer ventricular arrhythmia recurrences.79 Other treatment options like bilateral cardiac sympathectomy need to be studied further in order to investigate its optimal timing and use in ARVC/D management.80
Individuals who present with congestive heart failure are managed with diuretics and angiotensin‐converting enzyme (ACE) inhibitors or aldosterone inhibitors, with heart transplantation considered in terminal stages of the disease. Anticoagulation may be used in ARVC/D patients with large, hypokinetic RV and slow blood flow because of the risk of thrombosis.81 Management of family members of patients with ARVC/D is complex due to the incomplete penetrance and variable expressivity nature of the disease. This is beyond the scope of this review, and the reader is referred to the article here.44
6. PROGNOSTICATION
There are many factors that have been associated with adverse outcomes in patients with ARVC/D. These include clinical characteristics such as male gender, heart rate variability, and atrial arrhythmias.28, 82, 83, 84 Other important factors include electrocardiographic characteristics (atrioventricular block, T‐wave inversions),83, 85 echocardiographic parameters (RV diameter),86 and genetic factors (PKP2 carriers),87 all of which are associated with higher likelihood of ventricular arrhythmias, heart failure, and death. Biomarkers such as soluble ST2 have been associated with RV and LV functions in patients with ARVC/D and may aid in the determination of disease severity in this disease.88
7. CONCLUSION
ARVC/D is an inherited disease characterized by fibro‐fatty replacement of the right ventricular myocardium, which significantly increases the risk of paroxysmal ventricular arrhythmias and SCD. Diagnosis is based on the 2010 modified Task Force criteria, requiring clinical and family history, electrocardiography, and imaging. Diagnosis may be confirmed by endomyocardial biopsy, if the tissue is harvested from right ventricular wall or right interventricular septum and not from prominent right ventricular trabeculae. A further personalized approach in the management of the patients should be undertaken by specialists in high‐volume centers to enable practice of evidence‐based medicine in this condition with high morbidity and mortality.
CONFLICT OF INTERESTS
Authors declare no conflict of interests for this article.
ACKNOWLEDGEMENTS
Figures 1 and 2 provided courtesy of Dr. med. Georgi Wassilew of the Department of Pathology, Humboldt University, Berlin, Germany. GT and SW were supported by the Croucher Foundation of Hong Kong.
Li KHC, Bazoukis G, Liu T, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) in clinical practice. J Arrhythmia. 2018;34:11–22. https://doi.org/10.1002/joa3.12021
REFERENCES
- 1. Saffitz JE. The pathobiology of arrhythmogenic cardiomyopathy. Annu Rev Pathol. 2011;6:299–321. [DOI] [PubMed] [Google Scholar]
- 2. Calkins H. Arrhythmogenic right ventricular dysplasia. Curr Probl Cardiol. 2013;38:103–23. [DOI] [PubMed] [Google Scholar]
- 3. Protonotarios N, Tsatsopoulou A, Anastasakis A, et al. Genotype‐phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol. 2001;38:1477–84. [DOI] [PubMed] [Google Scholar]
- 4. Carvajal‐Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 1998;39:418–21. [DOI] [PubMed] [Google Scholar]
- 5. Sultan FA, Ahmed MA, Miller J, Selvanayagam JB. Arrhythmogenic right ventricular cardiomyopathy with biventricular involvement and heart failure in a 9‐year old girl. J Saudi Heart Assoc. 2017;29:139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki H, Sumiyoshi M, Kawai S, et al. Arrhythmogenic right ventricular cardiomyopathy with an initial manifestation of severe left ventricular impairment and normal contraction of the right ventricle. Jpn Circ J. 2000;64:209–13. [DOI] [PubMed] [Google Scholar]
- 7. Sen‐Chowdhry S, Syrris P, Prasad SK, et al. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–87. [DOI] [PubMed] [Google Scholar]
- 8. Lopez‐Moreno E, Jimenez‐Jaimez J, Macias‐Ruiz R, Sanchez‐Millan PJ, Alvarez‐Lopez M, Tercedor‐Sanchez L. Clinical profile of arrhythmogenic right ventricular cardiomyopathy with left ventricular involvement. Rev Esp Cardiol. 2016;69:872–4. [DOI] [PubMed] [Google Scholar]
- 9. Sen‐Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–20. [DOI] [PubMed] [Google Scholar]
- 10. Iyer VR, Chin AJ. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). Am J Med Genet C Semin Med Genet. 2013;163c:185–97. [DOI] [PubMed] [Google Scholar]
- 11. Ohno S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J Arrhythm. 2016;32:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vite A, Gandjbakhch E, Prost C, et al. Desmosomal cadherins are decreased in explanted arrhythmogenic right ventricular dysplasia/cardiomyopathy patient hearts. PLoS One. 2013;8:e75082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhurinsky J, Shtutman M, Ben‐Ze'ev A. Plakoglobin and beta‐catenin: protein interactions, regulation and biological roles. J Cell Sci. 2000;18:3127–39. [DOI] [PubMed] [Google Scholar]
- 14. Lewis JE, Wahl JK 3rd, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ. Cross‐talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol. 1997;136:919–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119–24. [DOI] [PubMed] [Google Scholar]
- 16. Fontaine G, Fontaliran F, Hebert JL, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med. 1999;50:17–35. [DOI] [PubMed] [Google Scholar]
- 17. Tse G, Lai ETH, Yeo JM, Tse V, Wong SH. Mechanisms of electrical activation and conduction in the gastrointestinal system: lessons from cardiac electrophysiology. Front Physiol. 2016;7:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tse G, Lai ET, Lee AP, Yan BP, Wong SH. Electrophysiological mechanisms of gastrointestinal arrhythmogenesis: lessons from the heart. Front Physiol. 2016;7:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tse G, Yeo JM. Conduction abnormalities and ventricular arrhythmogenesis: The roles of sodium channels and gap junctions. Int J Cardiol Heart Vasc. 2015;9:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age‐related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Opbergen CJ, Delmar M, van Veen TA. Potential new mechanisms of pro‐arrhythmia in arrhythmogenic cardiomyopathy: focus on calcium sensitive pathways. Neth Heart J. 2017;25:157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu Z, Zhu W, Wang C, et al. Genotype‐phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: a systematic review and meta‐analysis. Sci Rep. 2017;7:41387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoffmayer K, Scheinman M. Electrocardiographic patterns of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Front Physiol. 2012;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peters S. Refugee from Syria presents with chest pain due to arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2016;204:59–60. [DOI] [PubMed] [Google Scholar]
- 25. Ruwald AC, Marcus F, Estes NA 3rd, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung FP, Li HR, Chong E, et al. Seasonal variation in the frequency of sudden cardiac death and ventricular tachyarrhythmia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: the effect of meteorological factors. Heart Rhythm. 2013;10:1859–66. [DOI] [PubMed] [Google Scholar]
- 27. Wu L, Yao Y, Chen G, et al. Intracardiac thrombosis in patients with arrhythmogenic right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2014;25:1359–62. [DOI] [PubMed] [Google Scholar]
- 28. Camm CF, James CA, Tichnell C, et al. Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2013;10:1661–8. [DOI] [PubMed] [Google Scholar]
- 29. Mano H, Watanabe I, Okumura Y, et al. Atrial tachycardia in a patient with arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Arrhythm. 2013;29:238–41. [Google Scholar]
- 30. Burghouwt DE, Kammeraad JA, Knops P, du Plessis FA, de Groot NM. Bradyarrhythmias: first presentation of arrhythmogenic right ventricular cardiomyopathy? J Clin Med Res. 2015;7:278–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Antoniades L, Tsatsopoulou A, Anastasakis A, et al. Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin‐2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype‐phenotype relations, diagnostic features and prognosis. Eur Heart J. 2006;27:2208–16. [DOI] [PubMed] [Google Scholar]
- 34. Ward D, Syrris P, Sen‐Chowdhry S, McKenna WJ. Diagnosis; task force criteria including modifications for family members. In: Marcus FI, Nava A, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Milan, Italy: Springer Verlag, 2007; p. 87–96. [Google Scholar]
- 35. Strengths and weaknesses of the task force criteria ‐proposed modifications in arrhythmogenic right ventricular cardiomyopathy/dysplasia . In: Marcus FI, Nava A, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Milan, Italy: Springer Verlag, 2007; p. 97–104. [Google Scholar]
- 36. Femia G, Sy RW, Puranik R. Systematic review: impact of the new task force criteria in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2017;241:311–7. [DOI] [PubMed] [Google Scholar]
- 37. Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–50. [DOI] [PubMed] [Google Scholar]
- 38. Peters S. Prognostic value of epsilon waves in lead aVR in arrhythmogenic cardiomyopathy. Int J Cardiol. 2015;191:77–8. [DOI] [PubMed] [Google Scholar]
- 39. Protonotarios A, Anastasakis A, Tsatsopoulou A, et al. Clinical significance of epsilon waves in arrhythmogenic cardiomyopathy. J Cardiovasc Electrophysiol. 2015;26:1204–10. [DOI] [PubMed] [Google Scholar]
- 40. Saprungruang A, Tumkosit M, Kongphatthanayothin A. The presence of epsilon waves in all precordial leads (V1 ‐V6) in a 13‐year‐old boy with arrhythmogenic right ventricular dysplasia (ARVD). Ann Noninvasive Electrocardiol. 2013;18:484–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Platonov PG, Calkins H, Hauer RN, et al. High interobserver variability in the assessment of epsilon waves: implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2016;13:208–16. [DOI] [PubMed] [Google Scholar]
- 42. Peters S, Trümmel M. Diagnosis of arrhythmogenic right ventricular dysplasia‐cardiomyopathy: value of standard ECG revisited. Ann Noninvasive Electrocardiol. 2003;8(3):238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J, Yang B, Chen H, et al. Epsilon waves detected by various electrocardiographic recording methods in patients with arrhythmogenic right ventricular cardiomyopathy. Tex Heart Inst J. 2010;37:405–11. [PMC free article] [PubMed] [Google Scholar]
- 44. te Riele AS, James CA, Rastegar N, et al. Yield of serial evaluation in at‐risk family members of patients with ARVD/C. J Am Coll Cardiol. 2014;64:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Breithardt G, Cain ME, el‐Sherif N, et al. Standards for analysis of ventricular late potentials using high resolution or signal‐averaged electrocardiography. A statement by a Task Force Committee between the European Society of Cardiology, the American Heart Association and the American College of Cardiology. Eur Heart J. 1991;12:473–80. [DOI] [PubMed] [Google Scholar]
- 46. Tse G, Ali A, Prasad SK, Vassiliou V, Raphael CE. Atypical case of post‐partum cardiomyopathy: an overlap syndrome with arrhythmogenic right ventricular cardiomyopathy? BJR Case Rep. 2015;1:20150182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khaji A, Zhang L, Kowey P, Martinez‐Lage M, Kocovic D. Mega‐epsilon waves on 12‐lead ECG–just another case of arrhythmogenic right ventricular dysplasia/cardiomyopathy? J Electrocardiol. 2013;46:524–7. [DOI] [PubMed] [Google Scholar]
- 48. Dechering DG, Kochhauser S, Wasmer K, et al. Electrophysiological characteristics of ventricular tachyarrhythmias in cardiac sarcoidosis versus arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2013;10:158–64. [DOI] [PubMed] [Google Scholar]
- 49. Steckman DA, Schneider PM, Schuller JL, et al. Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012;110:575–9. [DOI] [PubMed] [Google Scholar]
- 50. Aquaro GD, Barison A, Todiere G, et al. Usefulness of combined functional assessment by cardiac magnetic resonance and tissue characterization versus task force criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2016;118:1730–6. [DOI] [PubMed] [Google Scholar]
- 51. Sen‐Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–53. [DOI] [PubMed] [Google Scholar]
- 52. te Riele AS, Tandri H, Bluemke DA. Arrhythmogenic right ventricular cardiomyopathy (ARVC): cardiovascular magnetic resonance update. J Cardiovasc Magn Reson. 2014;16:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated mutation carriers. Eur Heart J. 2015;36:847–55. [DOI] [PubMed] [Google Scholar]
- 54. Avella A, d'Amati G, Pappalardo A, et al. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 2008;19:1127–34. [DOI] [PubMed] [Google Scholar]
- 55. Kataoka S, Serizawa N, Kitamura K, et al. An overlap of Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Arrhythm. 2016;32:70–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maron BJ, Udelson JE, Bonow RO, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e273–80. [DOI] [PubMed] [Google Scholar]
- 57. Weiner RB, Baggish AL. Exercise‐induced cardiac remodeling. Prog Cardiovasc Dis. 2012;54:380–6. [DOI] [PubMed] [Google Scholar]
- 58. D'Ascenzi F, Pisicchio C, Caselli S, Di Paolo FM, Spataro A, Pelliccia A. RV remodeling in Olympic athletes. JACC Cardiovasc Imaging. 2017;10:385–93. [DOI] [PubMed] [Google Scholar]
- 59. Maron BJ, Pelliccia A. The heart of trained athletes: cardiac remodeling and the risks of sports, including sudden death. Circulation. 2006;114:1633–44. [DOI] [PubMed] [Google Scholar]
- 60. Romero J, Mejia‐Lopez E, Manrique C, Lucariello R. Arrhythmogenic right ventricular cardiomyopathy (ARVC/D): a systematic literature review. Clin Med Insights Cardiol. 2013;7:97–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dawson DK, Hawlisch K, Prescott G, et al. Prognostic role of CMR in patients presenting with ventricular arrhythmias. JACC Cardiovasc Imaging. 2013;6:335–44. [DOI] [PubMed] [Google Scholar]
- 62. Iriyama T, Kamei Y, Kozuma S, Taketani Y. Management of patient with arrhythmogenic right ventricular cardiomyopathy during pregnancy. J Obstet Gynaecol Res. 2013;39:390–4. [DOI] [PubMed] [Google Scholar]
- 63. Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29–37. [DOI] [PubMed] [Google Scholar]
- 64. Ermakov S, Scheinman M. Arrhythmogenic right ventricular cardiomyopathy – antiarrhythmic therapy. Arrhythm Electrophysiol Rev. 2015;4:86–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ermakov S, Gerstenfeld EP, Svetlichnaya Y, Scheinman MM. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2017;14:564–9. [DOI] [PubMed] [Google Scholar]
- 66. Schinkel AF. Implantable cardioverter defibrillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ Arrhythm Electrophysiol. 2013;6:562–8. [DOI] [PubMed] [Google Scholar]
- 67. Corrado D, Wichter T, Link MS, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36:3227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Silvano M, Corrado D, Kobe J, et al. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Herzschrittmacherther Elektrophysiol. 2013;24:202–8. [DOI] [PubMed] [Google Scholar]
- 69. Kirubakaran S, Bisceglia C, Silberbauer J, et al. Characterization of the arrhythmogenic substrate in patients with arrhythmogenic right ventricular cardiomyopathy undergoing ventricular tachycardia ablation. Europace. 2017;19:1049–62. [DOI] [PubMed] [Google Scholar]
- 70. Souissi Z, Boule S, Hermida JS, et al. Catheter ablation reduces ventricular tachycardia burden in patients with arrhythmogenic right ventricular cardiomyopathy: insights from a north‐western French multicentre registry. Europace. 2016. https://academic.oup.com/europace/advance-articleabstract/doi/10.1093/europace/euw332/2739018?redirectedFrom=fulltext [DOI] [PubMed] [Google Scholar]
- 71. Wei W, Liao H, Xue Y, et al. Long‐term outcomes of radio‐frequency catheter ablation on ventricular tachycardias due to arrhythmogenic right ventricular cardiomyopathy: a single center experience. PLoS One. 2017;12:e0169863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bai R, Di Biase L, Shivkumar K, et al. Ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy: arrhythmia‐free survival after endo‐epicardial substrate based mapping and ablation. Circ Arrhythm Electrophysiol. 2011;4:478–85. [DOI] [PubMed] [Google Scholar]
- 73. Romero J, Grushko M, Briceno DF, Natale A, Di Biase L. Radiofrequency ablation in arrhythmogenic right ventricular cardiomyopathy (ARVC). Curr Cardiol Rep. 2017;19:82. [DOI] [PubMed] [Google Scholar]
- 74. Philips B, te Riele AS, Sawant A, et al. Outcomes and ventricular tachycardia recurrence characteristics after epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2015;12:716–25. [DOI] [PubMed] [Google Scholar]
- 75. Jiang H, Zhang X, Yang Q, et al. Catheter ablation for ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: a systematic review and meta‐analysis. Acta Cardiol. 2016;71:639–49. [DOI] [PubMed] [Google Scholar]
- 76. Mussigbrodt A, Efimova E, Knopp H, et al. Epicardial ablation may not be necessary in all patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy and frequent ventricular tachycardia. Europace. 2017;19:2047–8. [DOI] [PubMed] [Google Scholar]
- 77. Santangeli P, Zado ES, Supple GE, et al. Long‐term outcome with catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8:1413–21. [DOI] [PubMed] [Google Scholar]
- 78. Mussigbrodt A, Efimova E, Knopp H, et al. Should all patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergo epicardial catheter ablation? J Interv Card Electrophysiol. 2017;48:193–9. [DOI] [PubMed] [Google Scholar]
- 79. Liao YC, Chung FP, Lin YJ, et al. The application of signal average ECG in the prediction of recurrences after catheter ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Int J Cardiol. 2017;236:168–73. [DOI] [PubMed] [Google Scholar]
- 80. Te Riele AS, Ajijola OA, Shivkumar K, Tandri H. Role of bilateral sympathectomy in the treatment of refractory ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2016;9:e003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wlodarska EK, Wozniak O, Konka M, Rydlewska‐Sadowska W, Biederman A, Hoffman P. Thromboembolic complications in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Europace. 2006;8:596–600. [DOI] [PubMed] [Google Scholar]
- 82. Battipaglia I, Scalone G, Macchione A, et al. Association of heart rate variability with arrhythmic events in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ J. 2012;76:618–23. [DOI] [PubMed] [Google Scholar]
- 83. Bhonsale A, James CA, Tichnell C, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol. 2013;6:569–78. [DOI] [PubMed] [Google Scholar]
- 84. Lin CY, Chung FP, Lin YJ, et al. Gender differences in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: clinical manifestations, electrophysiological properties, substrate characteristics, and prognosis of radiofrequency catheter ablation. Int J Cardiol. 2017;227:930–7. [DOI] [PubMed] [Google Scholar]
- 85. Kimura Y, Noda T, Matsuyama TA, et al. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: what are the risk factors? Int J Cardiol. 2017;241:288–94. [DOI] [PubMed] [Google Scholar]
- 86. Leren IS, Saberniak J, Haland TF, Edvardsen T, Haugaa KH. Combination of ECG and echocardiography for identification of arrhythmic events in early ARVC. JACC Cardiovasc Imaging. 2017;10:503–13. [DOI] [PubMed] [Google Scholar]
- 87. Bao J, Wang J, Yao Y, et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:552–6. [DOI] [PubMed] [Google Scholar]
- 88. Broch K, Leren IS, Saberniak J, et al. Soluble ST2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers. 2017;22:367–71. [DOI] [PubMed] [Google Scholar]
- 89. Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor‐beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–73. [DOI] [PubMed] [Google Scholar]
- 90. Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10:189–94. [DOI] [PubMed] [Google Scholar]
- 91. Severini GM, Krajinovic M, Pinamonti B, et al. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics. 1996;31:193–200. [DOI] [PubMed] [Google Scholar]
- 92. Taylor M, Graw S, Sinagra G, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy‐overlap syndromes. Circulation. 2011;124:876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Merner ND, Hodgkinson KA, Haywood AF, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right‐ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12‐p14. Am J Hum Genet. 2000;66:148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Klauke B, Kossmann S, Gaertner A, et al. De novo desmin‐mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19:4595–607. [DOI] [PubMed] [Google Scholar]
- 96. Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin‐2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–4. [DOI] [PubMed] [Google Scholar]
- 98. Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein‐2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–9. [DOI] [PubMed] [Google Scholar]
- 99. Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin‐2. Am J Hum Genet. 2006;79:978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Quarta G, Syrris P, Ashworth M, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–36. [DOI] [PubMed] [Google Scholar]
- 102. Erkapic D, Neumann T, Schmitt J, et al. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace. 2008;10:884–7. [DOI] [PubMed] [Google Scholar]
- 103. van Hengel J, Calore M, Bauce B, et al. Mutations in the area composita protein alphaT‐catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–10. [DOI] [PubMed] [Google Scholar]
- 104. Mayosi BM, Fish M, Shaboodien G, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10:pii: e001605. [DOI] [PubMed] [Google Scholar]