

Supplemental Digital Content is Available in the Text.
GM-CSF is a proinflammatory cytokine that plays a role in central pain pathways through the modulation of spinal glial cells.
Keywords: Granulocyte-macrophage colony-stimulating factor, Neuropathic pain, Inflammation, Microglia, Astrocyte, Blood–brain barrier, Cytokine
Abstract
With less than 50% of patients responding to the current standard of care and poor efficacy and selectivity of current treatments, neuropathic pain continues to be an area of considerable unmet medical need. Biological therapeutics such as monoclonal antibodies (mAbs) provide better intrinsic selectivity; however, delivery to the central nervous system (CNS) remains a challenge. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is well described in inflammation-induced pain, and early-phase clinical trials evaluating its antagonism have exemplified its importance as a peripheral pain target. Here, we investigate the role of this cytokine in a murine model of traumatic nerve injury and show that deletion of the GM-CSF receptor or treatment with an antagonizing mAb alleviates pain. We also demonstrate enhanced analgesic efficacy using an engineered construct that has greater capacity to penetrate the CNS. Despite observing GM-CSF receptor expression in microglia and astrocytes, the gliosis response in the dorsal horn was not altered in nerve injured knockout mice compared with wild-type littermate controls as evaluated by ionized calcium binding adapter molecule 1 (Iba1) and glial fibrillary acidic protein, respectively. Functional analysis of glial cells revealed that pretreatment with GM-CSF potentiated lipopolysaccharide-induced release of proinflammatory cytokines. In summary, our data indicate that GM-CSF is a proinflammatory cytokine that contributes to nociceptive signalling through driving spinal glial cell secretion of proinflammatory mediators. In addition, we report a successful approach to accessing CNS pain targets, providing promise for central compartment delivery of analgesics.
1. Introduction
Granulocyte-macrophage colony-stimulating factor is a small secreted hematopoietic growth factor required for the proliferation and differentiation of bone marrow precursor cells into distinct colonies comprised of granulocytes and macrophages. The GM-CSF receptor is a heterodimer that consists of a major ligand binding α-subunit and a common signal-transduction β-chain subunit, which in humans is shared with the interleukin (IL)-3 and IL-5 receptor.25 The α-subunit binds with high specificity and low affinity, and the β-chain subunit is responsible for Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, mitogen-activated protein kinase (MAPK) pathway, and the phosphoinositide 3-kinase (PI3K) pathway.20,25
Despite initially entering clinical practice for the treatment of chemotherapy-induced neutropenia,35 GM-CSF has since been proven to possess a broader range of biological activities within both innate and adaptive immunity.53 Pharmacological studies examining anti-ligand or anti-receptor antibodies in rodent models of inflammatory disease have shown profound effects on disease severity and progression.12,14,23 For example, in the antigen- and collagenase-induced models of arthritis, inhibition of signalling not only reduced cartilage destruction and synovitis12–14,23 but also attenuated pain.13,14 Likewise, in the complete Freund's adjuvant model of acute inflammatory pain13 and in a model of bone cancer,45 gene silencing of GM-CSF or its receptor was analgesic, whereas intraplantar administration of GM-CSF to the hind paw was painful.45
As administration of the nonsteroidal anti-inflammatory drug, indomethacin reversed pain but not the development of arthritis, it is apparent that separate downstream pathways are involved in GM-CSF–mediated pain and disease progression, as seen in the clinic.13,14 Although the precise mechanism for GM-CSF–dependent pain is unclear, the involvement of cyclo-oxygenase products implies an indirect effect on sensory neurons, possibly via prostaglandin.53 This notion is supported by reports of diverse ligand and receptor expression in the plasma, synovial fluid, and tissue of patients with rheumatoid arthritis (RA) as well as on circulating mononuclear cells,1,5,55 where it is believed that GM-CSF–driven modulatory effects on innate immune cells and other cell types induces cytokine secretion and the formation of proinflammatory cytokine networks that contribute to neuronal sensitization.25,35 Unsurprisingly, results from clinical trials targeting GM-CSF or its receptor in RA patients show significant analgesic efficacy, demonstrating its role as a pronociceptive cytokine.25 Granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR) expression has also been reported on peripheral nerves of healthy individuals and pancreatic cancer patients, as well as on peripheral nerves dispersed in the periosteum and throughout the bone matrix and in dorsal root ganglia sections of mice bearing calcaneus bone tumors, which suggests that GM-CSF may directly activate sensory neurons.45
To date, few studies have examined the role of GM-CSF in neuropathic pain and the potential mechanisms involved remain unclear.36,45 Here, we used an anti–GM-CSFR inhibitory mAb and GM-CSFR β-chain knockout (KO) mice to elucidate the role of the ligand in neuropathic pain pathways with a focus on the contribution of spinal glial cells.
2. Methods
2.1. In vivo study subjects
Studies were conducted on 8-week-old GM-CSFR β-chain deficient female mice on a C57/BL6 background, kindly provided by Jackson Laboratory (Bar Harbor, MA). Mice were housed in small groups in standard environmental conditions (12 hours light/dark cycle) with ad libitum access to food and water. Animal husbandry and the procedures used were in accordance with the guidelines of the AstraZeneca Animal Care Committee and complied with the Animals (Scientific Procedures) Act, 1986. All mice underwent insertion of transponders for identification purposes 5 days before the start of the study. For all studies, the experimenter was blinded to genotype and treatment, and animals were block randomised to treatment groups.
2.2. Paw pressure
In lightly restrained alert mice, noxious mechanical thresholds were examined using an Analgesymeter (7200; Ugo Basile, Monvalle VA, Italy).41 As previously described, the plantar surface of the hind paw was placed on a pedestal with a probe resting on the dorsal surface and increasing pressure was applied through the probe. The force at which a withdrawal response was observed was taken as the nociceptive threshold.41 Data were expressed as ipsilateral/contralateral ratios in grams.
2.3. Neuropathic pain model
Animals were anesthetized with 2% to 3% isoflurane (Abbott Animal Health, Maidenhead, United Kingdom), and the left hind paw was secured, shaved, and sterilized. A small incision was made midway of the left thigh to carefully expose and isolate approximately 1 cm of the sciatic nerve from neighbouring connective tissue. A 9-0 Virgin Silk suture (Ethicon, Livingston, United Kingdom) was inserted through the dorsal third of the nerve and tied tightly, as previously described.46 The incision was then closed using Vetbond, and the mice were allowed to recover for at least 3 days before commencement of testing. Sham-operated mice underwent the same procedure but the nerve was not ligated and the mice were sutured and allowed to recover.
Mice were tested on days 7 and 10 after surgery and were subsequently randomly allocated into several treatment groups with approximately equal ipsilateral/contralateral ratios as follows: sham + phosphate buffered saline (PBS), partial sciatic nerve ligation (PNL) + PBS, PNL + CAT004, PNL + CAM3003 (anti-mouse GM-CSFR–neutralising mAb, previously described in23), and PNL + Bbbt-CAM3003 (blood–brain barrier technology conjugated anti-mouse GM-CSFR mAb).
Mice were administered with either the isotype control, CAT004 (30 mg/kg subcut and 50 µg/5 µL intrathecal), anti-GM-CSFR mAb, CAM3003 (30 or 75 mg/kg subcut and 50 µg/5 µL intrathecal), Bbbt-CAM3003 (25, 50 or 100 mg/kg subcut), or with PBS vehicle on day 13 and were retested for changes in mechanical hyperalgesia 4 hours and on day 1, 2, 4, and 7 after dose. Intrathecal injections were performed under isoflurane anesthesia, and antibody or PBS was administered into the lumbar region of the spine (between the L5 and L6 vertebrae) using a 26 G 3/8-inch needle connected to a 25-µL Hamilton syringe.
2.4. Tissue preparation and immunohistochemistry
On completion of behavioural testing, mice were anesthetized with a lethal dose of sodium pentobarbital (Euthatal, Merial Animal Health Ltd, Woking, United Kingdom) and underwent transcardial perfusion with saline followed by fixation with 4% paraformaldehyde (PFA; VWR, Leicestershire, United Kingdom). Lumbar spinal cords were extracted and postfixed for 2 hours in PFA and cryoprotected in sucrose before embedding in optimum cutting temperature medium (VWR) and snap freezing with liquid nitrogen. Transverse spinal cord sections of the L4 and L5 lumbar region were cut on the cryostat and thaw mounted onto Superfrost plus microscope slides (VWR) and left to dry. As shown before, spinal cord sections were incubated overnight with primary antibody solution raised against Iba1 (rabbit anti-Iba1, 1:1000; Wako Chemicals, Neuss, Germany) to assess microgliosis or glial fibrillary acidic protein (GFAP) (rabbit anti-GFAP, 1:1000; DakoCytomation, Glostrup, Denmark) to assess astrogliosis followed by 2 hours incubation with the appropriate secondary antibody solution (1:1000; IgG conjugated Alexa Fluor 488 or 546; Invitrogen, United Kingdom). Slides were then cover slipped with vectashield mounting medium with DAPI (Vector Laboratories, Peterborough, United Kingdom), sealed with nail varnish and dried.9
2.5. Quantification of immunoreactivity
Images were visualised and captured on an Olympus microscope (Olympus KeyMed, Southend-On-Sea, United Kingdom). Analysis of Iba1 and GFAP immunoreactivity (IR) was performed by counting the number of positive profiles or measuring the IR signal, respectively, within 3 fixed 1 × 104 μm2 boxes in the lateral, central, and medial areas of the dorsal horn (DH). The nuclear marker DAPI was used to assist the identification of positive cell profiles, as previously described.8 Quantification was performed blind, and 3 sections per animal were assessed to obtain a mean value for both the ipsilateral and contralateral DHs.
2.6. Isolation and culture of neonatal glial cells
Primary cultures of mixed murine glial cells were prepared as previously described using a modified protocol.38,48 Briefly, brains from C57/BL6 P2 neonatal pups (purchased from Charles River, Saffron Walden, United Kingdom) were extracted, rolled across sterile filter paper to remove vasculature and meninges, and were mechanically dissociated. Cells were resuspended in 40 mL per 175 cm2 flask (Corning, Amsterdam, Netherlands) and were maintained in Dulbecco's Modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Invitrogen, Sigma Aldrich, Gillingham, United Kingdom) and incubated at 37°C (5% CO2/95% O2). A week later, 5 ng/mL of murine GM-CSF (R&D systems, Abingdon, United Kingdom) in fresh media was added to the flasks to expand yields, and the cells were maintained for a further week.48
Microglia were harvested by overnight shaking in an orbital shaker incubator (37°C, 180 rev/min), and the supernatant was collected from the flasks to obtain the dissociated microglia. Trypsin was subsequently added to lift the astrocytic layer. Microglia and astrocytes cells were plated as required for further experiments.
2.7. Verification of glial cell culture purity
2.7.1. Flow cytometry assessment
Microglia were collected from either a mixed glia T175 culture flask or purified by shaking as described above. Cell pellets were resuspended in FACS buffer (3% FCS/PBS) containing Mouse BD Fc Block (BD Biosciences, Wokingham, United Kingdom) with FITC rat anti-mouse CD11b or FITC rat IgG2b isotype control (BD Biosciences) and incubated for 30 minutes on ice. All antibodies had a final concentration of 1:50. Cells were washed by adding FACS buffer before being centrifuged (400g, 3 minutes) and analysed on the BD FACSCanto II system.
2.7.2. Immunocytochemistry assessment
As described above, microglia and astrocytes were harvested and plated into 24 well plates on glass slides and left to rest for 24 to 48 hours in DMEM with 10% FBS and 1% pen/strep at 37°C. Cells were fixed with 4% PFA for 10 minutes, washed with PBS, and incubated for 2 hours with primary antibody solution for the microglia marker Iba1 (rabbit anti-Iba1, 1:1000) or the astrocyte marker GFAP (rabbit anti-GFAP, 1:1000). Cells were then washed and incubated with the appropriate secondary antibody solution (1:1000; IgG conjugated Alexa Fluor 546) for 45 minutes. Glass slides were carefully mounted with Vectashield Mounting Medium with DAPI, and images were visualised and captured on an Olympus microscope (Olympus KeyMed). Positive cell profiles were identified by costaining with the nuclear marker DAPI.
2.8. Stimulation of neonatal glial cells and detection of cytokine release
Microglia and astrocytes cells were plated in 96 well plates at a density of 1 × 105 cells/well in DMEM with 10% FBS and 1% pen/strep. After 24 hours, the medium was replaced with FBS-free medium for a duration of 3 hours followed by 24 hours stimulation with increasing concentrations of lipopolysaccharide (LPS; Sigma Aldrich) +/−GM-CSF (5 ng/mL). Supernatants were collected and snap frozen with liquid nitrogen and stored at −80°C until further processing. The release of IL-1β, TNF-α, IL-6, and IL-10 was measured by an electrochemiluminescence immunoassay technique using the V-PLEX proinflammatory panel 1 mouse kit and analysed on a Sector Imager 6000 (Meso Scale Discovery, MD) according to manufacturer's guidelines.
2.9. Stimulation of neonatal glial cells and detection of phosphorylated extracellular signal-regulated kinase
Granulocyte-macrophage colony-stimulating factor–induced phosphorylated extracellular signal-regulated kinase (p-ERK) signals generated in neonatal microglia and astrocytes were measured using the p-ERK1/2 (Thr202/Tyr204) assay kit (Cisbio, Cedex, France) according to the 2-plate assay protocol detailed in the manufacturer's guide. Briefly, cells were stimulated for either 5, 15, or 30 minutes with GM-CSF (5 ng/mL) and the reaction was stopped by removing the media and adding supplemented lysis buffer to each well for 30 minutes at RT under shaking. After homogenisation, cell lysates were transferred to 384 well small volume white SV plates and premixed antibody solutions were added to each well and incubated for 2 hours. Between 2 and 24 hours, the fluorescence emission signal was read at 665 and 620 nm on an Envision plate reader (Perkin Elmer, Beaconsfield, United Kingdom). Phorbol 12-myristate 13-acetate (PMA, 100 ng/mL; Sigma Aldrich) was used as a positive control.
2.10. Development of the blood–brain barrier transmigrating anti–granulocyte-macrophage colony-stimulating factor receptor antibody
Development of the BBB-transporting antibody, Bbbt, is described in a separate article that is in preparation. Briefly, Bbbt was generated by phage display to select for single-chain variable fragments (scFvs) that bind mouse brain endothelial cells and cross the BBB in vitro and in vivo models. The bispecific Bbbt-CAM3003 antibody was expressed using a plasmid encoding the Bbbt antibody as a scFv fused using a (Gly4Ser)2 linker to the heavy chain of CAM3003 IgG1, that was cotransfected with a plasmid encoding the CAM3003 light chain. Antibodies were expressed as chimeric mouse IgG1 molecules in transiently transfected Chinese hamster ovary cells in serum-free media as described previously.17 Binding of CAM3003 mouse IgG1 and the Bbbt-CAM3003 bispecific to GM-CSFR was confirmed using a Biacore T100 system (GE Healthcare, Amersham, United Kingdom).
2.11. RNA extraction and quantification
Microglia and astrocytes cells were cultured as described above, and total RNA was purified from cell pellets using the Qiagen RNeasy plus mini kit according to manufacturer's instructions (RNeasy; Qiagen, Manchester, United Kingdom). After purification, total RNA was eluted using RNAse-free water, and the concentration and purity were measured using a NanoDrop ND-100 Spectrophotometer (Thermo Fisher Scientific, United Kingdom). In accordance with manufacturer's instructions, eluted RNA was subsequently reverse transcribed using the Life Tech VILO mastermix (Thermo Fisher Scientific). Relative mRNA expression levels were evaluated by Taqman real-time PCR using TaqMan Gene Expression Master Mix (Applied Biosystems, CA) and the Taqman Gene Expression assays for GM-CSF α-chain (Mm00438331_g1), β-chain (Mm00655745_m1), and eukaryotic 18S rRNA endogenous control (Thermo Fisher Scientific). Relative transcript levels were calculated using the ΔΔCT method, normalising against 18S. Control reactions using RNAse-free water as template or heat-inactivated reverse transcriptase produced no amplification signal.
2.12. Data and statistical analysis
All data were analysed using PRISM software. For multiple comparisons, one-way (immunohistochemical [IHC] data) or 2-way (behavioural and in vitro data) analysis of variance was used, followed by Tukey's or Sidak's post hoc test to determine individual group differences. In all cases, the data are presented as the mean ± SEM, and P < 0.05 was set as the level of statistical significance.
3. Results
3.1. Inhibition of granulocyte-macrophage colony-stimulating factor receptor signalling using an antireceptor mAb is analgesic in the partial sciatic nerve ligation model of neuropathic pain
To date, published literature has focused primarily on the peripheral inflammatory activity of GM-CSF in pain, and there has been limited exploration of a potential role in neuropathic pain.36,45 In light of this research gap and several studies showing expression of GM-CSFR in the CNS,16,43 we wanted to investigate the contribution of centrally mediated GM-CSF signalling in a nerve injury model of neuropathic pain.
It is well documented that the PNL model produces a robust mechanical and thermal hypersensitivity in the ipsilateral but not the contralateral hind paw, which may serve as a control. In agreement with this, nerve injured mice exhibited significant reductions in mechanical thresholds on days 7 and 10 compared with sham-operated controls (Fig. 1A). To test whether mechanical hyperalgesia induced by nerve injury was dependent on GM-CSF signalling, we investigated the effect of peripheral and central delivery of the inhibitory anti-mouse GM-CSFR mAb, CAM3003, previously described in.23 Peripheral administration of CAM3003 (Fig. 1A) had no effect at doses shown to be previously efficacious in the complete Freund's adjuvant model of inflammatory pain (data not shown) but significantly reversed mechanical hyperalgesia in nerve injured mice at 4 hours, 1 and 2 days when dosed intrathecally (Fig. 1B), increasing the ipsi/contra ratio from 53% to approximately 73% at 4 hours after dose and returning to baseline levels (∼58%) by day 4. The administration of isotype control, CAT004, and PBS vehicle, dosed subcutaneously or intrathecally, showed no reversal of this hyperalgesia at any time point, nor was there a reduction observed in the sham-operated groups or contralateral thresholds (Supplementary Figs. 1A and B, available online as supplemental digital content at http://links.lww.com/PAIN/A516) compared with pretreatment baseline levels, thus indicating that animal handling and drug administration procedures did not alter the response (Figs. 1A and B).
Figure 1.
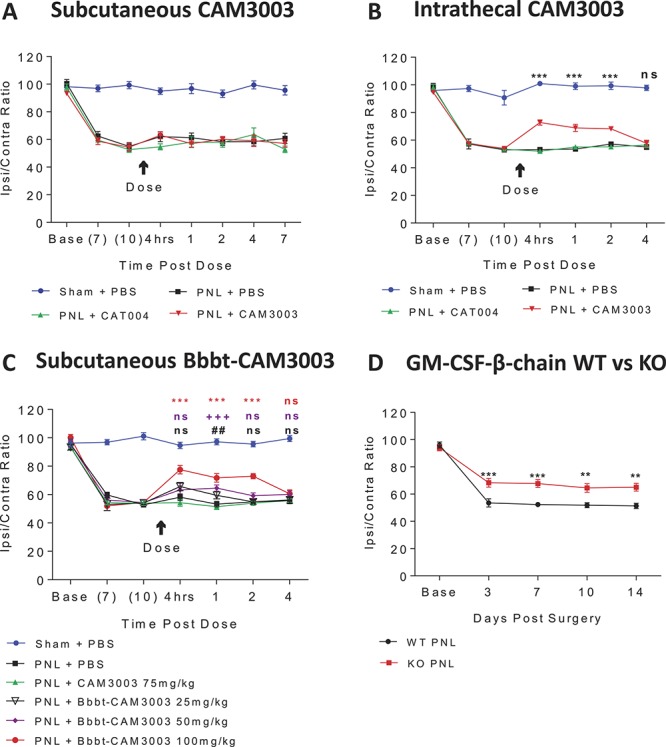
Inhibition of GM-CSFR signalling is analgesic in a mouse model neuropathic pain. Subcutaneous administration of anti–GM-CSFR mAb CAM3003 (30 mg/kg) or isotype control (CAT004, 30 mg/kg) had no effect on mechanical thresholds of PNL operated mice (A). Intrathecal administration of anti–GM-CSFR mAb CAM3003 (50 µg in 5 µL) attenuated mechanical hyperalgesia in PNL operated mice in contrast to isotype control (CAT004, 50 µg/5 µL). ***P < 0.001 PNL + CAT004 vs PNL + CAM3003 (B). Subcutaneous administration of anti–GM-CSFR mAb Bbbt-CAM3003 attenuated mechanical hyperalgesia in PNL-operated mice. ##P < 0.01 PNL CAM3003 vs PNL Bbbt 25 mg/kg, +++P < 0.001 PNL CAM3003 vs PNL Bbbt 50 mg/kg, ***P < 0.001 PNL CAM3003 vs PNL Bbbt 100 mg/kg (C). Nerve injured GM-CSFR β-chain KO mice (KO PNL) exhibited reduced mechanical hyperalgesia compared with WT littermate controls (WT PNL). **P < 0.01, ***P < 0.001 WT PNL vs KO PNL (D). All data are shown as the ratio of ipsilateral/contralateral weight bearing and are presented as the mean ± SEM, 2-way ANOVA with Tukey's multiple comparisons post hoc, n = 8 to 10. ANOVA, analysis of variance; GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; KO, knockout; PNL, partial sciatic nerve ligation; WT, wild-type
There is a growing agenda to enhance BBB penetration of CNS-targeted therapeutics and thus provide greater efficacy for a longer duration of time.52 To achieve delivery of a pharmacologic payload to the CNS, we generated a BBB-transporting antibody, Bbbt, which we have shown accumulates in the brain at up to 3% of the injected peripheral dose as opposed to non-BBB–targeted antibodies that typically accumulate at <0.1% of the injected dose (manuscript in preparation). Here, we are the first to re-engineer an anti–GM-CSFR mAb to increase its capacity to enter the CNS and have shown that subcutaneous administration of Bbbt-CAM3003 (100 mg/kg) strikingly alleviated mechanical hyperalgesia equivalent to that of intrathecal administration of CAM3003 (Fig. 1C). The ipsi/contra ratio was seen to increase from around 54% to 78% at 4 hours after dose and eventually return to 60% at day 4. As expected, lower doses of Bbbt-CAM3003 (25 and 50 mg/kg) had less analgesic efficacy for a shorter duration of time with mechanical thresholds beginning to decrease by day 2. Neither antibody nor PBS vehicle effected contralateral thresholds (Supplementary Fig. 1C, available online as supplemental digital content at http://links.lww.com/PAIN/A516).
As subcutaneous administration of CAM3003 lacked efficacy in contrast to Bbbt-CAM3003, these data suggest a centrally mediated mechanism for GM-CSF signalling in the neuropathic pain model as opposed to a peripherally mediated mechanism in the inflammation-induced pain model. However, a potential role at the level of the dorsal root ganglia cannot be unequivocally ruled out. In addition, we have demonstrated an exciting new approach to increase drug delivery access to the CNS and achieve greater therapeutic efficacy.
3.2. Nerve injured granulocyte-macrophage colony-stimulating factor receptor β-chain knockout mice exhibit attenuated mechanical hyperalgesia, which is not associated with an altered gliosis response
To consolidate the role of GM-CSFR–mediated signalling in neuropathic pain, we tested the mechanical thresholds of GM-CSFR β-chain KO mice after PNL surgery. Although the β-chain is still present, these mice show no signalling through the family of cognate cytokines, including GM-CSF, IL-3, and IL-5.27 Nerve injured β-chain KO mice exhibited significantly attenuated mechanical hyperalgesia compared with wild-type (WT) controls from day 3 to 14 with the ipsi/contra ratio averaging around 67% in the KO compared with 52% in the WT over the 12 testing days (Fig. 1D). The magnitude of the analgesic effect was notably consistent to that observed after intrathecal delivery of CAM3003 (Fig. 1B) and indicates that GM-CSFR deletion impairs the development and maintenance of behavioural hypersensitivity. These findings are in accordance with our mAb data and thus support our hypothesis of a pronociceptive role of GM-CSF. Notably, both genotypes exhibited equivalent baseline mechanical thresholds, and so GM-CSF may not be involved in steady-state nociception. There were no significant effects seen on contralateral thresholds (Supplementary Fig. 1D, available online as supplemental digital content at http://links.lww.com/PAIN/A516).
To determine, which cell types could be driving the centrally mediated pronociceptive effects of GM-CSF observed in our in vivo data, we profiled mRNA expression of its cognate receptor in glial cell cultures. In line with previous reports,42 Taqman data revealed both α- and β-chain subunit expression in neonatal microglia and astrocyte cultures (Figs. 2A and B).
Figure 2.
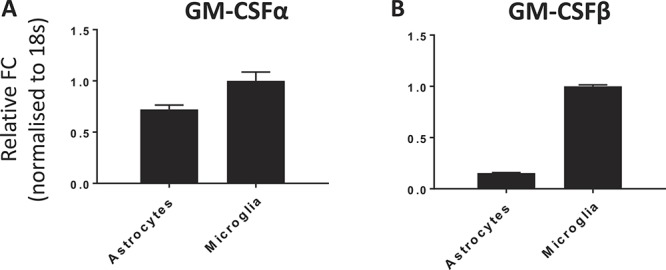
Verification of GM-CSFR mRNA expression. Taqman profiles confirm expression of both GM-CSFR α-chain (A) and common β-chain (B) subunits in astrocytes and microglia. GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor.
Since the role of microglia and astrocytes in pain pathways are well established28,56 and we saw glial expression of GM-CSFR, we sought to characterise nerve injury–induced gliosis in the KO mouse to determine whether this is altered with GM-CSFR deletion. Quantification of the Iba1 IR signal 14 days post PNL revealed that both genotypes showed a significant increase in Iba1-positive cells in the ipsilateral DH of the spinal cord compared with the contralateral DH (WT ipsi: 10.6 ± 1.2, KO ipsi: 9.7 ± 1.1 vs WT contra: 4.5 ± 0.8, and KO contra: 4.6 ± 0.5, Figs. 3A and B). There were no significant differences between genotypes, suggesting that microgliosis after nerve injury does not require GM-CSF signalling. Notably, KO microglia exhibited typical deramified and amoeboid morphology in response to injury to that seen in the WT.
Figure 3.
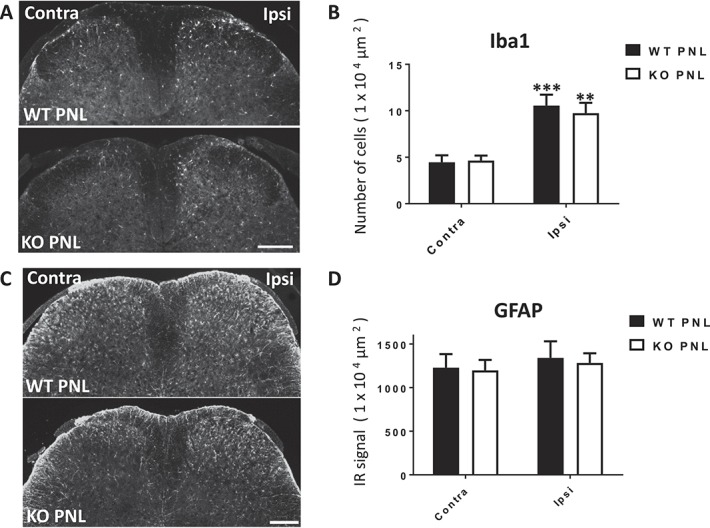
Nerve injury–induced gliosis is normal in GM-CSFR β-chain KO mice. At 14 days after PNL, there was a significant increase in Iba1-positive cells in the ipsilateral (Ipsi) DH of WT PNL and KO PNL mice, compared with the contralateral (Contra) DH (A), quantified in (B). There was bilateral expression of GFAP-positive cells in the DH of nerve injured WT and KO mice (C), quantified in (D). There were no significant differences between genotypes. Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001 vs contralateral, one-way ANOVA with Tukey's multiple comparisons post hoc, n = 3 to 6. Scale bar = 200 μm. ANOVA, analysis of variance; DH, dorsal horn; GFAP, glial fibrillary acidic protein; GM-CSFR, granulocyte-macrophage colony-stimulating factor receptor; Iba1, ionized calcium binding adapter molecule 1, KO, knockout; PNL, partial sciatic nerve ligation; WT, wild-type.
Quantification of the GFAP IR signal 14 days post PNL revealed that both genotypes exhibited a similar astrocytic response with a trend towards a higher ipsilateral signal compared with the contralateral DH. There were no significant differences between genotypes (WT ipsi: 1340.9 ± 189.7, KO ipsi: 1283 ± 119.4 vs WT contra: 1228.2 ± 157.0, and KO contra: 1198.1 ± 110.2, Figs. 3C and D). In summary, these data indicate that GM-CSF signalling contributes to pain-associated behaviour that is independent of a gliosis response. However, we must note that our evaluation was limited to the examination of 2 pan glial cell markers that only provide information about the number and morphology of the cells and so, based on these data alone, we cannot exclude the contribution of microglia and astrocytes.
3.3. Granulocyte-macrophage colony-stimulating factor receptor is functionally expressed in glial cells and is associated with an upregulation of lipopolysaccharide-induced cytokine release through a phosphorylated extracellular signal-regulated kinase pathway
As we found no immunophenotypic differences in the KO glial response, we sought to investigate whether there was a functional impact of GM-CSFR deletion by assessing the ability of microglia and astrocytes to launch an inflammatory response. This was based on the notion that an altered cytokine profile may correspond to an altered nerve injury–induced inflammatory response in the KO.
We, first, verified the purity of our primary microglia and astrocyte cultures through flow cytometry and immunocytochemistry for the glial-specific markers, CD11b/Iba1, and GFAP, respectively. In accordance with the literature, we demonstrated that our cultures consistently exceeded a purity of at least 95% (Supplementary Fig. 2, available online as supplemental digital content at http://links.lww.com/PAIN/A516). Activation of GM-CSFR is known to stimulate the JAK-STAT, MAPK, and PI3K signalling pathways.20,25 As p-ERK is a key MAPK involved in the transduction of nociceptive signalling and has been documented to be expressed in both microglia and astrocytes,30 we examined GM-CSF–induced p-ERK responses in these cells to confirm functional expression of the receptor. Phospho-ERK activation was demonstrated in neonatal microglial cultures (EC50 = 0.6 ng/mL; Fig. 4A), which was dose-dependently inhibited by CAM3003 (IC50 = 240 pM; Fig. 4C). Thus, confirming functional expression of GM-CSFR and antibody-dependent inhibition of signalling in microglia in accordance with our Taqman data. Isotype control, CAT004, had no significant effect in this system. In contrast, GM-CSF stimulation did not induce a p-ERK signal from neonatal astrocytes and although the baseline levels of p-ERK seemed high in these cultures, the reason for the lack of a GM-CSF effect remains unclear (Fig. 4B).
Figure 4.

Granulocyte-macrophage colony-stimulating factor (GM-CSF) induces p-ERK signalling in microglia, which is inhibited by antibody neutralisation. Neonatal mouse microglia (A) and astrocytes (B) were stimulated with GM-CSF for 5 to 60 minutes, and p-ERK activation was quantified. Granulocyte-macrophage colony-stimulating factor–induced (0.6 ng/mL) p-ERK activation in microglia could be inhibited by preincubation (30 minutes) with the anti-GM-CSFR mAb CAM3003 (C). Phorbol 12-myristate 13-acetate (100 ng/mL) was used as a positive control. Data are presented as mean ± SEM, n = 3.
There are several reports on the effects of GM-CSF on the release of mediators that may contribute directly or indirectly to nociceptive transduction.25,26 Based on this evidence, we screened the effects of GM-CSF on glial cell release of several putative cytokines in vitro. Cotreatment with recombinant mouse GM-CSF (5 ng/mL) significantly potentiated LPS-induced (1 µg/mL) IL-1β (1.7-fold), TNFα (1.7 fold), and IL-10 (1.3-fold) release from microglia as well as IL-6 release across several doses of LPS (0.06 µg/mL: 20.1-fold; 0.25 µg/mL: 2.0-fold; and 1 µg/mL: 1.6-fold; Fig. 5A), as previously shown.39 Granulocyte-macrophage colony-stimulating factor did not exhibit a significant effect on LPS-induced IL-1β, TNFα, and IL-10 release from astrocyte cultures but significantly potentiated IL-6 release by 1.6-fold (1 µg/mL LPS; Fig. 5B). Together these data support our hypothesis that GM-CSF promotes the release of proinflammatory cytokines that can directly and indirectly drive nociceptive transmission, which may be absent in the KO.
Figure 5.

Granulocyte-macrophage colony-stimulating factor potentiates LPS-induced cytokine release from neonatal microglia and astrocytes. Neonatal microglia (A) and astrocytes (B) were stimulated for 24 hours with increasing concentrations of LPS (0.06-1 µg/mL) −/+GM-CSF (5 ng/mL), and the protein expression of IL-1β, TNF-α, IL-6 and IL-10 released into the supernatant was measured by ELISA. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs control (−GM-CSF); 2-way ANOVA with Sidak's multiple comparisons test, n = 3 to 4. ANOVA, analysis of variance; IL, interleukin; LPS, lipopolysaccharide; TNF, tumor necrosis factor.
4. Discussion
Given the well-described roles of GM-CSF in peripheral inflammation and inflammation-induced pain, we hypothesized that it may also play a role in neuroinflammation associated with neuropathic pain. To date, there are very few studies examining GM-CSF in neuropathy, and to the best of our knowledge, we are the first to show that central inhibition of GM-CSFR signalling in the PNL model is analgesic, thus supporting a role of GM-CSF in neuropathic pain. In agreement with reports of mRNA and protein expression of GM-CSFR in the CNS,42,43 we confirmed transcript expression in glial cells and showed that nerve injured mice treated with a centrally delivered antagonizing mAb had attenuated mechanical hyperalgesia. By contrast, peripheral delivery was found to have no effect, thus implicating a centrally driven mechanism in line with successful demonstration of enhanced analgesic efficacy with a BBB penetrative construct that facilitates CNS delivery, as we have previously shown.52 In conjunction with these data, nerve injured KO mice had attenuated mechanical hyperalgesia compared with WT controls, equivalent to the magnitude of alleviation generated by CAM3003. Given that we observed functional expression of both GM-CSFR α- and β-chain subunits in microglia and astrocytes, we profiled the immunophenotypic response of these cells to nerve injury in the presence and absence of the receptor. Interestingly, we did not detect differences in spinal gliosis between genotypes but observed enhanced proinflammatory cytokine release from glial cell cultures after GM-CSF stimulation, thus implicating a GM-CSF–mediated neuroinflammatory mechanism.
Although GM-CSFR–positive macrophages may be driving peripheral sensitization in inflammation-induced pain, an analgesic effect in nerve injured mice was only exhibited by intrathecal administration of the inhibitory mAb, CAM3003, thus supporting a centrally mediated mechanism. A significant body of literature exists around a role of the receptor in the CNS, and studies to date have focused on models of MS (eg, EAE; reviewed in15) and stroke (reviewed in32). In EAE, GM-CSF has been widely described as pathogenic through actions on myeloid cells resulting in proinflammatory cytokine production. By contrast, GM-CSF has also been associated with beneficial neuroprotective effects in rodent models of stroke as a result of direct action on neurons.43 In the CNS, GM-CSFR expression has been described on microglia, astrocytes, neurons,22,33,42,43 and oligodendrocytes,4 but we only observed immunoreactivity that predominantly colocalised with glial-specific markers in the mouse spinal cord, and we saw no evidence for neuronal expression (data not shown). In vitro data confirmed functional expression of the receptor in microglia and astrocytes, so we focused our study on these cell types.
The role of glial cells in chronic pain mechanisms has been well characterised in several neuropathic and inflammatory pain paradigms, and many of the initial findings were based on studies using general glial inhibitors. A striking observation from our study was the significant gliosis response in the KO, despite the reduced pain behaviours compared to the WT. Although dissociation between gliosis and chronic pain pathways has been documented before,11,29,40 we acknowledge the fact that our studies were limited to gross immunophenotypic differences, and further work examining markers of activation may elucidate the contribution of these cells. Interestingly, it has been shown that direct administration of exogenous GM-CSF into the brain induces a rapid gliosis response,22 but it seems that in this model, GM-CSF was not the sole driver of gliosis. Nonetheless, the absence of overt change in the gliosis response does not preclude a glial-mediated effect of GM-CSF in this model.
The presence of proinflammatory mediators, particularly IL-1β, TNF, and IL-6, within the joints of patients with RA and osteoarthritis6,19 corroborates animal studies that have shown that inhibition of GM-CSF signalling reduces cytokine release in joint tissue.12,13 As we found no IHC evidence of an altered glial response in association with the KO behavioural phenotype, we sought to examine whether GM-CSFR deletion impairs the ability of microglia and astrocytes to launch an inflammatory response based on evidence that GM-CSF contributes to proinflammatory cytokine networks,25,26 and that we had shown glial expression of both the α- and β-chain receptor subunits. Here, we used a simplified in vitro model to measure protein expression changes in a selection of putative mediators subsequent to LPS ± GM-CSF stimulation. The rationale for this experimental paradigm is based on published and in-house data that in response to nerve injury, glial cells contribute to pain pathways through releasing a range of mediators such as TNF-α, IL-1β, and IL-6 that promote a proinflammatory environment. Lipopolysaccharide stimulation increases cytokine synthesis in a toll-like receptor-4 (TLR4) dependent manner, which is a key molecule involved in generating responses to pathogen-associated molecular patterns and endogenous molecules released from damaged tissue.34 Activation of TLR4 subsequently activates nuclear factor-κB and the transcription of proinflammatory mediators.34 Whilst the application of LPS to spinal cord induces pain behaviour, TLR4 inhibition attenuates pain-associated behaviours in models of nerve injury.47 Therefore, as we are proposing a glial-mediated mechanism in the GM-CSF signalling pathway, LPS-induced priming of these cells is a relevant in vitro paradigm to enable us to further elucidate the KO behavioural phenotype. Although GM-CSF did not seem to induce cytokine release in its own right, preincubation with GM-CSF significantly potentiated LPS-induced IL-1β, TNFα, IL-6, and IL-10 release from microglia as previously reported39 and IL-6 release from astrocytes.
A number of groups have demonstrated algesic effects of proinflammatory cytokines and have shown that whilst cytokine administration elicits the onset of mechanical and thermal hyperalgesia in rodents, this can be reversed using neutralising antibodies or inhibitors.2 Accordingly, in experimental models of neuropathic and inflammatory pain, cytokines and their cognate receptors are upregulated and the administration of neutralising antibodies or inhibitors alleviates pain-associated behaviours.2,37 Dysregulation of cytokines has also been reported in patients with painful neuropathies. For example, compared with healthy controls, cytokines such as IL-1β and TNFα were found to be elevated in both blood and CSF patient samples.3,49 Despite there being fewer studies showing reversal of pain with neutralising or inhibitory treatments, there is evidence of beneficial effects in some subsets of patients.10 In the spinal cord, proinflammatory cytokines secreted by glial cells have been shown to directly enhance neuronal excitability and synaptic transmission while simultaneously reducing inhibitory currents.18,31,50,54 This suggests that GM-CSF may not affect gliosis but instead elicit its pronociceptive effects indirectly through glial cell–mediated cytokine release, a response that may be impaired in the KO. Evidently, further study would be required to confirm this link in vivo.
Functional analysis revealed that GM-CSF–induced dose-dependent p-ERK responses in neonatal microglia could be inhibited by treatment with CAM3003. Unexpectedly, we did not detect p-ERK responses in astrocytes for reasons that remain unclear but could be related to high p-ERK basal levels. We did, however, observe significant potentiation of LPS-induced IL-6 release after GM-CSF pretreatment, which is indicative of a functional GM-CSFR signalling response in these cells despite being less marked than those seen in microglia. Literature reports of GM-CSFR expression and signalling in astrocytes are few,24 with the focus tending to be on GM-CSF secretion by astrocytes rather than their response to this factor. Nonetheless, astrocytes have been implicated in the maintenance of neuropathic pain,21 and our data suggests that they may indirectly contribute to the behavioural phenotype we observed.
In summary, we have identified for the first time, a centrally mediated mechanism for GM-CSFR signalling in the context of neuropathic pain that is driven primarily by glial cell mediator release. In light of these rodent data and the availability of a human anti–GM-CSFR antibody, mavrilimumab with demonstrated safety in the clinic,7 it is interesting to consider the potential of this molecule as a pain therapeutic. Moreover, reports showing increased GM-CSF in intervertebral disc associated with pain44 and increased serum GM-CSF in patients with disc herniation and pain,51 identify a potential population of patients who may benefit from anti–GM-CSFR antibody treatment. Finally, we have also demonstrated efficacy with a novel construct exemplifying an engineered solution to overcome traversing the endothelial cell–derived BBB and deliver proteins to the CNS.52 As previously shown, greater penetration of the CNS results in greater analgesia and prolonged exposure in the central compartment results in a longer duration of the analgesia.52 Certainly, this approach provides a promising avenue for centrally targeted entities not only for the treatment of neuropathic pain but also for other CNS diseases.
Conflict of interest statement
Authors who were employees of MedImmune or Astrazeneca at the time of the work have a theoretical conflict of interest through being employed by the organisation that both funded the work and has a potential commercial interest in the findings.
Supplementary Material
Acknowledgements
The authors thank all the staff of MedImmune's Biological Services unit for their invaluable help and technical assistance in performing the surgeries. All work described in this article was funded by MedImmune.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/A516.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J Immunol 1991;146:3365–71. [PubMed] [Google Scholar]
- [2].Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol 2010;229:26–50. [DOI] [PubMed] [Google Scholar]
- [3].Backonja MM, Coe CL, Muller DA, Schell K. Altered cytokine levels in the blood and cerebrospinal fluid of chronic pain patients. J Neuroimmunol 2008;195:157–63. [DOI] [PubMed] [Google Scholar]
- [4].Baldwin GC, Benveniste EN, Chung GY, Gasson JC, Golde DW. Identification and characterization of a high-affinity granulocyte-macrophage colony-stimulating factor receptor on primary rat oligodendrocytes. Blood 1993;82:3279–82. [PubMed] [Google Scholar]
- [5].Berenbaum F, Rajzbaum G, Amor B, Toubert A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw 1994;5:43–6. [PubMed] [Google Scholar]
- [6].Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, van den Berg WB. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum 2010;62:647–57. [DOI] [PubMed] [Google Scholar]
- [7].Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, Szombati I, Esfandiari E, Sleeman MA, Kane CD, Cavet G, Wang B, Godwood A, Magrini F. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis 2013;72:1445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain 2007;11:223–30. [DOI] [PubMed] [Google Scholar]
- [9].Clark AK, Staniland AA, Marchand F, Kaan TKY, McMahon SB, Malcangio M. P2X7 dependent release of interleukin 1β and nociception in the spinal cord following lipopolysaccharide. J Neurosci 2010;30:573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cohen SP, Bogduk N, Dragovich A, Buckenmaier CC, III, Griffith S, Kurihara C, Raymond J, Richter PJ, Williams N, Yaksh TL. Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety study of transforaminal epidural etanercept for the treatment of sciatica. Anesthesiology 2009;110:1116–26. [DOI] [PubMed] [Google Scholar]
- [11].Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol 1997;79:163–75. [DOI] [PubMed] [Google Scholar]
- [12].Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res 2001;3:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cook AD, Pobjoy J, Sarros S, Steidl S, Durr M, Lacey DC, Hamilton JA. Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann Rheum Dis 2013;72:265–70. [DOI] [PubMed] [Google Scholar]
- [14].Cook AD, Pobjoy J, Steidl S, Durr M, Braine EL, Turner AL, Lacey DC, Hamilton JA. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther 2012;14:R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Croxford AL, Spath S, Becher B. GM-csf in neuroinflammation: licensing myeloid cells for tissue damage. Trends Immunol 2015;36:651–62. [DOI] [PubMed] [Google Scholar]
- [16].Dame JB, Christensen RD, Juul SE. The distribution of granulocyte-macrophage colony-stimulating factor and its receptor in the developing human fetus. Pediatr Res 1999;46:358–66. [DOI] [PubMed] [Google Scholar]
- [17].Daramola O, Stevenson J, Dean G, Hatton D, Pettman G, Holmes W, Field R. A high-yielding CHO transient system: coexpression of genes encoding EBNA-1 and GS enhances transient protein expression. Biotechnol Prog 2014;30:132–41. [DOI] [PubMed] [Google Scholar]
- [18].DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. PAIN 2001;90:1–6. [DOI] [PubMed] [Google Scholar]
- [19].Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis 1993;52:870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 2005;25:405–28. [DOI] [PubMed] [Google Scholar]
- [21].Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010;7:482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Giulian D, Li J, Li X, George J, Rutecki PA. The impact of microglia-derived cytokines upon gliosis in the CNS. Dev Neurosci 1994;16:128–36. [DOI] [PubMed] [Google Scholar]
- [23].Greven DE, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, van Nieuwenhuijze A, Lewis A, Heasmen S, McCourt M, Corkill D, Dodd A, Elvin J, Statache G, Wicks IP, Anderson IK, Nash A, Sleeman MA, Tak PP. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis 2015;74:1924–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guillemin G, Boussin FD, Le Grand R, Croitoru J, Coffigny H, Dormont D. Granulocyte macrophage colony stimulating factor stimulates in vitro proliferation of astrocytes derived from simian mature brains. Glia 1996;16:71–80. [DOI] [PubMed] [Google Scholar]
- [25].Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008;8:533–44. [DOI] [PubMed] [Google Scholar]
- [26].Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov 2016;16:53–70. [DOI] [PubMed] [Google Scholar]
- [27].Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, Ramshaw H, Woodcock JM, Xu Y, Guthridge M, McKinstry WJ, Lopez AF, Parker MW. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 2008;134:496–507. [DOI] [PubMed] [Google Scholar]
- [28].Hansen RR, Malcangio M. Astrocytes–multitaskers in chronic pain. Eur J Pharmacol 2013;716:120–8. [DOI] [PubMed] [Google Scholar]
- [29].Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience 2000;98:585–98. [DOI] [PubMed] [Google Scholar]
- [30].Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev 2009;60:135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 2008;28:5189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lanfranconi S, Locatelli F, Corti S, Candelise L, Comi GP, Baron PL, Strazzer S, Bresolin N, Bersano A. Growth factors in ischemic stroke. J Cell Mol Med 2011;15:1645–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia 1994;12:309–18. [DOI] [PubMed] [Google Scholar]
- [34].Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 2008;42:145–51. [DOI] [PubMed] [Google Scholar]
- [35].Metcalf D. Hematopoietic cytokines. Blood 2008;111:485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mirski R, Reichert F, Klar A, Rotshenker S. Granulocyte macrophage colony stimulating factor (GM-CSF) activity is regulated by a GM-CSF binding molecule in Wallerian degeneration following injury to peripheral nerve axons. J Neuroimmunol 2003;140:88–96. [DOI] [PubMed] [Google Scholar]
- [37].Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev 2006;51:240–64. [DOI] [PubMed] [Google Scholar]
- [38].Ni M, Aschner M. Neonatal rat primary microglia: isolation, culturing and selected applications. Curr Protoc Toxicol 2010;12:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, Mizuno T, Suzumura A. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation 2012;9:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003;306:624–30. [DOI] [PubMed] [Google Scholar]
- [41].Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther 1957;111:409–19. [PubMed] [Google Scholar]
- [42].Sawada M, Itoh Y, Suzumura A, Marunouchi T. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci Lett 1993;160:131–4. [DOI] [PubMed] [Google Scholar]
- [43].Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, Gassler N, Aronowski J, Mier W, Kirsch F, Dittgen T, Bach A, Sommer C, Schneider A. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF). J Cereb Blood flow Metab 2008;28:29–43. [DOI] [PubMed] [Google Scholar]
- [44].Schroeder GD, Markova DZ, Koerner JD, Rihn JA, Hilibrand AS, Vaccaro AR, Anderson DG, Kepler CK. Are Modic changes associated with intervertebral disc cytokine profiles? Spine 2017;17:129–34. [DOI] [PubMed] [Google Scholar]
- [45].Schweizerhof M, Stosser S, Kurejova M, Njoo C, Gangadharan V, Agarwal N, Schmelz M, Bali KK, Michalski CW, Brugger S, Dickenson A, Simone DA, Kuner R. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med 2009;15:802–7. [DOI] [PubMed] [Google Scholar]
- [46].Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. PAIN 1990;43:205–18. [DOI] [PubMed] [Google Scholar]
- [47].Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 2005;102:5856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Ståhl S, Franssen EH, Dodd RB, Qamar S, Gomez Perez-Nievas B, Nicol LSC, Eketjäll S, Revell J, Jones C, Billinton A, St George-Hyslop PH, Chessell I, Crowther DC. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer's disease-associated H157Y variant. EMBO Mol Med 2017;9:1366–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Uceyler N, Rogausch JP, Toyka KV, Sommer C. Differential expression of cytokines in painful and painless neuropathies. Neurology 2007;69:42–9. [DOI] [PubMed] [Google Scholar]
- [50].Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci 2001;24:450–5. [DOI] [PubMed] [Google Scholar]
- [51].Weber KT, Satoh S, Alipui DO, Virojanapa J, Levine M, Sison C, Quraishi S, Bloom O, Chahine NO. Exploratory study for identifying systemic biomarkers that correlate with pain response in patients with intervertebral disc disorders. Immunol Res 2015;63:170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Webster CI, Hatcher J, Burrell M, Thom G, Thornton P, Gurrell I, Chessell I. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. PAIN 2017;158:660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol 2016;12:37–48. [DOI] [PubMed] [Google Scholar]
- [54].Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science 2000;288:1765–9. [DOI] [PubMed] [Google Scholar]
- [55].Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford) 2012;51:451–9. [DOI] [PubMed] [Google Scholar]
- [56].Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 2014;15:43–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.