

This review by Jain and Epstein discusses the developmental processes that influence cardiac lineage decisions and cellular competence and advances our understanding of cardiac cell specification, gene regulation, and chromatin organization and how they impact cardiac development.
Keywords: cardiac progenitor cells, gene regulation, lineage restriction, nuclear architecture
Abstract
The mature heart is composed primarily of four different cell types: cardiac myocytes, endothelium, smooth muscle, and fibroblasts. These cell types derive from pluripotent progenitors that become progressively restricted with regard to lineage potential, giving rise to multipotent cardiac progenitor cells and, ultimately, the differentiated cell types of the heart. Recent studies have begun to shed light on the defining characteristics of the intermediary cell types that exist transiently during this developmental process and the extrinsic and cell-autonomous factors that influence cardiac lineage decisions and cellular competence. This information will shape our understanding of congenital and adult cardiac disease and guide regenerative therapeutic approaches. In addition, cardiac progenitor specification can serve as a model for understanding basic mechanisms regulating the acquisition of cellular identity. In this review, we present the concept of “chromatin competence” that describes the potential for three-dimensional chromatin organization to function as the molecular underpinning of the ability of a progenitor cell to respond to inductive lineage cues and summarize recent studies advancing our understanding of cardiac cell specification, gene regulation, and chromatin organization and how they impact cardiac development.
Cardiac progenitors and progressive lineage restriction
During mammalian cardiac development, two distinct mesodermal pools of cardiac progenitor cells—the first and second heart field—give rise to the four major cell types of the mature heart (Meilhac et al. 2004). First heart field cells derive from anterior lateral plate mesoderm and initially form a crescent straddling the midline of the early embryo. Second heart field cells are located medial to the first heart field at the cardiac crescent stage. These cells are excluded from the early linear heart tube but migrate into the heart after looping of the heart tube has initiated, invading both the inflow and outflow tracts, contributing cells to the right ventricle and atria (Kelly et al. 2001; Mjaatvedt et al. 2001; Waldo et al. 2001; Cai et al. 2003). Expression of both Nkx2-5 (Lints et al. 1993) and Tbx5 (Bruneau et al. 1999) primarily marks first heart field cells, and Islet1 expression marks second heart field cells (Cai et al. 2003), although none of these are restricted entirely to one heart field or the other. The first and second heart field pools of progenitors are thought to be multipotent, although the molecular programs, timing, and position of differentiation into various lineages are distinct.
The specification of cardiac myocytes, endothelium, and smooth muscle from multipotent progenitors via stages of progressive lineage restriction is analogous to the process by which hematopoietic stem cells give rise to the various lineages of blood. Clonal studies using murine and human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) in culture have demonstrated that cardiac progenitor cells are multipotent and can give rise to the aforementioned derived cell types (Kattman et al. 2006, 2011; Moretti et al. 2006; Wu et al. 2006). It is likely that a more detailed understanding of the extrinsic and intrinsic factors that regulate the process of fate determination during cardiac development will provide therapeutic insights for the treatment of cardiac disease, just as our understanding of blood formation has yielded potent medications such as erythropoietin, granulocyte–macrophage-stimulating factor, and other colony-stimulating factors that are used clinically to treat human disease. The ability to model cardiac differentiation in vitro has led to the identification of factors that modulate cardiac lineage determination. These studies have been instrumental in defining the conditions and factors sufficient for differentiating ESCs into multipotent cardiac progenitor cells characterized by expression of Pdgfrα and Flk1 (Kattman et al. 2006, 2011). A recurrent theme of these studies is the critical roles for transient Wnt activation followed by Bmp signaling. It remains unclear whether some or all Pdgfα+/Flk1+ cardiac progenitor cells are competent to adopt all cardiac cell fates, and the plasticity, potential, and defining characteristics of the intermediary progenitors have yet to be fully defined.
Recent studies undertaken by the Bruneau (Devine et al. 2014) and Blainpain (Lescroart et al. 2014) laboratories address the question of whether and to what extent multipotent cardiac progenitors exist in vivo during cardiac development. These groups independently used complimentary genetic approaches to determine the fate of early murine mesodermal progenitors that express Mesp1. These mesodermal progenitors give rise to a broad range of tissues, including the heart, and are present in the embryo even before Pdgfrα+/Flk1+ cardiac progenitors are evident. Surprisingly, these studies revealed that most Mesp1+ cells are not tripotent or quadripotent but rather bipotent and even more frequently unipotent (Devine et al. 2014; Lescroart et al. 2014). Lineage tracing of an early subset of Mesp1+ cells, corresponding to the first heart field, suggests that these cells overwhelmingly give rise to myocytes, although a smaller number of clones were composed of endothelial cells (Lescroart et al. 2014). In both studies, only a small number of clones were composed of multiple cell types, suggesting that true multipotent progenitors are likely to be rare and short-lived in vivo. Single-cell data demonstrate heterogeneity of Mesp1+ cells, correlating with a large pool of unipotent lineage-restricted progenitors (Lescroart et al. 2014). The Bruneau laboratory (Devine et al. 2014) also demonstrated that a subset of Mesp1 cells labeled by expression of a specific enhancer of Smarcd3 (Baf60c) is lineage-restricted such that these cells can give rise only to cardiac myocytes even at very early time points before the delineation of first and second heart fields is apparent. Complementary studies focused on the first heart field found that a subset of these progenitors expresses Hcn4 and gives rise primarily to myocytes and the cardiac conduction system. Hcn4 is re-expressed in endothelium at later stages of cardiac development and is not expressed in second heart field progenitors (Spater et al. 2013). Thus, it is possible that the tripotent progenitor stage of cardiac lineage restriction is short-lived in vivo, although additional genetic tools, including additional inducible cre drivers specific for the first and second heart field coupled with clonal analyses in vivo, will be helpful for investigating this question in more detail. One important conclusion in common to these studies, however, is that some degree of lineage commitment occurs very early during cardiac development—earlier than previously appreciated.
Additional studies suggest that anatomical specification (e.g., atrial vs. ventricular myocytes) also occurs earlier than previously appreciated. The Dubois laboratory (Bardot et al. 2017) recently identified a progenitor pool marked by Foxa2 that contributes to ventricular myocardium but not atrial myocardium. Foxa2 cells give rise to cardiomyocytes, endothelium, smooth muscle, fibroblasts, and epicardium. Foxa2 progenitors are localized to the apex of the cardiac crescent, indicating that this region of the crescent is already specified to give rise to ventricular muscle. Atrial cardiomyocyte identity is regulated by Coup-TFII, which functions in part by repressing ventricular gene expression (Wu et al. 2013). However, the identity of a chamber-specific progenitor that gives rise to atrial cardiac myocytes remains elusive. In zebrafish, injury induces transdifferentiation of atrial to ventricular myocytes, although it is unclear whether this occurs in mammals or whether the transdifferentiation process involves reversion to chamber-specific progenitors or a direct conversion to a mature cell phenotype (Zhang et al. 2013). In addition, recent studies demonstrate that Six2 is dynamically expressed in a subset of second heart field cells (Zhou et al. 2017). Lineage tracing of early (embryonic day 6.0 [E6.0]) Six2+ cells indicates that this pool of progenitors gives rise primarily to the right ventricle, while cells expressing Six2 at E10.0 contribute primarily to the pulmonary trunk and ductus arteriosus. Finally, studies in the chick suggest that a specialized region of mesoderm termed the tertiary heart field contributes to pacemaker cells of the sino–atrial node (Bressan et al. 2013). It is yet to be determined whether specialized regions of mesoderm, distinct from the first and second heart fields, provide contributions to portions of the conduction system in mice or humans.
An active area of research in the cardiac development and regeneration fields involves the characterization of putative intermediate progenitor cell types that may be bipotent and those that may be restricted and committed to a specific lineage but are able to expand—analogous to the erythroblast, for example, in the red blood cell lineage. Recently, we identified the cardiomyoblast in the murine embryo that is entirely committed to the cardiac myocyte lineage but is proliferative and able to undergo clonal expansion (Jain et al. 2015b). The cardiomyoblast is characterized by expression of the atypical homeodomain-encoding gene Hopx and derives from both first and second heart field precursors. Lineage tracing using inducible cre recombinase knocked into the endogenous Hopx locus indicates that Hopx-expressing cardiomyoblasts give rise exclusively to cardiac myocytes and their derivatives, including components of the mature conduction system such as Purkinje cells and other specialized cells of the conduction system. Interestingly, although nearly all cardiac myocytes derive from Hopx-expressing cardiomyoblasts, a localized population of cardiac muscle surrounding the pulmonary veins does not derive from Hopx-expressing precursors. The cell of origin for these myocytes remains unknown.
In the first heart field, Hopx expression initiates very shortly after that of Nkx2-5, and, in the second heart field, Hopx expression follows that of Isl1 (Chen et al. 2002; Shin et al. 2002; Jain et al. 2015b). Fate mapping of Nkx2-5- or Isl1-expressing cells demonstrates that they give rise to endothelium, smooth muscle, cardiac fibroblasts, and cardiac myocytes (Moses et al. 2001; Stanley et al. 2002; Yang et al. 2006; Ma et al. 2008; Jain et al. 2015b). Thus, the onset of expression of Hopx correlates with lineage restriction to the cardiac myocyte fate—a fact that allows for the identification of the precise stage and location during development when commitment takes place. In the first heart field, this occurs at very early time points, even before the linear heart tube has formed, in agreement with the Mesp1 studies described above. In the second heart field, commitment takes place just as progenitor cells are entering the heart via either the inflow or the outflow tract, at approximately day 9.5 of murine embryonic development (E9.5). As these cells enter the heart, they are exposed to a localized domain of Bmp4 expression, and Hopx is expressed (correlating with commitment) just as active Wnt signaling is extinguished. Studies in vitro and in vivo using Hopx-null cells indicate that Hopx functions to integrate the local Bmp signals with repression of Wnt signaling within the progenitor cells—a cascade of events triggered by the presence of local environmental Bmp cues that culminates in lineage commitment and differentiation of the cardiomyoblast (Jain et al. 2015b). Other progenitor pools likewise have been shown to rely on a critical balance of Bmp and Wnt signaling to maintain tissue homeostasis. It will be of interest to determine whether Hopx plays a similar role in mediating cross-talk between niche signals in other stem cell and progenitor populations where it is expressed, including the brain, lung, hair follicle, hematopoietic system, and intestine (Takeda et al. 2011, 2013; Jain et al. 2015a; Li et al. 2015; Palpant et al. 2017).
Sophisticated lineage tracing approaches such as these have advanced our understanding of cell fate choices during cardiogenesis. Dual lineage tracing strategies have also been used and have provided impressive data on the spatio–temporal dynamics of different populations contributing to heart formation (Engleka et al. 2012; Devine et al. 2014). In addition, combining sophisticated multicolor lineage reporters with cre drivers, informed by single-cell and clonal data sets, will likely increase the resolution of lineage restriction and the definition of distinct subpopulations of progenitors. An emerging theme is that cellular competence and plasticity must be appropriately constrained in order for faithful adoption of fate to occur. Alternative fate pathways need to be effectively repressed in order for lineage identity to be properly established and maintained.
In a true “tour de force,” Loh and the Weissman group (Loh et al. 2016) provided a road map to human mesodermal lineage development using human ESC and murine models. A prominent theme in their studies is that it is necessary to block the ability of the cell to adopt a lineage alternate to the ultimate fate of the cell at multiple steps of lineage specification. This work highlighted, as is widely known in the field, the fact that a limited number of developmental morphogens is required reiteratively at multiple steps of cardiogenesis and mesoderm development in general and that the effect of these morphogens on responding cells is dependent on the characteristics of those cells and their competence to respond. Hence, outstanding questions are as follows: How is specificity of the effector molecules achieved? How are entire sets of lineage-specific gene programs activated or repressed to affect a lineage decision? These questions have direct relevance to our understanding of developmental “competence”; i.e., the dynamic ability of a cell to respond to specific environmental cues. We now focus on potential mechanisms that regulate cellular competence and through which lineage-specific programs of gene expression are established, including combinatorial assembly of transcription factor complexes, the role of noncoding RNAs (ncRNAs), and the potential role of chromatin packaging within the three-dimensional ecosystem of the nucleus.
‘Chromatin competence’: a nuclear architecture model for cellular competence
Conrad Waddington (Waddington 1940), often considered the “father of epigenetics,” wrote extensively about the concept of cellular competence, which he recognized as an intrinsic but transient property of a cell to respond to inductive signals. It was in the section on competence in his classic “Organisers and Genes” (Waddington 1940) that he introduced his famous “epigenetic landscape” to depict progressive lineage restriction and fate determination. Waddington (1940) recognized that much of the conundrum concerning the pleiotropic effects of common inducing agents could be explained by the intrinsic capacity of a receiving cell to respond to the inductive signal. Thus, mesenchymal stem cells respond to Bmp by forming bone, while cardiac progenitor cells respond to Bmp by forming cardiac muscle. What is the mechanism within the responding cell to account for such different outputs in response to a common morphogen? Competence can be partially explained by the expression—or lack of expression—of specific receptors for inducing factors or necessary components of downstream signaling pathways, including transcription factors capable of activating lineage-specific genes, without which a cell is unable to fully respond to an inductive signal. However, we propose that cellular competence may be largely explained and regulated by orchestrated and dynamic mechanisms for packaging and organizing the genome within the nucleus; i.e., by regulation of nuclear architecture (Fig. 1).
Figure 1.
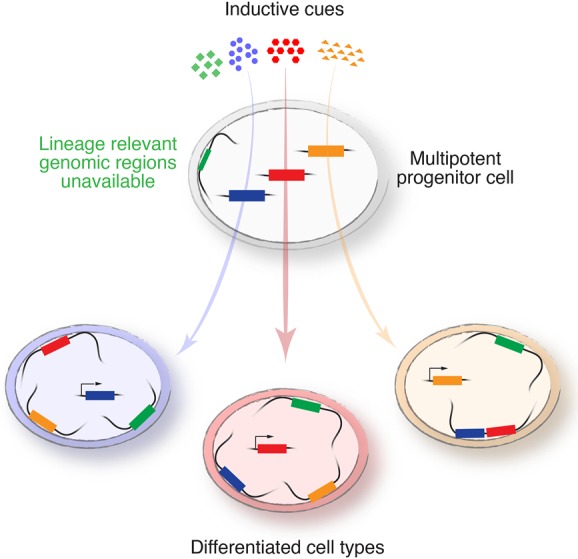
Proposed model of “chromatin competence.” We posit that spatial organization of the genome in three dimensions renders parts of the genome available for transcriptional activation, while other areas (e.g., those tethered to the nuclear lamina) are relatively less available. Inductive cues or morphogens can actively regulate only available regions of the genome in any given responding cell. Hence, high-order genome organization may regulate cellular competence; i.e., the ability of the cell to respond to a specific inductive cue. The combination of inductive cues and cellular competence shapes cellular identity.
This model, which we term “chromatin competence,” posits that the availability of regions of the genome containing critical lineage-specific regulators and gene programs is regulated through interactions of chromatin domains with the inner nuclear lamina and perhaps by other mechanisms as well. Regions of chromatin tightly associated with the nuclear lamina are termed lamina-associated domains (LADs) and are generally heterochromatic and tightly repressed (Guelen et al. 2008). We propose that these regions are relatively inaccessible for activation in response to inductive signals and that lineage programs associated with LADs must be released from their association with the nuclear periphery in order for the cell to be competent to activate those lineage programs. Likewise, when a lineage choice is made, such as when a cardiac progenitor cell becomes a cardiomyoblast, critical genes for alternate lineage programs will be sequestered in LADs and repressed for the duration of the lineage of that cell. Using murine and ESC models, we demonstrated that Hdac3 functions to tether chromatin to the nuclear periphery in a fashion that does not require its enzymatic deacetylase activity and that normal cardiac myocyte development from multipotent progenitor cells is associated with translocation of cardiac myocyte-specific genes from the nuclear periphery to the nucleoplasm, where they can be actively expressed. Precocious removal of Hdac3 in multipotent progenitors “releases” cardiac genes from LADs and leads to acceleration of cardiogenic differentiation (Poleshko et al. 2017). Thus, cellular identity and competence to respond to inductive signals may be at least partially defined by the “LAD map” of the cell, and disruption of LADs may enhance plasticity, restore competence, and increase the efficiency of transdifferentiation, reprogramming, or malignant transformation. The tethering of LADs to and release of LADs from the inner nuclear lamina thus emerge as a new mechanism of controlling gene expression and cellular competence that does not necessarily equate to gene activation or repression but rather to the availability of gene loci to be accessed by more traditional activation and repression complexes. The regulation of LAD tethering and release is an exciting area of active investigation.
The role of the nuclear lamina in the regulation of gene expression via interactions with chromatin is relevant to our understanding of a spectrum of human cardiac disorders known as the laminopathies, which include several types of muscular dystrophy often associated with cardiomyopathy. It will be of interest to determine whether manifestations of these human diseases are related to altered fate specification, plasticity, and competence as opposed to other commonly ascribed structural roles of the nuclear lamina not directly related to the regulation of gene expression.
Nuclear pore proteins as regulators of gene expression
Like the nuclear lamina, the nuclear pore has been implicated in the regulation of gene expression and interacts with chromatin. The nuclear pore complex is composed of ∼30 different nuclear pore proteins, and its primary ascribed function is nucleocytoplasmic transport. However, nontransport functions have been ascribed recently to nuclear pore components, including regulation of spatial organization of the genome and gene expression. Nuclear pore proteins can regulate transcription both “on pore” and “off pore” (Raices and D'Angelo 2012; Beck and Hurt 2017). Chromatin associates with the nuclear pore complex at the nuclear membrane (“on pore”), and this interaction serves, in part, to stabilize enhancer–promoter contacts (Pascual-Garcia et al. 2017). Unlike the repressed heterochromatin that interacts with the inner nuclear lamina, chromatin at the nuclear pore tends to be euchromatic and actively transcribed, consistent with the “gene gating” hypothesis (Blobel 1985). However, some nuclear pore proteins can also bind chromatin within the nucleoplasm (“off pore”), away from the nuclear membrane (Capelson et al. 2010; Kalverda et al. 2010). It remains unclear whether these individual nuclear pore proteins shuttle back and forth between the nuclear pore and the nucleoplasm or whether two different pools of these nuclear pore proteins exist. It is also unclear whether human diseases and cancers associated with mutations and translocations of nuclear pore components are caused by “on-pore” or “off-pore” effects. Of particular interest to cardiac biology are missense mutations in the nuclear pore protein NUP155 that have been identified in patients with atrial fibrillation. Global-null Nup155 mice die during embryogenesis, but heterozygous mice are viable and exhibit atrial fibrillation (Zhang et al. 2008). Studies from the Molkentin laboratory (Kehat et al. 2011) have shown a link between HDAC proteins and Nup155. In an elegant series of experiments, Kehat et al. (2011) demonstrated that Hdac4 physically interacts with Nup155, and Nup155 co-occupies a subset of Hdac4-bound regions of chromatin. Furthermore, abrogation of the interaction of Nup155 with Hdac4 results in relocalization of candidate loci, including Nppb, Cacna1c, Acta1, and Pln, away from the nuclear periphery, suggesting that the Nup155/Hdac4 complex normally tethers chromatin to the pore. This protein complex also appears necessary for Hdac4-dependent cardiac hypertrophy. Although the nuclear pore has been viewed as a complex conduit allowing passive and regulated movement across the nuclear membrane, it is now evident that more complex involvement of nuclear pore components in gene regulation exist, with implications for cardiac homeostasis and disease.
Higher-order genome organization
Genome compaction and organization are highly regulated processes, and causative links between high-order chromatin structure and function are being established. Chromosomes occupy specific locations within the nucleus and fold into defined territories. Territories are largely divided into two classes: the A compartment, which is frequently euchromatic and competent for transcription (Lieberman-Aiden et al. 2009), and the B compartment, which is usually late replicating, heterochromatic, and transcriptionally silent (Ryba et al. 2010). Chromatin contacts and chromatin loops occur more frequently within each compartment type than across compartments.
Smaller units of relatively conserved chromatin organization are termed topologically associated domains (TADs) that have been mapped using various chromosome capture techniques. TADs are relatively insulated from each other and largely invariant between cell types. However, much greater heterogeneity has been observed in sub-TAD architecture among different cell types. Chromatin looping—for example, between enhancers and promoters—is thought to be largely constrained such that they occur within individual TADs (Yu and Ren 2017). Emerging studies suggest important changes in chromatin interactions in the pathogenesis of congestive heart failure (Rosa-Garrido et al. 2017). The functional importance of the establishment and maintenance of proper TAD boundaries has also been highlighted by a series of recent studies. It has been reported recently that disruption of TADs contributes to aberrant gene expression during the progression of various cancers. Mutations in isocitrate dehydrogenase, which are associated with various brain cancers such as glioblastoma, result in hypermethylation of DNA at TAD boundaries, and reversal of this hypermethylation normalizes gene expression (Flavahan et al. 2016). Microdeletions are a common feature of T-cell acute lymphoblastic leukemia (T-ALL) and alter sub-TAD boundaries. This disruption in boundary formation results in precocious expression of proto-oncogenes, which could drive T-ALL progression (Hnisz et al. 2016). CCCTC-binding factor CTCF is a zinc finger nucleic acid-binding protein that can demarcate TAD boundaries. It has been demonstrated recently that the orientation of CTCF-binding sites is critical to forming correct loops, and incorrect orientation results in abnormal TAD formation and abnormal gene expression (Narendra et al. 2015). Nora et al. (2017) used a clever inducible approach to reversibly degrade CTCF in murine ESCs to demonstrate that CTCF insulates a subset of TADs. In addition, cohesin and related proteins have been found to be critical in chromatin loop formation and regulation of TAD stability (Haarhuis et al. 2017; Hansen et al. 2017; Rao et al. 2017; Schwarzer et al. 2017). Thus, there is a nascent but rapidly growing appreciation of the regulation of chromatin topology that is likely to impact gene expression, lineage stability, cellular competence, and cell fate. Future studies will undoubtedly leverage recent advances in sequencing and genomic technologies to understand how chromatin topology influences organogenesis in vivo.
ncRNAs
ncRNAs exist in many sizes and have enormous influence on a wide range of cellular functions, including gene expression. The important role of microRNAs in cardiac biology has been reviewed (Mendell and Olson 2012), and many exciting areas of ncRNA biology cannot be highlighted here due to space considerations. Rather, we focus on a few examples of the emerging role of long ncRNAs (lncRNAs) in cardiac development and homeostasis. lncRNAs have emerged as important regulators of various facets of cardiac biology, including lineage identity and differentiation. In some cases, they are thought to regulate nuclear architecture and can help to mediate long-range chromatin interactions and looping. lncRNAs (defined as noncoding transcripts >200 nucleotides in length) can be 5′ capped, alternatively spliced, and polyadenylated. The ever-increasing number of known ncRNAs is well into the thousands and includes specific examples of important regulators of cardiac development and homeostasis in the adult, and it seems certain that additional important roles will continue to emerge (Engreitz et al. 2016).
Braveheart, identified in the Boyer laboratory (Klattenhoff et al. 2013), is a cardiac lncRNA expressed from early time points in embryogenesis. Loss of Braveheart during mouse ESC differentiation results in a dramatic reduction in myogenesis. Transcriptome analysis suggests that Braveheart is upstream of Mesp1 and controls expression of critical regulators of cardiomyogenesis, including Hand1, Hand2, Nkx2-5, and Tbx20. Loss of Braveheart does not alter expression of neuronal-specific genes upon differentiation of mutant cultures into neural lineages, suggesting a specific function during cardiac development. RNAi-mediated knockdown of Braveheart in neonatal murine myocytes results in a loss of sarcomere gene expression. At the structural level, the 5′ asymmetric G-rich internal loop of Braveheart is necessary for proper cardiomyogenesis. This structural motif mediates interactions with transcription factors, including CCHC-type zinc finger nucleic acid-binding protein, which has been implicated in myotonic dystrophy (Xue et al. 2016). In vivo studies, including the identification of the specific cells that express Braveheart during embryogenesis, may yield additional insights into how this lncRNA modulates cardiac development and homeostasis.
Fendrr is another lncRNA that regulates cardiogenesis (Grote et al. 2013; Sauvageau et al. 2013). Fendrr contains seven exons and is transcribed upstream of the Foxf1 locus in mice. Genetic knockout of Fendrr results in embryonic lethality by E13.75, characterized by body wall defects, myocardial dysfunction, and myocardial hypoplasia. Fendrr is expressed by Eomes+ cardiac progenitor cells, and those lacking Fendrr express unusually high levels of Gata6 and Nkx2-5, transcription factors critical for body wall morphogenesis and cardiac development. Mechanistically, Fendrr binds PRC2 and TrxG/MLL complexes and controls histone modifications at loci relevant to body wall morphogenesis and cardiogenesis (Grote et al. 2013). Abnormalities in some endoderm-derived tissues, including the lung and gastrointestinal tract, have also been reported (Sauvageau et al. 2013).
Two prominent examples of lncRNAs controlling myocardial homeostasis in the adult have emerged. A cluster of alternatively spliced transcripts is transcribed from the Myh7 locus, antisense to Mhy7. The Chang laboratory (Han et al. 2014) termed these transcripts myosin heavy chain-associated RNA transcript (MyHEART and Mhrt). Mhrt transcripts are down-regulated ∼50% in murine models of pressure overload. Using a transgenic model, the Chang laboratory (Han et al. 2014) maintained expression of Mhrt during transaortic constriction (a model of pressure overload), and this intervention protected animals from cardiomyopathy and fibrosis. Overexpression of Mhrt 1–2 wk after transaortic constriction protected against cardiomyopathy but not to the same degree as overexpression starting prior to transaortic constriction. Mhrt binds the helicase domain of Brg1 and inhibits the ability of Brg1 to bind chromatinized targets in vitro. In pressure overload and other states of stress, there is a shift from Myh6 expression to Myh7 expression, which is mediated by Brg1. Therefore, taken together, the investigators suggest that Mhrt acts as a buffer to tone levels of Myh6 and Myh7 by acting as a “decoy” for Brg1. In states of stress, a reduction in the expression of Mhrt leads to an increased ability of Brg1 to drive expression of Myh6, potentially contributing to the deleterious consequences associated with pressure overload and cardiomyopathy.
More recently, a cardiac-enriched lncRNA called cardiac hypertrophy-associated epigenetic regulator (Chaer) was identified and characterized (Wang et al. 2016). Chaer is a two-exon lncRNA and lies upstream of Hopx on chromosome 5 in the mouse genome. Loss of Chaer in the adult attenuated cardiac hypertrophy in response to transaortic constriction, but no discernable phenotype was detected under basal conditions. Overexpression of Chaer also up-regulated genes typical of the hypertrophic response. Loss of Chaer increased total levels of H3K27me2 and H3K27me3, while overexpression decreased H3K27me3 levels. Chaer was found to interact with Suz12 and Ezh2, components of the PRC2 complex. The investigators demonstrated that this interaction was affected by mTOR signaling and responsible for maintaining H3K27me3 at regulatory regions of select cardiac genes.
Micropeptides
Assessment of some lncRNA function has been complicated recently by the exciting observation that some ncRNAs are actually coding micropeptides (e.g., small ORF peptides/proteins) that can have important functions in cardiac and skeletal muscle biology. For example, Myoregulin is a skeletal muscle-specific 46-amino-acid micropeptide discovered by the Olson laboratory (Anderson et al. 2015) when they were analyzing a presumed skeletal muscle lncRNA. Myoregulin localizes to the sarcoplasmic reticulum (SR) membrane in adult skeletal muscle, where it plays a role in the regulation of calcium uptake, affecting contractile function. Other micropeptides, including Dworf (Nelson et al. 2016) and Myomixer (Bi et al. 2017), have also emerged as important regulators of skeletal muscle function. Hence, at least some of the functions attributed to ncRNAs may in fact be due to surreptitiously encoded micropeptides.
Transcription factor cooperativity
Another significant concept in the regulation of cardiac gene expression relates to the combinatorial power of transcription factor cooperativity, which contributes in important ways to lineage-specific gene regulation. Pioneering studies demonstrated that transcription factors bind cooperatively to activate gene programs during skeletal myogenesis, particularly interactions between Mef2 family members and members of the myogenic family of basic helix–loop–helix (bHLH) proteins, including MyoD (Kaushal et al. 1994; Molkentin et al. 1995, 1996; Black and Olson 1998). Myogenic bHLH proteins can heterodimerize and bind to “E-boxes,” consensus CANNTG sequences. Mef2-binding sites are frequently positioned in close proximity to E-boxes in skeletal muscle genes. An elegant series of experiments demonstrated that a direct interaction between the MADS box of Mef2 proteins and the bHLH region of myogenic bHLH factors is required for activation of the myogeneic gene program (Molkentin et al. 1995, 1996). Multiple modes of binding DNA for these complexes have been described, including direct binding of both bHLH heterodimers and Mef2 simultaneously and independent binding via the bHLH or MADS domain alone (Molkentin and Olson 1996). Intriguingly, a recent study demonstrated an induction of Nup210 during skeletal myogenesis, which resulted in recruitment of Mef2C to the nuclear pore. The complex of Nup210 and Mef2C at the nuclear periphery regulates expression of critical genes required for skeletal myogenesis in zebrafish (Raices et al. 2017). It remains unclear whether this interaction requires bHLH factors (such as MyoD), classic transcriptional inducers of myogenesis, but it will be interesting to fully dissect subnuclear transcription factor localization in relation to spatial positioning of the genome. Nonetheless, the classic studies of transcriptional cooperativity between MEF2 and MyoD family members were seminal in understanding skeletal muscle differentiation and paved the way for future studies determining how cooperativity between core transcription factors regulates cardiogenesis.
Three cardiac transcription factors critical for cardiac development that have been intensively studied with regard to cooperative binding are Tbx5, Nkx2-5, and Gata4. The importance of these factors to the core transcriptional networks involved in cardiomyogenesis is highlighted by the fact that many strategies for direct transdifferentiation of fibroblasts into cardiac myocytes use these factors (Addis and Epstein 2013). Studies in Drosophila point to a regulatory circuit involving Nkx, Gata, T-box, Wnt, and Tgf-β family members (Bodmer and Frasch 1999). In addition, mutations in the genes encoding these factors have been implicated in the pathogenesis of various congenital heart defects. Mutations in the homeodomain of NKX2-5 were initially reported in patients with atrial septal defects and conduction abnormalities (Schott et al. 1998). More recently, mutations in NKX2-5 have been described in sequences encoding other domains of the protein, including the NK2 and transcriptional repression domains, and NKX2-5 mutations have been found in patients with complex conotruncal abnormalities, heterotaxy, and hypoplastic left heart syndrome (McElhinney et al. 2003; Chung and Rajakumar 2016). Autosomal dominant mutations in TBX5 are most commonly associated with Holt-Oram syndrome, which is characterized by upper limb abnormalities and congenital heart disease (Basson et al. 1997; Li et al. 1997). Mutations in GATA4 have been associated with a wide spectrum of congenital heart defects, including conotruncal abnormalities and septal defects (Pehlivan et al. 1999; Garg et al. 2003).
The T-box transcription factor Tbx5 and homeodomain transcription factor Nkx2-5 physically interact and synergistically activate transcription of cardiac genes in transcription reporter assays (Bruneau et al. 2001; Hiroi et al. 2001). Consistent with this, mice that are heterozygous null for both genes display more severe cardiac phenotypes than mice heterozygous for either gene alone, indicative of a genetic interaction (Moskowitz et al. 2007). Recent work by the Bruneau and Müller laboratories (Luna-Zurita et al. 2016) shed additional light on this interaction and how it regulates cardiogenesis. The group differentiated wild-type, Nkx2-5-null, Tbx-5-null, and double-null cells into cardiac myocytes and mapped DNA occupancy of the factors as well as Gata4 in each of these cell lines. They used a modification of chromatin immunoprecipitation (ChIP) with DNA sequencing (ChIP-seq) that improves the resolution of occupancy binding sites (a protocol called ChIP-exo) to confirm that Nkx2-5 and Tbx5 cobind regulatory regions and that transcription factor motifs for these factors in a given gene tend to occur on the same DNA strand in close proximity. They also confirmed that each factor can bind independently and interdependently. Moreover, they found that in the absence of each factor, the other factor bound DNA ectopically, perhaps because these proteins in complex with each other prevent establishment of less favored, weaker interactions when the proteins are present without their usual partners. Their conclusions were supported by the crystal structure of a fusion protein of the Nkx2-5 homeodomain and Tbx5 T-box domain bound to the Nppa promoter. This allowed the group to identify the exact residues of the heterodimeric complex that contact DNA and how the two proteins may interface with each other.
Analysis of a mutant form of GATA4 (G296S) found in human families with congenital heart disease revealed a potential interaction between Gata4 and Tbx5 (Garg et al. 2003). Echocardiograms from patients carrying this mutation revealed signs of cardiac dysfunction and abnormal morphogenesis of RV and LV muscle. Molecular gene reporter assays suggested that this mutant version of Gata4 is not able to activate gene expression as well as wild-type protein. Interestingly, patients with TBX5 mutations can present with similar phenotypes, leading the investigators to test the hypothesis that the two proteins interact and that the G296S GATA4 mutation diminishes this interaction. Overexpression studies in vitro supported this hypothesis (Garg et al. 2003). Later, murine studies revealed a genetic interaction between Gata4 and Tbx5, as compound heterozygous mice were found to display more frequent and severe structural heart defects than either heterozygote alone (Maitra et al. 2009). More recent studies using a mouse model in which the human GATA4 point mutation was knocked into the murine Gata4 locus confirmed this mutation as a driver of congenital heart disease in mice (Misra et al. 2012). Further studies have used human iPSCs from affected family members harboring the G296S mutation (Ang et al. 2016). Myocytes derived from these cells have relatively less “open” chromatin in the genomic regions containing cardiac genes. Interestingly, genes relevant to endothelial cell identity were more “open” when compared with control cells in which the mutation had been corrected. Genes relevant to endothelial cell identity were also ectopically expressed in these mutant myocytes. The group also found that GATA4 and TBX5 bind superenhancers, which are large highly conserved enhancers with extremely high occupancy of transcription factors and the Mediator complex that are important to cellular identity. The G296S GATA4 mutation renders the mutant form of GATA4 unable to bind superenhancers appropriately. TBX5 occupancy of superenhancers is reduced in the GATA4 mutant myocytes, and it is intriguing to speculate that a change in enhancer occupancy, secondary to a single point mutation, results in an “identity crisis.”
Additional studies have highlighted an interaction between Gata4, Tbx5, and Smardc3 in cardiac development and transdifferentiation of fibroblasts into cardiac myocytes. Inhibition of Nodal signaling in ESC differentiation induces Smardc3, which complexes with Tbx5 and Gata4 to regulate expression of Nkx2-5 by remodeling a critical enhancer (Cai et al. 2013). As mentioned above, cardiac transdifferentiation studies have relied primarily on cardiac transcription factors. Interestingly, in vivo reprogramming of noncardiac mesoderm as early as E6.5 into contracting myocytes required the addition of Smardc3 to a cocktail of Gata4 and Tbx5 (Takeuchi and Bruneau 2009). Smardc3-mediated remodeling of chromatin facilitated Gata4 occupancy. In addition, recent studies suggest that chemical blockade of the histone methyltrasferase G9a enhances transdifferentiation of human fibroblast into cardiac myocytes (Cao et al. 2016). Taken together, these studies highlight the critical interaction between genome accessibility and transcription factor occupancy in regulating lineage plasticity and cellular competence to adopt different fates.
Conclusions
The more we understand about how cardiac progenitor cells adopt specific lineage fates in the mature heart, the more we will be empowered to enhance and engineer regeneration of damaged cardiac tissue. The accepted paradigm of gene regulation that relies heavily on the potent activity of lineage-specific activators and repressors acting on enhancers to modulate gene expression is being rapidly updated by discoveries in the field. A critical role for the regulated organization of chromatin in three dimensions within the nucleus is becoming apparent, and a role for chromatin interactions with the nuclear periphery represents an exciting node for regulation of cellular competence and lineage stability. ncRNAs and complex interactions between RNA and transcription factor regulators of gene expression are demanding systems biology genome-wide approaches for understanding relative contributions to cellular phenotypes and identity. The pace of discovery regarding the regulation of cardiac lineage determination continues to accelerate, and the beat goes on.
Acknowledgments
We thank the Jain and Epstein laboratories for discussions and comments on the manuscript. We thank Cheryl Smith for critical reading of the manuscript. This work was supported by the National Institutes of Health (RO1 HL131611 and R01 HL071546 to J.A.E., and K08 HL119553 to R.J.), the Cotswold Foundation (to J.A.E.), the W.W. Smith endowed chair (to J.A.E.), the Spain Fund for Cardiovascular Research (to J.A.E.), the W.W. Smith Foundation (to R.J.), the Gilead Research Scholars Award (to R.J.), and the Burroughs Welcome Career Award for Medical Scientists (to R.J.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.308353.117.
References
- Addis RC, Epstein JA. 2013. Induced regeneration—the progress and promise of direct reprogramming for heart repair. Nat Med 19: 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, et al. 2015. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang YS, Rivas RN, Ribeiro AJ, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TM, Fu JD, Spencer CI, et al. 2016. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167: 1734–1749.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardot E, Calderon D, Santoriello F, Han S, Cheung K, Jadhav B, Burtscher I, Artap S, Jain R, Epstein J, et al. 2017. Foxa2 identifies a cardiac progenitor population with ventricular differentiation potential. Nat Commun 8: 14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. 1997. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15: 30–35. [DOI] [PubMed] [Google Scholar]
- Beck M, Hurt E. 2017. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18: 73–89. [DOI] [PubMed] [Google Scholar]
- Bi P, Ramirez-Martinez A, Li H, Cannavino J, McAnally JR, Shelton JM, Sanchez-Ortiz E, Bassel-Duby R, Olson EN. 2017. Control of muscle formation by the fusogenic micropeptide myomixer. Science 356: 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Olson EN. 1998. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 14: 167–196. [DOI] [PubMed] [Google Scholar]
- Blobel G. 1985. Gene gating: a hypothesis. Proc Natl Acad Sci 82: 8527–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R, Frasch M. 1999. Genetic determination of Drosophila heart development. In Heart development (ed. Harvey RP, Rosenthal N), pp. 65–90. Academic, San Diego. [Google Scholar]
- Bressan M, Liu G, Mikawa T. 2013. Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science 340: 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. 1999. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol 211: 100–108. [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. 2001. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106: 709–721. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. 2003. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5: 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Albini S, Wei K, Willems E, Guzzo RM, Tsuda M, Giordani L, Spiering S, Kurian L, Yeo GW, et al. 2013. Coordinate Nodal and BMP inhibition directs Baf60c-dependent cardiomyocyte commitment. Genes Dev 27: 2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu JD, Nie B, Xie M, Zhang M, Wang H, et al. 2016. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 352: 1216–1220. [DOI] [PubMed] [Google Scholar]
- Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. 2010. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Kook H, Milewski R, Gitler AD, Lu MM, Li J, Nazarian R, Schnepp R, Jen K, Biben C, et al. 2002. Hop is an unusual homeobox gene that modulates cardiac development. Cell 110: 713–723. [DOI] [PubMed] [Google Scholar]
- Chung IM, Rajakumar G. 2016. Genetics of congenital heart defects: the NKX2-5 gene, a key player. Genes 7: E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine WP, Wythe JD, George M, Koshiba-Takeuchi K, Bruneau BG. 2014. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. Elife 3: e03848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engleka KA, Manderfield LJ, Brust RD, Li L, Cohen A, Dymecki SM, Epstein JA. 2012. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ Res 110: 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Ollikainen N, Guttman M. 2016. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol 17: 756–770. [DOI] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. 2016. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. 2003. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424: 443–447. [DOI] [PubMed] [Google Scholar]
- Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, Macura K, Blass G, Kellis M, Werber M, et al. 2013. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951. [DOI] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. 2017. The cohesin release factor WAPL restricts chromatin loop extension. Cell 169: 693–707.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, Jin KK, Xu W, Lin CY, Lin CJ, et al. 2014. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X. 2017. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6: e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. 2001. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet 28: 276–280. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. 2016. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351: 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Barkauskas CE, Takeda N, Bowie EJ, Aghajanian H, Wang Q, Padmanabhan A, Manderfield LJ, Gupta M, Li D, et al. 2015a. Plasticity of Hopx+ type I alveolar cells to regenerate type II cells in the lung. Nat Commun 6: 6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, et al. 2015b. Heart development. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 348: aaa6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda B, Pickersgill H, Shloma VV, Fornerod M. 2010. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 140: 360–371. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Huber TL, Keller GM. 2006. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell 11: 723–732. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. 2011. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8: 228–240. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Schneider JW, Nadal-Ginard B, Mahdavi V. 1994. Activation of the myogenic lineage by MEF2A, a factor that induces and cooperates with MyoD. Science 266: 1236–1240. [DOI] [PubMed] [Google Scholar]
- Kehat I, Accornero F, Aronow BJ, Molkentin JD. 2011. Modulation of chromatin position and gene expression by HDAC4 interaction with nucleoporins. J Cell Biol 193: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. 2001. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell 1: 435–440. [DOI] [PubMed] [Google Scholar]
- Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. 2013. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152: 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescroart F, Chabab S, Lin X, Rulands S, Paulissen C, Rodolosse A, Auer H, Achouri Y, Dubois C, Bondue A, et al. 2014. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat Cell Biol 16: 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, et al. 1997. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15: 21–29. [DOI] [PubMed] [Google Scholar]
- Li D, Takeda N, Jain R, Manderfield LJ, Liu F, Li L, Anderson SA, Epstein JA. 2015. Hopx distinguishes hippocampal from lateral ventricle neural stem cells. Stem Cell Res 15: 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. 1993. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development 119: 419–431. [DOI] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, et al. 2016. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166: 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. 2016. Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164: 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Zhou B, Pu WT. 2008. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev Biol 323: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. 2009. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol 326: 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. 2003. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 42: 1650–1655. [DOI] [PubMed] [Google Scholar]
- Meilhac SM, Esner M, Kelly RG, Nicolas JF, Buckingham ME. 2004. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev Cell 6: 685–698. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. 2012. MicroRNAs in stress signaling and human disease. Cell 148: 1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra C, Sachan N, McNally CR, Koenig SN, Nichols HA, Guggilam A, Lucchesi PA, Pu WT, Srivastava D, Garg V. 2012. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet 8: e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. 2001. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol 238: 97–109. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN. 1996. Combinatorial control of muscle development by basic helix–loop–helix and MADS-box transcription factors. Proc Natl Acad Sci 93: 9366–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. 1995. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83: 1125–1136. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. 1996. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol 16: 2627–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. 2006. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127: 1151–1165. [DOI] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. 2001. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31: 176–180. [DOI] [PubMed] [Google Scholar]
- Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, et al. 2007. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 129: 1365–1376. [DOI] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. 2015. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347: 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, et al. 2016. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351: 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. 2017. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169: 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant NJ, Wang Y, Hadland B, Zaunbrecher RJ, Redd M, Jones D, Pabon L, Jain R, Epstein J, Ruzzo WL, et al. 2017. Chromatin and transcriptional analysis of mesoderm progenitor cells identifies HOPX as a regulator of primitive hematopoiesis. Cell Rep 20: 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Garcia P, Debo B, Aleman JR, Talamas JA, Lan Y, Nguyen NH, Won KJ, Capelson M. 2017. Metazoan nuclear pores provide a scaffold for poised genes and mediate induced enhancer-promoter contacts. Mol Cell 66: 63–76.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV. 1999. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet 83: 201–206. [PubMed] [Google Scholar]
- Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagan JE, et al. 2017. Genome–nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 171: 573–587.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M, D'Angelo MA. 2012. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol 13: 687–699. [DOI] [PubMed] [Google Scholar]
- Raices M, Bukata L, Sakuma S, Borlido J, Hernandez LS, Hart DO, D'Angelo MA. 2017. Nuclear pores regulate muscle development and maintenance by assembling a localized Mef2C complex. Dev Cell 41: 540–554.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. 2017. Cohesin loss eliminates all loop domains. Cell 171: 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih TT, Soehalim E, et al. 2017. High-resolution mapping of chromatin conformation in cardiac myocytes reveals structural remodeling of the epigenome in heart failure. Circulation 136: 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. 2013. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2: e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. 1998. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281: 108–111. [DOI] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, H Haering C, Mirny L, et al. 2017. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CH, Liu ZP, Passier R, Zhang CL, Wang DZ, Harris TM, Yamagishi H, Richardson JA, Childs G, Olson EN. 2002. Modulation of cardiac growth and development by HOP, an unusual homeodomain protein. Cell 110: 725–735. [DOI] [PubMed] [Google Scholar]
- Spater D, Abramczuk MK, Buac K, Zangi L, Stachel MW, Clarke J, Sahara M, Ludwig A, Chien KR. 2013. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat Cell Biol 15: 1098–1106. [DOI] [PubMed] [Google Scholar]
- Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. 2002. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3′UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int J Dev Biol 46: 431–439. [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. 2011. Interconversion between intestinal stem cell populations in distinct niches. Science 334: 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, Leboeuf MR, Padmanabhan A, Wang Q, Li L, Lu MM, Millar SE, Epstein JA. 2013. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6+ niche cells. Development 140: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Bruneau BG. 2009. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 459: 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. 1940. Organisers & genes. The University Press, Cambridge, England. [Google Scholar]
- Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. 2001. Conotruncal myocardium arises from a secondary heart field. Development 128: 3179–3188. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, Ren S, Wang X, Chen I, Wang H, et al. 2016. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med 22: 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Schultheiss TM, Orkin SH. 2006. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 127: 1137–1150. [DOI] [PubMed] [Google Scholar]
- Wu SP, Cheng CM, Lanz RB, Wang T, Respress JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ, et al. 2013. Atrial identity is determined by a COUP-TFII regulatory network. Dev Cell 25: 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Hennelly S, Doyle B, Gulati AA, Novikova IV, Sanbonmatsu KY, Boyer LA. 2016. A G-rich motif in the lncRNA braveheart interacts with a zinc-finger transcription factor to specify the cardiovascular lineage. Mol Cell 64: 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Cai CL, Lin L, Qyang Y, Chung C, Monteiro RM, Mummery CL, Fishman GI, Cogen A, Evans S. 2006. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 133: 1575–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Ren B. 2017. The three-dimensional organization of mammalian genomes. Annu Rev Cell Dev Biol 33: 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, et al. 2008. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, et al. 2013. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 498: 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Wang J, Guo C, Chang W, Zhuang J, Zhu P, Li X. 2017. Temporally distinct Six2-positive second heart field progenitors regulate mammalian heart development and disease. Cell Rep 18: 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]