

In this article, Tichon et al. studied conserved sequences in the lncRNA NORAD repeats, which are required for proper mitotic divisions in human cells. Their findings identify additional interacting partners and characterize the interaction between NORAD and the RNA-binding protein SAM68 (KHDRBS1), which is required for NORAD function in antagonizing Pumilio.
Keywords: long noncoding RNAs, post-transcriptional regulation, NORAD, Pumilio, SAM68
Abstract
The number of known long noncoding RNA (lncRNA) functions is rapidly growing, but how those functions are encoded in their sequence and structure remains poorly understood. NORAD (noncoding RNA activated by DNA damage) is a recently characterized, abundant, and highly conserved lncRNA that is required for proper mitotic divisions in human cells. NORAD acts in the cytoplasm and antagonizes repressors from the Pumilio family that bind at least 17 sites spread through 12 repetitive units in NORAD sequence. Here we study conserved sequences in NORAD repeats, identify additional interacting partners, and characterize the interaction between NORAD and the RNA-binding protein SAM68 (KHDRBS1), which is required for NORAD function in antagonizing Pumilio. These interactions provide a paradigm for how repeated elements in a lncRNA facilitate function.
The human genome encodes tens of thousands of long noncoding RNAs (lncRNAs) (Iyer et al. 2015), many of which have now been reported to be involved in a variety of cellular processes (Ulitsky and Bartel 2013). An increasing number of lncRNAs has been shown to be required for proper embryonic development or adult functions (Perry and Ulitsky 2016). Mechanisms of lncRNA action and in particular how lncRNA sequences and structures form interfaces between the lncRNA and other cellular factors remain largely unknown, in part due to the difficulties of studying relatively long (typically >1-kb) RNA molecules biochemically. Some of the best-studied lncRNAs act in the nucleus, but most have a sizable cytoplasmic presence (Derrien et al. 2012; Ulitsky and Bartel 2013; Cabili et al. 2015). Our group (Tichon et al. 2016) and Mendell's group (Lee et al. 2016) have recently described NORAD (noncoding RNA activated by DNA damage), an abundant and widely expressed lncRNA that is predominantly cytoplasmic. Similar to some other lncRNAs such as XIST and FIRRE (Hacisuleyman et al. 2016), the sequence of NORAD contains repeated elements—12 recognizable and sequence-similar “NORAD repeat units” (NRUs) that likely originated by tandem duplication at the rise of mammals and still share substantial sequence homology (Tichon et al. 2016). Most NRUs contain one or two binding sites for the two homologs of Pumilio (Pum) in mammals: PUM1 and PUM2. Overall, NORAD contains at least 17 such Pum recognition elements (PREs), which have the consensus sequence UGURUAUA (R = A/G). NORAD deletion or depletion in cell lines results in increased repression of genes carrying PREs in their 3′ untranslated regions (UTRs), and NORAD overexpression has an inverse effect (Lee et al. 2016; Tichon et al. 2016). NORAD-regulated Pum targets are enriched in genes involved in cell division (Tichon et al. 2016), and NORAD depletion results in defects in the mitosis and accumulation of cells with chromosome number instability (Lee et al. 2016). NORAD is thus emerging as a paradigm for a lncRNA that acts in the cytoplasm by binding to a substantial number of copies of a particular RNA-binding protein (RBP) family and affecting their ability to regulate their other targets through either competition or some lncRNA-dependent alteration of the RBP function. The repeated nature of the NORAD sequence provides an opportunity for a reductionist approach in which individual repeats can be studied independently, facilitating isolation of specific interaction partners.
There is strong evidence for an interaction between NORAD and Pum proteins that affects the potency of the regulation of Pum targets, but other aspects of NORAD's mode of action remain unclear. It is unknown whether the NORAD–Pum interaction is spatially or temporally regulated, beyond the reported approximately twofold p53-dependent increase in NORAD expression following DNA damage (Lee et al. 2016). Furthermore, PREs account for only ∼2.5% of the 5.3-kb NORAD sequence, and many other sequences in the 12 NRUs are highly conserved (including sequences that fold into conserved secondary structures) but have no known function. Those sequences and factors with which they interact can potentially regulate recruitment of Pum proteins to NORAD or affect Pum post-translationally.
In this study, we used a proteomic screen to identify proteins that interact with the two highly conserved NRUs and whose binding is affected by mutations in their various predicted sequence elements. Out of the novel interaction partners identified in this screen, we focus here on SAM68 (KHDRBS1), which is an abundant, multifunctional, and cell cycle-regulated RBP that we describe as a novel interaction partner of both NORAD and PUM2. We found that SAM68 is required for efficient recruitment of PUM2 to NORAD, regulation of Pum activity by NORAD, and proper chromosome segregation in mammalian cells.
Results
SAM68 binds NORAD repeats
We previously characterized the NRUs as ∼300-nucleotide (nt) sequences, each containing a combination of several recognizable elements (as illustrated in one of the most conserved repeats: NRU#7 in Fig. 1A). These elements are one or two PREs, a short hairpin of 4 base pairs (bp), a U-rich stretch, a longer hairpin of 8–9 bp, and an A/G-rich sequence. In order to identify proteins that specifically interact with the NRUs, we used synthetic oligos (Supplemental Table 1) for in vitro production of biotinylated RNAs. These RNAs included wild-type sequences of NRU#7 and NRU#9 (WT-7 and WT-9) and sequences carrying mutations in the PRE sites (mPRE-7, mPREv2-7, and mPRE-9), short hairpin (mSH-7), long hairpin (mLH-7 and mLHcom-7), U-rich region (mU-7), or A/G-rich region (mAG-7). We also used a shuffled sequence of NRU#7 (Shuffle-7) as a negative control. Pull-downs using U2OS cell extract with the different sequence variants followed by quantitative label-free mass spectrometry (MS) (Supplemental Data 1) identified 545 proteins supported by at least three unique peptides using WT-7 (two separate experiments) and WT-9 as bait. Of these, 146 were enriched in the pull-down using the WT-7 sequence compared with Shuffle-7 (intensity ratio ≥2). Since NORAD is localized predominantly in the cytoplasm (Lee et al. 2016; Tichon et al. 2016), we focused on 100 of those proteins with reported cytoplasmic localization (Binder et al. 2014). Twenty-five of these were recovered previously when the NRU#8 sequence was used as bait (Tichon et al. 2016), and six of them (HSPB1, XRCC6, EZR, HNRNPF, KHDRBS1, and RUVBL1) were also identified by Lee et al. (2016) as binding NRUs in HCT116 cell extract. Protein recovery was typically affected by mutations in NRU#7 and NRU#9, and the 100 cytoplasmic proteins were affected in their recovery in four mutant variants, on average (wild-type/mutant intensity ratio ≥2) (Supplemental Data 1). We focused on two candidates for further validation—RUVBL1 and SAM68 (KHDRBS1), which were affected by various mutations (Fig. 1B)—and performed RNA immunoprecipitation (RIP) using U2OS extract and antibodies specific to RUVBL1, SAM68, PUM2 (positive control), and GAPDH (negative control) followed by quantitative RT–PCR (qRT–PCR) for selected protein-coding and lncRNA genes (Fig. 1C). We observed 30-fold and 11-fold enrichment of NORAD using SAM68 and PUM2 antibodies, respectively (Fig. 1C), and therefore, in this study, we focus on the interaction between NORAD and SAM68, which was affected in the MS data by mutations in the PREs, hairpins, and A/G-rich region.
Figure 1.

SAM68 binds NORAD repeats. (A) Secondary structure of NRU#7 predicted by RNAfold (Hofacker 2003) with the elements marked. (B) MS intensities for the indicated proteins and the indicated sequence variants (all from the same experiment). (C) RIP of the indicated proteins quantified using qRT–PCR for the indicated genes (left; n ≥ 3, normalized to the input and the GAPDH mRNA; average ± SEM is shown) and visualized by Western blot (right). Negative control (NC) in Western blot is a lysis buffer to control for bands originating from the antibody. Twenty micrograms was loaded as input for the Western blot. (D) The distribution of SAM68-binding sites in the different NRUs and eCLIP (enhanced cross-linking immunoprecipitation) data of SAM68 and PUM2 from K562 cells (Van Nostrand et al. 2017) in NRU#7 and NRU#9. The color shadings highlight the different conserved elements. (Light green) PREs; (yellow) short hairpin; (green) U-rich region; (blue) long hairpin. (E) PhyloP conservation scores (based on the 100-way vertebrate alignment of the hg19 human genome in the University of California at Santa Cruz [UCSC] genome browser) for all of the bases in NORAD and just the bases overlapping the TAAA or TGTRTATA motifs. (F) Pull-down of four indicated fragments from NRU#8 and NRU#9 followed by Western blot for SAM68. The sequences of the fragments are annotated in Supplemental Figure 1B. (G) Pull-down of SAM68 using in vitro transcribed RNA from the indicated variants of NRU#7 (sequences in Supplemental Table 1).
NORAD contains multiple evolutionarily conserved SAM68-binding sites
SAM68 is a multifunctional RBP that is phosphorylated during mitosis and has known functions in the nucleus and cytoplasm (Lukong and Richard 2003). The RNA-binding preferences of SAM68 are quite well understood. SAM68 recognizes A/U-rich sequences and particularly UAAA motifs (Lin et al. 1997), with a preference for bipartite motifs separated by linkers of at least 15 bases (Galarneau and Richard 2009; Feracci et al. 2016). The human NORAD sequence contained 16 pairs of UAAA motifs separated by 15–35 bases—a 2.7-fold enrichment over the average number of such pairs in 200 random sequences (length and dinucleotide composition matched to NORAD). Similar enrichments were observed in the mouse and dog NORAD sequences (3.7-fold and 2.4-fold, respectively). Ten of the UAAA pairs in human NORAD are localized downstream from the PREs and the long hairpin in NRUs (sites are marked in bold in Fig. 1D), and these sites are conserved in mammals (cf. NRU#7 in Supplemental Fig. 1A). Overall, UAAA sites were significantly better conserved than the rest of the NORAD sequence, and their level of conservation was comparable with that of NORAD PREs (Fig. 1E). Four out of the 16 predicted UAAA pairs, including the three in NRU#9, overlapped high-confidence (enrichment greater than twofold; P < 0.01) SAM68-binding clusters in eCLIP (enhanced cross-linking immunoprecipitation) data from K562 cells from the ENCODE project (Fig. 1D; Van Nostrand et al. 2017), and eCLIP reads were also observed in other repeats (e.g., NRU#7 in Fig. 1D).
To further verify the interaction between SAM68 and NRUs in U2OS cells, we performed a pull-down using biotinylated RNAs of NRU#8 and NRU#9 (P8 and P9) (Fig. 1F) and adjacent sequences (C8 and C9, all from Tichon et al. 2016; marked in Fig. 1F; Supplemental Fig. 1B) followed by Western blots for SAM68. SAM68 was precipitated by NRU#8 and NRU#9 but not with C8 and C9 sequences (Fig. 1F; RNA bait control in Tichon et al. 2016). Pull-downs followed by Western blot using the sequence variants of NRU#7 used for MS experiments along with a sequence carrying mutations in three putative SAM68-binding sites (mSAM68-7) (Supplemental Table 1) showed mildly reduced binding by SAM68 to NRU#7 with mutated binding sites (Fig. 1G; Supplemental Fig. 1C). MS data from a pull-down by mSAM68-7 also showed substantially reduced binding by SAM68 (Supplemental Data 1), further supporting a sequence-specific interaction of SAM68 with the NRUs.
SAM68, NORAD, and Pum genes have opposing effects on gene expression
We showed previously that NORAD knockdown and overexpression specifically affect genes with multiple PREs in their 3′ UTRs (Tichon et al. 2016). We now performed RNA sequencing (RNA-seq) in U2OS cells following overexpression and knockdown of SAM68 using an siRNA pool (Dharmacon) (Supplemental Data 2). As knockdown and overexpression of both NORAD and Pum proteins generally had a mild but significant effect on expression of hundreds of genes (Lee et al. 2016; Tichon et al. 2016), we computed for each gene the ratio between the overexpression and knockdown effects (the difference between the log2 transformed fold changes) and compared groups of genes with varying numbers of PREs in their 3′ UTR sequences. As expected from previous studies, there was a strong positive correlation between PUM1 and PUM2 perturbations and a highly significant negative correlation between the effects of Pum and NORAD perturbations. Strikingly, for SAM68, we also observed a highly significant negative correlation with Pum perturbations that increased with the number of PREs (Fig. 2A). The correlation between perturbations of SAM68 and NORAD was significant but lower than that observed between SAM68 and Pum perturbations, suggesting that SAM68 may have an additional, NORAD-independent effect on Pum activity.
Figure 2.
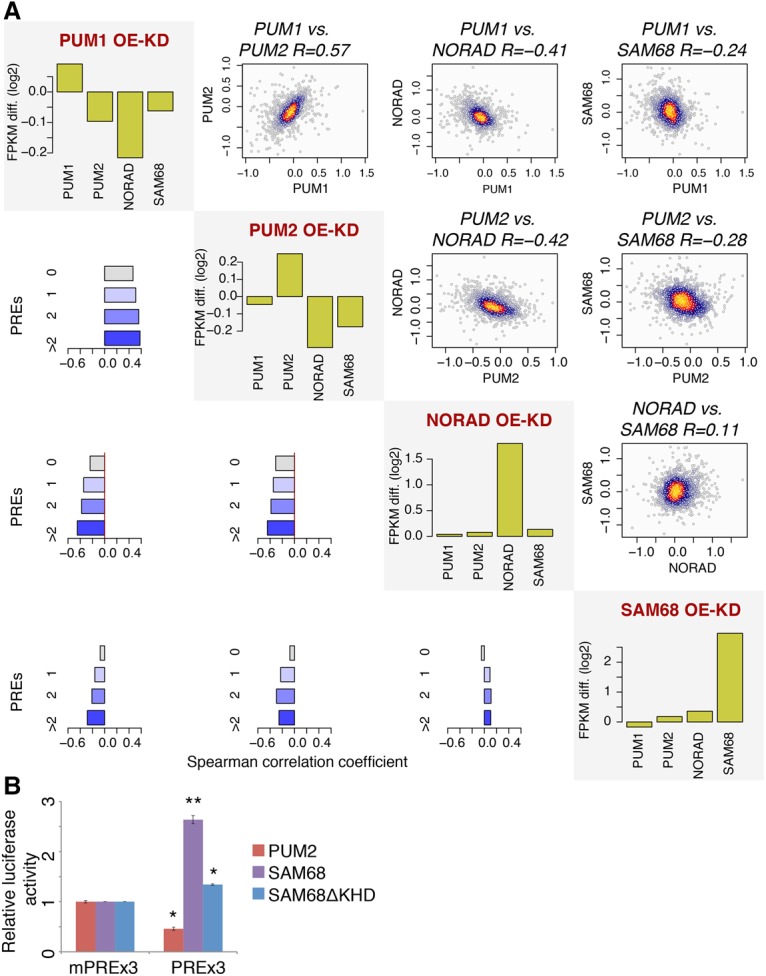
The effect of SAM68 on NORAD- and PUM2-regulated genes. (A) The effects of PUM1, PUM2, NORAD, and SAM68 perturbation on genes bearing PREs. For each protein, we computed fold changes in gene expression following overexpression and knockdown (two replicates each). The shown values are the differences between log2 transformed fold changes following overexpression and knockdown conditions (OE-KD). The diagonal plots show changes in RNA levels of the indicated genes in each of the perturbations. The scatter plots above the diagonal compare the indicated perturbations for genes having two or more PREs and report the Spearman R between the values. Colors indicate local point density. The bar charts below the diagonal show the Spearman correlation coefficients between the fold changes when considering genes with the indicated number of PREs. (B) The effects of the indicated perturbations on luciferase activity from psiCHECK1-PREx3 (psiCheck-1 containing 3× wild-type PREs) and psiCHECK1-mPREx3 (psiCheck-1 containing 3× mutated PREs) after transfection of an empty plasmid and plasmids for PUM2, SAM68, or SAM68 without the KH domain (SAM68ΔKHD). Average ± SEM is shown, based on at least three independent replicates. (*) P < 0.05; (**) P < 0.005, t-test.
SAM68 affects Pum repression of mRNAs with PREs in their 3′ UTRs
To confirm that the regulation of Pum targets by SAM68 is PRE-dependent, we quantified the levels of a luciferase reporter (Van Etten et al. 2013) with three consensus PREs (PREx3) in its 3′ UTR and a control vector with mutated PRE sites (mPREx3) in U2OS cells following overexpression of PUM2 or SAM68 (Supplemental Fig. 2A,B). PUM2 overexpression led to reduced luciferase activity in a PRE-dependent manner, while overexpression of SAM68 alleviated repression (Fig. 2B), consistent with the effects observed on endogenous Pum targets in the RNA-seq data.
SAM68 interacts with PUM2 and facilitates the interaction between PUM2 and NORAD
To test for a physical interaction between PUM2 and SAM68, we used reciprocal coimmunoprecipitation (co-IP) followed by Western blot using antibodies against SAM68 and PUM2. We observed an interaction between the two proteins in HCT116 and U2OS cells (Fig. 3A,B; Supplemental Fig. 3). The interaction was qualitatively NORAD-independent (Fig. 3A,B; Supplemental Fig. 2C; NORAD−/− cells from Lee et al. 2016) in HCT116 cells and RNase-insensitive in U2OS cells (RNase treatment) (see the Materials and Methods; Supplemental Fig. 3). Next, we tested whether SAM68 facilitates the interaction between NORAD and PUM2 by performing RIP with PUM2 antibodies in wild-type and SAM68−/− HCT116 cells with and without exogenous SAM68 expression. NORAD enrichment in PUM2 immunoprecipitation was significantly increased following SAM68 overexpression in both wild-type and SAM68 knockout cells and abolished in the SAM68−/− cells compared with wild type (PUM1 is a positive control for PUM2 binding) (Fig. 3C; Supplemental Fig. 4). This suggests that SAM68 is required for a detectable interaction between PUM2 and NORAD, and changes in SAM68 levels affect how efficiently PUM2 binds NORAD.
Figure 3.
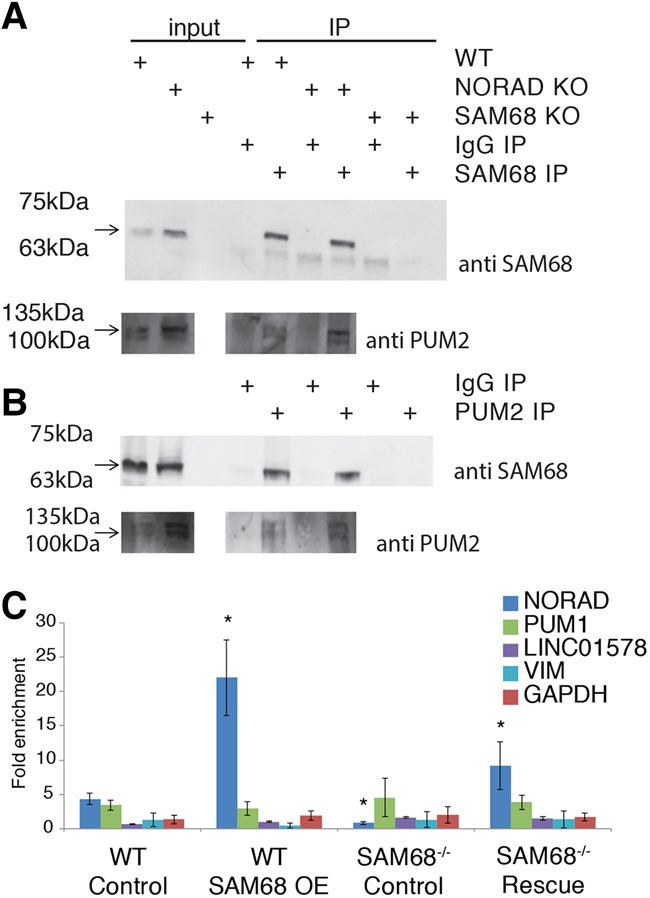
SAM68 interacts with PUM2 and facilitates the interaction between PUM2 and NORAD. (A,B) Interaction between SAM68 and PUM2 in lysates derived from HCT116 wild type and NORAD−/− was examined by co-IP using anti-SAM68 and PUM2 antibodies followed by Western blot. Five percent of the lysate used for immunoprecipitation was loaded as input. (C) RIP of PUM2 in HCT116 wild-type or SAM68−/− cells quantified using qRT–PCR for the indicated genes. n ≥ 3, normalized to the input and the IgG control. Average ± SEM. “Control” indicates cells transfected with an empty pcDNA3.1 plasmid, and “SAM68 OE” and “Rescue” indicate cells transfected with a SAM68 expression plasmid. Asterisks indicate P < 0.05 compared with the “wild-type control” sample (t-test).
PUM2 regulation by NORAD is SAM68-dependent
To test whether regulation of Pum targets by NORAD is dependent on SAM68, we quantified luciferase expression using PRE/mPRE reporter vectors in wild-type and SAM68−/− HCT116 cells overexpressing PUM2, NORAD, and SAM68 (Fig. 4A; Supplemental Fig. 5). As expected, PUM2 overexpression significantly reduced luciferase activity in all backgrounds, and NORAD overexpression alleviated repression (Fig. 4A). Strikingly, the effect of NORAD overexpression was significantly diminished in the SAM68−/− cells compared with wild type (Fig. 4A), indicating that SAM68 is required for regulation of Pum targets by NORAD. To evaluate the effect on regulation of endogenous targets, we knocked down NORAD using siRNAs in wild-type and SAM68−/− cells and performed RNA-seq. Baseline NORAD levels were similar in wild-type and SAM68−/− cells (log2 fold change −0.02; P = 0.86). As expected based on our results from U2OS cells and previous RNA-seq experiments in NORAD−/− HCT116 cells (Lee et al. 2016), we observed a specific down-regulation of Pum targets following NORAD knockdown in wild-type cells (Fig. 4B). In stark contrast, no down-regulation but rather up-regulation of Pum targets was observed in SAM68−/− cells (Fig. 4B). An intact SAM68 is therefore required for repression of Pum activity by NORAD.
Figure 4.

PUM2 regulation by NORAD is SAM68-dependent. (A) Regulation of expression of a luciferase with three consensus (PREx3) or mutated (mPREx3) PREs in its 3′ UTR in HCT116 cells with the indicated genotype following transfection of the indicated vector. Average ± SEM. n = 4 independent experiments. (*) P < 0.05; (**) P < 0.005, t-test. (B) Fold changes in expression of the indicated gene group following siRNA-mediated knockdown of NORAD in wild-type and SAM68−/− HCT116 cells. P-values were computed with Wilcoxon rank sum test.
Depletion of SAM68 and NORAD affects chromosome segregation
In order to evaluate the roles of SAM68 and NORAD in cell cycle progression, we used siRNAs to knock down SAM68, NORAD, or both genes in HeLa Kyoto cells expressing histone 2B-GFP (Neumann et al. 2010), which allows tracking of chromosome segregation errors (Fig. 5A). Both perturbations significantly increased the number of cells exhibiting aberrant events such as chromatin/anaphase bridges (Fig. 5B,C). Concurrent knockdown of both genes had an effect similar to individual knockdowns, suggesting that NORAD and SAM68 act in the same pathway. These results support the previous observations that NORAD deletion results in an increase in mitotic errors in HCT116 cells (Lee et al. 2016) and suggest that SAM68 is also required for proper progression through mitosis by buffering the repressive activity of Pum proteins.
Figure 5.
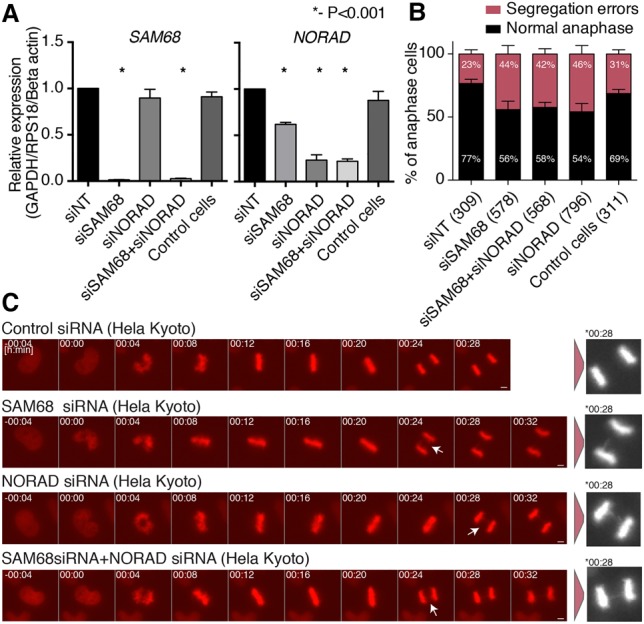
Chromosome segregation errors in the cells depleted of SAM68 and NORAD. (A) siRNA-mediated knockdown of SAM68 and NORAD in HeLa Kyoto cells. Expression levels were compared with the control nontargeting siRNA and normalized to the indicated controls. P-values were computed using two-sided t-test. (B) Quantification results of chromosome segregation errors from time-lapse microscopy of HeLa Kyoto cells progressing through mitosis after treatment with the indicated siRNAs. Each bar represents the mean of five independent experiments ±SEM. The numbers in parentheses indicate the number of anaphase cells analyzed per condition. (C) Representative still images from the time-lapse microscopy. White arrows depict chromosome segregation errors. Asterisk depicts magnified images at the indicated times. Bar, 5 µm.
Discussion
The antagonistic activity of NORAD on Pum genes is mediated by at least 17 PRE elements encoded in the NRUs. It has been proposed that other lncRNAs and mRNAs act in a similar fashion by binding other RBPs, most prominently the microRNA (miRNA)-loaded Argonaute (Ago) proteins, as part of the “competing endogenous RNA” (ceRNA) phenomenon (Salmena et al. 2011), the details and extent of which remain controversial (Denzler et al. 2014; Jens and Rajewsky 2015). A common concern about this mode of action for lncRNAs is that the number of RBP- or miRNA-binding sites presented by a single RNA species is typically small compared with the number of sites present in the rest of the transcriptome, and as lncRNAs are typically lowly expressed, they present too few sites to have a physiologically relevant impact. NORAD is abundant and has multiple PREs, and so NORAD molecules can provide thousands of binding sites for Pum proteins, a number comparable with that of Pum proteins in the cell (Lee et al. 2016; Tichon et al. 2016) but still small compared with the number of PREs in the entire transcriptome. Indeed, in the available PUM2 CLIP data from HEK293, HCT116, and K562 cells (Hafner et al. 2010; Lee et al. 2016; Van Nostrand et al. 2017), the number of reads in PUM2 pull-down experiments that map to NORAD is substantial but is typically <5% of all reads. Therefore, it seems unlikely that the effect of NORAD on Pum targets is through a global competition for Pum binding. Rather, it is possible that NORAD is privileged in the competition for Pum binding in a certain time or place in the cell or that NORAD-binding partners facilitate a particular post-translational regulation that represses Pum function. At present, there is no evidence for the former scenario, as NORAD distribution is quite uniform across the cytoplasm and does not appear to be altered by SAM68 knockdown (Supplemental Fig. 6), and NORAD levels across the cell cycle in the lines that we studied also appear to be quite stable (data not shown). Binding of SAM68, which is known to be regulated post-translationally during the cell cycle and following different stimuli (Lukong and Richard 2003) and which we now show regulates the ability of NORAD to recruit and affect Pum activity, can provide an important means for regulating Pum activity in time and space without altering NORAD levels or subcellular distribution.
Our hypothesis was that the conserved secondary structures found in NRUs immediately downstream from the PREs bind specific proteins that are important for NORAD function. Therefore, in our pull-down experiments, we used sequences that contained various mutations in different elements of those structures. SAM68 binds NRUs in the region that we previously referred to as “A/G-rich,” and its recovery in the MS data was indeed lower in this mutant as well as in several other mutants (Fig. 1B). Additional interesting candidates for binding the core NRU region are FAM120, an RBP that was identified in the Mendell laboratory MS data (Lee et al. 2016) and for which eCLIP data suggests binding in the PRE region of the NRUs (Supplemental Fig. 7A), and HNRNPF, which is one of the six shortlisted proteins with the strongest evidence from the proteomic screen and binding of which to NORAD is also evident in CLIP data (Supplemental Fig. 7B). The binding location of HNRNPF within NRUs is currently less clear, as HNRNPF was reported to bind U/G-rich sequences (Huelga et al. 2012) or GGG motifs (Van Nostrand et al. 2017), but its fly homolog, Glorund, recognizes both GGG and structured A/U-rich elements (Tamayo et al. 2017). Since SAM68 and HNRNPF binding in the MS data was affected by the mutations in multiple structured elements, an interesting possibility is that the conserved hairpin elements in the NRUs are conserved not because they serve as binding sites for a particular protein but rather because they help position the protein-binding sites at a favorable distance and orientation from each other.
We also describe here a previously unknown RNA-independent interaction between PUM2 and SAM68. Interestingly, an antagonistic interaction between a Pum homolog (FBF-1) and a SAM68-homolog (GLD-1) is well established in gonadal stem cells in Caenorhabditis elegans. The FBF-1 protein represses the GLD-1 RNA to control entry into meiosis, and GLD-1 in turn represses GLP-1/notch signaling, which is an fbf-1 activator (Kimble and Crittenden 2007). To the best of our knowledge, a physical interaction between GLD-1 and FBF-1 has not yet been described, but our results suggest that an antagonistic relationship between the two RBPs may have ancient roots.
The length of lncRNAs poses a substantial challenge for identifying and validating their protein interaction partners (Simon 2016). Since most RBPs bind short sequences of up to 8 bases, one molecule of a typical 1000-nt lncRNA can simultaneously accommodate hundreds of RBPs, and thus the results of pull-downs performed using full-length lncRNA sequences or using antisense probes capturing the full-length transcript are difficult to interpret. In order to address this challenge, we used here two attractive features of NORAD: the repeated nature that allows the study of several ∼300-nt fragments in lieu of the 5.3-kb transcript and the evolutionary conservation of NORAD among mammals that allows the identification of specific constrained elements and the study of the consequences of their mutation on the repertoire of in vitro binding partners. The repetitive nature of NORAD facilitates identification of binding partners but also makes it prohibitive to study the consequences of binding site loss. NORAD contains more than a dozen consensus PREs and consensus SAM68-binding sites, and, based on the eCLIP data, it is likely that additional suboptimal sites are present, making it very difficult to construct NORAD variants that will not be bound by those proteins. An interesting future direction would be to construct short “mini-NORAD” transcripts that can recapitulate the functionality of the full transcript but allow for efficient sequence manipulation. Such approaches combined with the resource of a set of NRU-binding proteins that we describe here (Supplemental Data 1) will shed further light on the mode of action of this fascinating lncRNA and potentially on other cytoplasmic lncRNAs.
Materials and methods
Cell culture
Human cell lines U2OS (osteosarcoma; obtained from American Type Culture Collection), HCT116 wild-type and NORAD−/− (colon carcinoma; obtained from Joshua T. Mendell, Southwestern University in Texas), and HCT116 SAM68−/− (obtained from Stéphane Richard from McGill University, Montreal, Canada) were routinely cultured in DMEM containing 10% fetal bovine serum (FBS) and 100 U of penicillin/0.1 mg mL−1 streptomycin at 37°C in a humidified incubator with 5% CO2. The HeLa Kyoto (EGFP-α-tubulin/ H2B-mCherry) cell line (Neumann et al. 2010) was obtained from Jan Ellenberg (EMBL, Heidelberg, Germany) and cultured in DMEM with 10% FBS.
MS
Proteolysis and MS data analysis are described in detail in the Supplemental Material. We analyzed two MS experiments using NRU#7 variants as bait and one MS experiment using NRU#9 variants as bait and combined the results with those reported previously using sense and antisense NRU#8 and the 3′ segment of NORAD (Tichon et al. 2016) as well as the 5′ and 3′ regions and pairs of NRU#1–#10 from Lee et al. (2016) (Supplemental Data 1). In each of the three new MS data sets, we considered proteins detected with at least three unique peptides identified when using the wild-type sequence.
RNA-seq and data analysis
Strand-specific mRNA-seq libraries were prepared from U2OS and HCT116 cells using the TruSeq stranded mRNA library preparation kit (Illumina) according to the manufacturer's protocol and sequenced on a NextSeq 500 machine to obtain at least 23 million 75-nt single- or paired-end reads. Reads were aligned to the human genome (hg19 assembly) using STAR Aligner (Dobin et al. 2013), expression levels were quantified using RSEM (Li and Dewey 2011), and differential expression fold changes were quantified using DESeq2 (Love et al. 2014). The raw read counts and the computed fold changes are in Supplemental Data 2.
RIP
Immunoprecipitation of endogenous ribonucleoprotein complexes from whole-cell extracts of U2OS cells was performed as described (Yoon et al. 2012). In brief, cells were incubated with lysis buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 0.5% Na-deoxycholate, 0.5% NP-40, Complete protease inhibitor cocktail [Sigma], 100 U/mL RNase inhibitor [EURx]) for 15 min on ice and centrifuged at 15,870g for 20–25 min at 4°C. Part of the supernatants was saved as total cell lysate input. The rest, containing 1–2 mg of protein extract, was incubated for 2 h at 4°C with gentle rotation with protein A/G magnetic beads (GeneScript). The beads were prewashed and coated with antibodies against GAPDH (diluted 1:1000; Santa Cruz Biotechnology, SC-32233), PUM2 and SAM68 (each diluted 1:1000; Bethyl Laboratories, A300-201A and A300-202A, respectively), and RUVBL1 (diluted 1:1000; Proteintech, 10210-2-AP) overnight at 4°C with gentle rotation. As a negative control, we incubated the magnetic bead–antibody complexes with lysis buffer. The beads were washed five times with lysis buffer, separated each time by magnetic force. The remaining mixture of magnetic bead–antibody–protein–RNA complexes was separated as follows: Three-quarters was mixed with sample buffer and boiled for 5 min at 95°C for further analysis by Western blot, and one-quarter was incubated with 1 mg mL−1 proteinase K for 30 min at 37°C with gentle shaking to remove proteins. The remaining RNA was extracted by TRI reagent. The RNAs isolated from the immunoprecipitation materials were further assessed by RT-qPCR analysis where immunoprecipitation material was normalized to total cell lysate. Western blot was used in order to verify that the desired protein was indeed precipitated. RIP and co-IP in HCT116 cells were performed as described in Lee et al. (2016).
Plasmids and siRNAs
All transfections were performed using Lipofectamine 3000 reagent (Thermo Fisher Scientific). NORAD and PUM2 plasmids were described previously (Tichon et al. 2016). For SAM68 overexpression, we used the pcDNA3-HA-SAM68 and pcDNA3-HA-SAM68-ΔKH plasmids, which encode the mouse SAM68 gene (a gift from David Shalloway; Addgene, nos. 17690 and 17688). As controls for overexpression experiments, we used pcDNA3.1(+) (Invitrogen). In transfection, 100 ng was used per 20,000 cells in 96-well plates for 48 h before cells were harvested. The luciferase experiments used the following plasmids: pGL4.13, psiCHECK-1 containing 3× wild-type PREs (which are underlined in the following sequence: 5′-TTGTTGTCGAAAATTGTACATAAGCCAA-3′), and psiCHECK-1 containing 3× mutated PREs (5′-TTGTTGTCGAAAATACAACATAAGCCAA-3′; a kind gift of Dr. Aaron Goldstrohm), as described previously (Van Etten et al. 2013). For luciferase experiments, we used 5 ng of pGL4.13 and 20 ng of different psiCHECK-1 plasmids per 20,000 cells in 96-well plates.
Knockdowns were performed using siRNAs directed against SAM68 and NORAD (from Dharmacon) (Supplemental Table 3). As a control, we used the mammalian nontargeting siRNA (Lincode nontargeting pool, Dharmacon) at a final concentration of 50 nM. Cells were harvested after 48 h for further experimental procedures.
RNA pull-down
Templates for in vitro transcription were generated by amplifying the desired sequences from cDNA or synthetic oligos (see Supplemental Table 1 for full oligo sequences) and adding the T7 promoter to the 5′ end for sense sequence (see Supplemental Table 2 for primer sequences). Biotinylated transcripts were produced using the MEGAscript T7 in vitro transcription reaction kit (Ambion) and Biotin RNA labeling mix (Roche). Template DNA was removed by treatment with DNaseI (Quanta). Cells were lysed in buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 0.5% Na-deoxycholate, and 0.5% NP-40 for 15 min on ice. The extract was cleared by centrifugation at 21,130g for 20 min at 4°C. Extract containing 0.5–2 mg of protein was incubated with 2–20 pmol of biotinylated transcripts. The pull-down products were analyzed by MS and/or Western blot. For the MS, the formed RNA–protein complexes were precipitated by Streptavidin-sepharose high-performance beads (GE Healthcare). Recovered proteins were then resolved on a 10% SDS–polyacrylamide gel and visualized by Coomassie blue staining. The entire lane was extracted and analyzed using MS analysis as described above.
Alternatively, the recovered proteins were separated on a 10% SDS–polyacrylamide gel and used for Western blot with anti-SAM68 antibody (Bethyl Laboratories). In addition, RNA was isolated using TRI reagent from an equal portion of the different protein–RNA pull-down complexes. This RNA was analyzed using qPCR for a loading control. RNaseA treatment in co-IP experiments was done with 50 U/mg protein for 30 min in 37°C.
Time-lapse microscopy
Ten-thousand HeLa Kyoto cells were plated in eight-well chamber slides (IbiTreat, Ibidi) and then transfected using Lipofectamine RNAiMax reagent (Thermo Fischer Scientific) following the manufacturer's instructions. The siRNAs (Thermo Fischer Scientific) were used at a final concentration of 50 nM, and time-lapse microscopy was done 48 h after transfection. Chromosome segregation errors, a category that includes lagging chromatids and chromatin bridges, were identified with H2B-mCherry signal. Time-lapse microscopy was performed using a Zeiss Axio Observer Z1 microscope equipped with a plan apo 0.95 NA 40× dry objective (Carl Zeiss Microscopy), a LED light source (Lumencor), and an Orca Flash 4.0 camera (Hamamatsu). Two positions were placed per well, and a z-stack was acquired at each position every 4 min for a total duration of 12 h. Voxel size was 0.325 µm × 0.325 µm × 2.5 µm. Zen software (Zeiss) was used for data collection and analysis. Throughout the experiment, the cells were maintained in a microscope stage incubator at 37°C in a humidified atmosphere of 5% CO2. The final representative images were obtained from a single Z-plane.
Statistics
All results are represented as an average ± SEM of at least three independent experiments. Statistics were performed as Student's t-test, Wilcoxon rank-sum test, or analysis of variance with Tuckey's post hoc test for three or more groups to be compared. In all results, P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). Plots were prepared using custom R scripts.
Data availability
All data presented in this work are available on request. All sequencing data have been deposited to the Gene Expression Omnibus database (accession no. GSE104856).
Supplementary Material
Acknowledgments
We thank members of the Ulitsky laboratory for helpful discussions and comments on the manuscript. We thank Tamar Ziv and the Smoler Proteomics Center at the Technion for help with proteomics and the resulting data analysis. We thank Fanni Gergely (Cancer Research UK Cambridge Institute) for advice on time-lapse microscopy and helpful comments on the manuscript. This research was supported by the Israeli Centers for Research Excellence (1796/12), Israel Science Foundation (1242/14 and 1984/14), European Research Council project lincSAFARI, Lapon Raymond, and the Abramson Family Center for Young Scientists. I.U. is incumbent of the Sygnet Career Development Chair for Bioinformatics. This work was supported by funding from Cancer Research UK C14303/A17043 to Fanni Gergely. L.S. acknowledges support from the University of Cambridge and Hutchison Whampoa Ltd.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.309138.117.
References
- Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O'Donoghue SI, Schneider R, Jensen LJ. 2014. Compartments: unification and visualization of protein subcellular localization evidence. Database 2014: bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, Rinn JL, Raj A. 2015. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol 16: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. 2014. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54: 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. 2012. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feracci M, Foot JN, Grellscheid SN, Danilenko M, Stehle R, Gonchar O, Kang HS, Dalgliesh C, Meyer NH, Liu Y, et al. 2016. Structural basis of RNA recognition and dimerization by the STAR proteins T-STAR and Sam68. Nat Commun 7: 10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A, Richard S. 2009. The STAR RNA binding proteins GLD-1, QKI, SAM68 and SLM-2 bind bipartite RNA motifs. BMC Mol Biol 10: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Shukla CJ, Weiner CL, Rinn JL. 2016. Function and evolution of local repeats in the Firre locus. Nat Commun 7: 11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL. 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. 2012. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1: 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. 2015. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jens M, Rajewsky N. 2015. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet 16: 113–126. [DOI] [PubMed] [Google Scholar]
- Kimble J, Crittenden SL. 2007. Controls of germline stem cells, entry into meiosis, and the sperm/oocyte decision in Caenorhabditis elegans. Annu Rev Cell Dev Biol 23: 405–433. [DOI] [PubMed] [Google Scholar]
- Lee S, Kopp F, Chang TC, Sataluri A, Chen B, Sivakumar S, Yu H, Xie Y, Mendell JT. 2016. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Taylor SJ, Shalloway D. 1997. Specificity and determinants of Sam68 RNA binding. Implications for the biological function of K homology domains. J Biol Chem 272: 27274–27280. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Richard S. 2003. Sam68, the KH domain-containing superSTAR. Biochim Biophys Acta 1653: 73–86. [DOI] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464: 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RB, Ulitsky I. 2016. The functions of long noncoding RNAs in development and stem cells. Development 143: 3882–3894. [DOI] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. 2011. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell 146: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD. 2016. Insight into lncRNA biology using hybridization capture analyses. Biochim Biophys Acta 1859: 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo JV, Teramoto T, Chatterjee S, Hall TMT, Gavis ER. 2017. The Drosophila hnRNP F/H homolog glorund uses two distinct RNA-binding modes to diversify target recognition. Cell Rep 19: 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichon A, Gil N, Lubelsky Y, Havkin Solomon T, Lemze D, Itzkovitz S, Stern-Ginossar N, Ulitsky I. 2016. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat Commun 7: 12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten J, Schagat TL, Goldstrohm AC. 2013. A guide to design and optimization of reporter assays for 3′ untranslated region mediated regulation of mammalian messenger RNAs. Methods 63: 110–118. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Blue SM, Dominguez D, Cody NAL, Olson S, Sundararaman B, et al. 2017. A large-scale binding and functional map of human RNA binding proteins. bioRxiv 10.1101/179648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. 2012. LincRNA–p21 suppresses target mRNA translation. Mol Cell 47: 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this work are available on request. All sequencing data have been deposited to the Gene Expression Omnibus database (accession no. GSE104856).