

Abstract
Objective
Previously, we developed the engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) technology, which isolates specific genomic regions while preserving their molecular interactions. In enChIP, the locus of interest is tagged with engineered DNA-binding molecules such as the clustered regularly interspaced short palindromic repeats (CRISPR) system, consisting of a catalytically inactive form of Cas9 (dCas9) and guide RNA, followed by affinity purification of the tagged locus to allow identification of associated molecules. In our previous studies, we used a 3xFLAG-tagged CRISPR system from Streptococcus pyogenes (S. pyogenes). In this study, to increase the flexibility of enChIP, we used the CRISPR system from Staphylococcus aureus (S. aureus) along with different epitope tags.
Results
We generated a plasmid expressing S. aureus dCas9 (Sa-dCas9) fused to a nuclear localization signal (NLS) and a 3xFLAG-tag (Sa-dCas9-3xFLAG). The yields of enChIP using Sa-dCas9-3xFLAG were comparable to those using S. pyogenes dCas9 fused with an NLS and a 3xFLAG-tag (3xFLAG-Sp-dCas9). We also generated another enChIP system using Sp-dCas9 fused with an NLS and a 2xAM-tag (Sp-dCas9-2xAM). We obtained high enChIP yields using this system as well. Our findings indicate that these tools will increase the flexibility of enChIP analysis.
Keywords: enChIP, Sa-dCas9, ChIP, Chromatin immunoprecipitation, CRISPR
Introduction
Identification of the molecules associated with a genomic region of interest in vivo is an essential step in understanding the regulatory mechanisms underlying its functions. To this end, we previously developed engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) technology to isolate genomic regions of interest while retaining their molecular interactions [1, 2]. In enChIP, engineered DNA-binding molecules such as transcription activator-like (TAL) proteins [3] or the clustered regularly interspaced short palindromic repeats (CRISPR) system [4, 5], which consists of a catalytically inactive form of Cas9 (dCas9) and guide RNA (gRNA), are used to tag the locus of interest, followed by affinity purification of the tagged locus to allow identification of associated molecules. For specific and efficient affinity purification, we usually use 3xFLAG-tag in conjunction with antibody (Ab) against the epitope tag. Locus-tagging can be done in the cell by expressing engineered DNA-binding molecules [1, 2, 6–9] or in vitro using recombinant or synthetic engineered DNA-binding molecules [10, 11]. Combination of enChIP with mass spectrometry (MS), RNA sequencing, and next-generation sequencing (NGS) has enabled us to identify proteins [1, 2, 6], RNAs [7], and genomic regions [9, 11] interacting with specific genomic regions of interest in a non-biased manner.
In our previous enChIP studies, we used the CRISPR system from Streptococcus pyogenes (S. pyogenes), the most extensively analyzed version of this system. However, because of its requirement for the protospacer adjacent motif (PAM) sequence (5′-NGG-3′), some DNA sequences cannot be targeted by this CRISPR system.
In addition to the S. pyogenes CRISPR system, those from other species have been used for genome editing or other purposes. Among others, the CRISPR system from Staphylococcus aureus (S. aureus) [12] has been used widely. Since the size of its Cas9 is smaller than that of the S. pyogenes Cas9, the length of its expression plasmid can be shorter than that of the S. pyogenes Cas9, enabling higher efficiency of transfection or transduction. In addition, since its PAM sequence (5′-NNGRRT-3′ or 5′-NNGRR(N)-3′) is distinct from the S. pyogenes system, DNA sequences difficult to be targeted by the S. pyogenes system can be targeted by the S. aureus system, increasing the flexibility of the enChIP technology.
To solve the potential problem of the S. pyogenes CRISPR system and increase the flexibility of enChIP analysis, we developed an enChIP system utilizing the 3xFLAG-tagged CRISPR system from S. aureus. In addition, we developed another enChIP system using the S. pyogenes CRISPR system fused to a different epitope tag. In combination, these tools might be used to constitute a sequential enChIP system with reduced background noise.
Main text
Materials and methods
Plasmids
3xFLAG-dCas9/pCMV-7.1 (Addgene #47948) was described previously [1]. To construct Sa-dCas9-NLS-3xFLAG/pcDNA3.1 (Addgene #98041), pcDNA3.1/myc-His(−) A (Invitrogen) was digested with NheI. After a blunting reaction, the plasmid was digested with NotI and treated with bacterial alkaline phosphatase (E. coli C75) (Takara Bio). The MSP2262 plasmid (Addgene #70703) [12] was digested with NcoI. After a blunting reaction, the plasmid was further digested with NotI. The cleaved pcDNA3.1/myc-His(−) A and the coding sequence of Sa-dCas9-3xFLAG from MSP2262 were purified by agarose gel electrophoresis and ligated.
To construct vectors for expression of gRNAs targeting the human interferon regulatory factor-1 (IRF-1) locus, two oligonucleotides for each gRNA were annealed, phosphorylated, and inserted into BsmBI-cleaved BPK2660 plasmid (Addgene #70709) [12] to yield hIRF-1 Sa gRNA #409 (Addgene #98134) and hIRF-1 Sa gRNA #351 (Addgene #105283). The nucleotide sequences were as follows: #409 (Addgene #98134), 5′-cacctctccagtgggaacactggg-3′, and 5′-aaaccccagtgttcccactggaga-3′; #351 (Addgene #105283), 5′-caccccagtgggatatcaagaagg-3′ and 5′-aaacccttcttgatatcccactgg-3′.
To construct pLenti_dCas9-2xAM (Addgene #92220), the coding region of Sp-dCas9 fused with an NLS and two copies of the AM-tag sequence (QDPQRKGNVILSQAY) was inserted into the CSII-U6-gRNA-CBh-3xFLAG-PA-dCas9-P2A-Puro plasmid (Addgene #83306) cleaved with BamHI and AgeI. To construct pLenti_dCas9-2xAM_hIRF-1 (Addgene #92221), two oligonucleotides (5′-caccgcgggggcgctgggctgtcc-3′ and 5′-aaacggacagcccagcgcccccgc-3′) were annealed, phosphorylated, and inserted into pLenti_dCas9-2xAM digested with BbsI.
Cell lines
The 293T cell line was derived from the 293 cell line isolated human embryonic kidneys (HEK) and transformed with the SV40 large T antigen [13]. The HT1080 cell line was derived from human fibrosarcoma [14] and purchased from ATCC (CCL-121). The 293T cell line, HT1080 cell line, and HT1080 derivatives were maintained in DMEM (Wako) supplemented with 10% fetal calf serum (FCS).
Transfection of Sa-dCas9-3xFLAG and gRNA
For transient expression of Sa-dCas9-3xFLAG, 3 µg of Sa-dCas9-NLS-3xFLAG/pcDNA3.1 was transfected into 1 × 106 293T cells using Lipofectamine 3000 (Life Technologies). For transient expression of 3xFLAG-Sp-dCas9 or Sa-dCas9-3xFLAG and their gRNA targeting the IRF-1 locus, 1.5 µg of 3xFLAG-Sp-dCas9/pCMV-7.1 or Sa-dCas9-NLS-3xFLAG/pcDNA3.1 in the absence or presence of 1.5 µg of the corresponding gRNA expression plasmid (gRNA-hIRF-1 #12 (Addgene #61079) for Sp-dCas9 or hIRF-1 gRNA #351 (Addgene #105283) and hIRF-1 gRNA #409 (Addgene #98134) for Sa-dCas9) was transfected into 1 × 106 293T cells using Lipofectamine 3000.
Transduction of pLenti_dCas9-2xAM and pLenti_dCas9-2xAM_hIRF-1
For transduction of pLenti_dCas9-2xAM and pLenti_dCas9-2xAM_hIRF-1, 5.1 µg of each plasmid was transfected into 1 × 106 293T cells along with pCAG-HIVgp (RIKEN BioResource Center RDB04394) and pCMV-VSV-G-RSV-Rev (RDB04393) [15] (3 µg each) using Lipofectamine 3000. Two days after transfection, viral supernatant was harvested and used for infection of HT1080 cells. Infected cells were selected in culture media containing puromycin (1 µg/ml).
Immunoblot analysis
Nuclear extracts (NE) were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). NE (10 µg) was subjected to immunoblot analysis with anti-FLAG M2 Ab (F1804, Sigma-Aldrich) or Ab against AM-tag (39715, Active Motif) as described previously [1].
enChIP-real-time PCR
enChIP-real-time PCR was performed as described previously [1]. Anti-FLAG M2 Ab (3 µg Ab/4 × 106 cells) or Ab against AM-tag (2 µg Ab/4 × 106 cells) were used. Primers used in the analysis are shown in Table 1.
Table 1.
Primers used in this study
| Number | Name | Sequence (5′ → 3′) | Experiments |
|---|---|---|---|
| 27222 | hSox2-prom-F | attggtcgctagaaacccatttatt | Real-time PCR in Figs. 1e and 2d (SOX2) |
| 27223 | hSox2-prom-R | ctgccttgacaactcctgatacttt | Real-time PCR in Figs. 1e and 2d (SOX2) |
| 27310 | hIRF1-prom-F | cgcctgcgttcgggagatatac | Real-time PCR in Figs. 1e and 2d (IRF1) |
| 27312 | hIRF1-prom-R1 + 2 | ctgtcctctcactccgccttgt | Real-time PCR in Figs. 1e and 2d (IRF1) |
Results and discussion
enChIP analysis using the S. aureus CRISPR system
To determine whether the S. aureus CRISPR system could be used for enChIP analysis (Fig. 1a, b), we constructed a mammalian expression plasmid (Sa-dCas9-NLS-3xFLAG/pcDNA3.1) encoding Sa-dCas9 fused with an NLS and the 3xFLAG-tag (Sa-dCas9-3xFLAG), and transiently transfected it into 293T cells. Expression of Sa-dCas9-3xFLAG was confirmed by immunoblot analysis (Fig. 1c).
Fig. 1.
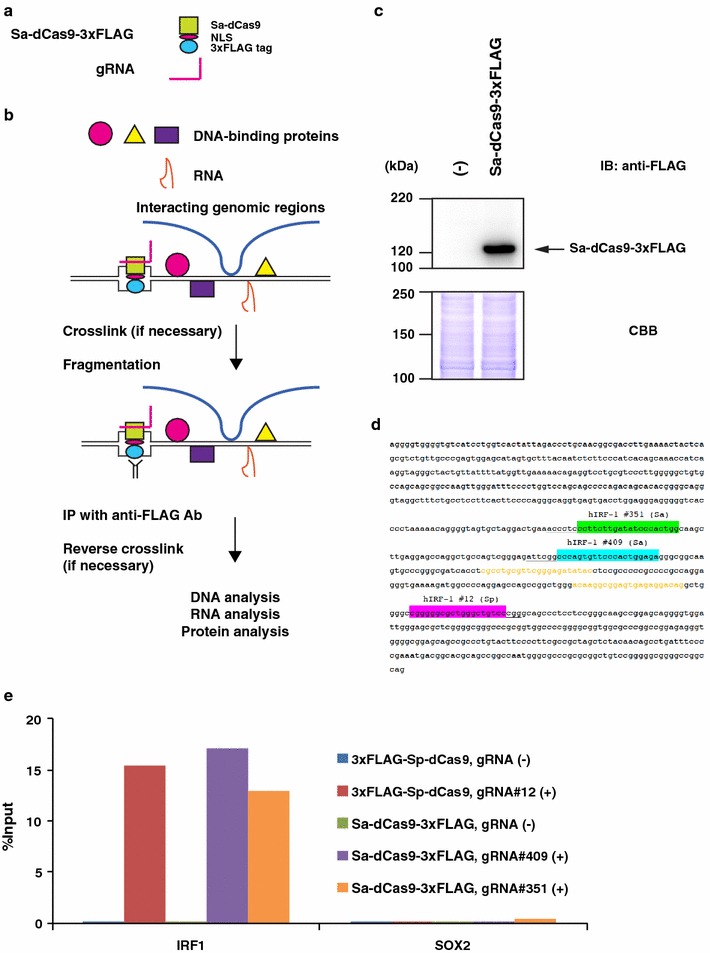
enChIP system using S. aureus CRISPR. a The S. aureus CRISPR system for enChIP. The system is composed of a fusion protein, Sa-dCas9-3xFLAG (consisting of Sa-dCas9, an NLS, and a 3xFLAG-tag) and a gRNA. b Scheme of the enChIP system using S. aureus CRISPR. The Sa-dCas9-3xFLAG and gRNA are expressed for locus-tagging in the target cells. The cells are crosslinked (if necessary), lysed, and fragmented by sonication or other methods. Chromatin complexes containing the CRISPR complex are immunoprecipitated with anti-FLAG Ab, and the crosslink (if used) is reversed. Molecules (DNA, RNA, proteins, etc.) associated with the target genomic region can be identified by downstream analyses (e.g., nucleic acids by next-generation sequencing, proteins by mass spectrometry). c Expression of Sa-dCas9-3xFLAG. Plasmid expressing Sa-dCas9-3xFLAG was transfected into 293T cells. Nuclear extracts were prepared and subjected to immunoblot analysis (IB) with anti-FLAG Ab. Coomassie Brilliant Blue (CBB) staining is shown as a protein loading control. d Positions of gRNAs in the IRF-1 promoter. Green highlight: hIRF-1 #351 (Sa) gRNA; blue highlight: hIRF-1 #409 (Sa) gRNA; magenta highlight: hIRF-1 #12 (Sp) gRNA; underlines: PAM sequences; orange letters: primers for enChIP-PCR analysis. e Isolation of the IRF-1 locus by enChIP using the S. aureus CRISPR system. Real-time PCR analysis of chromatin complexes isolated by enChIP is shown. An irrelevant locus (SOX2) was analysed as a negative control. The S. pyogenes CRISPR system was used as a positive control for enChIP
Next, for enChIP analysis, we transfected Sa-dCas9-NLS-3xFLAG/pcDNA3.1 alone or along with a plasmid expressing gRNA targeting the human IRF-1 promoter (hIRF-1 gRNA #351 or hIRF-1 gRNA #409) (Fig. 1d). As a positive control, we also transfected 3xFLAG-Sp-dCas9/pCMV-7.1 expressing 3xFLAG-Sp-dCas9, alone or with a plasmid expressing gRNA targeting the human IRF-1 promoter (gRNA-hIRF-1 #12) (Fig. 1d). Two days after transfection, cells were crosslinked with formaldehyde. Crosslinked chromatin was fragmented by sonication. Next, chromatin complexes containing Sa-dCas9-3xFLAG or 3xFLAG-Sp-dCas9 were immunoprecipitated with anti-FLAG Ab. For both Sp- and Sa-dCas9, real-time PCR showed that the IRF-1 promoter region was specifically detected only in the immunoprecipitants prepared from 293T cells transfected with gRNA targeting the IRF-1 promoter (Fig. 1e). The yields of enChIP for Sa-dCas9 were comparable with those for Sp-dCas9. These results showed that enChIP using the S. aureus CRISPR system can also specifically and efficiently isolate target genomic regions.
Generation of a lentiviral expression system for enChIP using S. pyogenes CRISPR and 2xAM-tag
The orthologues of Cas9 can be used for sequential enChIP, which might be useful for decreasing the background noise of either system alone. However, for sequential enChIP, CRISPR complexes of different species must be pulled down using different affinity-purification systems (e.g., purification using Abs against each dCas9 or different epitope tags). To this end, we generated a lentiviral expression system of S. pyogenes CRISPR in which Sp-dCas9 was fused with an NLS and the 2xAM-tag (Sp-dCas9-2xAM) (Fig. 2a). The lentiviral expression system allows quick establishment of stable transformants expressing the CRISPR complex even when the target cells are not proliferating. The coding region of Sp-dCas9-2xAM was inserted into the CSII-U6-gRNA-CBh-3xFLAG-PA-dCas9-P2A-Puro plasmid to generate pLenti_dCas9-2xAM. The gRNA dsDNA targeting the IRF-1 promoter was inserted into pLenti_dCas9-2xAM to generate pLenti_dCas9-2xAM_hIRF-1.
Fig. 2.
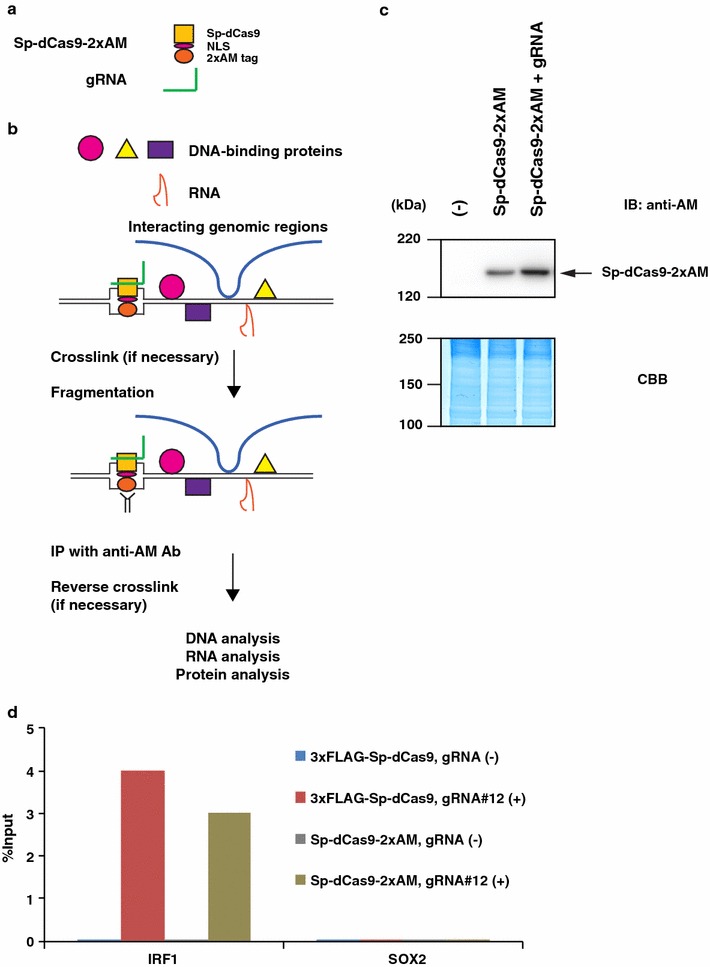
enChIP using the S. pyogenes CRISPR system tagged with the 2xAM-tag. a The S. pyogenes CRISPR system tagged with 2xAM-tag for enChIP. The system is composed of a fusion protein Sp-dCas9-2xAM (consisting of Sp-dCas9, an NLS, and a 2xAM-tag) and a gRNA. b Scheme of the enChIP system using S. pyogenes CRISPR tagged with the 2xAM-tag. After expression of Sp-dCas9-2xAM and a gRNA for locus-tagging in target cells, enChIP is performed using an Ab against the AM-tag, as shown in Fig. 1b. c Expression of Sp-dCas9-2xAM. pLenti_dCas9-2xAM or pLenti_dCas9-2xAM_hIRF-1 was transduced into HT1080 cells. After puromycin selection, expression of Sp-dCas9-2xAM was detected by immunoblot analysis with Ab against AM-tag. d Isolation of the IRF-1 locus by the S. pyogenes CRISPR system tagged with 2xAM-tag. Real-time PCR analysis was performed on chromatin complexes isolated by enChIP. An irrelevant locus (SOX2) was analysed as a negative control. The S. pyogenes CRISPR system tagged with the 3xFLAG-tag was used as a positive control for enChIP
To confirm that the system (Fig. 2b) works, pLenti_dCas9-2xAM or pLenti_dCas9-2xAM_hIRF-1 was transduced into a human fibrosarcoma cell line, HT1080. After puromycin selection, expression of Sp-dCas9-2xAM was confirmed by immunoblot analysis with Ab against the AM-tag (Fig. 2c).
Next, we examined yields of enChIP for the target IRF-1 promoter region. Cells expressing Sp-dCas9-2xAM in the absence or presence of gRNA targeting the IRF-1 promoter were crosslinked with formaldehyde. The crosslinked chromatin was fragmented by sonication, and complexes containing the CRISPR complexes were subjected to affinity purification using Ab against the AM-tag. The yields of enChIP in the presence of the gRNA were comparable with those obtained using HT1080 expressing 3xFLAG-dCas9 and gRNA targeting the IRF-1 promoter region [16] (Fig. 2d). These results showed that enChIP using the 2xAM-tag can also specifically and efficiently isolate target genomic regions. Because lentivirus can infect non-dividing cells, this enChIP system would also be useful for cells that are not proliferating.
We have not generated stable cell lines expressing Sa-dCas9-3xFLAG. Therefore, at this stage, we cannot compare the enChIP efficiencies between Sa-dCas9-3xFLAG and the lentiviral system of Sp-dCas9-2xAM. However, considering that the enChIP efficiencies between 3xFLAG-Sp-dCas9 and Sa-dCas9-3xFLAG using transient expression systems are comparable (Fig. 1e), and the enChIP efficiencies between 3xFLAG-Sp-dCas9 and Sp-dCas9-2xAM using stable transformants are also comparable (Fig. 2d), we speculate that the enChIP efficiencies between Sa-dCas9-3xFLAG and Sp-dCas9-2xAM might also be comparable.
Conclusions
In this study, we developed an enChIP system using S. aureus CRISPR. Due to its distinct PAM sequence (5′-NNGRRT-3′ or 5′-NNGRR(N)-3′), this system will increase the flexibility of the enChIP technology by increasing the proportion of genomic regions that can be targeted. In addition, we developed another enChIP system using S. pyogenes CRISPR fused with a different epitope tag different than the one used for S. aureus CRISPR. Both enChIP systems achieved high yields of the target locus, and it is expected that their yields would be comparable.
Limitations
Further studies might be necessary to assess the utility of the S. aureus system in enChIP combined with MS and NGS in order to allow comparisons with features of the S. pyogenes system such as background signals, off-target binding, etc. In addition, it would be of interest to determine whether sequential enChIP analysis could be performed using the S. pyogenes and S. aureus CRISPR systems.
Authors’ contributions
HF conceived the idea of potential use of Sa-dCas9 for enChIP and constructed expression plasmids. TF and HF designed and performed experiments and wrote the manuscript. MY performed experiments. HF directed and supervised the study. All authors read and approved the final manuscript.
Acknowledgements
We thank J. Keith Joung for MSP2262 (Addgene plasmid #70703) and BPK2660 (Addgene plasmid #70709), Tohru Kimura and Yoichi Sekita for CSII-U6-gRNA-CBh-3xFLAG-PA-dCas9-P2A-Puro (Addgene plasmid #83306), and Hiroyuki Miyoshi for pCAG-HIVgp (RIKEN BioResource Center RDB04394) and pCMV-VSV-G-RSV-Rev (RIKEN BioResource Center RDB04393).
Competing interests
T.F. and H.F. have patents on enChIP (Patent name: Method for isolating specific genomic region using molecule binding specifically to endogenous DNA sequence; Patent number: Japan 5,954,808; Patent Application Number: WO2014/125668). T.F. and H.F. are founders of Epigeneron, Inc.
Availability of data and materials
All data generated or analyzed during the current study are included in this published article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the Takeda Science Foundation (T.F.), Grant-in-Aid for Scientific Research (C) (#15K06895) (T.F.), and Grant-in-Aid for Scientific Research (B) (#15H04329) (T.F., H.F.), ‘Transcription Cycle’ (#15H01354) (H.F.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- enChIP
engineered DNA-binding molecule-mediated chromatin immunoprecipitation
- CRISPR
clustered regularly interspaced short palindromic repeats
- dCas9
a catalytically inactive form of Cas9
- NLS
nuclear localization signal
- gRNA
guide RNA
- TAL
transcription activator-like
- Ab
antibody
- MS
mass spectrometry
- NGS
next-generation sequencing
- PAM
protospacer adjacent motif
- NE
nuclear extracts
- IRF-1
interferon regulatory factor-1
Contributor Information
Toshitsugu Fujita, Email: toshitsugu.fujita@hirosaki-u.ac.jp.
Miyuki Yuno, Email: miyuki_yuno@biken.osaka-u.ac.jp.
Hodaka Fujii, Email: hodaka@hirosaki-u.ac.jp.
References
- 1.Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439:132–136. doi: 10.1016/j.bbrc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Fujita T, Asano Y, Ohtsuka J, Takada Y, Saito K, Ohki R, Fujii H. Identification of telomere-associated molecules by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) Sci Rep. 2013;3:3171. doi: 10.1038/srep03171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 4.Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright AV, Nuñez JK, Doudna JA. Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T, Fujii H. Identification of proteins interacting with genomic regions of interest in vivo using engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) Bio Protoc. 2014;4:e1124. doi: 10.21769/BioProtoc.1124. [DOI] [Google Scholar]
- 7.Fujita T, Yuno M, Okuzaki D, Ohki R, Fujii H. Identification of non-coding RNAs associated with telomeres using a combination of enChIP and RNA sequencing. PLoS One. 2015;10:e0123387. doi: 10.1371/journal.pone.0123387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita T, Yuno M, Fujii H. Allele-specific locus binding and genome editing by CRISPR at the p16INK4a locus. Sci Rep. 2016;6:30485. doi: 10.1038/srep30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujita T, Yuno M, Suzuki Y, Sugano S, Fujii H. Identification of physical interactions between genomic regions by enChIP-Seq. Genes Cells. 2017;22:506–520. doi: 10.1111/gtc.12492. [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Yuno M, Fujii H. Efficient sequence-specific isolation of DNA fragments and chromatin by in vitro enChIP technology using recombinant CRISPR ribonucleoproteins. Genes Cells. 2016;21:370–377. doi: 10.1111/gtc.12341. [DOI] [PubMed] [Google Scholar]
- 11.Fujita T, Kitaura F, Yuno M, Suzuki Y, Sugano S, Fujii H. Locus-specific ChIP combined with NGS analysis reveals genomic regulatory regions that physically interact with the Pax5 promoter in a chicken B cell line. DNA Res. 2017;24:537–548. doi: 10.1093/dnares/dsx023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/MCB.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080) Cancer. 1974;33:1027–1033. doi: 10.1002/1097-0142(197404)33:4<1027::AID-CNCR2820330419>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda H, Osakada F, Watanabe K, Mizuseki K, Haraguchi T, Miyoshi H, Kamiya D, Honda Y, Sasai N, Yoshimura N, et al. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc Natl Acad Sci USA. 2005;102:11331–11336. doi: 10.1073/pnas.0500010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujita T, Fujii H. Identification of proteins associated with an IFNγ-responsive promoter by a retroviral expression system for enChIP using CRISPR. PLoS One. 2014;9:e103084. doi: 10.1371/journal.pone.0103084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during the current study are included in this published article.