

Abstract
Background
Current therapies for HNSCC, especially platinum agents, are limited by their toxicities and drug resistance. This study evaluates a novel C-terminal Hsp90 inhibitor (CT-Hsp90-I) for efficacy and toxicity in vitro and in vivo in an orthotopic HNSCC model. Our hypothesis is that C-terminal inhibitors exhibit improved toxicity/efficacy profiles over standard therapies and may represent a novel group of anticancer agents.
Methods
MDA-1986 HNSCC cells were treated with doses of 17-AAG or KU363 (a CT-Hsp90-I) and compared for antiproliferation by GLO-Titer and trypan blue exclusion and for apoptosis by PARP cleavage and caspase-3 inactivation by Western analysis. In vivo studies in Nu/Nu mice examined an orthotopic model of MDA-1986 cells followed by drug dosing intraperitoneally for a 21-day period (mg/kg/dose: cisplatin = 3.5, low-dose KU363 = 5, high-dose KU363 = 25, 17-AAG = 175). Tumor size, weight, and toxicity (body score) were measured 3×/week.
Results
The IC50 levels for KU363 = 1.2–2 μM in MDA-1986. KU363 induces apoptosis at 1 μM with cleavage of PARP and inactivation of caspase-3 levels after 24 h. Client proteins Akt and Raf-1 were also downregulated at 1–3 μM of drug. In vivo, 100% of controls had progressive disease, while 100% of cisplatin animals showed some response, all with significant systemic toxicity. High-dose KU363 showed 88% of animals responding and low-dose KU363 showed 75% responding. KU363 animals showed significantly less toxicity (P < 0.01) than cisplatin or 17-AAG.
Conclusion
This novel CT-Hsp90-I KU363 manifests potent anticancer activity against HNSCC, showing excellent in vivo efficacy and reduced toxicity compared with standard agents justifying future translational evaluation.
Head and neck cancer accounted for more than 36,000 new cases in 2010 with nearly 8000 deaths in the United States, making it the 8th leading cause of new cancer cases among men.1 Worldwide, an estimated 644,000 new cases of head and neck cancers are diagnosed each year.2 Of these, head and neck squamous cell carcinoma (HNSCC) accounts for more than 90% of cases, with a median age for diagnosis in the 6th decade and a male predominance with a M:F incidence ratio of 3:1. Most patients with HNSCC present with advanced-stage locoregional disease, for which the standard of treatment is a multidisciplinary approach involving combinations of surgery, chemotherapy, and radiation.3 However, the overall 5-year survival rate from HNSCC is less than 50%, which has remained relatively unchanged for the past 2 decades.4 Also, the effectiveness of systemic chemotherapy, which is primarily platinum-based regimens, is limited by its toxicity and platinum-drug resistance in HNSCC patients.5,6 This indicates a need for additional treatment options that target the cancer more effectively and with reduced toxicity.
Heat shock protein 90 (Hsp90) is a molecular chaperone that has emerged in the last decade as a promising target for cancer therapy. While most current monotherapies, such as cisplatin, work by disrupting a single molecular function, Hsp90 is unique in that it modulates multiple oncogenic pathways simultaneously. As a molecular chaperone, it promotes the conformational maturation of “client” proteins, protecting them from degradation.7 Many of these “clients” are protein kinases (tyrosine kinases, Bcr-Abl, epidermal growth factor receptor [EGFR] family members, and serine/threonine kinases, Akt, Raf-1) and transcription factors (p53, Stat3) that are involved in multiple signal transduction pathways in HNSCC.8 Furthermore, Hsp90 is overexpressed in many human malignancies, such as HNSCC, and inhibition of Hsp90 allows for the development of small molecules that exhibit high differential selectivity.9–12 Therefore, inhibition of Hsp90 disrupts multiple signaling pathways that contribute to malignancy.13–15
Inhibitors of Hsp90 have been studied previously against HNSCC. Multiple studies have demonstrated that Hsp90 inhibition leads to degradation of client proteins and enhances tumor cell death. A geldanamycin (GA) derivative, 17-allylamino-17-demethoxygeldanamycin (17-AAG), is the most prevalent Hsp90 inhibitor used in preclinical studies and functions by binding to the N terminus of Hsp90.16–18 Recent clinical trials, however, have demonstrated that N-terminal HSP90 inhibitors were not therapeutically effective and that 17-AAG displays dose-limited toxicity and is somewhat difficult to formulate.19–21 In recent years, inhibitors that interact with the C terminus of Hsp90 have been investigated in several cancer models.22,23 Novobiocin, a member of the coumermycin family of antibiotics, has been shown to exhibit antitumor activity through inhibition of Hsp90 at the C terminus.24 KU363 and KU135, novobiocin-derived C-terminal Hsp90 inhibitors (CT-Hsp90-I) synthesized at the University of Kansas-Lawrence, have been shown to manifest antiproliferative activity against different cancer models.25,26 The aim of the present study is to investigate the efficacy of the novel C-terminal Hsp90 inhibitor KU363 against HNSCC both in vitro and in vivo for improved efficacy with reduced toxicity over N-terminal Hsp90 inhibitors and standard chemotherapeutic agents.
MATERIALS AND METHODS
Bioassay Materials
Culture media, fetal bovine serum (FBS), penicillin G, streptomycin, MEM-nonessential amino acids, ribonuclease A, and propidium iodide (PI) were obtained from Sigma-Aldrich (St. Louis, MO). MEM-vitamin solution was purchased from Life Technologies, Inc. (Grand Island, NY). Annexin V-FITC was from BD Bioscience (Bedford, MA). Primary antibodies against β-actin and secondary antibodies against mouse and rabbit antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies against phospho-Akt, total-Akt, Raf-1, ErB2b/Her2, Caspase-3, and PARP were obtained from Cell Signaling Technologies (Beverly, MA). BCA protein assay reagents were obtained from Pierce (Rockford, IL). Protease inhibitor mixture set II was obtained from Calbiochem (San Diego, CA). Novobiocin and 17-AAG were obtained from Sigma-Aldrich (St. Louis, MO), and KU363 was synthesized by the Department of Medicinal Chemistry at the University of Kansas (Lawrence, KS).
Cell Culture
The invasive oral squamous carcinoma cell lines JMAR and MDA-1986 were a gift from Dr. Jeffrey Myers (University of Texas, M.D. Anderson Cancer Center, Houston, TX), JHU-011 was a gift from Dr. Josef Califrano (Johns Hopkins University, Baltimore, MD), UM-SCC-2 was a gift from Dr. Scott Weed (University of West Virginia, Morgantown, WV), and the fibroblast cell line MRC-5 was purchased from the ATCC (Manassas, VA). Cell lines MDA-1986, JMAR, UM-SCC-2, and MRC-5 were grown in DMEM (Sigma-Aldrich), and JHU-011 was grown in RPMI (Sigma-Aldrich). Both media were supplemented with 10% FBS (Sigma-Aldrich), sodium pyruvate, nonessential amino acids, L-glutamine, a 2-fold MEM-vitamin solution, and 1% penicillin/streptomycin (100 IU/ml/100 μg/ml; Sigma-Aldrich). Adherent monolayer cultures were maintained in T-75 culture flasks and incubated at 37°C with 5% CO2 until they achieved 85% confluency. The cells were then trypsinized using 0.25% trypsin (Sigma-Aldrich) and passaged into T-75 flasks at a density of 1 × 106 cells. On the day of experiments, cells were trypsinized and counted via hemocytometer to determine the number of viable cells.
Cell Viability and Proliferation Assays
JMAR, MDA-1986, JHU-011, UM-SCC-2, and MRC-5 were plated in growth media on 384-well microtiter plates at a concentration of 2 × 103 cells/well and on 6-well plates at a concentration of 2 × 104 cells/well. Cells were incubated for 24 h at 37°C with 5% CO2. Cells were then treated with varying concentrations of 17-AAG (Sigma-Aldrich), Novobiocin (Sigma-Aldrich), or experimental drug KU363 (University of Kansas, Lawrence, KS) for 24 or 72 h. All drugs were dissolved in 100% dimethylsulfoxide (DMSO) (Sigma-Aldrich). The 384-well plates were analyzed according manufacturer’s directions for the CellTiterGlo Luminescent Cell Viability Assay (Promega, Madison, WI) to obtain IC50 values and were analyzed using a plate reader (Synergy 4, BioTek Instruments Inc., Winooski, VT) at an absorbance wavelength of 490 nM. The potency (IC50 value) and the efficacy (area under the dose-response curve [AUC]) of each drug were used as the response measures to compare the treatment groups. The IC50 is defined as the concentration of drug at which 50% of the cells are inhibited by the drug, and AUC was determined using data points bounded by the highest and lowest concentrations and the highest and lowest percent viability for each curve. Nonlinear regression and sigmoidal dose-response curves (GraphPad Prism, La Jolla, CA) were used to calculate IC50 values, and AUC was calculated using the trapezoid rule before normalizing the data. The 6-well plates were counted on a hemocytometer with trypan blue staining to determine cell proliferation under different levels of drug treatment. All samples were analyzed in triplicate.
Fluorescence-Activated Cell Sorting Analysis
MDA-1986 cells in T-75 flasks were treated with varying concentrations of KU363 for 24 h. Cells were trypsinized, washed with 0.9% NaCl, and fixed with 70% cold ethanol for 30 min at room temperature. They were then collected by centrifugation (700g for 5 min) and stained with PI (50 mg/ml in phosphate-buffered saline [PBS]) for 30 min and then treated with DNAse-free RNAse (1 mg/ml) for 30 min and analyzed by flow cytometry. Evaluation for induction of apoptosis was performed using Annexin V/PI costaining with flow cytometry (BD LSRII, Becton Dickerson, San Diego, CA). An analysis of phosphatidylserine (PS) on the outer leaflet of apoptotic cell membranes was performed using Annexin V-FITC and PI to distinguish between apoptotic and necrotic cells. After treatment, 1 × 105 cells/ml were washed with 1× PBS and trypsinized. They were then stained with Annexin-V/PI according to the manufacturer’s instruction (BD-Pharmingen, San Diego, CA) and analyzed by flow cytometry.
Western Blot Analysis
MDA-1986 cells were treated for 24 h in T-75 flasks with varying concentrations of KU363. After treatment, cells were lysed (0.5% Nonidet P-40,100 mM, 10 mMTris [pH 7.5], 1:500 protease inhibitor mixture set II, 1 mMNaF, and 1 mM sodium orthovanadate), and lysates were placed on ice for 20 min. Clear lysates were obtained by centrifugation (14,000g for 20 min). Equal amounts of protein were separated by SDS-PAGE and electrotransferred onto a Hybond nitrocellulose membrane (Amersham). The membranes were blocked and probed with the appropriate dilution of primary antibody overnight at 4°C. The blots were washed three times in PBS–Tween-20 for 10 min and then incubated in horseradish peroxidase-conjugated secondary antibody in PBS–Tween-20 at room temperature for 1 h. After washing in PBS–Tween-20, the proteins were visualized by enhanced chemiluminescence reagent (Amersham) and captured on Kodak XAR-5 film (Eastman Kodak, Rochester, NY). Where indicated, the blots were reprobed with antibody against β-actin to ensure equal loading and transfer of proteins.
In Vivo Tumor Model
All animal studies were done in accordance with IACUC guidelines. MDA-1986 cells were prepared in a 1× PBS solution at a concentration of 1 × 106 cells/100 μl. Cells (100 μl) were injected under isoflurane anesthesia into the retromandibular buccal mucosa of 4- to 6-week-old Nu/Nu mice using a 25G needle (20–25 g, Charles River Laboratories, Wilmington, MA). Tumor size was measured three times weekly using a digital caliper. Tumor volume was calculated using:
When tumors reached a minimum volume of 50 mm3, mice were randomized into control (PBS) or 1 of 4 treatment groups (17-AAG at 175 mg/kg/dose, cisplatin at 3.5 mg/kg/dose, high-dose KU363 at 25 mg/kg/dose, and low-dose KU363 at 5 mg/kg/dose). All drugs were administered intraperitoneally (ip). Controls, low-dose, and high-dose KU363 and 17-AAG treatment groups were treated every other day, and the cisplatin group received daily injections. All mice were treated for a total of 21 days. Mice were euthanized prior to completion of the experiment if the tumor reached >20 mm in any direction or body score fell below 2.
Statistical Analysis
Comparisons of differences between 2 or more means were determined by unpaired t test (2 means) and Fisher exact test. More than 2 means were analyzed by 2-way ANOVA followed by Duncan’s multiple range test (2+ means) and Bonferroni post hoc testing via a standard statistical analysis software package (SPSS version 17.0, SPSS Inc., Chicago, IL). Significance was defined for P < 0.05.
RESULTS
KU363 Reduces Cell Viability and Proliferation in HNSCC Cells
To investigate the biological effect of Hsp90 inhibition by KU363 in HNSCC cells, we incubated 4 HNSCC cell lines (JMAR, MDA-1986, JHU-011, and UM-SCC-2) and MRC-5, a fibroblast cell line as a comparison for normal cells with increasing concentrations of KU363, novobiocin, and 17-AAG. Viability was determined by the CellTiterGlo Luminescent Cell Viability Assay. KU363 inhibited cell viability in a dose-dependent manner in all 4 HNSCC cell lines (Fig. 1a). KU363 IC50 was determined to be 1.42, 1.24, 0.89, 1.11, and 3.91 μM for MDA-1986, JMAR, UM-SCC-2, JHU-011, and MRC-5 cells, respectively, at 72 h, which are comparable to the 17-AAG IC50 values and are 30-fold lower than those of novobiocin (Fig. 1b). Also, KU363 was noted to manifest an IC90 of <5 μM. Trypan blue staining was used to investigate cell proliferation and cell death. MDA-1986, JMAR, UM-SCC-2, and JHU-011 cells were treated with varying concentrations of KU363 at 24- and 72-hour time points. KU363 was shown to reduce cell proliferation and induces cell death in MDA-1986 in a concentration-dependent manner. Treatment of MDA-1986 cells with KU363 reduced cell proliferation by 3- and 7-fold, respectively, at IC50 and 2 × IC50 concentrations. In addition, the percentage of trypan blue positive cells increased from 5% in untreated cells to approximately 30% at IC50 and 70% at 2 × IC50 concentrations of HNSCC cells treated with KU363 (raw data not shown).
FIG. 1.

a GLO-titer cell-viability curve for KU363 in JMAR, UMSCC-2, MDA-1986, and JHU-011 head and neck squamous cell cancer cells in vitro. KU363 is cytotoxic with IC50 levels 0.8–2 μM and KU363 with IC90 level <5 μM. b Comparison of in vitro cell viability with C- and N-terminal Hsp90 inhibitors using GLO-titer assay with IC50 levels reported in μM concentrations
KU363 Induces Apoptosis and Inhibition Through Hsp90 Pathways
In order to evaluate induction of apoptosis, we examined MDA-1986 cells treated with KU363 by flow cytometry using AnnexinV/PI staining. After treatment with 3 μM concentration of KU363, the MDA-1986 show a shift of cells staining with AnnexinV, indicating that the majority of the cells are in an apoptotic state (Fig. 2a). To confirm the induction of apoptosis, we evaluated the activation of caspase-3 upon treatment of HNSCC cells with KU363. MDA-1986 cells were treated with increasing concentrations of KU363. The activation of caspase was observed by increased cleavage of the caspase-3 substrate PARP as well as an increase in the levels of cleaved caspase-3 (Fig. 2b). PARP cleavage was detected at concentrations as low as 1 μM after 24 h of KU363 treatment. Caspase-3 activation was also observed starting at 1 μM, which is consistent with KU363 IC50 data. These results demonstrate that caspase-3 activation begins as early as 24 h after treatment with KU363.
FIG. 2.

a Annexin V/PI staining of MDA-1986 cells untreated (top) versus treated with KU363 (bottom) for 24 h. b Western Blot analysis of MDA-1986 cells at 24-hour treatment with KU363 for PARP, cleaved PARP, cleaved caspase 3, and β-actin. c Western Blot analysis of MDA-1986 cells at 24-hour treatment with KU363 for p-Akt and β-actin with densitometry measurements in blue. d Western Blot analysis of MDA-1986 cells at 24-hour treatment with KU363 or 17-AAG for Raf-1, total Akt, ErbB2/Her2, and β-actin with densitometry measurements in blue
We next examined the effect of KU363 on the PI3 kinase/Akt signaling pathway as well as on the Hsp90 client protein pathway of Raf-1 and ErbB2/Her2. These results showed that KU363 reduced Ser473 phosphorylation of Akt and total Akt levels in HNSCC cells after 24 h of treatment in a dose-dependant manner (Fig. 2c, d). Densitometry was performed on these blots, indicating a quantitative reduction in p-Akt and total Akt levels with KU363 treatment (p-Akt expression is downregulated starting at 2.5 μM KU363 [approximately twice IC50], while total Akt levels are downregulated at only 1 μM of drug). Treatment of MDA-1986 cells with KU363 also showed reduction of ErbB2/Her2 and Raf-1 in a concentration-dependent fashion starting as low as 1 μM for the reduction of ErbB2/Her2, again confirmed by densitometry. With regard to 17-AAG treatment, although there was a reduction noted in levels of total Akt, no reduction was seen in the Hsp90 client proteins ErbB2/Her2 and Raf-1 (Fig. 2d).
In Vivo Efficacy of KU363
To examine the efficacy of KU363 in vivo, an orthotopic tumor model of HNSCC was created through injection of the buccal mucosa of female 4- to 6-week Nu/Nu mice. When a tumor volume of 50 mm3 was obtained, the mice were randomized into 1 of 5 groups (control, 17-AAG, cisplatin, high-dose KU363, and low-dose KU363). There were eight animals placed in each group for a total of 40 animals. The mice were monitored for tumor volume, weight, and body score 3 × /week for 6 weeks. Tumor volume growth curves of the mice can be seen in Fig. 3.
FIG. 3.

Tumor volume and survival curves of the MDA-1986 Nu/Nu mice for a the control group (PBS ip), b cisplatin (3.5 mg/kg/dose ip), c 17-AAG (175 mg/kg/dose ip), d low-dose KU363 (5 mg/kg/dose ip), and e high-dose KU363 (25 mg/kg/dose ip)
The cisplatin treatment group demonstrated the best overall tumor response rate, with 100% of the mice showing some level of response (six complete response [CR] and two partial responders [PR]; >30% reduction). The next most efficacious therapy was the high-dose (25 mg/kg/dose) KU363 group, which had 88% of the animals with some response to treatment (three CR [37%], four PR [50%], and one with stable disease [SD]). Next came the low-dose (5 mg/kg/dose) KU363 treatment group, in which 75% of the animals had a response to therapy (one CR [12%], five PR [63%], and two SD [25%]). Following this was the 17-AAG treatment group, which had 5 of 8 animals (63%) showing a partial response to therapy. In this group there were no CRs, two SDs (25%), and one animal with progressive disease (PD) (12%). Finally, the control group had the worst outcomes with 0% of animals showing a response and 100% of animals showing PD (Table 1). Upon completion of 3 weeks of treatment, the tumor size in the control group compared with the complete response in the KU363 high dose group can easily be seen (Fig. 4).
TABLE 1.
MDA1986 Nu/Nu treatment response based on modified RECIST criteria
| Tx group | Animal no. 1 | Animal no. 2 | Animal no. 3 | Animal no. 4 | Animal no. 5 | Animal no. 6 | Animal no. 7 | Animal no. 8 |
|---|---|---|---|---|---|---|---|---|
| Control | PD | PD | PD | PD | PD | PD | PD | PD |
| 5 mg/kg KU363 | PR | CR | PR | PR | PR | PR | SD | SD |
| 25 mg/kg KU363 | CR | CR | PR | SD | PR | CR | PR | PR |
| 17-AAG | PR | PR | PD | PR | PR | SD | PR | SD |
| Cisplatin | PR | PR | CR | CR | CR | CR | CR | CR |
Percentages are obtained by comparison of final tumor volume to tumor volume at time of first treatment
CR complete response, PR partial response (>30% reduction in tumor volume), SD stable disease (<30% reduction in tumor volume or <30% growth in tumor volume), PD progressive disease (>30% growth in tumor volume)
FIG. 4.
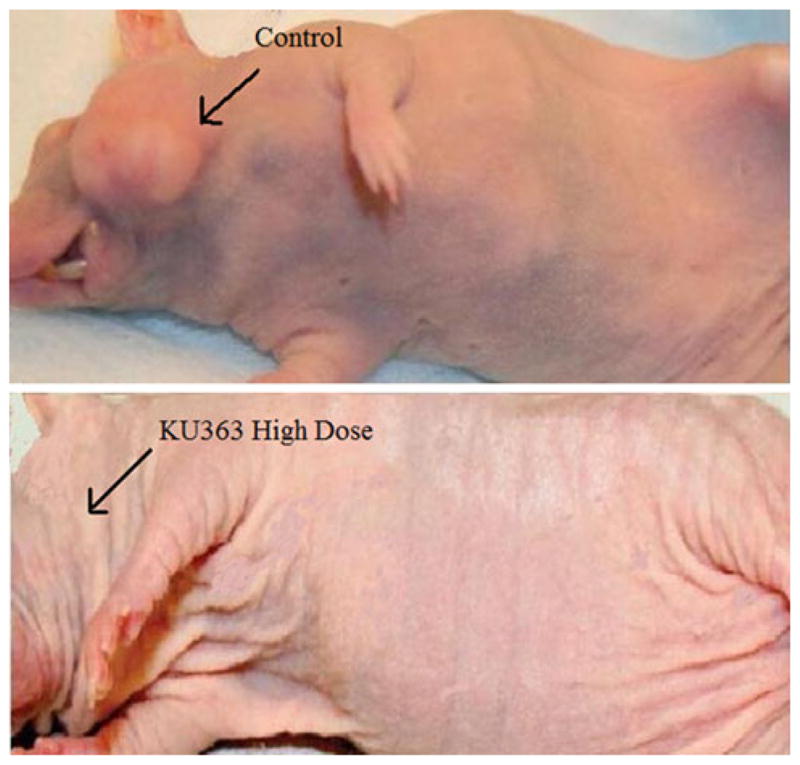
Tumor response in Nu/Nu mice
Although cisplatin had an overall better efficacy than KU363 or 17-AAG, it also resulted in a significantly worse toxicity profile. Overt signs of drug toxicity were seen in 100% of the animals as evidenced by a 20% or greater weight loss at the end of the 3-week treatment period that was sustained throughout the duration of the trial. In comparison, only one animal each in the low-dose and high-dose KU363 groups (12%) showed any significant sustained weight loss, which was statistically significant (P = 0.0003 for high dose and P = 0.0067 for low dose) (Fig. 5). It should be noted, however, that the KU363 high-dose group did have transient weight loss around the time of treatment that was recovered once the treatment was concluded. This was not seen in the cisplatin group. With regard to body score, the cisplatin animals all had body scores of two at the completion of the study compared with only one animal in the KU363 high-dose group and no animals in the KU363 low-dose group. Upon gross pathologic analysis of the mice at the completion of the study, it was seen that there was some evidence of intra-abdominal adhesion development in three of the mice that were treated in the high-dose KU363 group. This was not seen in the low-dose KU363 group or in the cisplatin or 17-AAG groups.
FIG. 5.

Average weight loss per treatment group of MDA-1986 Nu/Nu mice. P values are calculated in comparison to cisplatin
DISCUSSION
Small-molecule kinase-targeted drugs have been at the forefront of a new class of cancer therapeutics. However, cancer cells routinely develop drug resistance to these targeted therapies. Therefore, there is a need to be able to identify novel targets that regulate multiple proteins and signal transduction pathways, such Hsp90. In recent pre-clinical studies, N-terminal Hsp90 inhibitors have been used to target HNSCC; however, these inhibitors have been shown to be less successful in other cancer lines in human clinical trials.16,17,20,21
KU363, however, functions by binding at the C terminus of Hsp90, thus resulting in inhibition of its chaperone activity. In vitro, it was observed to reduce cell viability and proliferation in four different HNSCC cell lines. It also triggered significant induction of apoptosis that was seen with flow cytometry and confirmed by caspase-3 activation. The Western data also revealed downregulation of Hsp90 client proteins ErbB2/Her2 and Raf-1. In recent studies, it has been shown that ErbB2/Her2 is one of the more sensitive client proteins of Hsp90.27
In the in vivo experiments, KU363 was shown to be efficacious. Although cisplatin demonstrated a higher level of efficacy than KU363, it also had a significantly worse toxicity profile that was observed in the decreased body scores and substantial weight loss of the mice. It is also important to note that the low-dose KU363 displayed comparable levels of efficacy with that of the high dose but without the overt evidence of systemic toxicity that was observed on gross pathology in some of the high-dose mice. Both groups, however, were noted to be more efficacious than 17-AAG, which has been used in clinical trials in cancer patients.
Overall, KU363, a novel C-terminal Hsp90 inhibitor, exhibited potent anticancer activity against HNSCC. These early data indicate that C-terminal Hsp90 inhibitors, such as KU363, are a viable chemotherapeutic alternative to N-terminal Hsp90 inhibitors. Although KU363 as a monotherapy was not superior to cisplatin in this study, its lower toxicity profile and comparable efficacy provide support to incorporate its use as an adjunct to current treatments, allowing for antitumor activity through a different, complementary pathway. Although the results obtained were significant, future studies examining larger numbers of mice in vivo with longer treatment schedules would provide a more complete evaluation of the drug’s efficacy and long-term toxicity profile. These data, however, provide early preclinical support for the development of CT-Hsp90-Is, such as KU363, as novel agents for the treatment of HNSCC, especially in cases where cisplatin resistance warrants the use of alternatives with comparable efficacy.
Acknowledgments
The authors would like to acknowledge grant support for this project from NIH 2U01-CA-120458-04 (B.S.J. Blagg and M.S. Cohen).
Footnotes
DISCLOSURE: No disclosures to report.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489–501. doi: 10.4065/83.4.489. [DOI] [PubMed] [Google Scholar]
- 3.Kim L, King T, Agulnik M. Head and neck cancer: changing epidemiology and public health implications. Oncology (Williston Park) 2010;24:915–9. [PubMed] [Google Scholar]
- 4.Ragin CC, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res. 2007;86:104–14. doi: 10.1177/154405910708600202. [DOI] [PubMed] [Google Scholar]
- 5.Cooper JS, Pajak TF, Forastiere AA, Jacobs J, Campbell BH, Saxman SB, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350:1937–44. doi: 10.1056/NEJMoa032646. [DOI] [PubMed] [Google Scholar]
- 6.Goon PK, Stanley MA, Ebmeyer J, Steinsträsser L, Upile T, Jerjes W, et al. HPV & head and neck cancer: a descriptive update. Head Neck Oncol. 2009;1:36. doi: 10.1186/1758-3284-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morimoto RI, Kline MP, Bimston DN, Cotto JJ. The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997;32:17–29. [PubMed] [Google Scholar]
- 8.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med. 2004;82:488–99. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 9.Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer. 2006;13:S125–35. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 10.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 11.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562:11–5. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 12.Brandt GE, Blagg BS. Alternate strategies of Hsp90 modulation for the treatment of cancer and other diseases. Curr Top Med Chem. 2009;9:1447–61. doi: 10.2174/156802609789895683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. 2008;15:2702–17. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop SC, Burlison JA, Blagg BS. Hsp90: a novel target for the disruption of multiple signaling cascades. Curr Cancer Drug Targets. 2007;7:369–88. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- 15.Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–57. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Yin X, Zhang H, Burrows F, Zhang L, Shores CG. Potent activity of a novel dimeric heat shock protein 90 inhibitor against head and neck squamous cell carcinoma in vitro and in vivo. Clin Cancer Res. 2005;11:3889–96. doi: 10.1158/1078-0432.CCR-04-2272. [DOI] [PubMed] [Google Scholar]
- 17.Yin X, Zhang H, Lundgren K, Wilson L, Burrows F, Shores CG. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Int J Cancer. 2010;126:1216–25. doi: 10.1002/ijc.24815. [DOI] [PubMed] [Google Scholar]
- 18.Smith V, Sausville EA, Camalier RF, Fiebig HH, Burger AM. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethox-ygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother Pharmacol. 2005;56:126–37. doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- 19.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2:131–8. [PubMed] [Google Scholar]
- 20.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–7. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. doi: 10.1007/s10637-010-9493-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 22.Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem. 2009;284:35381–9. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura H, Yukitake H, Tajima Y, Suzuki H, Chikatsu T, Morimoto S, et al. ITZ-1, a client-selective Hsp90 inhibitor, efficiently induces heat shock factor 1 activation. Chem Biol. 2010;17:18–27. doi: 10.1016/j.chembiol.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92:242–8. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 25.Nirmalanandhan VS, Duren A, Hendricks P, Vielhauer G, Sittampalam GS. Activity of anticancer agents in a three-dimensional cell culture model. Assay Drug Dev Technol. 2010;8:581–90. doi: 10.1089/adt.2010.0276. [DOI] [PubMed] [Google Scholar]
- 26.Shelton SN, Shawgo ME, Matthews SB, Lu Y, Donnelly AC, Szabla K, et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent anti-proliferative effects in human leukemic cells. Mol Pharmacol. 2009;76:1314–22. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tillotson B, Slocum K, Coco J, Whitebread N, Thomas B, West KA, et al. Hsp90 (heat shock protein 90) inhibitor occupancy is a direct determinant of client protein degradation and tumor growth arrest in vivo. J Biol Chem. 2010;285:39835–43. doi: 10.1074/jbc.M110.141580. [DOI] [PMC free article] [PubMed] [Google Scholar]